")
Back to Journals » OncoTargets and Therapy » Volume 7
Targeted drug delivery via folate receptors in recurrent ovarian cancer: a review
Authors Marchetti C, Palaia I, Giorgini M, De Medici C, Iadarola R, Vertechy L, Domenici L, Di Donato V, Tomao F , Muzii L, Benedetti Panici P
Received 8 January 2014
Accepted for publication 8 April 2014
Published 10 July 2014 Volume 2014:7 Pages 1223—1236
DOI https://doi.org/10.2147/OTT.S40947
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Claudia Marchetti, Innocenza Palaia, Margherita Giorgini, Caterina De Medici, Roberta Iadarola, Laura Vertechy, Lavinia Domenici, Violante Di Donato, Federica Tomao, Ludovico Muzii, Pierluigi Benedetti Panici
Department of Gynecology, Obstetrics and Urological Sciences, Sapienza University of Rome, Rome, Italy
Abstract: Ovarian cancer is the most common cause of gynecological cancer-related mortality, with the majority of women presenting with advanced disease; although chemotherapeutic advances have improved progression-free survival, conventional treatments offer limited results in terms of long-term responses and survival. Research has recently focused on targeted therapies, which represent a new, promising therapeutic approach, aimed to maximize tumor kill and minimize toxicity. Besides antiangiogenetic agents and poly (ADP-ribose) polymerase inhibitors, the folate, with its membrane-bound receptor, is currently one of the most investigated alternatives. In particular, folate receptor (FR) has been shown to be frequently overexpressed on the surface of almost all epithelial ovarian cancers, making this receptor an excellent tumor-associated antigen. There are two basic strategies to targeting FRs with therapeutic intent: the first is based on anti-FR antibody (ie, farletuzumab) and the second is based on folate–chemotherapy conjugates (ie, vintafolide/etarfolatide). Both strategies have been investigated in Phase III clinical trials. The aim of this review is to analyze the research regarding the activity of these promising anti-FR agents in patients affected by ovarian cancer, including anti-FR antibodies and folate–chemotherapy conjugates.
Keywords: ovarian cancer, targeted therapies, folate receptor, antifolate, farletuzumab, vintafolide
Introduction
Ovarian cancer is the ninth most common cancer in the female population and the second-most-common gynecological cancer after cancer of corpus uteri, with 21,980 new cases in the United States in 20141 and 67,000 new cases in Europe in 2008.2 In the last decades, survival of women with epithelial ovarian cancer has improved, thanks especially to more-aggressive surgical techniques aimed to achieve optimal cytoreduction and to the introduction of platinum-based treatment.3 Nonetheless, approximately 60% of patients with advanced disease at primary diagnosis will experience recurrent disease within 5 years from diagnosis,4 and some will develop resistance to chemotherapy. In detail, patients who relapse after 24 months will usually show response rates similar to those reported in the initial setting; conversely, when ovarian cancer recurs within 6 months, the overall response rate to chemotherapy is very poor.
In this setting, the most active agents have shown an overall response rate of about 10%, with a progression-free survival (PFS) <4 months and an overall survival of approximately 1 year.5 With the aim to obtain a better overall response and to counteract the occurrence of mechanisms of resistance to chemotherapy, particularly in patients who develop recurrent disease, new therapeutic approaches are required. Targeted therapies have recently gained great attention.6 These agents, which interfere exclusively with specific molecular targets, promise greater selectivity and lower toxicities than traditional cytotoxic drugs; indeed, it is noteworthy to consider that prolonged toxicity may decrease the efficiency of an antitumor treatment.7 Furthermore, even if cure is not yet an objectively valid goal of therapy, the emerging strategies should essentially be designed to focus equally on the quality of life as well as on the length of survival.
Several molecules have been evaluated and used in ovarian cancer, including agents that target vascular endothelial growth factor, vascular endothelial growth factor receptor, epidermal growth factor receptor, poly (ADP-ribose) polymerase tumor suppressor gene, and phosphate tensin homolog.8 Of these, bevacizumab has recently been included in the National Comprehensive Cancer Network guidelines for treatment of ovarian cancer, and more target agents are likely to appear in these guidelines soon.9,10
Recently, folate and its membrane-bound receptor have also emerged as a potential novel targets. In fact, it has been demonstrated that folate metabolism for replication of DNA is primary for cancer cells, which are therefore particularly sensitive to drugs that inhibit folate metabolism.11,12 In addition, cancer cells can develop folate receptors (FRs) that are not overexpressed in normal tissues but are highly expressed in more than 90% of ovarian cancer cells, retaining expression in both metastatic and recurrent disease.10 Therefore, FRs could represent a target for therapy that minimizes toxicity and maximizes antitumor efficacy.
This report will review the clinical activity of the FR-targeting agents in the treatment of recurrent ovarian cancer, with a special focus on anti-FR antibodies and folate–chemotherapy conjugates.
Folate, folate receptors, and cancer
Folate (vitamin B9) is a water-soluble vitamin used as a one-carbon donor in biosynthesis of purines and thymidylate essential for the de novo production of RNA and DNA. Folates are also needed for vitamin B12-dependent synthesis of methionine, required for methylation of DNA, histones, lipids, and neurotransmitters.13 Mammals must obtain their folates from their diets: folic acid can be synthesized and reduced by the normal bacterial flora of the small intestine and, through a highly specific transport mechanism known as the proton-coupled folate transporter, delivered to the liver via the hepatic portal vein.14
The terms “folic acid” and “folate” are often used interchangeably, but more appropriately, “folic acid” refers to the fully oxidized synthetic compound (pteroylmono-glutamic acid) used in dietary supplements and in food fortification, whereas “folate” refers to the various tetrahydrofolate derivatives naturally present in foods. Reduced folates are found as the partially reduced form 7,8-dihydrofolate or the reduced species 5,6,7,8-tetrahydrofolate (THF). In humans, the primary circulating reduced folate is 5-methyl-THF (5-CH3-THF). 5,10-CH2-THF is the cofactor necessary for the reductive methylation activity of thymidylate synthase that catalyzes the conversion of 2-deoxyuridine monophosphate to 2-deoxythymidine monophosphate.15
Folate is a nutrient required by all living cells, and it is essential for cellular division. Folate can enter the cell through two different routes: 1) by the reduced folate carrier (RFC), a bidirectional anion-exchange mechanism, which has low affinity for folate; and 2) by the FR pathway, which has high-affinity for folate. The RFC is a transport protein that is expressed ubiquitously in healthy tissues and tumors and is the major folate transporter, which transports folate cofactors from the blood into cells of peripheral tissues.16 In human tissues, highly elevated human RFC transcripts are detected in placenta and liver, with significant levels in other tissues, including leukocytes, kidney, lung, bone marrow, intestine, and portions of the central nervous system and brain.17 In contrast, rapidly dividing cancer cells overexpress the high-affinity FR. The FR is not significantly expressed on most normal tissues. Lung, brain, small intestine, kidney, and activated macrophages are the normal tissues known to express the FR. In the lung, brain, and small intestine the FR does not face the bloodstream, thus these folate receptors are not accessible to most folate-targeted drugs.
FRs capture folate from outside the cell; FR-bound folate is then transported intracellularly by FR-mediated endocytosis, by engulfing it within a vesicle called the endosome. This endogenous process of cellular entry can be exploited by linking the folic acid molecule to a chemotherapy agent. The chemotherapeutic agent bound to folic acid will then enter the cell inside the early endosome. Once internalized, a proton pump lowers the pH inside the endosome to allow the release of the folic acid–drug compound for the receptor. The FR protein may then cycle back to the plasma membrane, allowing it to begin a new cycle. Meanwhile, the drug is released intracellularly to exhibit its toxic effect.
