Back to Journals » Biologics: Targets and Therapy » Volume 8
Targeted alpha anticancer therapies: update and future prospects
Authors Allen B, Huang C, Clarke R
Received 17 February 2014
Accepted for publication 6 May 2014
Published 10 November 2014 Volume 2014:8 Pages 255—267
DOI https://doi.org/10.2147/BTT.S29947
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Barry J Allen,1,2 Chen-Yu Huang,3 Raymond A Clarke2
1Faculty of Physics, University of Sydney, Sydney, NSW, Australia; 2Faculty of Medicine, Ingham Institute, University of Western Sydney, Liverpool, NSW, Australia; 3Central Clinical School, University of Sydney, Sydney, NSW, Australia
Abstract: Targeted alpha therapy (TAT) is an emerging option for local and systemic cancer treatment. Preclinical research and clinical trials show that alpha-emitting radionuclides can kill targeted cancer cells while sparing normal cells, thus reducing toxicity. 223RaCl2 (Xofigo®) is the first alpha emitting radioisotope to gain registration in the US for palliative therapy of prostate cancer bone metastases by indirect physiological targeting. The alpha emitting radioisotopes 211At, 213Bi, 225Ac and 227Th are being used to label targeting vectors such as monoclonal antibodies for specific cancer therapy indications. In this review, safety and tolerance aspects are considered with respect to microdosimetry, specific energy, Monte Carlo model calculations, biodosimetry, equivalent dose and mutagenesis. The clinical efficacy of TAT for solid tumors may also be enhanced by its capacity for tumor anti-vascular (TAVAT) effects. This review emphasizes key aspects of TAT research with respect to the PAI2-uPAR complex and the monoclonal antibodies bevacizumab, C595 and J591. Clinical trial outcomes are reviewed for neuroendocrine tumors, leukemia, glioma, melanoma, non-Hodgkins lymphoma, and prostate bone metastases. Recommendations and future directions are proposed.
Keywords: biodosimetry, microdosimetry, mutagenesis, PAI2, bevacizumab, C595, J591, tumors, cancer, metastases
Background
Monoclonal antibodies (MAbs) raised against cancer antigens may mediate antibody-dependent cell-mediated cytotoxicity.1,2 This form of cancer control arises from cytolysis of a target cell by effector lymphocytes, such as cytotoxic T lymphocytes or natural killer cells. However, most of these antibodies have low to moderate efficacy in tumor control. Antibodies targeting hormone receptors expressed by the cancer have shown greater tumor control compared with other cell membrane targets.
Labeling cancer targeting antibodies with a toxin can potentiate antibody efficacy in tumor control. In this way the antibody becomes an invaluable targeting vector for delivery of the toxin to the cancer cells. The toxin–antibody complex is called the immunoconjugate (IC). Different molecules, chemicals or radioisotopes (RIs) can serve as toxins provided their activity can be maintained during their conjugation to antibodies, which in the case of RIs involves the use of special chelating agents. Toxins may have long half-lives in the body, eg, ricin, which increases the toxicity to both the cancer and normal tissues. However, the different RIs have a wide range of half-lives and radiation decay properties to suit different applications.
Nuclear imaging uses long lived gamma emitters, which allow blood clearance as the tumor increases its uptake of the conjugate over time, so improving contrast. I-131 and I-123, Tl-201, Ga-67, and In-111 are some reactor and cyclotron produced RIs for this purpose. Beta emitting RIs, predominantly I-131, have had only modest success in radioimmunotherapy.2 More recently, high linear energy transfer (LET) radiation in the form of alpha particles has been studied. Alpha radiation is ideal for killing isolated cancer cells in transit in the vascular and lymphatic systems and regressing tumors by disruption of tumor capillary networks by targeting and killing tumor capillary endothelial cells (ECs). The LET is typically ~100 keV/μm, giving a higher probability of causing double strand breaks (the ratio of single strand break/double strand break, ~20, compared with 60 for low LET radiation).3
Apoptosis is the dominant form of cell death resulting from high LET radiation.4 A programmed sequence of events leads to the elimination of cells without releasing cytotoxic substances into the surrounding tissues. Apoptosis plays a crucial role in developing and maintaining healthy tissues by eliminating old cells, unnecessary cells, and unhealthy cells.
Over the past 20 years the development of alpha-immunoconjugates (AICs) has enabled targeted alpha therapy (TAT) to progress from in vitro studies, through in vivo experiments and on to clinical trials.5 The dose to normal tissues always provides a limitation to the injected dose and that received by the tumor. However, TAT can achieve cancer regression within the maximum tolerance dose (MTD) for normal tissue. TAT was originally thought to be an ideal therapy for “liquid” cancers, eg, leukemia and micro-metastases, as the short half-lives of the RIs were sufficient to target these blood borne cancer cells and the short range ensured that the targeted cancer cells received the highest radiation dose.
The difficulty of proving efficacy against micro-metastatic disease cannot be underestimated.6 However, Phase I trials of end-stage acute myeloid leukemia cancer patients have established the tolerance dose and provided evidence of efficacy.7
Tumor anti-vascular alpha therapy (TAVAT)8 offers the potential of disrupting tumor capillaries, thus limiting blood supply to the tumor and so inhibiting tumor growth. This effect has been ascribed to tumor regressions observed in a Phase I trial of TAT for metastatic melanoma targeting the melanoma-associated chondroitin sulfate proteoglycan (MCSP).9
Further, direct targeting of tumor specific receptors and antigens is possible. For example, the J591 antibody targets the prostate specific membrane bound antigen, which is upregulated in the tumor vasculature of many different tumor types.10 Short range alpha therapy can lead to endothelial cell death and the closure of tumor capillaries.
