")
Back to Journals » International Journal of Nanomedicine » Volume 17
Targeted Delivery of Nanovaccine to Dendritic Cells via DC-Binding Peptides Induces Potent Antiviral Immunity in vivo
Authors Lu Y , Liu ZH, Li YX, Xu HL, Fang WH, He F
Received 8 January 2022
Accepted for publication 22 March 2022
Published 5 April 2022 Volume 2022:17 Pages 1593—1608
DOI https://doi.org/10.2147/IJN.S357462
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Farooq A. Shiekh
Ying Lu,* Ze-Hui Liu,* Ying-Xiang Li, Hui-Ling Xu, Wei-Huan Fang, Fang He
Department of Veterinary Medicine, Institute of Preventive Veterinary Medicine & Zhejiang Provincial Key Laboratory of Preventive Veterinary Medicine, College of Animal Sciences, Zhejiang University, Hangzhou, 310058, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fang He, Department of Veterinary Medicine, Institute of Preventive Veterinary Medicine & Zhejiang Provincial Key Laboratory of Preventive Veterinary Medicine, College of Animal Sciences, Zhejiang University, 866 Yuhangtang Road, Hangzhou, 310058, People’s Republic of China, Email [email protected]
Background: Dendritic cell (DC) targeted antigen delivery is a promising strategy to enhance vaccine efficacy and delivery of therapeutics. Self-assembling peptide-based nanoparticles and virus-like particles (VLPs) have attracted extensive interest as non-replicating vectors for nanovaccine design, based on their unique properties, including molecular specificity, biodegradability and biocompatibility. DCs are specialized antigen-presenting cells involved in antigen capture, processing, and presentation to initiate adaptive immune responses. Using DC-specific ligands for targeted delivery of antigens to DCs may be utilized as a promising strategy to drive efficient and strong immune responses.
Methods: In this study, several candidates for DC-binding peptides (DCbps) were individually integrated into C-terminal of porcine circovirus type 2 (PCV2) Cap, a viral protein that could self-assemble into icosahedral VLPs with 60 subunits. The immunostimulatory adjuvant activity of DC-targeted VLPs was further evaluated in a vaccine model of PCV2 Cap.
Results: With transmission electron microscopy (TEM), E. coli expressed Cap-DCbp fusion proteins were observed self-assembled into highly ordered VLPs. Further, in dynamic light scattering (DLS) analysis, chimeric VLPs exhibited similar particle size uniformity and narrow size distribution as compared to wild type Cap VLPs. With a distinctly higher targeting efficiency, DCbp3 integrated Cap VLPs (Cap-DCbp3) displayed enhanced antigen uptake and increased elicitation of antigen presentation-related factors in BM-DCs. Mice subcutaneously immunized with Cap-DCbp3 VLPs exhibited significantly higher levels of Cap-specific antibodies, neutralizing antibodies and intracellular cytokines than those with other DCbp integrated or wild type Cap VLPs without any DCbp. Interestingly, Cap-DCbp3 VLPs vaccine induces robust cellular immune response profile, including the efficient production of IFN-γ, IL-2 and IL-10. Meanwhile, the improved proliferation index in lymphocytes with Cap-DCbp3 was also detected as compared to other VLPs.
Conclusion: This study described the potential of DC-binding peptides for further improved antigen delivery and vaccine efficacy, explainning nanovaccine optimization in relation to a range of emerging and circulating infectious pathogens.
Keywords: dendritic cell-targeted delivery, self-assembling peptide-based nanoparticles, virus-like particles, nanovaccine, enhanced immunogenicity
Introduction
Dendritic cells (DCs), as the most efficient antigen-presenting cells (APCs), represent the interface of innate and adaptive immunity.1 Scattered throughout the body, DCs constitute the first line of defense against invading pathogens.2 Innate immune recognition by DCs is based on the recognition of microbial motifs by specialized receptors. Following interaction with antigens (Ags), DCs undergo a maturation process resulting in the up-regulated expression of co-stimulatory, adhesion and MHC molecules enhancing their capacity to present peptides to naive T cells.3 In this process, DCs shape the adaptive immune response by internalizing and processing antigens through MHC class I (MHC-I) and class II (MHC-II) pathways and presenting these peptides to CD4+ and CD8+ T lymphocytes. The ability to present peptides derived from exogenous antigens on MHC-I, also known as cross-presentation, is critical for CD8+ T cell immunity against intracellular pathogens, especially for those that do not primarily infect DCs like influenza A virus.
Previous research demonstrated that antitumor vaccines which are derived from in vitro cultured DCs coupled with tumor antigen can be successfully returned to the body for autologous transplantation. Although this method has achieved phased success, it is limited due to the uncertain side effects of the complex.4 Subsequently, a simpler and more effective method was proposed to directly present antigens to DC via DC-restricted surface molecules, including toll-like receptors (TLRs), C-type lectin receptors (CLRs) such as DEC-205, Clec9A, the mannose receptor, and dendritic cell inhibitory receptor 2 (DCIR2);5,6 for example, the studies of fusion expression of antigens and Fc fragment of IgG, DC receptor-specific monoclonal antibody or single chain antibody fragments (scFvs).7,8 In this way, the fusion protein is internalized by DCs, propagated and degraded in the endocytic pathway. As a consequence, antigen peptides are loaded into MHC-I and or MHC-II. There is also an in situ approach of delivering antigens with DCbps using nanoparticles to encapsulate both antigens and adjuvants.9,10 However, in many researches, results indicated varied efficacy with DC targeting strategies, which may be attributed to the varied loading or encapsulation efficiency of antigens into nanoparticles.
In contrast to the whole Ab or ScFv molecules guided DC-targeting strategy, DC-binding peptides offer an outstanding advantage based on their small size, which significantly improves the efficiency to cross biological barriers and greatly reduces production costs.11 For this purpose, several DC-targeting peptides were identified from a 12-mer peptide phage display library, which could bind to human DCs and recognize the conserved region of the ligand on avian, canine, equine, and feline DCs. It was found that the genetic fusion of the isolated DC-targeting peptide with the C terminus of hepatitis C virus (HCV) NS3 enhanced DC activation, resulting in the increased expression of IFN-γ and TNF-α in CD4+ and CD8+ T cells from HCV-infected patients.11 On this basis, a mucosal, DC-targeted approach was employed in lactobacilli vectored vaccine to enhance protective immunity against B. anthracis infection, as evidenced by local IgA secretion, T cell immunity, and high titers of specific antibodies.12 Currently, DC-binding peptides have attracted increasing attention with their potential to develop more efficacious vaccines.
Porcine circovirus type 2 (PCV2) is a major swine virus which causes post-weaning multi-systemic wasting syndrome (PMWS) and lymphadenopathy in weanling piglets, along with a range of clinical signs including jaundice, nephropathy, reproductive and respiratory disorders, collectively known as porcine circovirus associated diseases, or PCVAD. Inactivated vaccines and Cap-based subunit vaccines are currently major strategies in clinical use for the prevention and control of PCVAD. Vaccination can significantly reduce PCV2-related lymphatic tissue damage and reduce viremia, thereby alleviating clinical symptoms and reducing economic losses. However, traditional inactivated vaccines also have some limitations regarding safety and scale-up, restricting the application of vaccines. Cap subunit vaccine has gradually become the favored choice against PCV2. Conventional Cap antigens effectively induce humoral immune responses, but fail to induce sufficient cellular immunity, which is essential for the elimination of intracellular pathogens.13 A series of strategies has been proposed to enhance the immunogenicity of the subunit vaccine, including the employment of cytokine adjuvants,14–16 the development of Cap VLP-based vaccine17,18 or chimeric Cap VLP containing small molecules as adjuvants.19,20 However, deepened studies in Cap VLP delivery and the related efficacy mechanism are not widely available.
VLPs resemble native virus in terms of the size and the orderly repetitive antigen array, which induces a stronger immune response than monomer immunogen. In vaccine design, the self-adjuvanticity of VLPs is regarded as an outstanding advantage.21 Compared to soluble antigens, VLPs have been shown to bind to DCs and be internalized efficiently. Besides, antigen internalization is of crucial importance to achieve significant MHC-I cross-presentation and effective T and B cell responses.21,22 Previous studies demonstrated that PCV2 cap derived either from baculovirus, E. coli or yeast systems forms VLPs and functions as effective vaccines. PCV2 Cap is compatible to C-terminal inserts without any disturbance in particle assembling, which allows surface display of epitopes, and therefore serves as foreign epitope carriers.23
In the present study, we explored whether the potency of VLP-based nanovaccines would be further elevated via DC targeting technology. To this end, a series of DC-binding peptides was screened and evaluated in terms of the capacity to promote antigen uptake and presentation in DCs. The peptide candidate DCbp3 was selected and fused to C-terminal PCV2 Cap to generate DC targeting PCV2 VLPs for further studies. We hypothesize that PCV2 VLP vaccines can be efficiently delivered via the combination of VLPs and DCbps, and therefore this DC-targeting technology would boost immunological competence compared to conventional subunit vaccines and nanoparticulate vaccines. Further, the efficacy of this novel vaccine candidate against PCV2 was tested regarding evoking effective protection in mice.