There are three membrane-bound isoforms of folate (α, β, and γ), and each has its own tissue-specific distribution.18,19 The α and β isoforms are glycosylphosphatidylinositol-anchored, whereas the γ isoform is a soluble protein constitutively secreted by lymphoid cells.20–23 The most widely studied isoform is FRα, which is encoded for by the FOLR1 gene.24 The genes coding for FR, FOLR1–FOLR4, are located on the long arm of chromosome 11 (q11.3–q13.5).25–27
FRs are significantly different in their relative affinities for folate compounds and antifolates although they bind folic acid with a uniformly high affinity.28 This different affinity allows the creation of drugs against these receptors that are even more tissue-specific. Studying the distribution of specific mouse monoclonal antibodies named MOv18 and MOv19 in normal and malignant tissues, it was possible to reconstruct the tissue distribution of FR.29 Clinical studies on radioimmunoscintigraphy using 131 I-MOv18 were carried out in ovarian cancer patients and showed some efficacy.30 MOv19/interleukin-2 fusion protein was evaluated as an immunotherapy agent against a preclinical model of an FR+ murine tumor and was shown to be effective.31
FRα is not expressed in the majority of normal tissue and its expression is limited to epithelial cells in the choroid plexus, proximal kidney tubules, fallopian tube, uterus, epididymis, submandibular salivary, bronchial gland, acinar cells of the breast, type I and type II pneumocytes in the lung, and trophoblasts of the placenta.32–34 Cancer types such as endometrial, cervix, ovary, testicular choriocarcinoma, lung, colorectal, pediatric ependymomas, mesotheliomas, and renal cell carcinomas show FRα expression.34–36 It has been shown that elevated FRα expression may be a negative prognostic factor for chemotherapy resistance for at least breast, ovarian, and endometrial cancers.37 It has also been shown that FRα has a low expression on the apical surface of most normal cells. This difference in expression makes FRα a very attractive therapeutic target for novel anticancer agents that would have limited toxicity on normal tissues.38,39
Approximately 80% of epithelial ovarian cancers express FRα, and its expression is associated with parameters of biological aggressiveness;32,40–42 indeed, the highest FRα expression level is correlated with poorly differentiated tumors.32,43 Furthermore, the selective upregulation of FRα on tumor compared with normal tissue suggests FRα as a therapeutic target in epithelial ovarian cancer.9 In fact, in a clinical trial utilizing in vivo scans, it was demonstrated that in approximately half of tumors that overexpress FRα, all index lesions were positive.44
FRβ, which shares ~70% sequence homology with FRα, is most frequently found in a nonfolate-binding isoform on normal granulocytes, possibly due to an alternative posttranslational modification.3 FRβ is expressed in normal myelopoiesis and in placenta, spleen, and thymus.45,46 Functional FRβ is found in myeloid leukemia and in activated macrophages associated with inflammation and malignant tumor.20,47–52 Therefore, FRβ is potentially useful as a marker for myeloid leukemia, for chronic inflammatory diseases such as rheumatoid arthritis, and for tumor-associated macrophages.49,50,53,54 FRβ expression is regulated by retinoid receptors and can be upregulated by all-trans retinoic acid, particularly in combination with histone deacetylase inhibitors.53,55
FRγ has been detected in normal and malignant hematopoietic cells, as well as in carcinomas of the ovary, endometrium, and cervix.18,21,47
There are two known strategies for targeting therapeutics to the FR. The first is based on anti-FR antibody and the second is based on folic acid as a high-affinity receptor ligand. Significant progress has been made following both these strategies.
Farletuzumab and ovarian cancer (monoclonal antibody FRα-targeted antagonists)
Preclinical data
Farletuzumab (MORAb-003) is an improved humanized version of a murine antibody produced in Chinese hamster ovary (CHO-K1) cell and constructed by grafting complementarity-determining regions of a murine antibody into a human IgG1/κ backbone.56
In preclinical studies with human and primate tissues, farletuzumab showed strong affinity for FRα, which is overexpressed in virtually all epithelial ovarian cancer cells, and very limited cross-reactivity to normal tissue.57 It does not block FRα of folates and antifolates and minimally retards both folate delivery via FRα-mediated transport and cell growth in vitro.58
MORAb-003 was effective in preclinical xenograft models of ovarian cancer. It was shown to inhibit phosphorylation of proteins by the Lyn kinase (a member of the Src family of kinases) and to inhibit the growth of FRα cells in low-folate conditions. In xenograft studies a synergistic effect of MORAb-003 with taxanes was demonstrated.59 Farletuzumab has demonstrated cytotoxic effects on ovarian cancer cell growth through both mechanisms of antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity. Binding of mAb to FRα also results in a decreased cellular proliferation through the inhibition of Lyn kinase substrate phosphorylation. It has also been shown that, in several cell lines naturally expressing FRα, farletuzumab did not block binding of folic acid, 5-methyltetrahydrofolate, pemetrexed, and other anti-folates and only minimally altered FRα-mediated folate uptake and cell growth in vitro. Because of the differential expression of FRα on ovarian cancer cells and normal tissues, it has been shown that folate can still penetrate normal cells naturally through the reduced folate carrier.
Clinical development
Safety of farletuzumab
A Phase I study demonstrated the safety and tolerability of farletuzumab in the management of heavily pretreated patients with epithelial ovarian cancer.60 The primary objective was to determine the safety of farletuzumab and to establish the maximum tolerated dose. The secondary objective was to determine the serum and in vivo pharmacokinetics of farletuzumab, as well as to detect any human antihuman antibodies (HAHA).
In this trial, farletuzumab was administered to 25 patients (median age, 56 years) with platinum-refractory or platinum-resistant epithelial ovarian cancer at the escalation dose of 12.5–400.0 mg/m2 intravenously, following a modified Fibonacci sequence on day 1, day 8, day 15, and day 22 of a 5-week cycle. Safety data were recorded throughout the treatment period and for 2 weeks after the last dose. Most patients (23/25) received at least four weekly doses of farletuzumab. The remaining two patients had clinical disease progression and were withdrawn from the trial.
No dose-limiting toxicity was observed up to 400 mg/m2. There were no treatment-related adverse events classified as serious or severe (grade $3) and no treatment-related myelotoxicity or neurotoxicity. All adverse events were grade 2 or lower, except for grade 3 fatigue reported by single patients in the 12.5 mg/m2 and 400.0 mg/m2 cohorts. The most common treatment-related adverse events were hypersensitivity reactions (15 patients, 60%), fatigue (12 patients, 48%), and diarrhea (four patients, 16%).
Drug hypersensitivity reactions that occurred no more than 2 days after a farletuzumab infusion were experienced by 15/25 patients (60%); however, all were mild (grade 1 or 2) and easily controlled with antipyretics and/or antihistamines. The most commonly reported drug hypersensitivity adverse events were pyrexia (eight patients, 32%) and chills (five patients, 20%). Twelve of 15 hypersensitivity reactions were grade 1. No treatment discontinuation due to drug-related adverse events or deaths was observed during the study period.
HAHA and pharmacokinetic analyses were performed. Most patients did not exhibit anti-MORAb-003 antibodies at any point. Increased levels of HAHA were observed in two patients (one at baseline and one on day 15). The patient with elevated HAHA at baseline experienced grade 1 hypersensitivity reaction during the first infusion. Premedication prevented hypersensitivity reaction during subsequent infusions.60
Systemic exposure to farletuzumab, usually shown as Cmax and AUC0–24, increased in an approximately dose-proportional manner across the dose range of 12.5–400.0 mg/m2, following single and multiple weekly infusions. The t½ (terminal half-life of farletuzumab), ranging from 121–260 hours, was consistent with slow clearance of the drug. The pharmacokinetics of farletuzumab and the biodistribution of the radiolabeled antibody were consistent with other (radiolabeled) humanized/chimeric antibodies.56 Without the maximum tolerated dose, a precise recommendation of dose for Phase II trials could not be determined. The tolerable dose of 400.0 mg/m2 established in the Phase I study of farletuzumab corresponded to dosing of 10–12 mg/kg. It was recommended that dosing for Phase II trials should be in the range 2.5–10.0 mg/kg.
An open-label, multicenter Phase II study (MORAb-003)61 was conducted in platinum-sensitive epithelial ovarian cancer patients at first relapse in order to determine the efficacy of farletuzumab as a single agent or in combination with platinum and taxane. The most frequent serious adverse events were febrile neutropenia, thrombocytopenia, abdominal pain, diarrhea, large intestinal obstruction, subileus, urosepsis, and hyperglycemia (two patients; 3.7% each). No patients died during active treatment with MORAb-003. No additive toxicity was observed with prolonged use of farletuzumab, as a single agent or in combination with platinum-based chemotherapy.61
Finally, an open-label, multicenter, Phase I study (MORAb-003–005),62 assessed the safety of weekly farletuzumab in combination with carboplatin and pegylated liposomal doxorubicin (PLD) in 15 patients with platinum-sensitive ovarian cancer in first or second relapse. As expected, anemia, fatigue, and hand–foot syndrome were the most common adverse events, occurring in 14 patients (93%), eight patients (53%), and eight patients (53%), respectively, consistent with the toxicity profile of the carboplatin/PLD regimen. No grade 4 or 5 toxicity was documented. No clinically significant changes in cardiac ejection fraction were noted. Preliminary data from this study support further investigation into the efficacy of this combination.62
Interesting results, both in terms of toxicity and efficacy, have been found with the use of farletuzumab in the clinical setting, leading to the development of several recently concluded Phase III trials (Table 1).
