")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Targeted delivery of chemically modified anti-miR-221 to hepatocellular carcinoma with negatively charged liposomes
Authors Zhang W, Peng F, Zhou T, Huang Y, Zhang L, Ye P, Lu M, Yang G, Gai Y, Yang T, Ma X, Xiang G
Received 19 December 2014
Accepted for publication 30 April 2015
Published 29 July 2015 Volume 2015:10(1) Pages 4825—4836
DOI https://doi.org/10.2147/IJN.S79598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Wendian Zhang,1 Fangqi Peng,1 Taotao Zhou,1 Yifei Huang,2 Li Zhang,3 Peng Ye,4 Miao Lu,1 Guang Yang,5 Yongkang Gai,1 Tan Yang,1 Xiang Ma,1 Guangya Xiang1
1School of Pharmacy, Tongji Medical College, 2Department of Pharmacy, 3Department of Ultrasound, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 4Department of Pharmacy, Wuhan University, Renmin Hospital, 5School of Medicine, Jianghan University, Wuhan, People’s Republic of China
Abstract: Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related death. Gene therapy was established as a new strategy for treating HCC. To explore the potential delivery system to support the gene therapy of HCC, negatively charged liposomal delivery system was used to deliver miR-221 antisense oligonucleotide (anti-miR-221) to the transferrin (Tf) receptor over expressed HepG2 cells. The liposome exhibited a mean particle size of 122.5 nm, zeta potential of -15.74 mV, anti-miR-221 encapsulation efficiency of 70%, and excellent colloidal stability at 4°C. Anti-miR-221-encapsulated Tf-targeted liposome demonstrated a 15-fold higher delivery efficiency compared to nontargeted liposome in HepG2 cells in vitro. Anti-miR-221 Tf-targeted liposome effectively delivered anti-miR-221 to HepG2 cells, upregulated miR-221 target genes PTEN, P27kip1, and TIMP3, and exhibited greater silencing efficiency over nontargeted anti-miR-221 liposome. After intravenous injection into HepG2 tumor-bearing xenografted mice with Cy3-labeled anti-miR-221 Tf-targeted liposome, Cy3-anti-miR-221 was successfully delivered to the tumor site and increased the expressions of PTEN, P27kip1, and TIMP3. Our results demonstrate that the Tf-targeted negatively charged liposome could be a potential therapeutic modality in the gene therapy of human HCC.
Keywords: transferrin, gene, HCC, target delivery system, anionic liposome
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors worldwide. The morbidity of HCC was ranked the fourth among all types of cancers, and it is the second leading cause of cancer-related death in People’s Republic of China.1 Over the years, chemotherapy remained the most used systemic approach for the treatment of HCC. However, the conventional chemotherapy has not been demonstrated to prolong survival rate of patients with locally advanced or metastatic HCC. Sorafenib, an oral multiple kinase inhibitor, was developed recently as the only molecular targeting medicine approved for advanced HCC. However, sorafenib was beneficial to only about 30% of advanced HCC patients. And most patients, who initially respond to sorafenib, would develop tumor recurrence and progression after a few months of sorafenib therapy, and no alternative is available after the sorafenib failure.2 The lack of survival benefits of treatment with conventional drugs and sorafenib makes the searching of new agents and novel therapeutic strategies an urgent need.3
In the past decades, studies have focused on investigating the genes and proteins underlying the development of HCC.4,5 Many reports have described a new class of small regulatory RNA molecules termed microRNAs (miRNAs) that were implicated in HCC development and progression.6 miRNA is a kind of noncoding RNA that can regulate the expression of target gene by translational repression through the RNA-induced silencing complex. Many studies have shown that miRNAs expression was dysregulated in HCC and played an important role in tumor development and progression. To name a few, miR-26a exhibited lower expression in both human and murine liver tumors than in normal adult liver and could reverse HCC progression in a liver cancer mouse model.7 miR-105 expression was downregulated significantly in both HCC cell lines and clinical HCC tissues compared to normal human hepatocytes and adjacent noncancerous tissues, and it could suppress cell proliferation and inhibit PI3K/AKT signaling in HCC.8 The miR-545/374a cluster was overexpressed in hepatitis B virus related HCC and promoted tumorigenesis and tumor progression.9 The rapid progress in miRNA studies has led to translational studies for applying the miRNA-targeted therapy in cancer treatment. However, there are many challenges in developing miRNA-based therapeutics, including minimizing premature degradation in blood, achieving sufficient cellular uptake, enhancing tissue-specific delivery, and minimizing systemic toxicity and off-target effects.10 Chemical modification provided some solutions to increase the stability of the miRNA therapeutics. For example, a phosphodiester linkage was introduced at the sugar 2′-position to confer enhanced serum stability to RNA; phosphorothioate and 2′-OMe substitutions at three terminal nucleotides resulted in an increase in serum stability.11 While chemical modification cannot solve all those problems like cellular uptake efficiency and off-target effects, carrier systems were then applied. Carrier systems such as nanoparticles were able to protect oligonucleotide from premature degradation in blood and deliver RNA to its target sites and facilitate cellular uptake.10 Many kinds of delivery systems have been applied in gene therapy such as virus-based systems,12 liposome,13 inorganic nanoparticle,14 dendrimer,15 cationic micelle,16 and polymeric nanoparticle.17
Among all kinds of delivery systems, many investigators have demonstrated that liposome-based nanoparticle carrier systems may be a promising approach to deliver oligonucleotide with high degree of biocompatibility.18 Chen et al developed formulation of liposome-polycation-hyaluronic acid nanoparticle modified with a tumor-targeting human monoclonal antibody to systemically deliver small interfering RNA (siRNA) and miRNA into murine B16F10 melanoma model.19 Zhang et al reported a lactosylated gramicidin-containing lipid nanoparticle to deliver anti-miR-155 to HCC cells.20 Hatakeyama et al developed a pH-sensitive anti-miRNA oligonucleotide liposome to successfully reduce the level of miR-122 in mice.21
In the current study, a traditional negatively charged liposome, which was composed of hydrogenated soybean phospholipid (HSPC)/cholesterol (CHOL)/1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-methoxy(polyethylene glycol) (mPEG), was used to deliver miRNAs in an attempt to enhance the cellular uptake and the pharmacological effectiveness. The composition of the liposome was clinically proved safe, and it was also the formulation for the liposomal drug Doxil. We selected miR-221 antisense oligonucleotide (anti-miR-221), a potential tumor suppressor in HCC, as a candidate for miRNA delivery. miR-221 has been shown to be overexpressed in HCC, and correlated with cirrhosis, tumor size, and tumor stage.22 Most importantly, high-level expression of miR-221 was related with poor prognosis of HCC patients.23,24 In order to enhance the transfection efficiency of negatively charged liposomes, transferrin (Tf) was used as liposomes-targeting moiety.25,26 Tf, a well-known blood plasma protein, is responsible for the transport of iron into the cells by binding to the Tf receptor (TfR) on the cell membrane. Because TfRs are overexpressed in many HCC cells, TfR is expected to be a good cellular marker for delivery of miR-221 to the TfRs-elevated HCC cells.27 We demonstrated in this work that the encapsulation efficiency (EE) of the Tf-targeted anti-miR-221 liposome (Tf-RL) was high up to 70%, and the liposome can efficiently deliver the anti-miR-221 to produce the antitumor effect both in vitro and in vivo. Thus, Tf-RL could be a potential candidate for miRNA delivery system.