Materials and Methods
Cells and Virus
PK-15 cells (ATCC, CCL-33) were cultured in DMEM containing 10% FBS, 100 IU/mL penicillin and 100 mg/L streptomycin at 37°C in a 5% CO2 atmosphere. PCV2d JH strain (GenBank Accession Number MG_245867.1) was propagated in PK-15 cells and utilized for virus neutralizing test (VNT).
Cloning and Expression of Cap-DCbp Particles
DNA sequence coding for 5 different peptides, including DCbpcon, DCbp1, DCbp2, DCbp3 and DCbp4,12,24–26 were synthesized respectively and cloned into pET28a to generate pET28a-Cap-DCbps. Flexible linker (4×GlyGlySer) was incorporated between Cap and DCbps (Figure 1) to avoid steric hindrance and allow proper folding. Recombinant plasmids were transformed into E. coli BL21 (DE3) for protein expression. A single colony was placed into a 10 mL starter culture of LB medium containing 50 µg/mL kanamycin and incubated for 16 h at 37°C, with shaking at 200 rpm. The culture was then diluted into 400 mL 2×YT containing 50 µg/mL kanamycin and incubated at 37°C, with shaking at 200 rpm. When the absorbance reached 0.8, cultures were mixed with 0.6 mM IPTG and grown for 14 h, with shaking at 180 rpm at 25°C. Bacterial pellet was lysed with ultra-sonication, and supernatant containing target proteins were collected and loaded on an HisSep Ni-NTA Agarose Resin (Yeasen, China). The protein was subsequently eluted with PBS buffer (PH 7.4) supplemented with 500 mM imidazole.
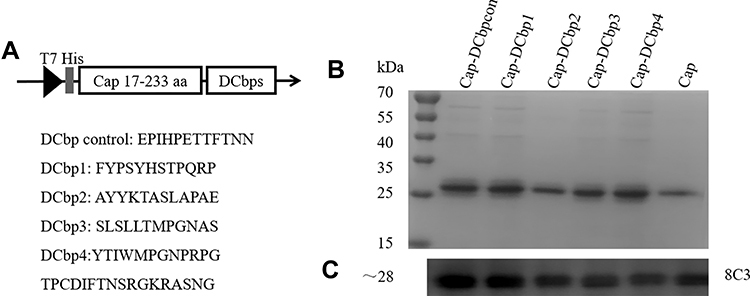 |
Figure 1 Construction and expression of DC-binding peptide-fused Cap proteins. Notes: (A) The construction schematic of recombinant expression vector. Amino acid sequences corresponding to different DC-binding peptides (DCbps) were inserted into C terminal of Cap. The expression of fusion sequence was under the control of T7 promoter. SDS-PAGE (B) and Western blots (C) of recombinant proteins expression. 8C3: mouse anti-Cap MAb. |
Characterization of Cap-DCbp Particles
Purified Cap-DCbp fusion proteins (designated as Cap-DCbpcon, Cap-DCbp1, Cap-DCbp2, Cap-DCbp3 and Cap-DCbp4) were filtered using 0.22 μM syringe filter, and subsequently dialyzed against assembly solution (200 mM NaCl, 50 mM Tris, 0.1% Tween-20, pH 8.0) at 16°C overnight. Dynamic light scattering (DLS) and a transmission electron microscope (TEM) were then performed to evaluate self-assembly of VLPs, as previously described.27
Isolation of Murine Bone Marrow-Derived Dendritic Cells (BM-DCs)
BM-DCs were obtained from femoral bone marrow of C576BL male mice and washed twice with RPMI-1640. Red blood cells (RBC) were lysed in RBC Lysis Buffer (Beyotime, China) and the remaining cells were collected. Subsequently, cells were resuspended in 6-well cell plates with RPMI-1640 complete medium (containing 10% FBS, 10 ng/mL mouse GM-CSF, 10 ng/mL mouse IL-4) at a density of 106 cells/well. Medium was refreshed every 2 days. After culturing for 7 days, most cells developed typical dendritic morphology.
Cellular Uptake
Internalization of Cap VLPs by BM-DCs was determined using the following steps. In brief, cells were pre-seeded on 6-well cell culture plates at 2×105 cells/mL and cultured overnight; 100 μg/mL Cap VLPs (Cap-DCbpcon, Cap-DCbp1, Cap-DCbp2, Cap-DCbp3 or Cap-DCbp4) were then added, respectively, and incubated for 12 h. After incubation, cultured cells were washed twice with PBS to remove unbound particles and cells were fixed with 80% cold acetone. Fixation was blocked with FBS (100 μL/well) and washed with PBS. Cells were sequentially incubated with Cap monoclonal antibody (8C3) for 1 h and fluorescent secondary antibody for 1 h at 37°C. Cells were then kept at room temperature and stained with DAPI for 10 min before immunofluorescence signals corresponding to Cap internalization were detected with confocal laser scanning microscopy (CLSM) IX81-FV1000 (Olympus).
Analysis of Marker Genes and Cytokine Expression in BM-DC
Maturation and cytokine expression in BM-DCs with Cap VLPs were investigated. After exposure to 100 μM of Cap VLPs for 4 h, BM-DCs were subject to total RNA isolation with an Easy RNA Kit according to the manufacturer’s instructions, and then converted to cDNA using a commercial product (Vazyme, R212-01, China). Quantitative real-time PCR (qPCR) was performed using ChamQ Universal SYBR qPCR Master Mix according to the manufacturer’s instructions (Vazyme, Q711-02, China). Primers used for qPCR were synthesized by Sangon (Shanghai, China) based on target sequences reported in NCBI, and details are shown in Table 1. β-actin was amplified as an internal control. The relative expression of target gene was detected using 2−ΔΔct method.
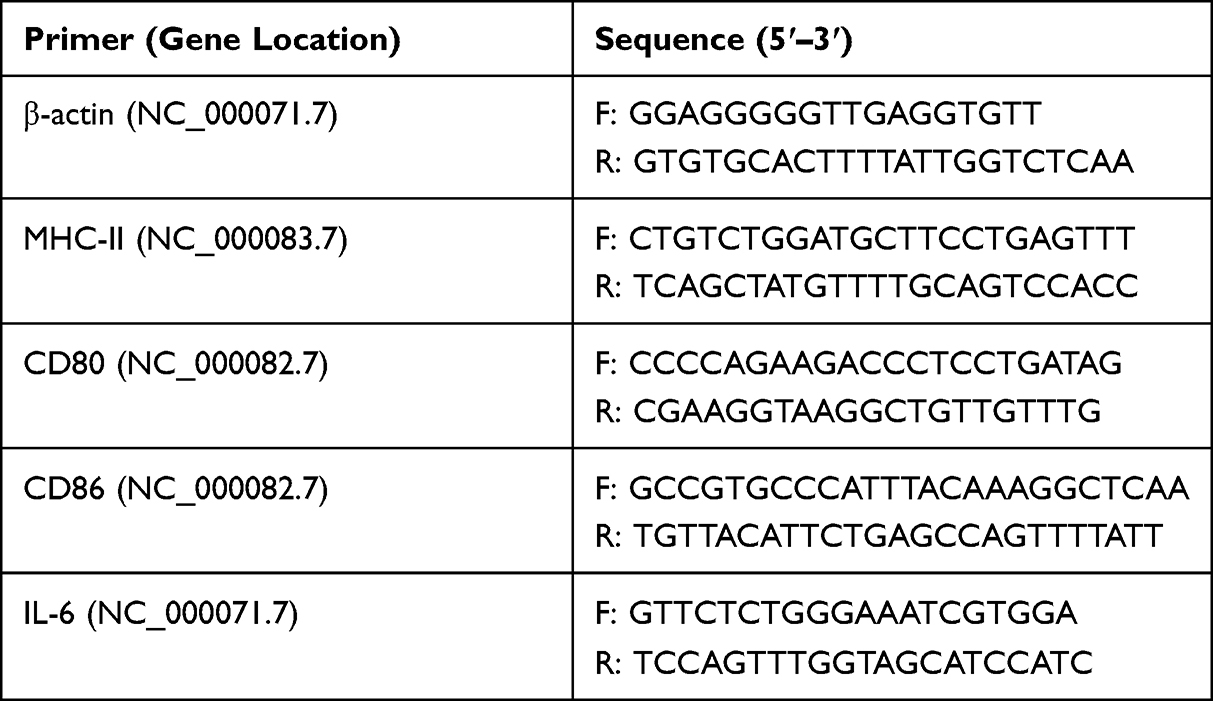 |
Table 1 The Primer Sequences used for detecting marker genes and cytokine expression in BM-DCs |
Immunization
Animal experimental procedures were supervised and approved by Laboratory Animal Care and Use Committee, Zhejiang University. Guideline for Ethical Review of Laboratory Animal welfare (GB/T 35892–2018), Guiding Opinions on Treating Laboratory Animals Kindly (2006–398) and Laboratory Animal Management Regulations (2017) were followed. The corresponding ethical application code is No. 17243.