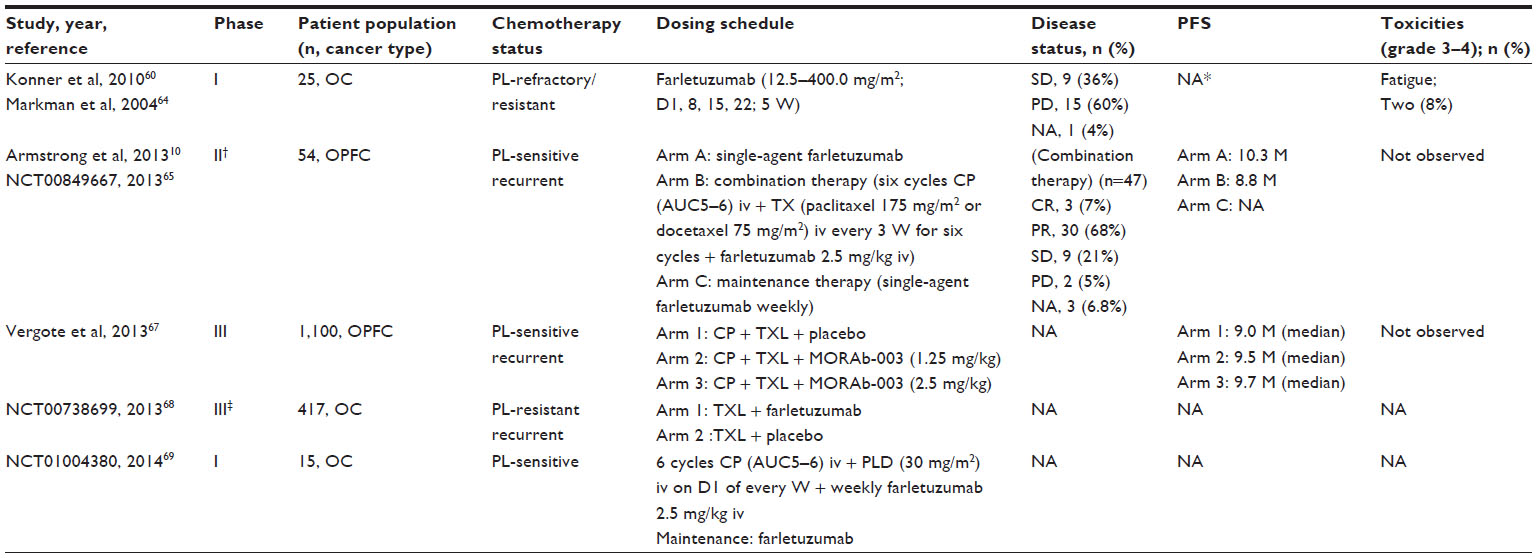 | Table 1 Clinical trials of farletuzumab (MORAb-003) in ovarian cancer |
Efficacy of farletuzumab
In the same Phase I study60 performed on 25 patients, after one cycle of treatment, computerized tomography evaluation by Response Evaluation Criteria in Solid Tumors (RECIST) identified nine patients with stable disease; four patients had decreased CA-125 levels. On the basis of the response seen after one treatment cycle, three patients went on to receive farletuzumab as extended therapy. One patient exhibited a 48% decrease in CA-125 after receiving two additional cycles. Radiolabeled tracer studies were conducted in three patients, showing significant tumor uptake of labeled farletuzumab.60
Farletuzumab was subsequently investigated in patients with platinum-sensitive recurrent ovarian cancer in an open-label, Phase II study.10,61 The trial studied the clinical activity of farletuzumab, both as single agent and in combination with platinum (carboplatin AUC5–6) and taxane (paclitaxel 175 mg/m2 or docetaxel 75 mg/m2) chemotherapy. Patients were enrolled and divided into two arms: arm A included 28 patients with nonsymptomatic relapse (elevated CA-125 only) who received single-agent farletuzumab; arm B included 26 women with symptomatic relapse who received platinum/taxane chemotherapy plus farletuzumab directly. Twenty-one of 28 patients in arm A (75%) who experienced symptomatic relapse or progression then received platinum/taxane and farletuzumab, and 36 patients (67% of all patients) who attained a complete or partial response received single-agent farletuzumab maintenance therapy.
A total of 47 patients received farletuzumab in combination with chemotherapy, and all were considered evaluable for efficacy response. In 38/47 patients (81%; 95% confidence interval [CI] =67%–91%) treated with farletuzumab in combination with chemotherapy, CA-125 normalized, according to Rustin criteria.63 Combination therapy provided clinically complete (7%) or partial (68%) RECIST response in 33 patients (75%; 95% CI =60%–87%) and showed stable disease in an additional 9 patients (21%). Only two patients (5%) had progressive disease as best response. The duration of each patient’s second progression-free interval was analyzed and compared with that patient’s first-response interval. With this analysis, the authors found that in 9/42 evaluable subjects (21%), the progression-free interval was greater or equal to the first-response interval, more durable than the historical documented rate of 3% for platinum-based therapy alone shown by Markman et al.64
There was a high response rate among patients with a first-response interval <12 months (75%), comparable to that in patients with a first-response interval ≥12 months (84%). This led the authors to conclude that farletuzumab with carboplatin and taxane could enhance the response rate and duration of response in patients with platinum-sensitive ovarian cancer with first relapse after remission of 6–18 months.
These results provided a rationale for the beginning of a three-arm randomized, double-blind, placebo-controlled, Phase III study investigating farletuzumab (FAR 131/MORAb-003) in combination with carboplatin and a taxane in 1,100 patients with platinum-sensitive epithelial ovarian cancer in first relapse.65–67 Paclitaxel and carboplatin at standard dosages were given with either placebo (arm 1) or farletuzumab at two different dose levels, 1.25 mg/kg (arm 2) and 2.5 mg/kg (arm 3). The primary outcome was PFS evaluated according to RECIST criteria; secondary endpoints included 1) assessment of the safety or tolerability of weekly doses of 1.25 mg/kg or 2.5 mg/kg of farletuzumab in combination with carboplatin/taxane chemotherapy and 2) the effects of farletuzumab on overall survival, CA-125–defined PFS and serologic response (Rustin criteria),63 length of second remission versus that of first remission, tumor response (RECIST criteria), patient quality of life, and resource utilization.66 Length of first remission was 6 months to ≤12 months for 45% of patients, 12 months to ≤18 months for 35% of patients, and 18 months to ≤24 months for 19% of patients and was similar in the three arms of treatment. The most commonly reported adverse events across arms were those known to be associated with the study chemotherapy agents. Median PFS was 9.0 months in the placebo group (arm 1), 9.5 months in arm 2, and 9.7 months in arm 3, with no statistically significant difference between arms (hazard ratio [HR] =0.86, 95% CI =0.70–1.06 for 2.5 mg/kg versus placebo). Therefore, the study did not meet its primary endpoint of PFS. The post hoc exploratory analysis presented at the 18th International Meeting of the European Society of Gynaecological Oncology in 2013 showed, however, a trend toward improved PFS in some patient subsets. In particular, it has been found that exposure to higher doses of farletuzumab may be correlated with PFS improvement; furthermore, patients with CA-125 ≤3 times the upper limit of normal may significantly benefit from receiving farletuzumab, corroborating the hypothesis that CA-125 may inhibit the potential antibody-dependent cellular cytotoxicity effect of farletuzumab.67
Another Phase III trial (FAR-122)68 explored the role of this monoclonal antibody associated with paclitaxel in platinum-resistant ovarian cancer patients, compared with paclitaxel plus placebo. However, the study was prematurely closed in 2012 because it did not meet the prespecified criteria for continuation following an interim futility analysis.
As is apparent from the data, both randomized trials in platinum-sensitive and platinum-recurrent ovarian cancer did not confirm the results of the Phase II trial that conversely demonstrated the safety and feasibility of farletuzumab as an agent in ovarian cancer. Additionally, some immune-mediated events were observed. Interestingly, the manufacturer of MORAb-003 (Morphotek, Inc., Exton, PA, USA) has announced that it is actively developing a companion diagnostic assay to identify patients with high FRα expression, because these patients may receive more benefit from farletuzumab therapy than those with low FRα expression. This suggests the possibility that future trial designs may incorporate stratification of patients based on their tumor FR expression status.