Materials and methods
Materials
HSPCs were purchased from Shanghai Advanced Vehicle Technology L.T.D. Co (Shanghai, People’s Republic of China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-mPEG2000) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-mPEG2000-NH2) was purchased from Laysan Bio, Inc (Arab, AL, USA). CHOL was obtained from J&K Chemical, Ltd. (Beijing, People’s Republic of China). 3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), Holo-Transferrin, and Sepharose CL-4B chromatograph media were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). N-Hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethyllaminopropyl) carbodiimide hydrochloride (EDC) were purchased from Gracia Chemical Technology Co. Ltd (Chengdu, People’s Republic of China). Annexin V–fluorescein isothiocyanate (FITC) apoptosis detection kit, micro BCA protein assay kit, and apoptosis and cell cycle analysis kit were purchased from Beyotime Biotechnology Co. Ltd (Wuhan, People’s Republic of China). All reagents were of analytical grade and were used without any further purification.
Anti-miR-221 was synthesized and purchased from RiboBio Ltd. (Guangzhou, People’s Republic of China). The sequence of anti-miR-221 used is as follows: 5′-GAAACCCAGACAGACAAUGUAGCU-3′. Oligonucleotide Cy3-anti-miR-221 with or without 5′-CHOL modification was chemically conjugated with a phosphorothioate linkage, 2′-OMe, and 3′-inverted Cy3.28
Preparation of liposome
Liposome was prepared by thin-film dispersion method, followed by polycarbonate extrusion. The lipid composition of nontargeted liposome was HSPC/CHOL/mPEG-DSPE at a molar ratio of 55:40:5. For Tf-RL, the composition was HSPC/CHOL/mPEG-DSPE/DSPE-mPEG2000-NH2 at molar ratios of 55:40:4:1. Briefly, the mixture of lipids was dissolved in CHCl3 and dried to a thin film in a round-bottomed flask on a rotary evaporator under reduced pressure for 1 hour at 37°C. The dried lipid mixture was then rehydrated in 2 mL of phosphate-buffered saline (PBS; pH 7.4) which contained anti-miR-221 at 60°C for 1 hour. The resulting suspension of vesicles was extruded through the 200 nm and 100 nm pore-size polycarbonate membranes at least five times using a Lipex extruder (Northern Lipids Inc., Vancouver, BC, Canada), and then purified by size exclusion chromatography on a Sepharose CL-4B column to remove free anti-miR-221.
Tf-conjugated liposomes were prepared by coupling Tf to drug-loaded liposome containing 1% NH2-mPEG2000-DSPE. Briefly, a solution of Tf in PBS was added to a stirring solution of liposome (containing 1% NH2-mPEG2000-DSPE) at the ratio of 100 μg Tf conjugate per micromole of total lipid. EDC and NHS were then added into the mixture, and the reaction was allowed to proceed at room temperature for 12 hours. Tf-conjugated liposomes were purified by a Sepharose CL-4B column to remove free Tf.
To calculate the binding efficiency of Tf-targeted liposome, we used calcein, a green fluorescent, as a model formulation. Calcein liposomes were prepared using the above-described procedures, except that the lipid film was hydrated in 50 mM calcein. The lipid compositions of Tf-targeted liposome loaded with calcein (Tf-CL) and nontargeted control liposome loaded with calcein (CL) were the same as the liposome containing anti-miR-221.
Characterization of liposome
The concentration of calcein in the liposome was determined by measuring absorbance at 492 nm on a ultraviolet–visible spectrometer (Shanghai Spectrum Instruments Co. Ltd, Shanghai, People’s Republic of China) after the liposome was lysed in 10% Triton X-100. The concentration of anti-miR-221 in the liposome was determined by measuring the fluorescence of Cy3 at 530/590 nm on a Synergy™ HT multidetection microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) after the liposome was lysed in 10% Triton X-100. Drug EE (%) was calculated using the following equation:
EE (%) = W1/W0×100 |
where W1 is the weight of encapsulated drug and W0 is the weight of feed drug.
The mean diameter and polydispersity index of liposomes were determined by a Nano Brook Zeta PALS (Brookhaven Instruments Corporation, Holtsville, NY, USA), based on the dynamic light-scattering principle technique. Briefly, liposomes were diluted to optimal concentration and measured at 25°C. The conjugating efficiency of Tf to NH2-mPEG2000-DSPE was determined by micro BCA protein assay kit.
Cells and animals
The human HCC cell line HepG2 was a generous gift from Professor Zhiping Zhang’s laboratory. HepG2 cells were grown in Dulbecco’s Modified Eagle’s Medium, supplemented with 10% (v/v) heat-inactivated fetal bovine serum, and maintained in a humidified atmosphere containing 5% CO2 at 37°C. Cells were maintained within their exponential growth phase.
Female Kunming mice were obtained from the animal care facilities of Huazhong University of Science and Technology (Wuhan, People’s Republic of China). Six-week-old female BALB/c-nu nude mice (Beijing HFK Bioscience Co. Ltd, Beijing, People’s Republic of China) were maintained in the animal care facilities of Huazhong University of Science and Technology at 22°C±2°C with a time-regulated period of light and provided with water and food ad libitum. The experiment protocol was approved by the ethical committee of animal experiment of Huazhong University of Science and Technology.
Cell-binding efficiency
Binding and internalization efficiency of the liposomes in HepG2 cells and Hep3B cells were examined by fluorescence microscope. Cells were seeded in six-well plates at the density of 5×104 cells per well. After 24 hours, they were treated with CL or Tf-CL, and Tf-CL with 100 μg/mL of free Tf. After 4 hours, cells were washed with PBS and then examined under an Olympus IX71 fluorescence microscope (PerkinElmer Inc., Waltham, MA, USA). For the quantitative analysis of cellular uptake after co-incubating cells with various liposomes for 4 hours in plates, cells were gently trypsinized and collected in tubes for flow cytometry detection using a BD-LSRFortessa™ flow cytometer (BD Biosciences, San Jose, CA, USA).
MTT assay
HepG2 cells were seeded in 96-well plates at a density of 5×103 cells per well. After 24 hours, cells were treated with various concentration of Tf-RL. After 72 hours of co-incubation, media were removed, and cells were washed twice with PBS, then replaced with 200 μL of medium containing 20 μL of MTT (5 mg/L), and cultured for an additional 4 hours at 37°C in a 5% CO2 atmosphere. After media were removed, the blue formazan was dissolved in 200 μL of dimethyl sulphoxide, and the optical density value was determined by the microplate reader (Multiskan MK3; Thermo Fisher Scientific, Atlanta, GA, USA) at 492 nm.