According to the manufacturer’s instructions, antigens were emulsified with ISA-206 adjuvant (Seppic, France) at a ratio of 50:50 (w/w). Eight-week-old female BALB/c mice were randomly divided into 7 groups (n=5). Groups A, B, C, D and E were subcutaneously inoculated with 40 μg of Cap VLPs (Cap-DCbpcon, Cap-DCbp1, Cap-DCbp2, Cap-DCbp3 or Cap-DCbp4, respectively). PBS was immunized as negative controls. A commercial subunit vaccine (Pulike, Luoyang, China) served as a positive control. Mice were boosted at 2-week intervals.
Humoral Response Evaluation
Titers of Cap-specific IgG antibodies were determined in ELISA with sera collected after the second immunization. Flat-bottomed 96-well plates were coated with 5 µg/mL Cap protein in 50 mM carbonate buffer (pH 9.6) overnight. Plates were washed with PBST and blocked with 1% Casein. Serial dilutions of serum were performed in PBST, and plates were incubated for 2 h at 37°C. Plates were then washed again and incubated for 1 h at 37°C with HRP-goat anti-mouse IgG, IgG1 and IgG2a (1:5000, ABclonal, Wuhan, China), after being rinsed with PBST to remove unbound antibodies. The reaction was developed by adding 100 μL tetramethylbenzidine (TMB) for 10 min at room temperature before being stopped by adding 50 μL 2 M H2SO4. The absorbance at 450 nm (OD450) was read using a microplate reader (BioTek).
To determine the titers of anti-PCV2 neutralizing antibodies (NAbs) in serum samples, VNT was performed as described previously.19 Briefly, 4×105 PK-15 cells were pre-seeded into 96-well plates and cultured in DMEM containing 10% FBS at 37°C for 24 h. Heat-inactivated serum samples were 2-fold serially diluted to 1:256. 100 μL of each diluted sample was mixed with an equal volume of PCV JH (200 TCID50) and added to the cells, and allowed to interact for 1 h at 37°C. After washing with Hanks solution, 200 μL DMEM containing 2% FBS was added to each well. After 72 h, cells were fixed with 4% paraformaldehyde, and then exposed to PCV2 monoclonal antibody 8C3 (1:500), followed by FITC-labelled goat anti-mouse IgG (1:1500) for immunofluorescence assay. NAb titers were determined and expressed as the log2 of the reciprocal of the highest serum dilution that was able to completely block PCV2 infection.
Lymphocyte Proliferation Assay
Mice were sacrificed on day 42 after the first immunization. Spleen cells were collected and analyzed for the proliferation in response to specific antigens. Splenocytes were re-suspended in RPMI 1640 supplemented with 10% FBS and seeded into 96-well flat-bottom microtiter plate at a density of 2×106 cells/mL. Cap antigen (10 µg/mL) was added. Wells with medium only were used as a control. Plates were first incubated for 36 h before cell proliferation was evaluated using the CCK8 method. Stimulation index (SI) was calculated based on the formula: SI = OD value of stimulated cells/OD value of unstimulated cells.
Cytokine Production by Splenocytes
Spleens were extracted from immunized mice on day 42 post euthanasia and meshed by physical homogenization. After treatment in RBC lysis buffer for 10 min, cells were washed with RPMI 1640 by centrifugation at 1200 rpm for 5 min. Then, 106 cells were suspended in 1 mL medium in the presence of 10 μg/mL Cap antigen and cultured for 36 h at 37°C in a CO2 incubator. Cells were subject to total RNA extraction as described above. IFN-γ, IL-2 and IL-10 mRNA was quantified in qPCR using corresponding primer pairs, as shown in Table 2. The relative expression of target gene was detected using 2−ΔΔct method.
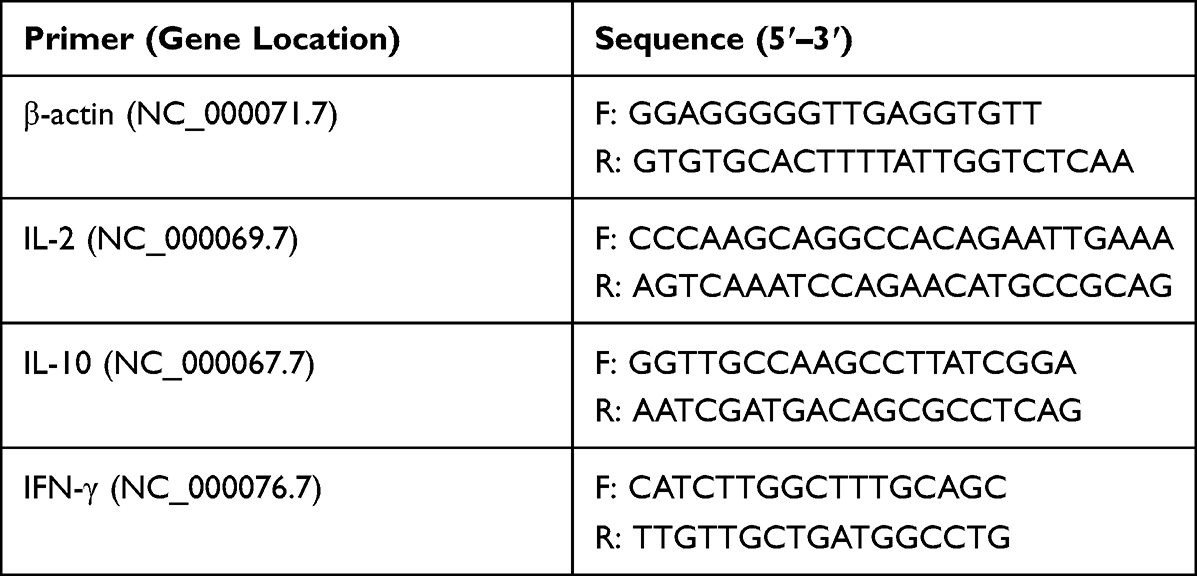 |
Table 2 The Primer Sequences used for detecting Cytokine production by splenocytes |
Statistical Analysis
Student’s t-test was used to analyze the significant of differences between the two groups. P-values of <0.05 were considered statistically significant. Data were expressed as mean ± standard error of mean.
Results
Design and Expression of Cap DC-Binding Peptides
It is well-documented that PCV2 Cap N-terminal contains a nuclear localization signal (NLS). NLS consists of multiple arginine residues and is mostly encoded with rare codons in E. coli.28 To promote the soluble expression, a truncated Cap (deleting N-terminal 1–16 amino acids) was selected for further study. DCbps fused cap expression plasmids (Figure 1A) were transformed into E. coli BL21 (DE3) and subjected to Ni-NTA chromatography purification. Results indicated that fusion proteins were inconsistent with an expected molecular weight of ~28 kDa (Figure 1B), with a purity of about 90%. Moreover, Western blotting confirmed DCbps fused Cap proteins (designated as Cap-DCbpcon, Cap-DCbp1, Cap-DCbp2, Cap-DCbp3 and Cap-DCbp4) developed significant reactivity with PCV2 Cap monoclonal antibody 8C3 (Figure 1C).
Successful Assembling of Cap-DCbp Fusion Proteins into Stable VLPs
Purified proteins were subject to dialysis against PBS buffer and then analyzed under a TEM. Results revealed that Cap-DCbps was successfully assembled into uniform VLPs with a diameter of ~20 nm (Figure 2A). DLS results indicate that the dominant particle diameter of Cap-DCbp VLPs is 20 ± 4.8 nm of greater than 99% (Figure 2B). The sizes of Cap-DCbps VLPs observed using the two methods were consistent, indicating that Cap-DCbps VLPs exhibited considerable integrity and narrow size distribution.