An advantage of the anti-FR antibody strategy is that tumor targeting is not affected by the level of FR saturation by folate found in the circulation and that the antibody is presumably FR subtype-specific. A potential drawback of this strategy is that a humanized antibody may still elicit a host immune response in the form of a neutralizing anti-idiotypic antibody that may ultimately undercut its therapeutic efficacy. Nevertheless, in clinical practice other therapeutic monoclonal antibodies, such as trastuzumab and cetuximab, are routinely utilized, suggesting that the overall approach of antibody therapy is potentially noteworthy.
On the basis of the results of the reported clinical trials, the toxicity profile of farletuzumab seems to be acceptable, although data on the efficacy of this drug are still conflicting. Additional studies are ongoing to further investigate the full potential of this innovative approach.69
Vintafolide (EC145) and ovarian cancer
Folate conjugation is another versatile strategy for FR-targeted drug delivery. The rationale underlying FRα-targeted drug delivery lies in the substrate specificity of folic acid versus FRα. In this type of approach, folate can be linked to various therapeutic agents, namely low-molecular-weight chemotherapeutic agents; in fact, folate is amenable to chemical conjugation with other molecules through its γ-carboxyl group, without decreasing its binding affinity to the FR. As a low-molecular-weight physiological ligand, folate is also presumably nonimmunogenic and is easily derivatized.70 These desirable properties, combined with its convenient availability, have made folate one of the most studied ligands in targeted drug delivery.50 Several small-molecule drug conjugates to target all the FR isoforms have been developed, including folate conjugates of platinum,71 fluorodeoxyuridine monophosphate oligomer,72 paclitaxel,73 maytansinoids,74 mitomycin C, vinca alkaloid, and a prodrug of thiolate histone deacetylase inhibitor.75
One of the most promising of these innovative agents is vintafolide (originally known as EC145), a conjugate of the vitamin folate that is coupled chemically to the vinca alkaloid; in detail, it is a small water-soluble molecule drug conjugate (SMDC) consisting of a folate conjugate of desacetylvinblastine monohydrazide (DAVLBH), which is a potent microtubule-destabilizing agent.76,77 An advantage of this drug is that the linker mechanism of the SMDC allows for drug stability in the blood. Furthermore, vintafolide is a unique type of targeted therapy, as the active DAVLBH “warhead” is released upon endocytosis.78 It binds specifically to the FR with a high affinity79 and has been selected as the first folate–drug conjugate to enter into clinical trials as a therapeutic agent, along with etarfolatide (EC20; Endocyte, Inc., West Lafayette, IN, USA), a technetium-labeled, companion imaging agent for FR status determination.
The two molecules (folate and vinca) that constitute vintafolide are connected in a regioselective manner via a hydrophilic peptide spacer and a self-immolative group based on disulfides as the cleavable linkage. Desacetylvinblastine hydrazide is prepared from vinblastine-free base by reaction with anhydrous hydrazine, whereas the targeting and spacer components are prepared by assembly, using standard fluorenylmethyloxycarbonyl-based solid-phase peptide synthesis. The second step involves inserting the disulfide cleavable linkage on desacetylvinblastine hydrazide by reaction with the heterobifunctional reagent (2-[benzotriazole-1-yl-(oxycarbonyloxy)-ethyldisulfanyl]-pyridine). The final reaction comprises a mild thiol-disulfide exchange reaction between the components.
Beside vintafolide, etarfolatide is a corresponding folate-targeted companion imaging agent that can anatomically recognize FRα-positive tumors,80 remaining bound to the receptor to identify cells that express FR without entering the cytosol. In addition, etarfolatide can be used as an imaging agent in chronic inflammatory diseases, such as rheumatoid arthritis, based on targeting FRβ.81
Both these drugs share a similar mechanism of action. A generic FR-targeted conjugate is composed by folate (element 1) that binds to the drug (element 4) through a spacer (element 2), which confers specific properties to the complex folate-drug that allow the entry into the cell. The last component, element 3, works like a cleavable or stable bond based on the functional role of element 4. If the goal is the release of a drug, the bond between element 3 and element 4 will lysate via endocytosis to ensure the action of the substance into the cell. Alternatively, if element 4 is an imaging agent and its release is not desired, element 3 will have a non-cleavable bond, as in the case of 99mTc-folate, which remains bound to the receptor without entering the cytosol, to identify cells that express FR (Figure 1).44
 | Figure 1 Diagram of generic folate receptor–target conjugate. |
Exogenously added folate–drug conjugates bind specifically to the folate receptor protein with high affinity. The FR-mediated system internalizes folate–drug conjugates via a membrane-bound glycoprotein.81–83 The ligand–receptor complex is introduced into the cell through endosomes coated with a molecule called clathrin. As the lumen of the maturing endosome acidifies to pH ~5, the receptor changes conformation and releases the conjugate in the cytosol while the receptor resets to a new cycle of uptake (Figure 2). The reduced folate carrier, which unlike the FR is an anion transporter, can only shuttle unconjugated folate molecules into the cell.
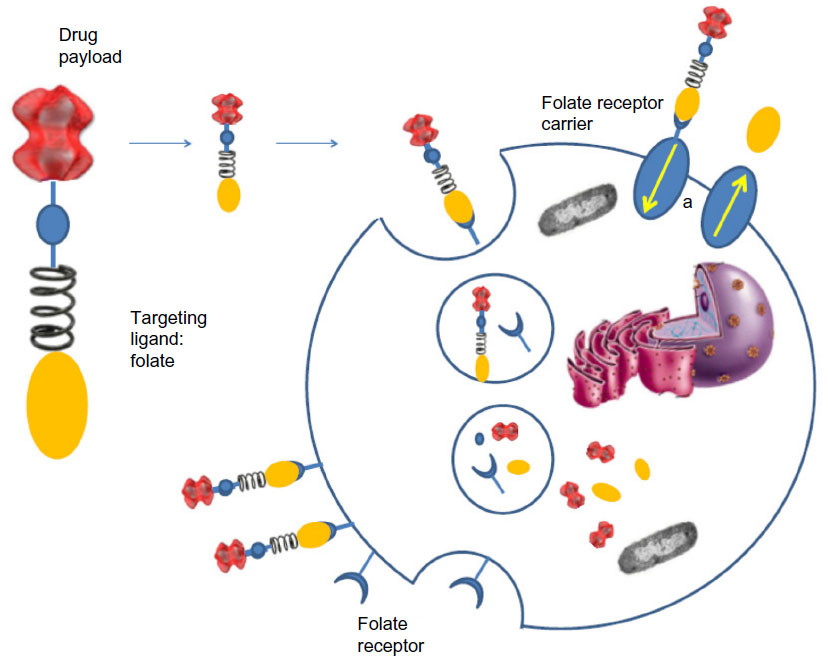 | Figure 2 Folate receptor-mediated endocytosis of folate drug conjugates. |
Preclinical data
Vintafolide has been tested in a number of different in vivo models, including M109 mouse lung adenocarcinoma, a KB tumor xenograft model, and aggressive FR-positive J6456 lymphoma. In one study, brief treatment of nude mice bearing approximately 100 mm3 FR+ human xenografts led to complete response in 5/5 mice and cures, without a relapse for >90 days post-tumor implantation in 4/5 mice.82 Similarly, complete response and cures were also noted when EC145 was used to treat mice initially bearing tumors as large as 750 mm3. Furthermore, similar results (cures in 5/5 mice) were achieved after treatment in an aggressive FR+ J6456 lymphoma model. No noticeable weight loss or major organ tissue degeneration was registered. The enhanced therapeutic index due to folate conjugation was also evidenced by the fact that the unconjugated drug (desacetylvinblastine monohydrazide) was found to be completely inactive when administered at nontoxic dose levels and only marginally active when given at highly toxic dose levels. Similar to the results described above, these experiments demonstrated excellent activity with little toxicity for EC145. EC145 was able to eradicate large, well-established tumors. By contrast, there was significant toxicity and relatively little activity for DAVLBH. These findings of both increased activity and decreased toxicity of EC145 in comparison with the unconjugated vinca alkaloid (DAVLBH) provide proof of principle that the approach both increases the selectivity of the drug for tumor cells in comparison with normal tissues and enhances cytotoxicity. These preclinical findings provide the basis for the introduction of this drug in clinical-trials setting (Table 2).