Apoptosis
HepG2 cells were seeded overnight in six-well plates at the density of 5×104 cells per well. They were treated with Tf-RL and nontargeted liposome containing anti-miR-221 (RL) of 200 nM. After 24 hours, cells were gently trypsinized and collected in tubes. Annexin-V–FITC (5 μL) and propidium iodide (PI) (10 μL) were added to the cells, and the stained cells were analyzed by flow cytometry, using a BD-LSRFortessa™ flow cytometer (BD Biosciences) within 1 hour.
Cell cycle
To determine the proportion of cells in each phases, we conducted cell cycle assay by flow cytometry. The HepG2 cells were seeded in six-well plates at the density of 5×105 cells per well. After 24 hours, cells were treated with various liposomal formulations for 24 hours. The cells were then gently trypsinized and washed with PBS, and fixed for 24 hours in 3 mL of 70% ethanol at 4°C. The fixed HepG2 cells were washed with PBS and incubated with RNase A (0.1 mg/mL) for 1 hour at 37°C. After stained with PI (0.1 mg/mL) for 30 minutes in the dark at 37°C, the DNA content of the cells was measured by BD-LSR cytometer (BD Biosciences), and the data were evaluated using the ModFit software.
In vitro gene-silencing study
The gene-silencing effects by anti-miR-221 liposome on miR-221 target genes P27kip1, PTEN, and TIMP3 were measured at both mRNA and protein levels. For the real-time polymerase chain reaction (RT-PCR) study, HepG2 cells were plated in six-well plates at a density of (5×106 cells per well. After 24 hours, the plated cells were incubated with Tf-RL and RL containing 200 nM of anti-miR-221 for 24 hours. Total mRNA was then extracted using the TRIzol® reagent (Invitrogen, Grand Island, NY, USA) and purified following the standard protocol. RT-PCR was conducted using SYBR Green I Master Mix and a LightCycler 2.0 (Roche Applied Science, Mannheim, Germany). The primer was obtained from Invitrogen Biotechnology Co., Ltd.
For Western blotting assay, lysate of whole cells was prepared at 48 hours after the incubation of cells with 200 nM of various liposomal formulations. Cells were added with lysis buffer on ice. Proteins in lysate, quantified by micro BCA protein assay kit as described by the manufacturer, were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 10% gels. Western blotting was performed using antibodies specific for P27kip1 (1:1,000), PETN (1:1,000), TIMP3 (1:1,000), and β-actin (1:2,500). Bands were visualized using a horseradish peroxidase-conjugated AffiniPure goat anti-rabbit IgG.
In vivo biodistribution
Female BALB/c-nu mice and female Kunming mice were given Cy3-anti-miR-221-containing Tf-targeted liposome (Tf-RL-Cy3-anti-miR-221) or nontargeted liposome (RL-Cy3-anti-miR-221) intravenously at a dose of 1 mg/kg. After 4 hours, mice were sacrificed and their tissues were harvested. Collected tissues were fixed in 4% paraformaldehyde for 12 hours at 4°C. Fluorescent Tf-RL-Cy3-anti-miR-221 and RL-Cy3-anti-miR-221 were used for measuring in vivo distribution in different tissues by IVIS imaging. Cy3 fluorescent signals of whole tissue were measured using Xenogen IVIS-200 optical in vivo imaging system (Caliper Life Sciences, Hopkinton, MA, USA).
In vivo gene-silencing study
Tf-RL and RL were given to female BALB/c-nu mice by intravenous (iv) injection at a dose of 1.2 mg/kg. After 48 hours, mice were anesthetized, and tumors were collected and immediately frozen in liquid nitrogen. RNA extraction and RT-PCR were performed as described earlier.
Statistical analysis
All data are shown as means ± standard deviation unless specifically noted. Student’s t-test or one-way analyses of variance was performed. A P-value <0.05 was considered to be statistically significant.
Results
Synthesis and characterization of liposomes
The particle size and zeta potential of RL and Tf-RL were evaluated by a Nano Brook Zeta PALS and are presented in Table 1. Mean size distributions of both kinds of liposomes were about 120 nm in a suitable homogeneity, and zeta potentials of RL and Tf-RL were −14.35 mV and −15.74 mV, respectively. The mean entrapment efficiency of anti-miR-221 modified with CHOL was about 70%, while the mean entrapment efficiency of non-modified anti-miR-221 was only 20%. Thus, CHOL-modified anti-miR-221 was selected for the following investigation. The liposomes showed good stability when stored in PBS at 4°C. The negative zeta potential of the formulations contributed to the stability because the negative charge could make the particles repel each other to prevent the interparticle aggregation; thus, the liposomes can be maintained for a long period of time. The conjugating efficiency of Tf to NH2-mPEG2000-DSPE is 60% which was determined by micro BCA protein assay kit.
 | Table 1 Particle size and zeta potential of liposomes at room temperature |
Cell-binding efficiency
In order to demonstrate that Tf-RL can be efficiently taken up by HepG2 cells via a Tf-mediated endocytosis, liposome containing a green fluorescent dye calcein was prepared, and the cellular uptake by HepG2 cells was quantitatively analyzed using flow cytometry and visualized by fluorescence microscopy. As shown in Figure 1B, the cellular uptake of Tf-CL was about 15 times higher than that of CL, and the uptake decreased when free Tf was added. Meanwhile, the fluorescence intensity of the Tf-CL group was significantly higher than that of the CL group (Figure 1A). The study indicates that Tf-targeted liposome can be more efficiently taken up by the Tf-positive cells than the nontargeted liposomes, and the Tf-mediated endocytosis can be blocked by free Tf (Figure 1). Tf-RL can also be efficiently taken up by Hep3B cells via a Tf-mediated endocytosis (Figure 1C).