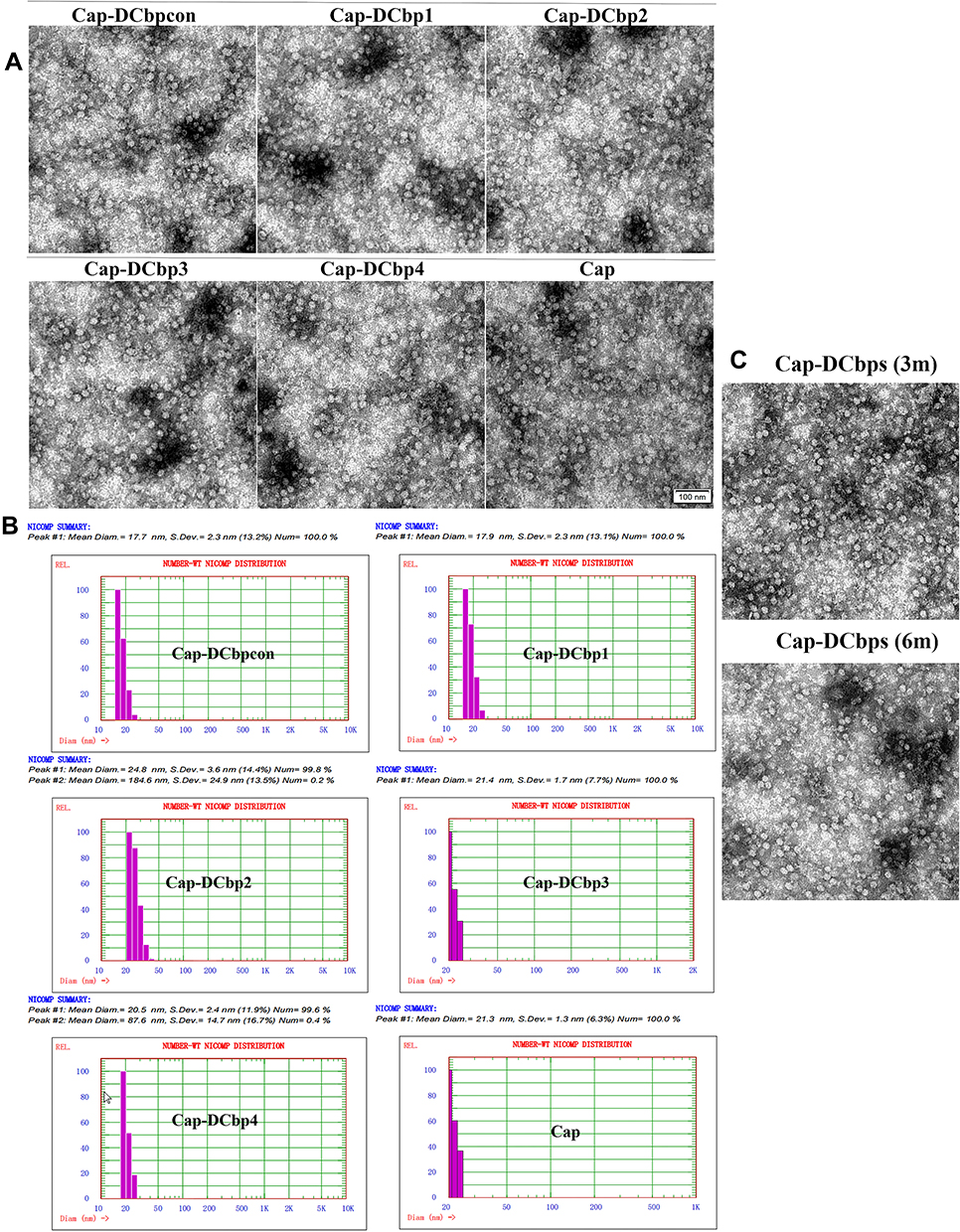 |
Figure 2 Physicochemical characterization of Cap-DCbp VLPs. Notes: (A) Morphological characterization of Cap-DCbp VLPs under a TEM (×25,000). (B) Particle diameter determination of Cap-DCbp VLPs by DLS. (C) Morphological detection of Cap-DCbp VLPs after storage under a TEM (×25,000). |
As low-temperature storage is usually required to sustain vaccine activity, a protocol was designed to maintain Cap-DCbps VLPs. To assess the long-term stability and solubility in 4°C, VLPs stored for 3 and 6 months were evaluated in SDS-PAGE and BCA. Samples were spun at 16,000 rpm for 30 min at 4°C to remove aggregates before the tests. Results indicated that, after storage at 4°C for 3 or 6 months, respectively, no significant change was observed in VLP morphology and homogeneity (Figure 2C). As shown in Table 3, up to 61.5% and 52.9% of VLPs remained in the soluble fraction, indicating that Cap-DCbp VLPs were highly stable as vaccine candidates.
 |
Table 3 Storage Efficiency of Cap VLPs |
Enhanced Cellular Uptake and BM-DC Maturation by Cap-DCbp VLPs
To determine whether DC-binding peptides enhance the uptake of Cap VLPs in APCs, BM-DCs were prepared from mice for the test. When suspensions of bone marrow were cultured in the presence of 10 ng/mL mouse GM-CSF, 10 ng/mL mouse IL-4 for 7 days, most cells displayed dendritic appearance with satellite and slightly branched morphology (Figure 3A). BM-DCs were incubated with 100 μg/mL Cap-DCbp VLPs or blank medium for 12 h. As shown in Figure 3B, a strong green fluorescence signal was observed in BM-DCs in the Cap-DCbp3 VLPs group, while much weaker FITC signals were detected in the Cap-DCbp4 VLPs group, followed by Cap-DCbpcon, Cap-DCbp1 and Cap-DCbp2. Cells treated with medium only developed a non-specific fluorescence signal.
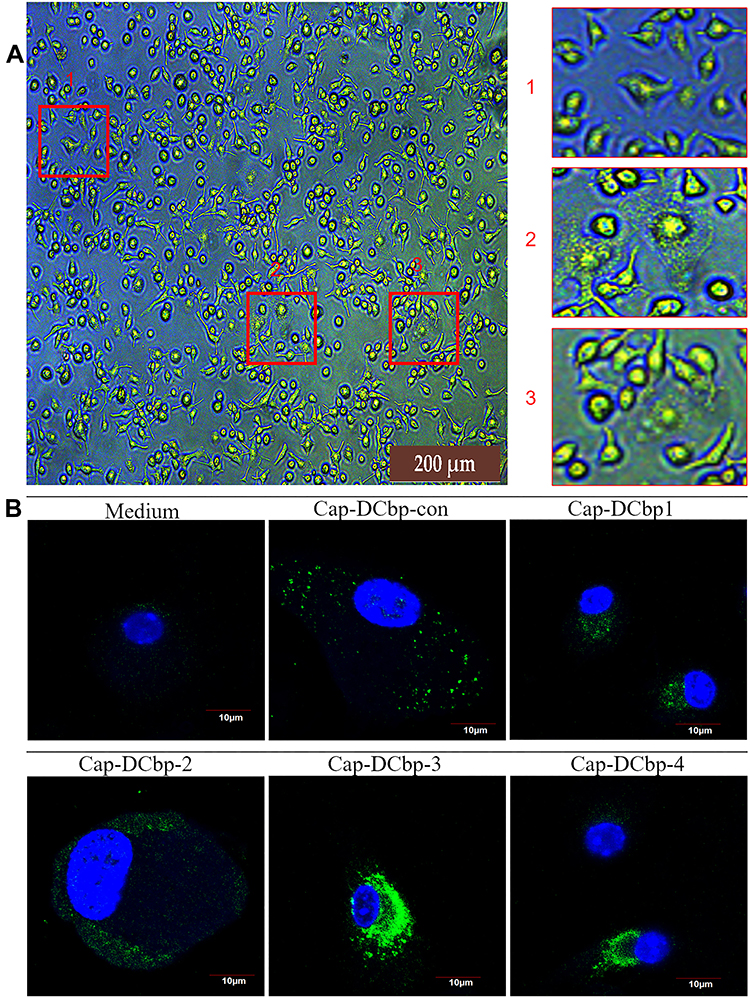 |
Figure 3 DC-binding peptides enhanced uptake of Cap VLPs in BM-DCs. Notes: (A) Differentiated BM-DCs. Suspensions of bone marrow were cultured with GM-CSF and IL-4. (B) Cap internalization. BM-DCs were incubated with Cap VLPs or medium. Primary antibody: mouse 8C3 mAb; secondary antibody: FITC-coupled goat anti-mouse IgG. Signals of cell nuclei (blue) and Cap (green) were detected by CLSM. |
The ability of Cap-DCbps VLPs was investigated in qPCR, to induce the expression of the surface marker MHC-II and co-stimulatory molecules CD80, CD86. It was found that the expression levels of MHC-II (Figure 4A) CD80 (Figure 4B) and CD86 (Figure 4C) remarkably increased upon the incubation of Cap-DCbp3 VLPs (Figure 4). The levels of MHC-II in the Cap-DCbp3 VLPs were 3.56-, 5.77-, 3.32-, 2.75- and 1.55-fold higher than those of Cap-DCbpcon, Cap-DCbp1, Cap-DCbp2, Cap-DCbp4 and native Cap, respectively. Similar results were observed in CD80 and CD86, and the level of CD80 in the Cap-DCbp3 VLPs was 3.91-, 6.24-, 2.72-, 1.51- and 5.96-fold higher than those of Cap-DCbpcon, Cap-DCbp1, Cap-DCbp2, Cap-DCbp4 and native Cap, respectively. The level of CD86 in the Cap-DCbp3 VLPs was 4.64-, 10.57-, 3.08-, 1.93- and 10.80-fold higher than those of Cap-DCbpcon, Cap-DCbp1, Cap-DCbp2, Cap-DCbp4 and native Cap, respectively. IL-6 secreted from DCs can induce T and B cell activation, proliferation and differentiation, and the levels of IL-6 in the Cap-DCbp3 VLPs were 2.71-, 10.42-, 1.56-, 2.07- and 2.74-fold higher than those of Cap-DCbpcon, Cap-DCbp1, Cap-DCbp2, Cap-DCbp4 and native Cap, respectively (Figure 4D). Taken together, our results showed that Cap-DCbp3 VLPs could not only enhance antigens uptake but also upregulate the expression of related molecules of antigen presentation in vitro.