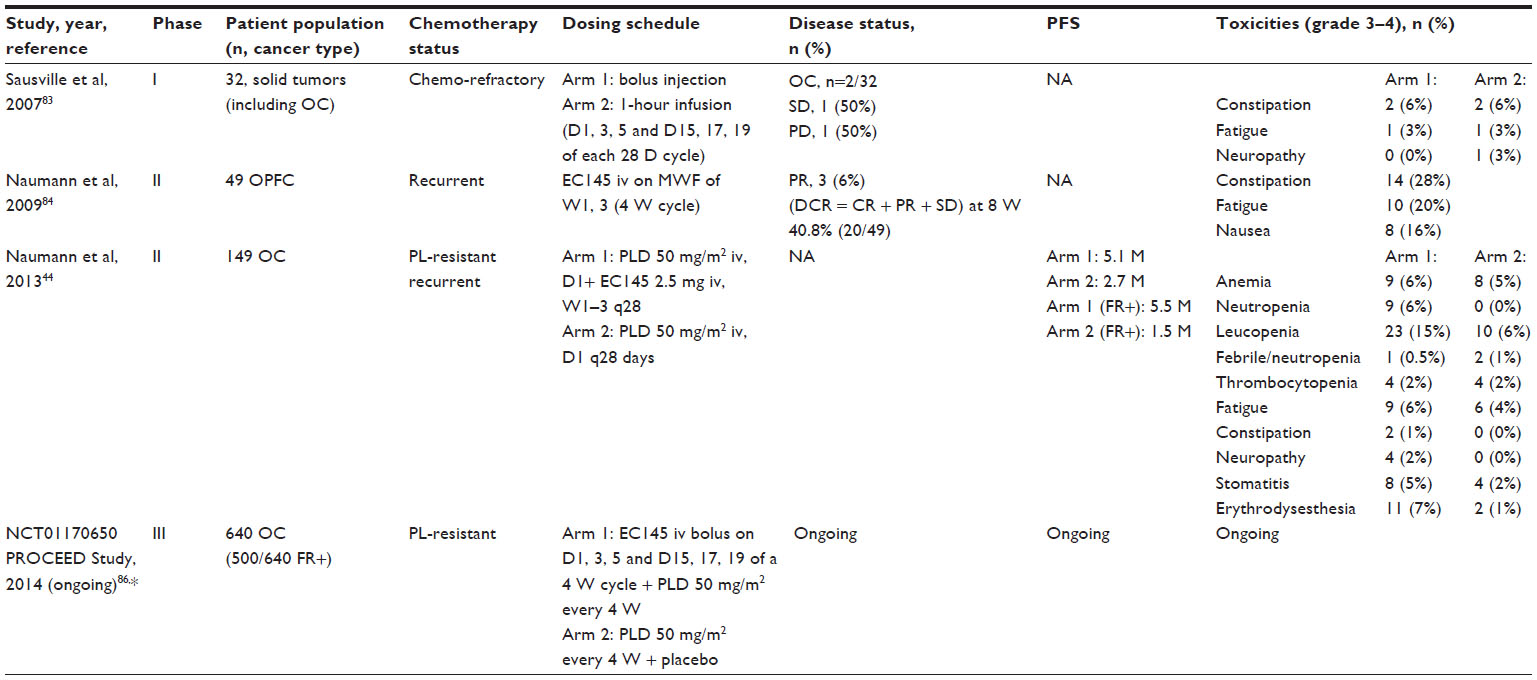 | Table 2 Clinical trials of vintafolide (EC145) in ovarian cancer |
Clinical development: safety and efficacy
A Phase I study proved the safety of EC145 administered with flat escalation dose to patients with refractory solid tumors. EC145 was generally well tolerated at bolus doses of 2.5 mg, with one patient continuing on study for six cycles. Overall, the most common side effects were fatigue (n=9), constipation (n=7), and neuropathy (n=6).83
A Phase II nonrandomized trial investigated the efficacy and safety of EC145 in patients with advanced ovarian, fallopian tube, or primary peritoneal carcinoma.84 EC145 was administered intravenously at the dose of 2.5 mg on day 1, day 3, and day 5 of week 1 and week 3 of a 4-week schedule. The most frequent drug-related >grade 2 adverse events included constipation (14/49; 28%), fatigue (10/49; 20%) and nausea (8/49; 16%). The disease-control rate at 8 weeks for all study patients was 41% (20/49). For patients with a history of ≤4 prior therapies, the 8-week disease-control rate was 50%, less when compared to that for patients with exposure to <3 prior chemotherapeutic regimens (59%). In addition to the reported rate of disease control, three patients exhibited partial response according to RECIST criteria.
Another clinical study evaluated the safety and efficacy of vintafolide in a nonrandomized Phase II clinical trial in patients with advanced ovarian, fallopian tube, or primary peritoneal carcinoma, after identification of FR expression using etarfolatide (n=47, median age 61 years).85 The trial, conducted from 2007–2012, examined two different doses of vintafolide administered three times a week on week 1 and week 3 of a 4-week cycle. The disease-control rate (complete response + partial response + stable disease) at 8 weeks in patients receiving vintafolide as third-line or fourth-line intravenous therapy was 75%, compared (historically) with a rate of 47% in women receiving second-line or third-line PLD. There were also three partial responses. From this study, it appeared that vintafolide was very well tolerated, with minimal toxicity. Fatigue was the most common grade 3 toxicity, occurring in 8.2% of patients.85
On the basis of these data, EC145 was evaluated as part of the international randomized PRECEDENT trial.44 This study compared PFS in subjects with platinum-resistant ovarian cancer who received combination therapy with EC145 and PLD (PLD/Doxil® [Janssen Products, LP, Titusville, NJ, USA]/Caelyx® [Janssen-Cilag Pty Ltd, NSW, Australia]) versus PLD alone. FR status (10%–100% of target lesions positive for FR expression) was assessed using 99mTc nuclear SPECT imaging with etarfolatide before initiation of therapy. Patients were randomized to receive vintafolide (2.5 mg on day 1, day 3, and day 5 and day 15, day 17, and day 19 of each 4-week cycle) plus PLD (50 mg/m2 intravenously, on day 1 of each 4-week cycle) or PLD alone (at the same dosage/schedule) until disease progression or death or until the maximum allowable cumulative dose of PLD had been given (550 mg/m2 of doxorubicin HCl). Vintafolide plus PLD was the first combination to show a statistically significant increase in PFS for women with platinum-resistant ovarian cancer. The greatest benefit was observed in patients with 100% of lesions positive for FR, with median PFS of 5.5 months compared with 1.5 months for PLD alone (HR =0.38; 95% CI =0.17–0.85; P=0.013). The group of patients with FR+ disease (10%–90%) experienced some PFS improvement (HR =0.873; 95% CI =0.33–2.28; P=0.79), in contrast to patients with FR– neoplasia (HR =1.806; 95% CI =0.37–8.83; P=0.468).44
On the basis of these promising results, a randomized double-blind Phase III trial comparing EC145 and PLD (PLD/Doxil®/Caelyx®) in combination versus PLD alone in participants with platinum-resistant ovarian cancer (the PROCEED study) is currently recruiting participants.86 Patients are randomized to vintafolide with or without PLD. PLD 50 mg/m2 is administered on day 1 of a 4-week cycle and vintafolide 2.5 mg or placebo is administered on days 1, 3, 5, 15, 17, and 19 of a 4-week cycle. Treatment can continue for up to 20 cycles or until unacceptable toxicity or disease progression. The primary outcome of this study is PFS, and the secondary outcomes are overall survival and incidence of adverse events. Enrollment of 640 patients, including approximately 500 that are folate receptor positive, is planned. All participants will undergo imaging with the FR-targeting investigational diagnostic agent EC20 during the screening period to assess binding of the imaging agent EC20 to tumors.
Table 2 describes clinical trials with vintafolide in ovarian cancer.
Other anti-FR agents in ovarian cancer
Several other agents targeting folate receptors have recently been investigated in Phase I–II clinical trials.
Folate Immune is a novel immunotherapy comprising a vaccine EC90 with GP-0100 adjuvant and a folate-conjugated vaccine (EC17) that targets FR+ tumor cells to enhance tumor-cell removal by cytokine-stimulated immune cells. EC17 acts as an adapter molecule that redirects anti-fluorescein isothiocyanate-conjugated hapten antibody produced by the patient to FR-expressing tumor cells; the safety of this strategy was demonstrated in a Phase I trial including one ovarian cancer patient and renal cancer patients.87 This approach has certain potential advantages over the farletuzumab strategy, because EC17, which activates the therapeutic mechanism, is a small molecule and is therefore very inexpensive and rapidly clearing, and thus can potentially achieve high tumor-to-normal targeting ratios.