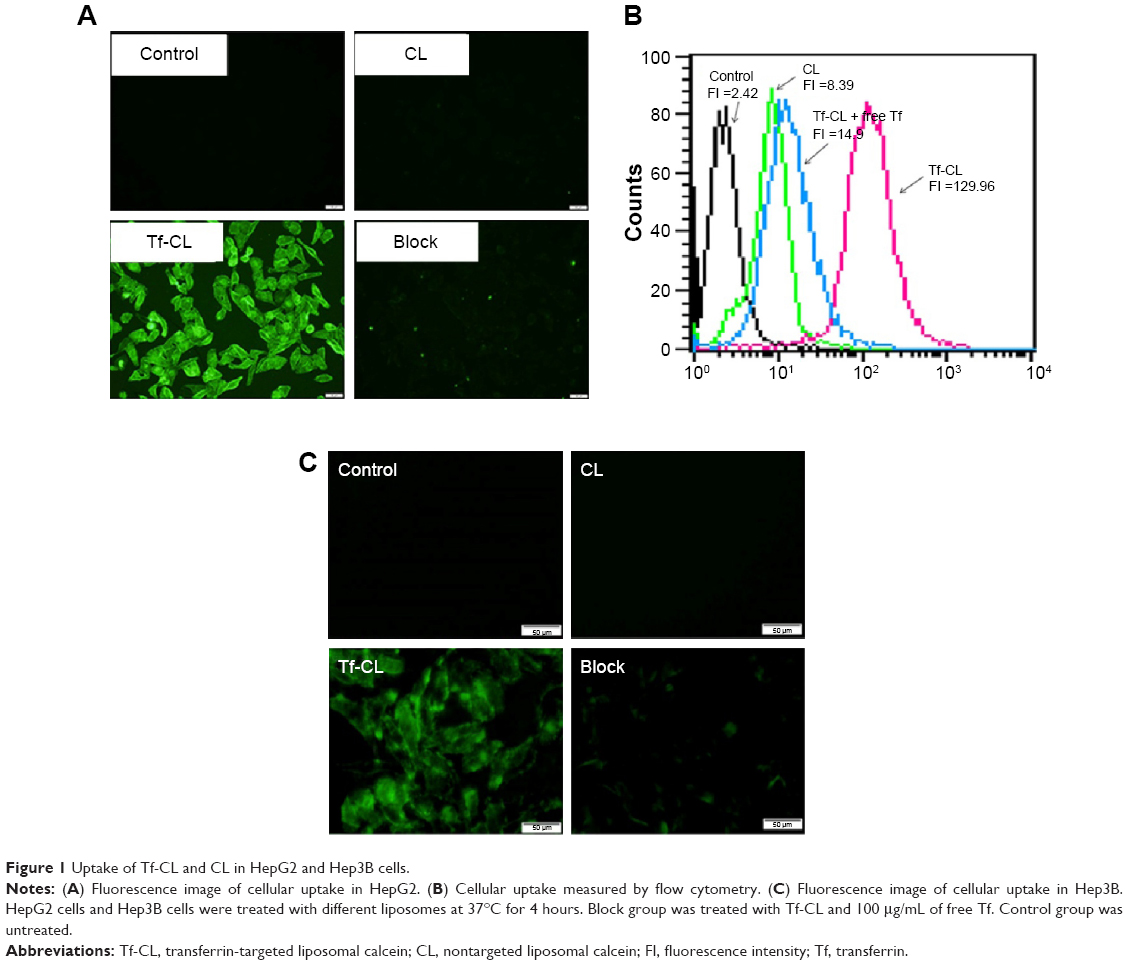 | Figure 1 Uptake of Tf-CL and CL in HepG2 and Hep3B cells. |
Flow cytometry and fluorescence microscopy were then used to study the cellular uptake of Tf-RL-Cy3-anti-miR-221 and RL-Cy3-anti-miR-221. As shown in Figure 2, the fluorescence signal of cells transfected with Tf-RL was much higher than that of those transfected with nontargeted liposomes. These results suggest that Tf-targeted liposomes were able to deliver anti-miR-221 more efficiently and thus provided better efficacy than nontargeted liposome in the HepG2 cells through the Tf-mediated endocytosis process.
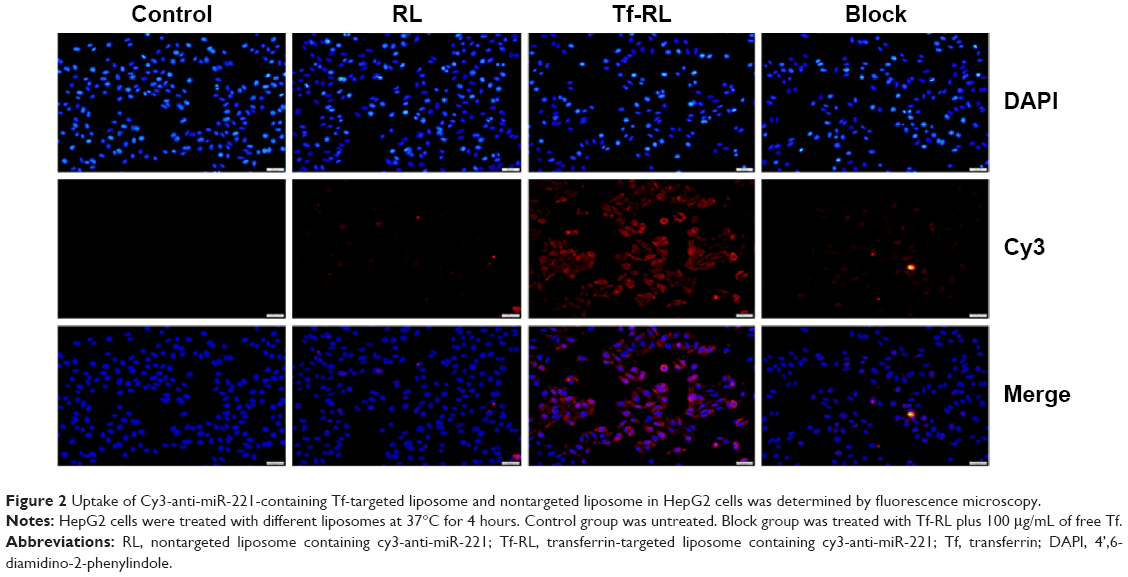 | Figure 2 Uptake of Cy3-anti-miR-221-containing Tf-targeted liposome and nontargeted liposome in HepG2 cells was determined by fluorescence microscopy. |
MTT assay
To determine to effect of Tf-RL on the proliferation of HepG2, MTT assay was performed on HepG2 cells. As shown in Figure 3, the cell survival rate of HepG2 cell was 80% when the concentration was up to 500 nM which is a really high concentration.
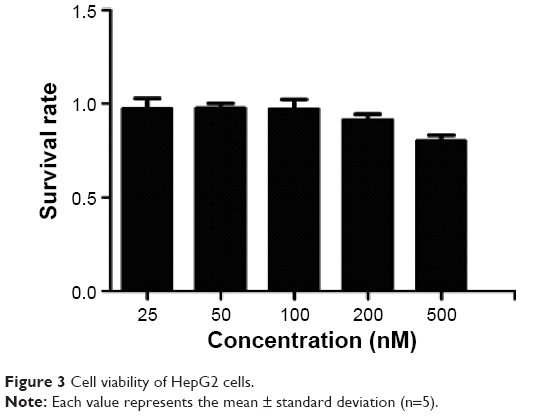 | Figure 3 Cell viability of HepG2 cells. |
Apoptosis
Annexin V–FITC/PI double staining was used for determining whether Tf-RL has greater apoptotic effects than RL has. The HepG2 cells were treated with blank control and both liposomal solutions for 24 hours, and then subjected to simultaneous staining and analyzed by flow cytometry. As shown in Figure 4A, the ratio of viable cells decreased and apoptotic cells increased when HepG2 cells were treated with both liposomes. The Tf-RL was more effective in activation of apoptosis of HepG2 cells (the percentage of apoptotic cells was 12.7%) than RL was (the percentage of apoptotic cells was 8.7%).