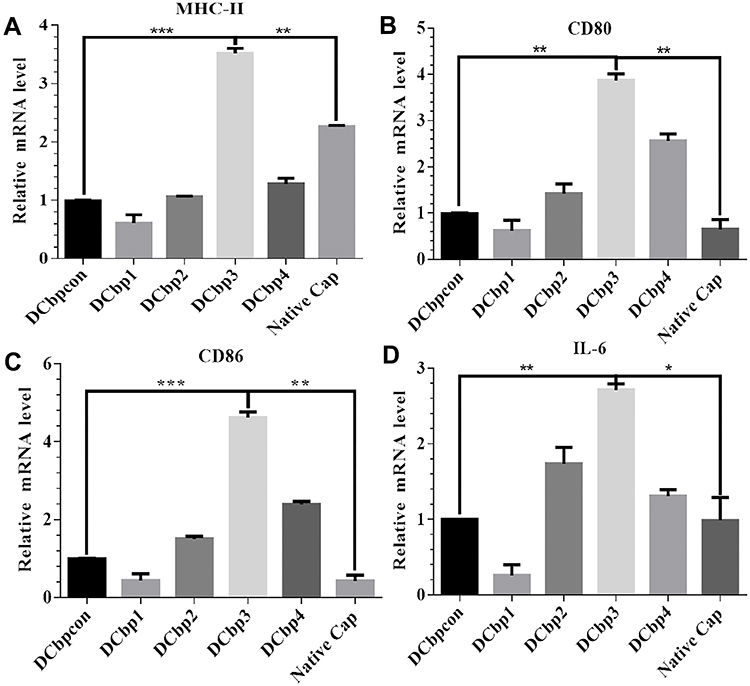 |
Figure 4 Assessment of antigen uptake and immunostimulatory effects in BM-DCs. Notes: BMDCs were exposed to 100 μg/mL antigens for 12 h. MHC-II (A), CD80 (B), CD86 (C) and IL-6 (D) were then investigated by qPCR. (*p<0.05, **p<0.01,***p<0.001). |
Potent Humoral Immunity Stimulated by DC-Targeted Cap VLPs
Serum samples from all the groups were tested in ELISA to determine the levels of anti-Cap antibodies. As shown in Figure 5, Cap-specific antibodies increased with the immunizations of Cap-related antigens. Significantly higher IgG levels were detected in sera of mice immunized with Cap-DCbp3 VLPs than in sera of the other groups either at 35 dpi (Figure 5A, p<0.05) or 42 dpi (Figure 5B, p<0.05). And OD450 corresponding to IgG in Cap-DCbp3 VLPs was 1.55-fold higher than the positive control (commercial vaccine Yuankexin), a conventional CAP VLP vaccine without any DC-binding peptide. As expected, the negative control group with PBS developed non-significant antibody responses.
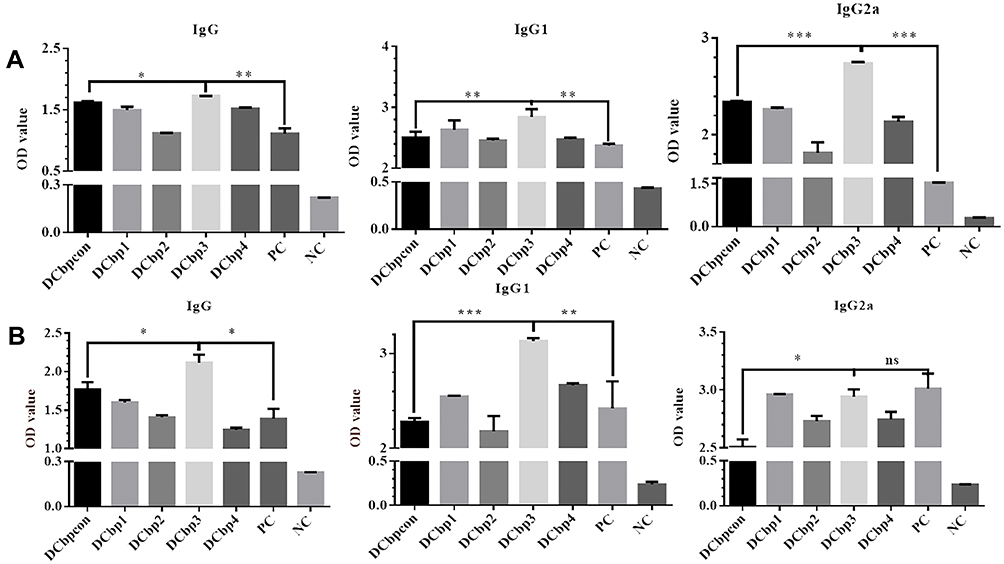 |
Figure 5 Detection of Cap-specific antibodies in sera. Notes: Specific antibodies including IgG, IgG1 and IgG2a against Cap were measured in serum samples by ELISA at 35 dpi (A) and 42 dpi (B). (*p<0.05; **p<0.01; ***p<0.001). |
The distribution of IgG subclasses IgG1 and IgG2a was further analyzed in the sera of mice immunized with different Cap VLPs. Significantly higher IgG1 levels were detected in the serum of mice immunized with Cap-DCbp3 VLPs than in the sera of other control Cap-DCbp VLPs either at 35 dpi (Figure 5A, p<0.01) or 42 dpi (Figure 5B, p<0.001). OD450 corresponding to IgG1 in the Cap-DCbp3 VLPs was 1.2-fold higher than in the positive control. In contrast, there is a remarkable difference between Cap-DCbp3 VLPs and Cap-DCbpcon VLPs immunized group in the level of IgG2a at 35 dpi (p<0.001). And OD450 corresponding to IgG2a in the Cap-DCbp3 VLPs was 1.8-fold higher than in the positive control. At 42 dpi, OD450 corresponding to IgG1 in Cap-DCbp3 VLPs was 1.3-fold higher than in the positive control. No difference was detected between Cap-DCbp3 VLPs and the positive control. These results indicated that, as compared to conventional Cap VLPs, Cap-DCbp3 VLPs are more efficient in eliciting humoral immune responses in vivo.
Generally, it has been shown that viral neutralizing antibodies (NAb) against PCV2 correlate well with in vivo protection.29,30 For this reason, we therefore performed neutralization assay to detect NAbs upon Cap-DCbp immunization (Figure 6). Mice from each group were sacrificed at 35 dpi and serum samples were assayed for NAbs against PCV2. No PCV2-specific NAb was detected in the negative control. In contrast, a significant increase in NAb was detected in all of the other experimental groups. In Cap-DCbp3 VLPs groups, the average NAb titer was (1:55.72), which was significantly (p<0.05) higher than those in other experimental groups (1:12.13 in Cap-DCbpcon, 1:16.80 in Cap-DCbp1, 1:11.55 in Cap-DCbp2, 1:11.88 in Cap-DCbp4 VLPs, 1:22.16 in PC). The titer of Cap-DCbp3 VLPs was 2.5-fold higher than PC. Therefore, it was concluded that the employment of Cap-DCbp3 for targeted vaccine offered an alternative strategy for optimized Cap VLP vaccine.
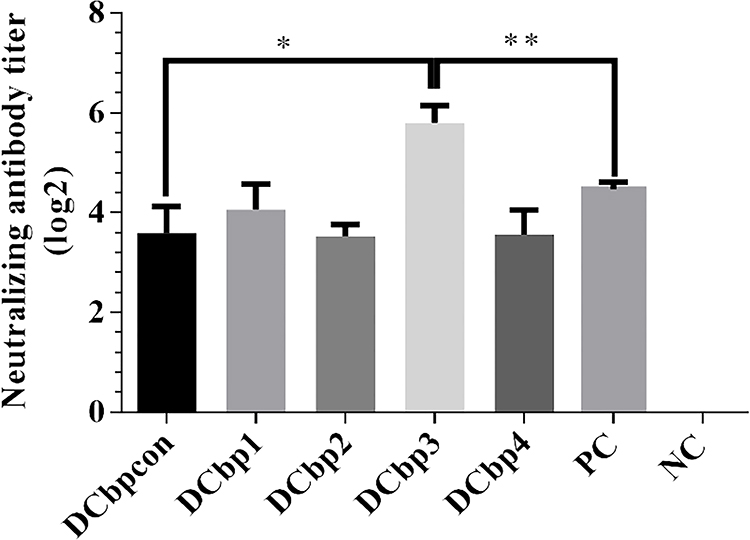 |
Figure 6 Detection of PCV2-specific NAbs in sera. Notes: Neutralizing antibodies against PCV2 JH strain were measured in serum samples at 42 dpi. NAb titers were calculated and expressed as the log2 of the reciprocal of the highest serum dilution that was able to completely block PCV2-infection in PK-15 cells. (*p<0.05, **p<0.01). |
Significantly Elevated Cellular Immune Response via DC-Binding Peptide
It is clear that the level of lymphocyte proliferation is an important indicator to evaluate the cellular immune response. Effects of spleen lymphocyte proliferation were analyzed by CCK8 assay and the data is shown in Figure 7A. The stimulating effects of Cap-DCbp VLPs and positive control were significantly enhanced compared with the negative control group (p<0.001), the proliferation stimulation index (SI) of Cap-DCbp3 VLPs was 2- and 1.5-fold higher than those of Cap-DCbpcon and PC, respectively. There was a significant difference in SI between Cap-DCbp3 VLPs and other experimental groups (Cap-DCbp VLPs) with Cap antigen stimulus, while no statistical difference was detected among experimental groups except Cap-DCbp3 VLPs. These results indicated that Cap-DCbp3 VLPs enhance significantly the immunogenicity of Cap VLPs, and serve as a stronger inducer of spleen lymphocyte proliferation.