Due to vintafolide’s potential for success, recent efforts have focused on determining the impact of modifying the spacer, linker, or drug payload on the antitumor activity. In fact, preclinical studies were conducted to determine the impact of 1) substituting the DAVLBH vinca alkaloid unit in vintafolide with analogues of vincristine, vindesine, vinorelbine, or vinflunine; 2) substituting the naturally (S)-configured Asp-Arg-Asp-Asp-Cys peptide spacer in vintafolide with other hydrophilic spacers; and 3) varying the composition of the linker module to evaluate the impact of having a readily releasable, self-immolative linker system (eg, disulfi debased) versus a more stable linker system (eg, a thioether). In these studies, substitutions of some or all of the natural amino acids within vintafolide’s hydrophilic spacer module did not significantly change the in vitro or in vivo potency of the SMDCs.
Furthermore, following vintafolide, several additional agents have also been studied in clinical trials recently. EC0225, a folate conjugated to two different agents (a vinca alkaloid and mitomycin), has completed a Phase I trial.88 BMS-753493 (epofolate) is a folate conjugate of epothilone A, a microtubule-stabilizing agent; it is under investigation in a Phase I/II clinical trial sponsored by Bristol-Myers Squibb in advanced cancer.89,90 Another agent being tested in a Phase I trial in patients with refractory or metastatic tumors is EC0489, an analogue of vintafolide, with reduced hepatic clearance.91 Nonetheless, vintafolide remains one of the most potent folate conjugates produced to date, and continued clinical development is warranted.
Discussion
Anti-FR drugs represent a new viable option for treatment of recurrent ovarian cancer. Folate conjugates target both FRα and FRβ. This enables the possibility of targeting tumors that are low in FRα but high in FRβ (eg, tumors that are highly infiltrated by tumor-associated macrophages that are FRβ+).26 On the other hand, this reduces the selectivity for tumor cells. Nonetheless, this could expand the range of those patients in the cancer population who may respond to FR-targeted therapy. Furthermore, differently from antibodies, a low-molecular-weight folate conjugate is rapidly distributed into tumor tissues and is rapidly cleared from systemic circulation, reducing concentration in the plasma and in nontarget tissues and is typically not immunogenic. This leads to a higher tumor-to-normal tissue ratio.45 Finally, it is possible to produce these agents by total synthesis as a single pure chemical entity, which is not always possible for macromolecular conjugates like antibodies.
The potential disadvantages of folate conjugates include interference with FR targeting by the circulating folate, influenced by the patient’s diet, and high and persistent accumulation in the kidneys due to FRα expression in the apical membrane of the promixal tubules.58 While this has not adversely affected clinical translation of folate conjugates, it may reduce the prospect of clinical application of low-molecular-weight folate conjugate-based radiopharmaceuticals for targeted radiotherapy. It is worth noting that renal toxicity has not been reported in clinical trials of folate conjugate-based chemotherapies.
Regarding the clinical trial strategies used, there are some important distinctions between the trials of farletuzumab and those using vintafolide. In farletuzumab trials, the drug was used in combination with platinum/taxane and mostly focused on patients with platinum-sensitive early-stage ovarian cancer. Meanwhile, in vintafolide trials, the drug was combined with Doxil®/PLD and focused on patients with platinum-resistant advanced disease, using etarfolatide imaging to identify patients with high FR expression. It is interesting that, in contrast to vintafolide, farletuzumab failed to show benefit in patients with advanced disease, who presumably are more likely to be FR+. Nevertheless, it should be underlined that in the vintafolide trials, great benefit was obtained in those patients who were confirmed to express FRα after etarfolatide’s exposition. Furthermore, the developer of farletuzumab, Morphotek, Inc., is seeking to develop an FR assay to screen patients for FR expression status. It would be interesting to see the effect of incorporating this assay into a future trial on the clinical efficacy of farletuzumab.
Conclusion
The recent movement of farletuzumab and vintafolide into Phase III trials suggests that FR targeting is finally reaching a critical point and that the strategy of identifying subpopulations of cancer patients with high FR expression could be very promising. Meanwhile, continued research is warranted, for example, on developing a serum-based FR assay kit and FR isoform selectivity. Appropriate preidentification of susceptible patients who may benefit from this therapeutic approach is paramount. Next trials should also focus on the investigation of more FR-targeted drugs, either as a therapeutic combination with existing therapy or even as monotherapy. FR targeting can, therefore, potentially become the first example in which scientific knowledge and innovative biomedical technology achieve significant results in ovarian cancer treatment.
Disclosure
The authors report no conflicts of interest in this work. The authors received no funding for this research.
References
National Cancer Institute. Ovarian cancer [homepage on the Internet]. Bethesda, MD: National Cancer Institute; 2014. Available from: http://www.cancer.gov/cancertopics/types/ovarian. Accessed April 26, 2014. | |
Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer. 2010;46(4):765–781. | |
Vaughan S, Coward JI, Bast RC Jr, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11(10):719–725. | |
Burke TW, Morris M. Secondary cytoreductive surgery for ovarian cancer. Obstet Gynecol Clin North Am. 1994;21(1):167–178. | |
Naumann RW, Coleman RL. Management strategies for recurrent platinum-resistant ovarian cancer. Drugs. 2011;71(11):1397–1412. | |
Armstrong DK, Bundy B, Wenzel L, et al; Gynecologic Oncology Group. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34–43. | |
Chatelut E, Delord JP, Canal P. Toxicity patterns of cytotoxic drugs. Invest New Drugs. 2003;21(2):141–148. | |
Carroll AR, Coleman RL, Sood AK. Therapeutic advances in women’s cancers. Front Biosci (Schol Ed). 2011;3:82–97. | |
Walters CL, Arend RC, Armstrong DK, Naumann RW, Alvarez RD. Folate and folate receptor alpha antagonists mechanism of action in ovarian cancer. Gynecol Oncol. 2013;131(2):493–498. | |
Armstrong DK, White AJ, Weil SC, Phillips M, Coleman RL. Farletuzumab (a monoclonal antibody against folate receptor alpha) in relapsed platinum-sensitive ovarian cancer. Gynecol Oncol. 2013;129(3):452–458. | |
Kelemen LE, Goodman MT, McGuire V, et al; Australian Cancer Study (Ovarian Cancer) Study Group; Australian Ovarian Cancer Study Group; Ovarian Cancer Association Consortium. Genetic variation in TYMS in the one-carbon transfer pathway is associated with ovarian carcinoma types in the Ovarian Cancer Association Consortium. Cancer Epidemiol Biomarkers Prev. 2010;19(7):1822–1830. | |
Choi SW, Mason JB. Folate and carcinogenesis: an integrated scheme. J Nutr. 2000;130(2):129–132. | |
Stokstad, ELR. Historical perspective on key advances in the biochemistry and physiology of folates. In: Picciano MF, Stokstad ELR, Gregory JF, editors. Folic Acid Metabolism in Health and Disease. New York: John Wiley & Sons, Incorporated; 1990:1–21. | |
Zhao R, Matherly LH, Goldman ID. Membrane transporters and folate homeostasis: intestinal absorption and transport into systemic compartments and tissues. Expert Rev Mol Med. 2009;11:e4. | |
Gonen N, Assaraf YG. Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist Updat. 2012;15(4):183–210. | |
Matherly LH, Hou Z, Deng Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007;26(1):111–128. | |
Whetstine JR, Flatley RM, Matherly LH. The human reduced folate carrier gene is ubiquitously and differentially expressed in normal human tissues: identification of seven non-coding exons and characterization of a novel promoter. Biochem J. 2002;367(Pt 3):629–640. | |