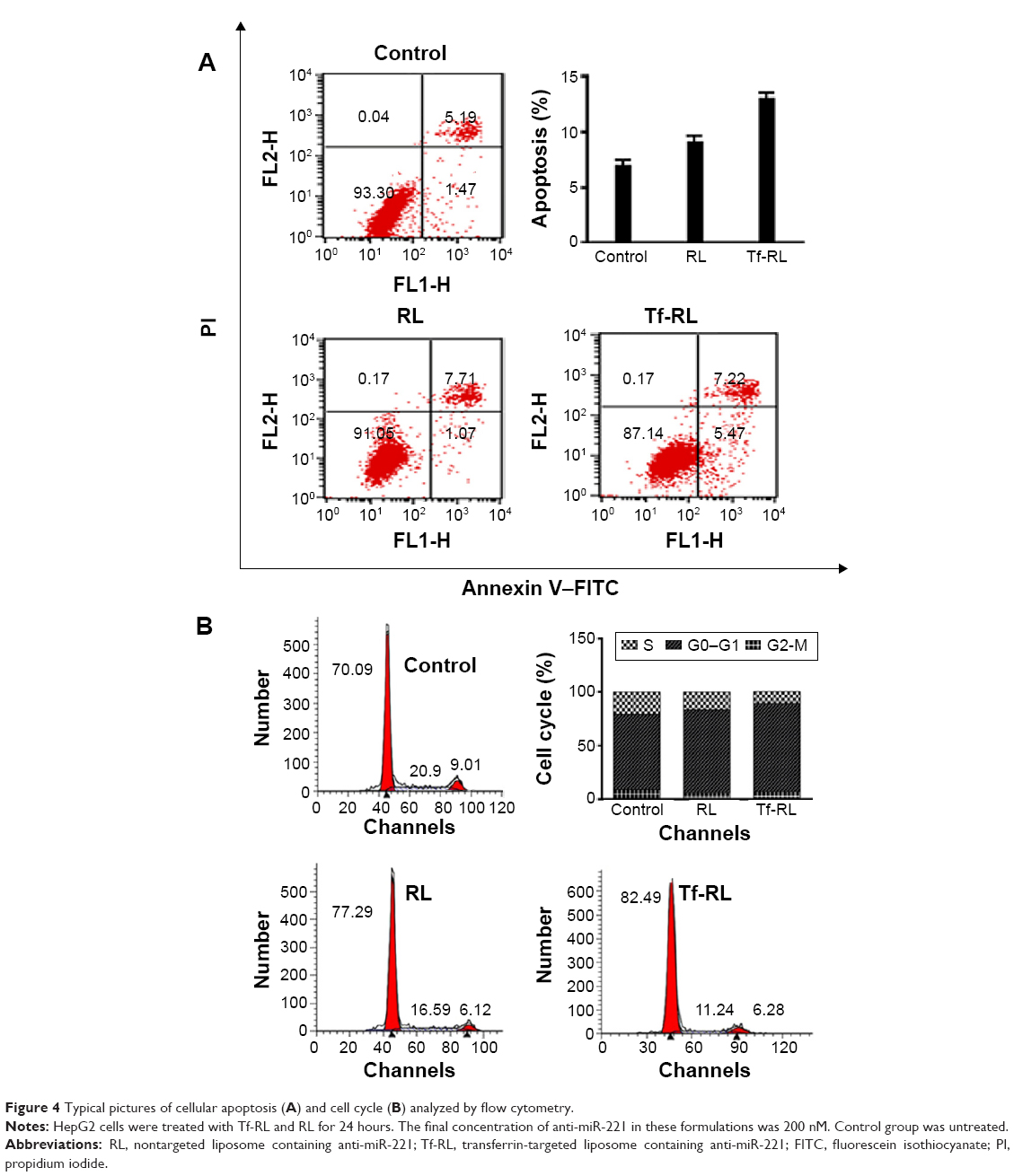 | Figure 4 Typical pictures of cellular apoptosis (A) and cell cycle (B) analyzed by flow cytometry. |
Cell cycle
The cell cycle distributions of HepG2 cells were determined by flow cytometry. As shown in Figure 4B, the percentages of the cells in G0–G1 and S phase were 70.09% and 20.9%, respectively, in untreated control group. However, RL and Tf-RL group treatments of the HepG2 cells induced differences in the proportions of G0–G1 (77.29% and 16.59%, respectively) and S phase (82.49% and 11.24%, respectively). These results demonstrate that inhibition of miR-221 expression by anti-miR-221 could cause more cells to be arrested in G0–G1 phase, and Tf-RL has more significant effect than RL has.
In vitro gene-silencing study
Before evaluating the efficiency of the presently described anti-miR-221 liposomes in silencing the expression levels of target gene and protein, we checked the in vitro toxicity of the blank liposome using MTT assay. The result showed that high percentage of viable cells (>85%) was observed with up to 25 mg/mL of lipids-incubated HepG2 cells for 72 hours.
The effects of Tf-RL on expression of downstream target genes PTEN, P27kip1, and TIMP3 were then evaluated in HepG2 cells using RT-PCR (Figure 5A) and Western blotting (Figure 5B). The expression of PTEN, P27kip1, and TIMP3 was increased when cells were transfected with both Tf-RL and RL. Significant change in target proteins expression was also observed when Tf-RL or RL was used. Both studies demonstrated that Tf-RL is more efficiently delivering the anti-miR-221 to the targets because of the higher silencing efficiency and protein expression. These results suggest posttranscriptional regulation of PTEN, P27kip1, and TIMP3 by anti-miR-221.
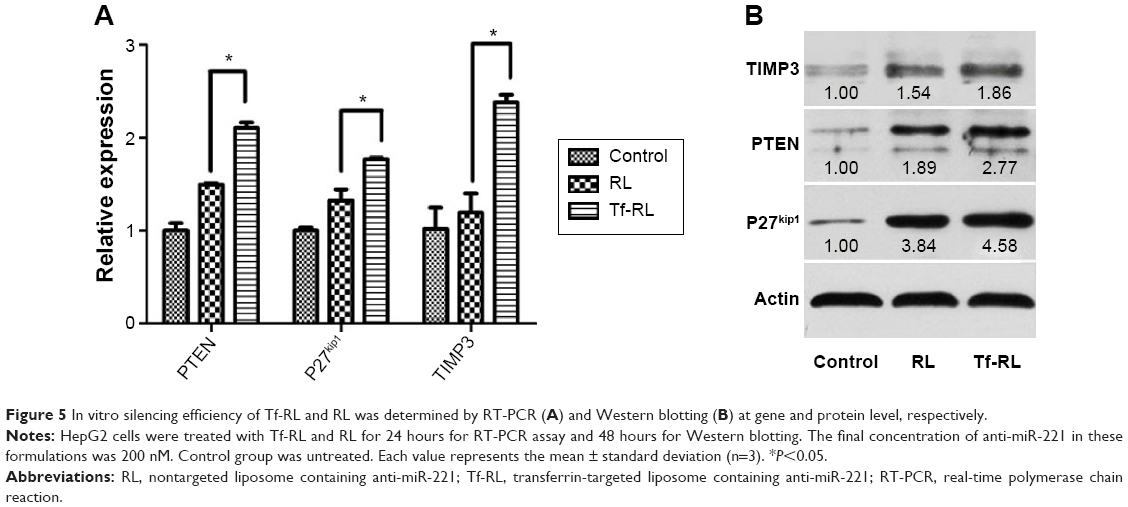 | Figure 5 In vitro silencing efficiency of Tf-RL and RL was determined by RT-PCR (A) and Western blotting (B) at gene and protein level, respectively. |
In vivo biodistribution
The biodistribution of Tf-RL and RL was studied in female Kunming mice and a xenograft model of HepG2 human liver cancer in female BALB/c-nu mice by using Cy3-labeled anti-miR-221. As shown in Figure 6, the biodistribution of RL and Tf-RL was almost the same. However, there was a significant difference between Tf-RL and RL in the xenograft model of HepG2 human liver cancer in female BALB/c-nu mice (Figure 7). The intensity of Tf-RL in liver was lower than RL was, while higher than RL in tumor. The result demonstrated that Tf-RL did change the biodistribution of anti-miR-221 in vivo, and accumulated more drug in tumor.