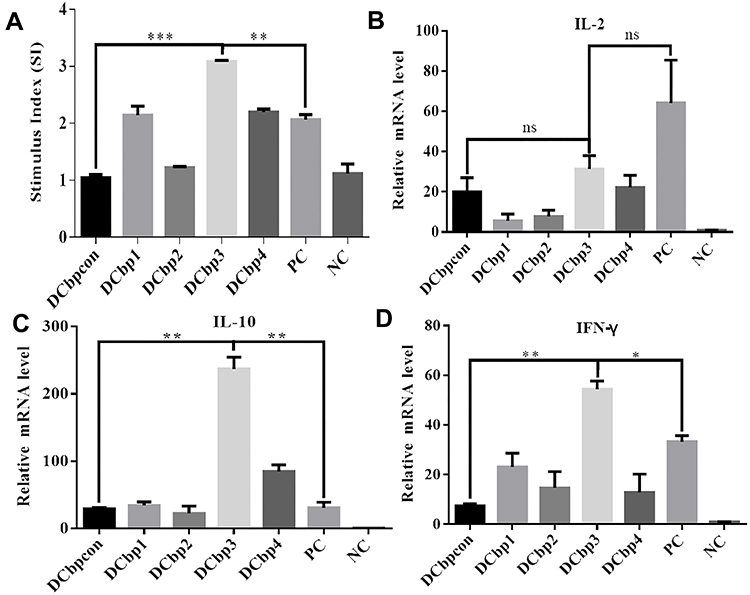 |
Figure 7 Lymphocyte proliferation and cytokine induction in response to specific antigen. Abbreviation: ns, not significant. Notes: (A) Results of lymphocyte proliferation. Plates were incubated with 10 μg/mL Cap for 36 h before their proliferation stimulation index (SI) was evaluated by CCK8 method. Relative mRNA fold changes of IL-2 (B), IL-10 (C) and IFN-γ (D) were detected by qPCR. (*p<0.05; **p<0.01; ***p<0.001, p>0.05). |
To further investigate the initiation of cellular immunity, the relative cytokine secretion levels of IL-2, IL-10 and IFN-γ from Cap-stimulated splenocytes were determined in qPCR (Figures 7B–D). Results showed that mice with Cap-DCbp3 VLPs or PC triggered high levels of cytokines. The levels of IL-10 in the group of Cap-DCbp3 VLPs were 8.06- and 7.67-fold higher than those of Cap-DCbpcon and PC, respectively. And the levels of IFN-γ with Cap-DCbp3 VLPs were 7.35- and 1.63-fold higher than those of Cap-DCbpcon and PC. These results indicate that vaccination with Cap-DCbp3 VLPs was more efficient in eliciting cellular immune response.
Discussion
In recent years, DC-targeting strategies have emerged as one of the focuses in research as DCs play a significant role in the induction and regulation of immune responses.5,31–33 Much progress has been achieved in human DC-targeting vaccines, whereas DC-targeting swine vaccines have been rarely reported. Ags purely used for vaccines are often poorly immunogenic and require specific reagents, termed adjuvants, to enhance the induction of Ag-specific immune responses, as indicated by antibody production and effector T cell functions.34,35 As a swine vaccine, additional practical issues should be taken into consideration to generate E. coli derived PCV2 VLPs, including production cost, dosage and efficacy. The DC-targeting strategy has become a desirable option;36 hence, we utilized DC-binding peptides to deliver VLPs for more reasonable and effective immune responses.
Several studies have demonstrated the potential to allow insertion or substitution of small fragments of foreign peptides of C terminal of PCV2 Cap without apparent negative effect on formation of VLPs.19,37 In the present study, different DC-binding peptides were introduced into C-terminal of PCV2 Cap, and fusion proteins were expressed in soluble form. After one-step Ni-NTA affinity purification, Cap-DCbp fusion proteins form highly organized VLPs with similar morphology. These findings were consistent with previous results. For an effective vaccine, one of the important points should be noted is stability, because intact VLPs were closely related to the immunogenicity and effective protection in both experimental and field conditions.38 We found that Cap VLPs could maintain 61.5% and 52.9% of the starting concentration after storage at 4°C for 3 months and 6 months, respectively, without apparent change in VLP morphology and homogeneity. These results revealed that Cap-DCbp VLPs could retain stable and functional forms after long-term storage, making them suitable for vaccination purposes.
DCs are professional antigen-presenting cells (APCs) and key modulators of adaptive immunity mainly owing to their superior ability to take up and present antigens (Ags).39 To increase the bioavailability and antigen presentation of the Cap VLPs, DC-targeting peptides were screened in this study. Highly purified BM-DCs were prepared from mice.40,41 Using this model, we found that DCbp3 exhibited a distinctly higher targeting efficiency, and Cap-DCbp3 VLPs were efficiently internalized by BM-DCs and mainly located in the cytoplasm. It is well accepted that, upon exposure to microbial stimuli or Ags, immature DCs phagocytose Ags and undergo a maturation process characterized by the increased expression of co-stimulatory molecules, the production of pro-inflammatory cytokines and the presentation of Ags to T cells.39,42 In this study, significantly higher expression levels of surface molecules MHC II, co-stimulatory molecules CD80 and CD86 were observed in Cap-DCbp3 VLPs compared to other VLPs, which likely contributed to the fact that Cap-DCbp3 VLPs with multiple copies of DCbp3 on the surface resulted in more efficient cell entry and internalization. This was consistent with the results in previous studies, indicating that antigen delivery via specific ScFv enhances antigen internalization and up-regulates the expression of surface molecules and co-stimulatory molecules in DCs.43 As a result, Cap-DCbp3 VLPs also significantly increased the level of IL-6 following internalization. As IL-6 can activate naive CD4+ T cells and accelerate the differentiation of CD4+ cells into Th1 and Th2 in lymphoid tissues,44,45 Cap-DCbp3 VLPs also initiate an innate immune response after entering BM-DCs with high efficiency. Based on these results, it can be concluded that DCbp3 significantly enhances antigen presentation and initiates subsequent immune response via up-regulating internalization, serving as an ideal small molecule adjuvant for the development of subunit vaccines.
After immunization in a mouse model, it was found that the efficient internalization of Cap-DCbp3 indeed correlated with a more effective humoral immune response, especially a significant increase in IgG1 and IgG2a. Of note, as compared to PC, Cap-DCbp3 VLPs triggered dramatically stronger or comparable IgG2a immune responses at 35 dpi and 45 dpi, respectively. IgG2a production suggests a predominant Th1 type response, which plays an indispensable role in protective efficiency against viral infection.46 Therefore, it can be concluded that Cap-DCbp3 VLPs have great potential to enhance Th1 response and further improve protection.
Neutralizing antibodies are an important indicator of the quality of vaccines. Generally, neutralizing antibodies can bind to antigens on the surface of pathogenic microorganisms, preventing adherence to target cell receptors and cell invasion.47 Compared with Cap-DCbpcon VLPs, Cap-DCbp3 VLPs can increase the level of PCV2-specific neutralizing antibodies by 2.5 times. Importantly, they can reach equivalent or even higher levels of neutralizing antibodies with commercial vaccines.