Kelemen LE. The role of folate receptor alpha in cancer development, progression and treatment: cause, consequence or innocent bystander? Int J Cancer. 2006;119(2):243–250. | |
Shen F, Wang H, Zheng X, Ratnam M. Expression levels of functional folate receptors alpha and beta are related to the number of N-glycosylated sites. Biochem J. 1997;327 (Pt 3):759–764. | |
Shen F, Ross JF, Wang X, Ratnam M. Identification of a novel folate receptor, a truncated receptor, and receptor type beta in hematopoietic cells: cDNA cloning, expression, immunoreactivity, and tissue specificity, Biochemistry. 1994;33(5):1209– 1215. | |
Shen F, Wu M, Ross JF, Miller D, Ratnam M. Folate receptor type gamma is primarily a secretory protein due to lack of an efficient signal for glycosylphosphatidylinositol modification: protein characterization and cell type specificity. Biochemistry. 1995;34(16):5660–5665. | |
Wang H, Ross JF, Ratnam M. Structure and regulation of a polymorphic gene encoding folate receptor type gamma/gamma’. Nucleic Acids Res. 1998;26(9):2132–2142. | |
Rijnboutt S, Jansen G, Posthuma G, Hynes JB, Schornagel JH, Strous GJ. Endocytosis of GPI-linked membrane folate receptor-alpha. J Cell Biol. 1996;132(1–2):35–47. | |
Ramamoorthy K, Potala S, Verma RS. Insilco analysis of functionally important residues in folate receptors. Bioinformation. 2007;2(4):157–162. | |
Yuan Y, Nymoen DA, Dong HP, et al. Expression of the folate receptor genes FOLR1 and FOLR3 differentiates ovarian carcinoma from breast carcinoma and malignant mesothelioma in serous effusions. Hum Pathol. 2009;40(10):1453–1460. | |
Spiegelstein O, Eudy JD, Finnell RH. Identification of two putative novel folate receptor genes in humans and mouse. Gene. 2000;258(1–2):117–125. | |
Ragoussis J, Senger G, Trowsdale J, Campbell IG. Genomic organization of the human folate receptor genes on chromosome 11q13. Genomics. 1992;14(2):423–430. | |
Wang X, Shen F, Freisheim JH, Gentry LE, Ratnam M. Differential stereospecificities and affinities of folate receptor isoforms for folate compounds and antifolates. Biochem Pharmacol. 1992;44(9):1898–1901. | |
Miotti S, Canevari S, Ménard S, et al. Characterization of human ovarian carcinoma-associated antigens defined by novel monoclonal antibodies with tumor-restricted specificity. Int J Cancer. 1987;39(3):297–303. | |
Kalofonos HP, Karamouzis MV, Epenetos AA. Radioimmunoscintigraphy in patients with ovarian cancer. Acta Oncol. 2001;40(5):549–557. | |
Melani C, Figini M, Nicosia D, et al. Targeting of interleukin 2 to human ovarian carcinoma by fusion with a single-chain Fv of antifolate receptor antibody. Cancer Res. 1998;58(18):4146–4154. | |
Toffoli G, Cernigoi C, Russo A, Gallo A, Bagnoli M, Boiocchi M. Overexpression of folate binding protein in ovarian cancers. Int J Cancer. 1997;74(2):193–198. | |
Elnakat H, Ratnam M. Distribution, functionality and gene regulation of folate receptor isoforms: implications in targeted therapy. Adv Drug Deliv Rev. 2004;56(8):1067–1084. | |
Chancy CD, Kekuda R, Huang W, et al. Expression and differential polarization of the reduced-folate transporter-1 and the folate receptor alpha in mammalian retinal pigment epithelium. J Biol Chem. 2000;275(27):20676–20684. | |
Weitman SD, Frazier KM, Kamen BA. The folate receptor in central nervous system malignancies of childhood. J Neurooncol. 1994;21(2):107–112. | |
Garin-Chesa P, Campbell I, Saigo PE, Lewis JL Jr, Old LJ, Rettig WJ. Trophoblast and ovarian cancer antigen LK26. Sensitivity and specificity in immunopathology and molecular identification as a folate-binding protein. Am J Pathol. 1993;142(2):557–567. | |
Toffoli G, Russo A, Gallo A, et al. Expression of folate binding protein as a prognostic factor for response to platinum-containing chemotherapy and survival in human ovarian cancer. Int J Cancer. 1998;79(2):121–126. | |
Lorusso PM, Edelman MJ, Bever SL, et al. Phase I study of folate conjugate EC145 (Vintafolide) in patients with refractory solid tumors. J Clin Oncol. 2012;30(32):4011–4016. | |
Bellati F, Napoletano C, Gasparri ML, et al. Monoclonal antibodies in gynecological cancer: a critical point of view. Clin Dev Immunol. 2011;2011:890758. | |
Hartmann LC, Keeney GL, Lingle WL, et al. Folate receptor overexpression is associated with poor outcome in breast cancer. Int J Cancer. 2007;121(5):938–942. | |
Kalli KR, Oberg AL, Keeney GL, et al. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol Oncol. 2008;108(3):619–626. | |
Brown Jones M, Neuper C, Clayton A, et al. Rationale for folate receptor alpha targeted therapy in “high risk” endometrial carcinomas. Int J Cancer. 2008;123(7):1699–1703. | |
Wu M, Gunning W, Ratnam M. Expression of folate receptor type alpha in relation to cell type, malignancy, and differentiation in ovary, uterus, and cervix. Cancer Epidemiol Biomarkers Prev. 1999;8(9):775–782. | |
Naumann RW, Coleman RL, Burger RA, et al. PRECEDENT: a randomized phase II trial comparing vintafolide (EC145) and pegylated liposomal doxorubicin (PLD) in combination versus PLD alone in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2013;31(35):4400–4406. | |
Ratnam M, Marquardt H, Duhring JL, Freisheim JH. Homologous membrane folate binding proteins in human placenta: cloning and sequence of a cDNA. Biochemistry. 1989;28(20);8249–8254. | |
Ross JF, Chaudhuri PK, Ratnam M. Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer. 1994;73(9):2432–2443. | |
Salazar MD, Ratnam M. The folate receptor: what does it promise in tissue-targeted therapeutics? Cancer Metastasis Rev. 2007;26(1):141–152. | |
Puig-Kröger A, Sierra-Filardi E, Domínguez-Soto A, et al. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009;69(24):9395–9403. | |
Ross JF, Wang H, Behm FG, et al. Folate receptor type beta is a neutrophilic lineage marker and is differentially expressed in myeloid leukemia. Cancer. 1999;85(2):348–357. | |
Zhao X, Li H, Lee RJ. Targeted drug delivery via folate receptors. Expert Opin Drug Deliv. 2008;5(3):309–319. | |
Sadasivan E, da Costa M, Rothenberg SP, Brink L. Purification, properties, and immunological characterization of folate-binding proteins from human leukemia cells. Biochim Biophys Acta. 1987;925(1):36–47. | |
Sadasivan E, Rothenberg SP, da Costa M, Brink L. Characterization of multiple forms of folate-binding protein from human leukemia cells. Biochim Biophys Acta. 1986;882(3):311–321. | |
Wang H, Zheng X, Behm FG, Ratnam M. Differentiation-independent retinoid induction of folate receptor type beta, a potential tumor target in myeloid leukemia. Blood. 2000;96(10):3529–3536. | |
Paulos CM, Turk MJ, Breur GJ, Low PS. Folate receptor-mediated targeting of therapeutic and imaging agents to activated macrophages in rheumatoid arthritis. Adv Drug Deliv Rev. 2004;56(8):1205–1217. | |
Pan XQ, Zheng X, Shi G, Wang H, Ratnam M, Lee RJ. Strategy for the treatment of acute myelogenous leukemia based on folate receptor beta-targeted liposomal doxorubicin combined with receptor induction using all-trans retinoic acid. Blood. 2002;100(2):594–602. | |
Morphotek. Safety, tolerability and pharmacokinetics of MORAb-003 in subjects with advanced ovarian cancer. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2013 [updated November 5, 2013]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00428766. NLM identifier: NCT00428766. Accessed April 26, 2014. | |
Ebel W, Routhier EL, Foley B, et al. Preclinical evaluation of MORAb-003, a humanized monoclonal antibody antagonizing folate receptor-alpha. Cancer Immun. 2007;7:6. | |
Kamen BA, Smith AK. Farletuzumab, an anti-folate receptor α antibody, does not block binding of folate or anti-folates to receptor nor does it alter the potency of anti-folates in vitro. Cancer Chemother Pharmacol. 2012;70(1):113–120. | |
Armstrong D. Farletuzumab (MORAb-003) in platinum-sensitive ovarian cancer patients experiencing a first relapse. Community Oncol. 2010;7(2)Suppl 1:1–4. | |
Konner JA, Bell-McGuinn KM, Sabbatini P, et al. Farletuzumab, a humanized monoclonal antibody against folate receptor alpha, in epithelial ovarian cancer: a phase I study. Clin Cancer Res. 2010;16(21):5288–5295. | |