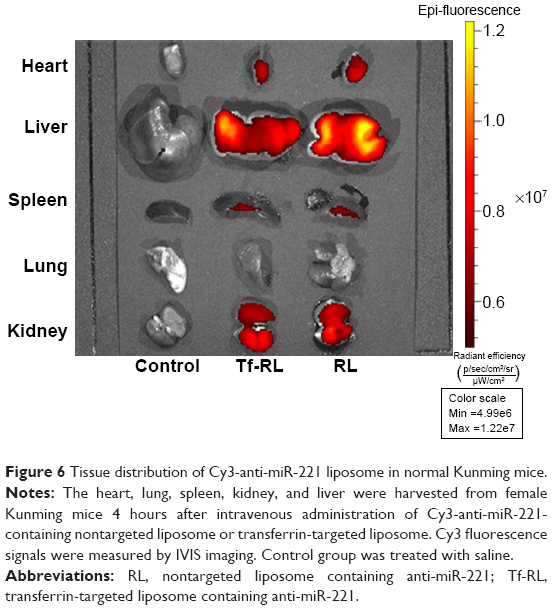 | Figure 6 Tissue distribution of Cy3-anti-miR-221 liposome in normal Kunming mice. |
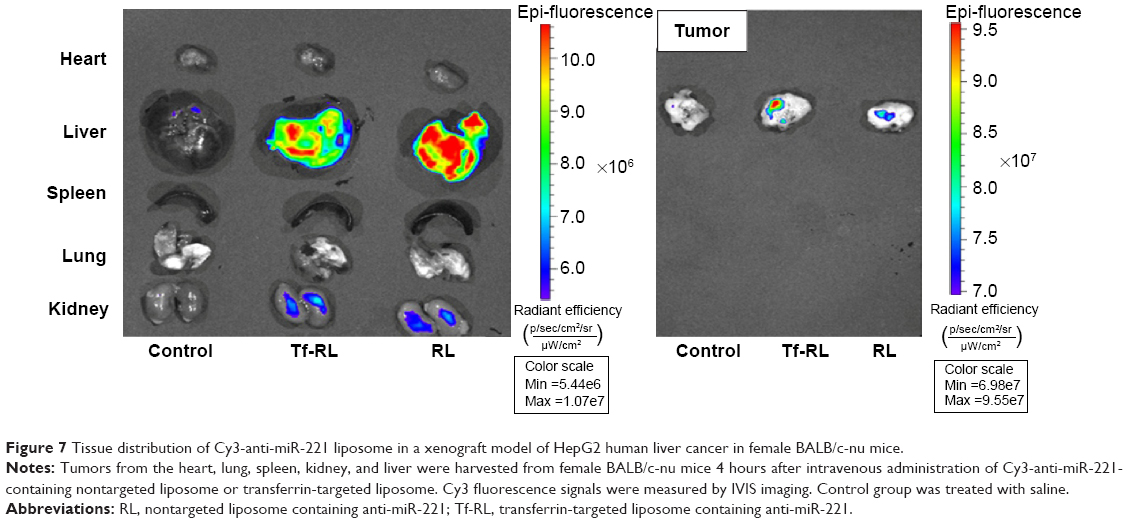 | Figure 7 Tissue distribution of Cy3-anti-miR-221 liposome in a xenograft model of HepG2 human liver cancer in female BALB/c-nu mice. |
In vivo gene-silencing study
In order to determine the delivery efficiency of Tf-RL in vivo, the expressions of target genes PTEN, P27kip1, and TIMP3 were evaluated in vivo by RT-PCR. Tf-RL and RL were given to each mouse at a single dose of 1.2 mg/kg through tail vein iv injection. After 48 hours postadministration, the mice were sacrificed and their tissues were harvested. The expressions of target genes PTEN, P27kip1, and TIMP3 were evaluated by RT-PCR (Figure 8). As expected, the expressions were increased in Tf-RL- and RL-treated groups, compared to the untreated group, while Tf-RL group had a higher increase than RL group. These data demonstrate superior delivery efficiency by Tf-RL, and corroborate the results of our in vitro experiments.
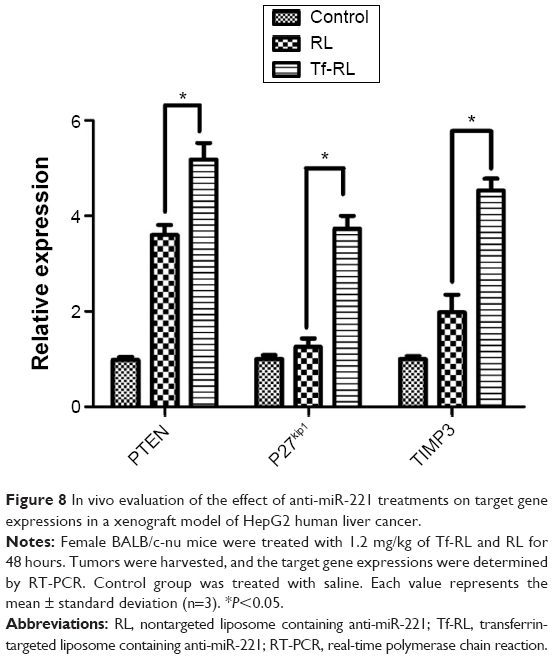 | Figure 8 In vivo evaluation of the effect of anti-miR-221 treatments on target gene expressions in a xenograft model of HepG2 human liver cancer. |
Discussion
Since Fire et al first discovered that the RNA interference (RNAi) could silence target gene expression in Caenorhabditis elegans in 1998,29 RNAi research which includes siRNA and miRNA has been an important and fast developing part in cancer treatment. Particularly, miRNAs are evolutionarily conserved small noncoding RNAs that regulate approximately up to one-third of genome. In liver cancer, miRNAs play important roles in tumor development, progression, and metastasis. Some of the specific miRNA expression signatures may serve as potential targets for tumor therapeutics,30 and there has been many research about miR-221 which proved that it could be one of the potential targets in the HCC.28,31,32 However, many challenges were behind in the development of miRNA-based drugs in tumor treatment, including tissue-specific delivery, efficient cellular uptake, and minimization of off-target effects in systemic administration of miRNA. Here, we developed a novel Tf-targeted drug delivery system of nanosized liposomes encapsulating anti-miR-221, an miRNA that directly targets miR-221, and characterized its properties and uptake, the targeting specificity, and delivery efficiency in vitro and in vivo.