The level of cellular immune response is crucial for viral vaccines. Previous studies have confirmed that the DC targeting strategy can significantly increase the level of cellular immunity induced by anti-tumor vaccines via up-regulated cellular immunity-related cytokines and CTL responses.48 Lymphocytes are an important cellular component of the immune response and the main executor of almost all immune functions of the lymphatic system. After recognizing a specific antigen, T cells expand clonally, proliferate, migrate to specific sites, differentiate and acquire effector functions such as the ability to kill infected target cells, and synchronously secrete cytokines to coordinate immune responses.49 To explore whether elevated cellular immunity via DC-binding peptides occurs, we have validated both immune cells and cytokines. Proliferation is one of the effector functions of T lymphocytes, and proliferation assays are reliable, simple, and easy to perform, and have been widely used to assess the overall immune competence of T cells.50 As shown in Figure 3, the level of stimulated lymphocyte proliferation is an important index of cellular immunity. DCbp3 targeting vaccine induced a high level of proliferation when splenocytes from Cap-DCbp3 vaccinated mice were exposed to cap antigen. In cellular immunity, CD4 T cells can differentiate into Th1 or Th2 types based on the regulation of different immune factors participating in different immune pathways. CD8 T cells usually differentiate into cytotoxic T lymphocytes after activation and specifically kill target cells. The production of the cytokine IFN-γ is mediated by Th1 cells involved in mediating intracellular killing of multiple infectious pathogens of cellular immune responses,51,52 while IL-10 is mediated by Th2 cells associated with humoral immune responses. In a recent study, IL-10 increased granzyme B expression in Th2 cells and led to increased Th2 cell death, which provided robust evidence that IL-10 has direct effects on Th2 cells differentiation, regulating the survival of Th2 cells and severity of Th2-mediated allergic airway inflammation.53 IL-2 is a potent immunostimulatory molecule that plays a key role in T and NK cell activation and expansion; activated naive CD8 T cells expanded by IL-2 immunocomplexes are able to establish a robust population of functional memory cells.54,55 Based on these researches, the relative cytokine secretion levels of IL-2, IL-10 and IFN-γ were determined as shown in Figure 7. Results prove that mice with Cap-DCbp3 VLPs triggered high levels of cytokines, including IL-2, IL-10 and IFN-γ. These results indicate that vaccination with Cap-DCbp3 VLPs was more efficient in eliciting cellular immune response. On this basis, a DC-targeting strategy was believed to effectively elicit a robust cellular immune response.
DCs possess a broad spectrum of cell surface receptors involved in the initiation, promotion and execution of immune responses.56 Receptors including TLR, scavenger receptors and C-type lectin receptors are important pattern recognition receptors (PRR), recognizing PAMP on pathogens and, indeed, antigens, adjuvants and vaccines essential for the development of innate immune responses and defense.57 With PAMP binding to PRR on the DC surface, they also offer potential as targets for improved vaccine delivery. Recently, many studies have focused on DC-binding peptides for targeting delivery, mainly by selecting phage displayed peptide library towards in vitro isolated DCs. This strategy has demonstrated that DC-binding peptide-based vaccines facilitated antigen presentation to enhance antigen-specific responses,58 but these may induce side effects by targeting undefined receptors because various surface markers on DCs play multiple roles in immune system and cell life cycle. Therefore, it is necessary to select specific ligands for more reasonable DC targeting vaccine design. Phage display and combinatorial peptide design strategies proposed in previous studies59,60 could provide a series of targeting ligands, based on their affinity with the extracellular domain of specific DC markers as a bait. These more defined peptides would accelerate DC-targeting vaccines in clinical applications.
Conclusion
In conclusion, we have developed a targeted antigen delivery method by utilizing the specificity and high affinity binding properties of DC-binding peptides targeting DCs. DC-binding peptides were chosen over the whole antibody or ScFv molecules for antigen targeting since the former offer a small size and thus improve efficiency in crossing biological barriers. Here, VLP-based nanovaccine of PCV2 was decorated with Dcbp3. This strategy using DCbp3 targeting led to enhanced VLP antigen uptake and increased expression of antigen presentation-related factors in BM-DCs. Comparatively, vaccination of mice with DC targeting vaccine (Cap-DCbp3 VLPs) elicited higher specific antibodies (IgG, IgG1 and IgG2a) and neutralizing antibodies compared to the untargeted vaccine (Cap-DCbpcon VLPs or wild-type Cap VLPs). The higher immune stimulating ability of DCbp3 was also reflected in cellular immunity, whereby DCbp3-based targeting vaccine induced elevated ex-vivo splenocyte stimulation index and higher levels of cytokines (IFN-γ, IL-2 and IL-10) compared to untargeted vaccine. Overall, the findings vindicate the use of DC-binding peptides as a powerful tool to further boost antigen presentation in DCs, which has implications for efficacy enhancement applicable to VLP-based and nanoparticle-based vaccines against emerging diseases.
Acknowledgments
This work was supported by 100-Talent Program of Zhejiang University. We appreciated the technical support for the microscopy from the core facilities, College of Animal Science, Zhejiang University.
Disclosure
The authors declare no competing financial interests and no conflicts of interest for this work.
References
1. Owen JL, Sahay B, Mohamadzadeh M. New generation of oral mucosal vaccines targeting dendritic cells. Curr Opin Chem Biol. 2013;17(6):918–924. doi:10.1016/j.cbpa.2013.06.013
2. Moll H. Dendritic cells and host resistance to infection. Cell Microbiol. 2003;5(8):493–500. doi:10.1046/j.1462-5822.2003.00291.x
3. Demangel C, Zhou J, Choo AB, Shoebridge G, Halliday GM, Britton WJ. Single chain antibody fragments for the selective targeting of antigens to dendritic cells. Mol Immunol. 2005;42(8):979–985. doi:10.1016/j.molimm.2004.09.034
4. Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7(10):790–802. doi:10.1038/nri2173
5. Caminschi I, Shortman K. Boosting antibody responses by targeting antigens to dendritic cells. Trends Immunol. 2012;33(2):71–77. doi:10.1016/j.it.2011.10.007
6. Apostolopoulos V, Thalhammer T, Tzakos AG, Stojanovska L. Targeting antigens to dendritic cell receptors for vaccine development. J Drug Deliv. 2013;2013:869718. doi:10.1155/2013/869718
7. Keler T, He L, Ramakrishna V, Champion B. Antibody-targeted vaccines. Oncogene. 2007;26(25):3758–3767. doi:10.1038/sj.onc.1210375
8. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. doi:10.1038/nrc3258
9. Cruz LJ, Tacken PJ, Pots JM, Torensma R, Buschow SI, Figdor CG. Comparison of antibodies and carbohydrates to target vaccines to human dendritic cells via DC-SIGN. Biomaterials. 2012;33(16):4229–4239. doi:10.1016/j.biomaterials.2012.02.036
10. Chen P, Liu X, Sun Y, Zhou P, Wang Y, Zhang Y. Dendritic cell targeted vaccines: recent progresses and challenges. Hum Vaccin Immunother. 2016;12(3):612–622. doi:10.1080/21645515.2015.1105415
11. Vives E, Schmidt J, Pelegrin A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim Biophys Acta. 2008;1786(2):126–138. doi:10.1016/j.bbcan.2008.03.001
12. Mohamadzadeh M, Duong T, Sandwick SJ, Hoover T, Klaenhammer TR. Dendritic cell targeting of Bacillus anthracis protective antigen expressed by Lactobacillus acidophilus protects mice from lethal challenge. Proc Natl Acad Sci U S A. 2009;106(11):4331–4336. doi:10.1073/pnas.0900029106
13. Dunachie SJ, Hill AV. Prime-boost strategies for malaria vaccine development. J Exp Biol. 2003;206(Pt 21):3771–3779. doi:10.1242/jeb.00642
14. Wang YP, Liu D, Guo LJ, et al. Enhanced protective immune response to PCV2 subunit vaccine by co-administration of recombinant porcine IFN-gamma in mice. Vaccine. 2013;31(5):833–838. doi:10.1016/j.vaccine.2012.11.062
15. Mao Q, Zhang W, Ma S, et al. Fusion expression and immune effect of PCV2 cap protein tandem multiantigen epitopes with CD154/GM-CSF. Vet Sci. 2021;8(10). doi:10.3390/vetsci8100211