Coleman RL, Armstrong DK, White AJ, et al. A novel monoclonal antibody against folate receptor-alpha, exhibits clinical efficacy in platinum-sensitive 1st relapse of ovarian cancer subjects. Proc Eur Soc Gynecol Oncol. 2009;Abstract 90. | |
Kim KH, Jelovac D, Armstong DK, et al. Phase I safety study of farletuzumab, carboplatin, and pegylated liposomal doxorubicin (PLD) in patients with platinum-sensitive epithelial ovarian cancer (EOC). American Society of Clinical Oncology 48th Annual Meeting; June 1–5, 2012; Chicago, IL. J Clin Oncol. 2012;30(Suppl 18): abstract 5062. | |
Rustin GJ. Use of CA-125 to assess response to new agents in ovarian cancer trials. J Clin Oncol. 2003;21(10 Suppl):187s–193s. | |
Markman M, Markman J, Webster K, et al. Duration of response to second-line, platinum-based chemotherapy for ovarian cancer: implications for patient management and clinical trial design. J Clin Oncol. 2004;22(15):3120–3125. | |
Morphotek. Efficacy and safety of MORAb-003 in subjects with platinum-sensitive ovarian cancer in first relapse. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2013 [updated November 18, 2013]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00849667. NLM identifier: NCT00849667. Accessed April 26, 2014. | |
PRNewswire. Morphotek announces top-line results of a phase III study of farletuzumab in patients with relapsed platinum-sensitive epithelial ovarian cancer [press release]. Exton, PA: Morphotek Inc.; 2013 [Jan 10]. Available from: http://www.prnewswire.com/news-releases/morphotek-announces-top-line-results-of-a-phase-iii-study-of-farletuzumab-in-patients-with-relapsed-platinum-sensitive-epithelial-ovarian-cancer-186384621.html. Accessed April 26, 2014. | |
Vergote I, Armstrong D, Scambia G, et al. Phase 3 double-blind, placebo-controlled study of weekly farletuzumab with carboplatin/taxane in subjects with platinum-sensitive ovarian cancer in first relapse. European Soceity of Gynaecological Oncology 18th International Meeting; October 19–22, 2013 ; Liverpool, UK. Int J Gynecol Cancer. 2013;23(8 Suppl 1):11. | |
Morphotek. An efficacy and safety study of MORAb-003 in platinum-resistant or refractory relapsed ovarian cancer (FAR-122). In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2013 [updated November 18, 2013]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00738699. NLM identifier: NCT00738699. Accessed April 26, 2014. | |
Morphotek. Safety Study of farletuzumab, carboplatin and pegylated liposomal doxorubicin (PLD) to treat platinum-sensitive ovarian cancer. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2013 [updated November 5, 2013]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01004380. NLM identifier: NCT01004380. Accessed April 26, 2014. | |
Zwicke GL, Mansoori GA, Jeffery CJ. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012;3. | |
Aronov O, Horowitz AT, Gabizon A, Gibson D. Folate-targeted PEG as a potential carrier for carboplatin analogs. Synthesis and in vitro studies. Bioconjug Chem. 2003;14(3):563–574. | |
Liu J, Kolar C, Lawson TA, Gmeiner WH. Targeted drug delivery to chemoresistant cells: folic acid derivatization of FdUMP[10] enhances cytotoxicity toward 5-FU-resistant human colorectal tumor cells. J Org Chem. 2001;66(17):5655–5663. | |
Majoros IJ, Myc A, Thomas T, Mehta CB, Baker JR. PAMAM dendrimer-based multifunctional conjugate for cancer therapy: synthesis, characterization, and functionality. Biomacromolecules. 2006;7(2):572–579. | |
Ladino CA, Chari RV, Bourret LA, Kedersha NL, Goldmacher VS. Folate-maytansinoids: target-selective drugs of low molecular weight. Int J Cancer. 1997;73(6):859–864. | |
Suzuki T, Hisakawa S, Itoh Y, et al. Design, synthesis, and biological activity of folate receptor-targeted prodrugs of thiolate histone deacetylase inhibitors. Bioorg Med Chem Lett. 2007;17(15):4208–4212. | |
Vlahov IR, Santhapuram HK, Kleindl PJ, Howard SJ, Stanford KM, Leamon CP. Design and regioselective synthesis of a new generation of targeted chemotherapeutics. Part 1: EC145, a folic acid conjugate of desacetylvinblastine monohydrazide. Bioorg Med Chem Lett. 2006;16(19):5093–5096. | |
Weitman SD, Lark RH, Coney LR, et al. Distribution of the folate receptor GP38 in normal and malignant cell lines and tissues. Cancer Res. 1992;52(12):3396–3401. | |
Vlahov IR, Leamon CP. Engineering folate-drug conjugates to target cancer: from chemistry to clinic. Bioconjug Chem. 2012;23(7):1357–1369. | |
Li J, Sausville EA, Klein PJ, et al. Clinical pharmacokinetics and exposure-toxicity relationship of a folate-Vinca alkaloid conjugate EC145 in cancer patients. J Clin Pharmacol. 2009;49(12):1467–1476. | |
Fisher RE, Siegel BA, Edell SL, et al. Exploratory study of 99mTc-EC20 imaging for identifying patients with folate receptor-positive solid tumors. J Nucl Med. 2008;49(6):899–906. | |
Matteson EL, Lowe VJ, Prendergast FG, et al. Assessment of disease activity in rheumatoid arthritis using a novel folate targeted radiopharmaceutical Folatescan. Clin Exp Rheumatol. 2009;27(2):253–259. | |
Reddy JA, Dorton R, Westrick E, et al. Preclinical evaluation of EC145, a folate-vinca alkaloid conjugate. Cancer Res. 2007;67(9):4434–4442. | |
Sausville E, LoRusso P, Quinn M, et al. A phase I study of EC145 administered weeks 1 and 3 of a 4-week cycle in patients with refractory solid tumors. American Society of Clinical Oncology 43rd Annual Meeting; June 1–5, 2007; Chicago, IL. J Clin Oncol. 2007;25(18 Suppl): abstract 2577. | |
Naumann RW, Morris R, Harb W, et al. Protocol EC-FV-02: A phase II study of EC145 in patients with advanced ovarian cancer. European Society of Gynaecological Oncology 16th International Meeting; October 11–14, 2009; Belgrade, Serbia. Int J Gynecol Cancer. 2009;19(Suppl 2): abstract 1181. | |
Merck Sharp and Dohme Corp. Study of EC145 in patients with advanced ovarian and endometrial cancers. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2013 [updated September 19, 2013]. Available from: http://clinicaltrials.gov/show/NCT00507741. NLM identifier: NCT00507741. Accessed April 26, 2014. | |
Merck Sharp and Dohme Corp. Study for women with platinum resistant ovarian cancer evaluating EC145 in combination with Doxil® (PROCEED). In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2014 [updated April 17, 2014]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01170650. NLM identifier: NCT01170650. Accessed April 26, 2014. | |
Amato RJ, Engel J, Lu J, Low P, Naveed F. Phase I Trial of EC90 (keyhole-limpet hemocyanin fluorescein isothiocyanate conjugate) with GPI-0100 adjuvant followed by EC 17 (folate-fluorescein isothiocyanate conjugate) in patients with metastatic renal cell carcinoma and ovarian cancer. American Society of Clinical Oncology 40th Annual Meeting; June 5–8, 2004; New Orleans, LA. J Clin Oncol. 2004;22(Suppl 14): abstract 4754. | |
Endocyte. Study of EC0225 for the treatment of refractory or metastatic tumors. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2012 [updated March 8, 2012]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00441870. NLM identifier: NCT00441870. Accessed April 26, 2014. | |
Bristol-Myers Squibb. A phase 1/2 study of epofolate (BMS-753493) in subjects with advanced cancer (schedule 1). In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2011 [updated January 24, 2011]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00546247. NLM identifier: NCT00546247. Accessed April 26, 2014. | |
Bristol-Myers Squibb. A phase 1/2 study of epofolate (BMS-753493) in subjects with advanced cancer (schedule 2). In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2011 [updated January 24, 2011]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00550017. NLM identifer: NCT00550017. Accessed April 26, 2014. | |
Endocyte. Study of EC0489 for the treatment of refractory or metastatic tumors. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2012 [updated August 7, 2012]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00852189. NLM identifier: NCT00852189. Accessed April 26, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.