We know that, miRNA has a negative charge on it due to the phosphate groups on the backbone of its structure. The negative charge makes it difficult to be entrapped into negatively charged liposomes. It is one of the reasons that limited the application of anionic liposome in RNA delivery. Negatively charged liposomes have been widely used in chemical drug delivery. For example, Doxil, the doxorubicin hydrochloride encapsulated in STEALTH® liposomes for iv administration, was approved by the US Food and Drug Administration in clinic. The STEALTH® liposomes have the formulation of HSPC/CHOL/DSPE-mPEG, which was exploited in our study for the miRNA delivery. In this liposome formulation, we used HSPC and CHOL as the basic structure composition, with the help of DSPE-mPEG2000 to make the liposome stable and long circulating in vivo. HSPC is a neutral lipid which has been used a lot in liposome preparation.33–35 CHOL was a common component in miRNA carriers, and introducing CHOL as a component of certain DNA/RNA carriers was reported to be a factor in improving gene transfection in vivo.36 In our study, CHOL played a dual role in the delivery of miRNA. Besides being incorporated in the liposomal formulation to facilitate cell fusion or endosomal internalization, the CHOL was conjugated to miRNA to act as a targeting entity and protector, improving cellular uptake and transfection and decreasing miRNA degradation.37 In addition, mPEG2000-DSPE was added to the formulation to increase the stability and circulation time of liposomes. PEG has long been used to modify the surface of liposomes for improving pharmacokinetics after iv administration and for minimizing protein binding to escape surveillance by the reticulendothelial system, which together with the diameter could help the liposomes to exhibit the enhanced permeability and retention effect. The lipid composition of nontargeted liposome was HSPC/CHOL/mPEG2000-DSPE at a molar ratio of 55:40:5. DSPE-mPEG2000-NH2 has been used as a linker for conjugating Tf.38–40 Using the passive entrapment procedure and thin-film dispersion method, CHOL-anti-miR-221 was encapsulated at an efficiency of 70% which is much higher than that of anti-miR-221 without CHOL modification (20%). In the preparation, Tf-RL had an average particle size of 122 nm with a narrow size distribution (low polydispersity index values). The uniform nanoscale size of the liposomes could give them a consistent kinetic profile in vivo. Meanwhile, both RL and Tf-RL possessed slight negative zeta potentials, which might be due to the presence of the negatively charged lipid mPEG2000-DSPE in the formulation, and this also helps the liposomes get a good physical stability against aggregation and fusion.35
The efficacy of liposome was further examined in vitro in HepG2 and Hep3B cell lines, which are the Tf receptor over-expressed HCC cell lines. As expected, our Tf-CL was more efficiently taken up by the HepG2 cells, which was confirmed by the CL uptake study. The uptake could be blocked by 100 μg of free Tf, suggesting that internalization of Tf-CL was specifically mediated by a TfR-mediated endocytosis process. The higher internalization efficiency demonstrated that liposomes conjugated with Tf could effectively target the HepG2 cells through the TfR, which had previously been proved by reports on different lipid compositions or different cell lines.39,41–43 The similar results can be seen in Hep3B cells. In vivo biodistribution of RNA liposomes was tested in normal Kunming female mice and female BALB/c-nu mice bearing HepG2 HCC tumor; the distribution Tf-RL almost has no difference with RL in normal Kunming mice. There was an obvious difference between Tf-RL and RL, and Tf-RL can accumulate more miRNA in tumors compared to RL in tumors-bearing mice, which suggest that the targeting of miRNA in vivo could be enhanced by drug carriers. Consistent with the cellular uptake test, TfR dependence was demonstrated in an apoptosis test. The results of the apoptosis test showed that Tf-RL can induce more apoptosis in HepG2 cells through active targeting, and cell cycle assay showed that growth of more cells was inhibited and blocked in G0–G1 phase in Tf-RL group compared to RL group.
Tf-RL exhibited an efficient silencing efficiency in HepG2 cells. In PCR and Western blotting assay, Tf-RL can upregulate the expression of PTEN, P27kip1, and TIMP3 at gene and protein level more efficiently; among them, PTEN is involved in the regulation of the cell cycle, preventing cells from growing and dividing too rapidly, and functions as a tumor suppressor by negatively regulating Akt/PKB signaling pathway.23 P27kip1 is cyclin-dependent kinase (CDK) inhibitor 1B. It is often referred to as a cell cycle inhibitor protein because its major function is to stop or slow down the cell division cycle. It encodes a protein which belongs to the Cip/Kip family of CDK inhibitor proteins. The encoded protein binds to and prevents the activation of cyclin E–CDK2 or cyclin D–CDK4 complexes, and thus controls the cell cycle progression at G1 which is just as shown in cell cycle assay.44,45 TIMP3 is a tumor suppressor gene which inhibits tumor growth, angiogenesis, invasion, and metastasis and induces apoptosis.23 The upregulation of these genes and protein was consistent with the result of cell apoptosis and cell cycle assay. Tf-RL can exhibit a good silencing efficiency and better than RL which may be owing to the TfR-induced targeting effect as shown in cellular uptake assay and in vivo biodistribution assay. We built a delivery system of TfR-targeted liposomes with encapsulated anti-miR-221 in this study. From the results, the drug delivery system in our study is in the appropriate nanoscale size range and allows it to accumulate in tumor tissues through the passive targeting effect; it can specifically target to tumor cells and induce more cell apoptosis, and exhibit a good gene-silencing efficiency in vitro and in vivo. These results suggest that Tf-RL possesses the basic characteristics of targeted delivery systems, and the further application of this delivery system with miRNA will be deeply researched in the future.
Conclusion
The preparation of this liposome is very simple and robust, the composition of this liposome is relatively safer than most cationic lipids, and this system can achieve a good transfection efficacy. Taking these into consideration, we believe that the liposome studied in this paper is a promising gene delivery system. In the meanwhile, we also realize that the liposome loaded only with anti-miR-221 has limited effect on tumors. We will consider studying co-delivery system in the future.
Acknowledgments
This work was supported by grants from Natural Science Foundation of China (No 81301235), Natural Science Foundation of Hubei Province (No 2014CFB405), and Independent Innovation Foundation of HUST (No 2014YGYL013).
Disclosure
The authors report no conflicts of interest in this work.