16. Chang XX, Fan K, Meng W, et al. Truncated diphtheria toxin DT390 enhances the humoral immunogenicity of porcine circovirus Type 2 capsid antigen in mice. Viral Immunol. 2021;34(7):448–456. doi:10.1089/vim.2020.0339
17. Wu PC, Lin WL, Wu CM, Chi JN, Chien MS, Huang C. Characterization of porcine circovirus type 2 (PCV2) capsid particle assembly and its application to virus-like particle vaccine development. Appl Microbiol Biotechnol. 2012;95(6):1501–1507. doi:10.1007/s00253-012-4015-2
18. Xi X, Mo X, Xiao Y, et al. Production of Escherichia coli-based virus-like particle vaccine against porcine circovirus type 2 challenge in piglets: structure characterization and protective efficacy validation. J Biotechnol. 2016;223:8–12. doi:10.1016/j.jbiotec.2016.02.025
19. Liu X, Liu Y, Zhang Y, Zhang F, Du E. Incorporation of a truncated form of flagellin (TFlg) into porcine circovirus type 2 virus-like particles enhances immune responses in mice. BMC Vet Res. 2020;16(1):45. doi:10.1186/s12917-020-2253-6
20. Li W, Wang X, Bai J, et al. Construction and immunogenicity of recombinant porcine circovirus-like particles displaying somatostatin. Vet Microbiol. 2013;163(1–2):23–32. doi:10.1016/j.vetmic.2012.11.045
21. Mohsen MO, Zha L, Cabral-Miranda G, Bachmann MF. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin Immunol. 2017;34:123–132. doi:10.1016/j.smim.2017.08.014
22. Silva AL, Rosalia RA, Varypataki E, Sibuea S, Ossendorp F, Jiskoot W. Poly-(lactic-co-glycolic-acid)-based particulate vaccines: particle uptake by dendritic cells is a key parameter for immune activation. Vaccine. 2015;33(7):847–854. doi:10.1016/j.vaccine.2014.12.059
23. Li X, Meng X, Wang S, et al. Virus-like particles of recombinant PCV2b carrying FMDV-VP1 epitopes induce both anti-PCV and anti-FMDV antibody responses. Appl Microbiol Biotechnol. 2018;102(24):10541–10550. doi:10.1007/s00253-018-9361-2
24. Curiel TJ, Morris C, Brumlik M, et al. Peptides identified through phage display direct immunogenic antigen to dendritic cells. J Immunol. 2004;172(12):7425–7431. doi:10.4049/jimmunol.172.12.7425
25. Ye C, Choi JG, Abraham S, Shankar P, Manjunath N. Targeting DNA vaccines to myeloid cells using a small peptide. Eur J Immunol. 2015;45(1):82–88. doi:10.1002/eji.201445010
26. Ye C, Choi JG, Abraham S, et al. Human macrophage and dendritic cell-specific silencing of high-mobility group protein B1 ameliorates sepsis in a humanized mouse model. Proc Natl Acad Sci U S A. 2012;109(51):21052–21057. doi:10.1073/pnas.1216195109
27. Liu ZH, Xu HL, Han GW, et al. Self-assembling nanovaccine enhances protective efficacy against CSFV in pigs. Front Immunol. 2021;12:689187. doi:10.3389/fimmu.2021.689187
28. Marcekova Z, Psikal I, Kosinova E, Benada O, Sebo P, Bumba L. Heterologous expression of full-length capsid protein of porcine circovirus 2 in Escherichia coli and its potential use for detection of antibodies. J Virol Methods. 2009;162(1–2):133–141. doi:10.1016/j.jviromet.2009.07.028
29. Fort M, Olvera A, Sibila M, Segales J, Mateu E. Detection of neutralizing antibodies in postweaning multisystemic wasting syndrome (PMWS)-affected and non-PMWS-affected pigs. Vet Microbiol. 2007;125(3–4):244–255. doi:10.1016/j.vetmic.2007.06.004
30. Meng XJ. Porcine circovirus type 2 (PCV2): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci. 2013;1:43–64. doi:10.1146/annurev-animal-031412-103720
31. Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27(1):74–95. doi:10.1038/cr.2016.157
32. Ueno H, Klechevsky E, Schmitt N, et al. Targeting human dendritic cell subsets for improved vaccines. Semin Immunol. 2011;23(1):21–27. doi:10.1016/j.smim.2011.01.004
33. Sehgal K, Dhodapkar KM, Dhodapkar MV. Targeting human dendritic cells in situ to improve vaccines. Immunol Lett. 2014;162(1Pt A):59–67. doi:10.1016/j.imlet.2014.07.004
34. Dubensky TJ, Reed SG. Adjuvants for cancer vaccines. Semin Immunol. 2010;22(3):155–161. doi:10.1016/j.smim.2010.04.007
35. Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33(4):492–503. doi:10.1016/j.immuni.2010.10.002
36. McCullough KC, Summerfield A. Targeting the porcine immune system–particulate vaccines in the 21st century. Dev Comp Immunol. 2009;33(3):394–409. doi:10.1016/j.dci.2008.07.015
37. Ding P, Zhang G, Chen Y, et al. Reasonable permutation of M2e enhances the effect of universal influenza nanovaccine. Int J Biol Macromol. 2021;173:244–250. doi:10.1016/j.ijbiomac.2021.01.132
38. Wang N, Zhang Y, Lei X, et al. Optimized conditions for preserving stability and integrity of porcine circovirus type2 virus-like particles during long-term storage. J Virol Methods. 2017;243:146–150. doi:10.1016/j.jviromet.2017.01.021
39. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi:10.1038/32588
40. Liu ZH, Xu HL, Han GW, et al. A self-assembling nanoparticle: implications for the development of thermostable vaccine candidates. Int J Biol Macromol. 2021;183:2162–2173. doi:10.1016/j.ijbiomac.2021.06.024
41. Chiang CY, Chen YJ, Wu CC, Liu SJ, Leng CH, Chen HW. Efficient uptake of recombinant lipidated survivin by antigen-presenting cells initiates antigen cross-presentation and antitumor immunity. Front Immunol. 2018;9:822. doi:10.3389/fimmu.2018.00822
42. Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5(4):296–306. doi:10.1038/nri1592
43. Lakhrif Z, Moreau A, Herault B, et al. Targeted delivery of toxoplasma gondii antigens to dendritic cells promote immunogenicity and protective efficiency against toxoplasmosis. Front Immunol. 2018;9:317. doi:10.3389/fimmu.2018.00317
44. Yang J, Sakai J, Siddiqui S, et al. IL-6 impairs vaccine responses in neonatal mice. Front Immunol. 2018;9:3049. doi:10.3389/fimmu.2018.03049
45. Ballesteros-Tato A, Randall TD. Priming of T follicular helper cells by dendritic cells. Immunol Cell Biol. 2014;92(1):22–27. doi:10.1038/icb.2013.62
46. Hannestad K, Scott H. A nonadjuvanted IgG2a monoclonal antibody against nucleosomes elicits potent T cell-dependent, idiotype-specific IgG1 responses and glomerular IgG1/IgG2a deposits in normal mice. J Immunol. 2017;199(2):489–500. doi:10.4049/jimmunol.1600718
47. Ali MG, Zhang Z, Gao Q, Pan M, Rowan EG, Zhang J. Recent advances in therapeutic applications of neutralizing antibodies for virus infections: an overview. Immunol Res. 2020;68(6):325–339. doi:10.1007/s12026-020-09159-z
48. Fu C, Zhou L, Mi QS, Jiang A. DC-based vaccines for cancer immunotherapy. Vaccines. 2020;8(4). doi:10.3390/vaccines8040706
49. Dimeloe S, Burgener AV, Grahlert J, Hess C. T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology. 2017;150(1):35–44. doi:10.1111/imm.12655
50. Heinzel S, Marchingo JM, Horton MB, Hodgkin PD. The regulation of lymphocyte activation and proliferation. Curr Opin Immunol. 2018;51:32–38. doi:10.1016/j.coi.2018.01.002
51. Xu X, Huang H, Wang Q, et al. IFN-gamma-producing Th1-like regulatory T cells may limit acute cellular renal allograft rejection: paradoxical post-transplantation effects of IFN-gamma. Immunobiology. 2017;222(2):280–290. doi:10.1016/j.imbio.2016.09.012
52. Zhang Y, Zhang Y, Gu W, Sun B. Erratum to: th1/Th2 cell differentiation and molecular signals. Adv Exp Med Biol. 2014;841:E1–2.
53. Coomes SM, Kannan Y, Pelly VS, et al. CD4(+) Th2 cells are directly regulated by IL-10 during allergic airway inflammation. Mucosal Immunol. 2017;10(1):150–161. doi:10.1038/mi.2016.47
54. Tomala J, Chmelova H, Mrkvan T, Rihova B, Kovar M. In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J Immunol. 2009;183(8):4904–4912. doi:10.4049/jimmunol.0900284
55. Becker JC, Pancook JD, Gillies SD, Furukawa K, Reisfeld RA. T cell-mediated eradication of murine metastatic melanoma induced by targeted interleukin 2 therapy. J Exp Med. 1996;183(5):2361–2366. doi:10.1084/jem.183.5.2361
56. Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111(7):927–930. doi:10.1016/S0092-8674(02)01201-1
57. Pulendran B. Modulating vaccine responses with dendritic cells and Toll-like receptors. Immunol Rev. 2004;199:227–250. doi:10.1111/j.0105-2896.2004.00144.x
58. Ma S, Qiao X, Xu Y, et al. Screening and identification of a chicken dendritic cell binding peptide by using a phage display library. Front Immunol. 2019;10:1853. doi:10.3389/fimmu.2019.01853
59. Agnew HD, Coppock MB, Idso MN, et al. Protein-Catalyzed Capture Agents. Chem Rev. 2019;119(17):9950–9970. doi:10.1021/acs.chemrev.8b00660
60. Chan KH, Lim J, Jee JE, Aw JH, Lee SS. Peptide-peptide co-assembly: a design strategy for functional detection of C-peptide, A biomarker of diabetic neuropathy. Int J Mol Sci. 2020;21(24):9671. doi:10.3390/ijms21249671
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.