References
Wei KR, Yu X, Zheng RS, et al. Incidence and mortality of liver cancer in China, 2010. Chin J Cancer. 2014;8:388–394. | ||
Siegel AB, Olsen SK, Magun A, Brown RS. Sorafenib: where do we go from here? Hepatology. 2010;52(1):360–369. | ||
Li H, Piao L, Yu B, et al. Delivery of calf thymus DNA to tumor by folate receptor targeted cationic liposomes. Biomaterials. 2011;32(27):6614–6620. | ||
Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol. 2013;87(2):227–247. | ||
Aravalli RN, Steer CJ, Cressman EN. Molecular mechanisms of hepatocellular carcinoma. Hepatology. 2008;48(6):2047–2063. | ||
Huang S, He X. The role of microRNAs in liver cancer progression. Br J Cancer. 2011;104(2):235–240. | ||
Landen CN Jr, Chavez-Reyes A, Bucana C, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65(15):6910–6918. | ||
Shen G, Rong X, Zhao J, et al. MicroRNA-105 suppresses cell proliferation and inhibits PI3K/AKT signaling in human hepatocellular carcinoma. Carcinogenesis. 2014;35(12):2748–2755. | ||
Zhao Q, Li T, Qi J, Liu J, Qin C. The miR-545/374a cluster encoded in the Ftx lncRNA is overexpressed in HBV-related hepatocellular carcinoma and promotes tumorigenesis and tumor progression. PLoS One. 2014;9(10):e109782. | ||
Gao K, Huang L. Nonviral methods for siRNA delivery. Mol Pharm. 2009;6(3):651–658. | ||
Choung S, Kim YJ, Kim S, Park HO, Choi YC. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem Biophys Res Commun. 2006;342(3):919–927. | ||
Yildiz I, Lee KL, Chen K, Shukla S, Steinmetz NF. Infusion of imaging and therapeutic molecules into the plant virus-based carrier cowpea mosaic virus: cargo-loading and delivery. J Control Release. 2013;172(2):568–578. | ||
Rothdiener M, Müller D, Castro PG, et al. Targeted delivery of SiRNA to CD33-positive tumor cells with liposomal carrier systems. J Control Release. 2010;144(2):251–258. | ||
Sokolova V, Epple M. Inorganic nanoparticles as carriers of nucleic acids into cells. Angew Chem Int Ed Engl. 2008;47(8):1382–1395. | ||
Taratula O, Garbuzenko OB, Kirkpatrick P, et al. Surface-engineered targeted PPI dendrimer for efficient intracellular and intratumoral siRNA delivery. J Control Release. 2009;140(3):284–293. | ||
Zhu C, Jung S, Luo S, et al. Co-delivery of siRNA and paclitaxel into cancer cells by biodegradable cationic micelles based on PDMAEMA-PCL-PDMAEMA triblock copolymers. Biomaterials. 2010;31(8):2408–2416. | ||
Patel A, Patel M, Yang X, Mitra AK. Recent advances in protein and peptide drug delivery: a special emphasis on polymeric nanoparticles. Protein Pept Lett. 2014;21(11):1102–1120. | ||
Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release. 2014;194c:238–256. | ||
Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther. 2010;18(9):1650–1656. | ||
Zhang M, Zhou X, Wang B, et al. Lactosylated gramicidin-based lipid nanoparticles (Lac-GLN) for targeted delivery of anti-miR-155 to hepatocellular carcinoma. J Control Release. 2013;168(3):251–261. | ||
Hatakeyama H, Murata M, Sato Y, et al. The systemic administration of an anti-miRNA oligonucleotide encapsulated pH-sensitive liposome results in reduced level of hepatic microRNA-122 in mice. J Control Release. 2013;173:43–50. | ||
Fornari F, Gramantieri L, Ferracin M, et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27(43):5651–5661. | ||
Garofalo M, Di Leva G, Romano G, et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16(6):498–509. | ||
Medina R, Zaidi SK, Liu CG, et al. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008;68(8):2773–2780. | ||
Ippoliti R, Lendaro E, D’Agostino I, et al. A chimeric saporin-transferrin conjugate compared to ricin toxin: role of the carrier in intracellular transport and toxicity. FASEB J. 1995;9(12):1220–1225. | ||
Li X, Ding L, Xu Y, Wang Y, Ping Q. Targeted delivery of doxorubicin using stealth liposomes modified with transferrin. Int J Pharm. 2009;373(1–2):116–123. | ||
Sakurai K, Sohda T, Ueda S, et al. Immunohistochemical demonstration of transferrin receptor 1 and 2 in human hepatocellular carcinoma tissue. Hepatogastroenterology. 2014;61(130):426–430. | ||
Park JK, Kogure T, Nuovo GJ, et al. miR-221 silencing blocks hepatocellular carcinoma and promotes survival. Cancer Res. 2011;71(24):7608–7616. | ||
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. | ||
Zhang G, Wang Q, Xu R. Therapeutics based on microRNA: a new approach for liver cancer. Curr Genomics. 2010;11(5):311–325. | ||
Li J, Wang Y, Yu W, Chen J, Luo J. Expression of serum miR-221 in human hepatocellular carcinoma and its prognostic significance. Biochem Biophys Res Commun. 2011;406(1):70–73. | ||
Yuan Q, Loya K, Rani B, et al. MicroRNA-221 overexpression accelerates hepatocyte proliferation during liver regeneration. Hepatology. 2013;57(1):299–310. | ||
Huang Y, Yang T, Zhang W, et al. A novel hydrolysis-resistant lipophilic folate derivative enables stable delivery of targeted liposomes in vivo. Int J Nanomedicine. 2014;9:4581–4595. | ||
Xiang G, Wu J, Lu Y, Liu Z, Lee RJ. Synthesis and evaluation of a novel ligand for folate-mediated targeting liposomes. Int J Pharm. 2008;356(1–2):29–36. | ||
Ye P, Zhang W, Yang T, et al. Folate receptor-targeted liposomes enhanced the antitumor potency of imatinib through the combination of active targeting and molecular targeting. Int J Nanomedicine. 2014;9:2167–2178. | ||
Liu Y, Mounkes LC, Liggitt HD, et al. Factors influencing the efficiency of cationic liposome-mediated intravenous gene delivery. Nat Biotechnol. 1997;15(2):167–173. | ||
Rossi JJ. Medicine: a cholesterol connection in RNAi. Nature. 2004;432(7014):155–156. | ||
Gaspar MM, Radomska A, Gobbo OL, Bakowsky U, Radomski MW, Ehrhardt C. Targeted delivery of transferrin-conjugated liposomes to an orthotopic model of lung cancer in nude rats. J Aerosol Med Pulm Drug Deliv. 2012;25(6):310–318. | ||
Derycke AS, Kamuhabwa A, Gijsens A, et al. Transferrin-conjugated liposome targeting of photosensitizer AlPcS4 to rat bladder carcinoma cells. J Natl Cancer Inst. 2004;96(21):1620–1630. | ||
Hao ZF, Cui YX, Li MH, et al. Liposomes modified with P-aminophenyl-alpha-D-mannopyranoside: a carrier for targeting cerebral functional regions in mice. Eur J Pharm Biopharm. 2013;84(3):505–516. | ||
Chiu RY, Tsuji T, Wang SJ, Wang J, Liu CT, Kamei DT. Improving the systemic drug delivery efficacy of nanoparticles using a transferrin variant for targeting. J Control Release. 2014;180:33–41. | ||
Eltayeb SE, Su Z, Shi Y, Li S, Xiao Y, Ping Q. Preparation and optimization of transferrin-modified-artemether lipid nanospheres based on the orthogonal design of emulsion formulation and physically electrostatic adsorption. Int J Pharm. 2013;452(1–2):321–332. | ||
Wallace DF, Summerville L, Subramaniam VN. Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology. 2007;132(1):301–310. | ||
Visone R, Russo L, Pallante P, et al. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr Relat Cancer. 2007;14(3):791–798. | ||
Galardi S, Mercatelli N, Giorda E, et al. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J Biol Chem. 2007;282(32):23716–23724. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.