Alpha radiation from 233RaCl2 (called Alpharadin® or Xofigo®) has had recent success in extending life and reducing pain in prostate cancer patients with bone metastases due to its preferential uptake by the osteoblasts in bone (as a calcium analogue).11 However, alpha radiation is most effective when delivered directly to cancer cells by the AIC using cancer specific antibodies. With energies from 2–8 MeV and ranges of 20–80 μm, alpha emitting RIs can deliver high LET radiation with increased rates of double strand breaks that can induce DNA damage and apoptosis in cells within the alpha particle range. However, if mismatch DNA repair occurs, then alpha radiation has a much higher probability than photons (×20) of causing genetic damage and carcinogenesis.
The primary objectives of alpha-immunotherapy are to kill targeted cancer cells, limit tumor progression, and where possible eradicate the cancer. This is achieved by improved delivery and specificity of the targeting vector. Delivery is improved by selecting antibodies with the highest specificity, which in turn limits damage to normal tissues. Cancer management begins with surgery to remove solid tumors, radiotherapy to control local tumors and reduce local recurrence (as in post-lumpectomy radiotherapy for breast cancer), and chemotherapy or hormonal oblation therapy to control systemic disease or for palliation. TAT is a next generation approach with potential to make a major contribution to the management of cancer.
Alpha-immunotherapy is best suited for the following specific applications:
- Targeting isolated cancer cells circulating in the lymphatic and vascular systems.
- Regression of metastatic cancer cell clusters.
- Disrupting the vasculature of solid tumors.
Each application requires a different approach, as discussed in the following sections.
Therapeutic applications
Cancer cells in transit in the vasculature and lymphatics
These cells have special problems relating to cell cycle and bioavailability, including:
- Circulating cancer cells that are not dividing may be relatively insensitive to routine radiotherapy and chemotherapy.
- Systemic cancer targeting is required, but only a small fraction of the dose will reach its target so short range cytotoxic action is essential to reduce normal tissue damage, which will be the dose limiting factor.
- High labeling efficiency is required to reduce unlabeled antibody saturation of targeted antigens.
- Short radionuclide half-life is preferred to reduce normal tissue toxicity.
- Lymphatic administration may be required to eliminate cells in transit from primary lesions.
- Therapeutic response to TAT is difficult to evaluate.6 However, comparing changes in circulating cancer cell counts before and after therapy may be possible using magnetic microspheres coated with the targeting MAb.12
Cell clusters
Cancer cells in transit can reach distant sites where the cell attaches and again enters the cell cycle leading to the development of micro-metastases that are too small to be detected by imaging. In the early stages of metastasis, the endothelial cell growth factors expressed by cancer cells are yet insufficient to stimulate vascular extensions into the lesion. As such, micro-metastases would be located adjacent to the circulatory system that transported the cancer cells to the new site. In this context, the short range effects of alphas are desirable to limit normal tissue toxicity. Moreover, TAT has demonstrated efficacy in effective targeting of cancer cell spheroids in vitro,13,14 but there are limitations to efficacy for larger cell clusters (Huang, unpublished data, 2014).
Solid tumors
Tumor capillary permeability is an important parameter that determines in part the bio-availability of cancer cells within a tumor. Leaky neogenic capillaries allow the extravasation of the IC into the perivascular space to saturate the targeted antigens expressed by contiguous and adjacent cancer cells.
- Long range cross fire effect gives beta radiation an important advantage and reduces the effect of heterogeneous uptake of the beta-IC in the tumor.
- Intralesional injections with AICs can overcome this problem.15
- Chemotoxins will suffer from bioavailability and the lack of a cross fire effect.
- The potential role of the bystander effect16 could be of benefit here. This radiation-induced phenomenon causes non-irradiated cells to exhibit effects as a result of signals received from nearby irradiated cells, causing the mutation of these cells. In vitro studies show that when the medium containing irradiated cells is transferred to non-irradiated cells, these cells show bystander responses when assayed for clonogenic survival and oncogenic transformation.
- Tumor capillary permeability causes a high density of ICs targeted to antigens around the capillaries, with a rapid drop off with distance from the capillaries. Whereas this is a drawback for beta-emitting ICs, it enhances the toxicity of AICs to the capillary ECs. The short range (~80 μm) of the emitted alpha radiation ensures a high radiation dose to the endothelial cell nucleus, inducing apoptosis. The capillary will shut down and if enough such capillaries also close, the tumor will regress. We call this process TAVAT.8 It explains the tumor regressions observed in the Phase I clinical trial of systemic TAT of metastatic melanoma.9 This process could be assisted by tumor vascular disruption agents that increase tumor capillary permeability.
Safety and tolerability
Systemic therapies are always handicapped by the measurement of dose to the normal tissues and tumors. Chemotherapy and immunotherapy dose is normalized to body surface area and radiopharmaceutical activity is given per unit body mass. Nuclear imaging agents can allow dose to the tumor and organs to be determined. High energy gamma rays are emitted from alpha emitters, which also allow dose estimates for organs and tumors. The MTD for AICs has been determined in many animal models.
Bi-213 AIC
Mice have acute activity tolerances in the region of 890–1,330 MBq/kg (24–36 mCi/kg) for systemic (intraperitoneal) injections. However, long-term toxicity (~6 months in mice) in the form of delayed radiation nephrosis, reduces the MTD to ~330 Mbq/kg (9 mCi/kg) in mice and between 110 and 330 MBq/kg (3 and 9 mCi/kg) in rabbits.17 While these limits are useful in preparing for human therapy, Phase I trials are mandatory to establish safe operating conditions.
Bi-213 PAI2
The long-term MTD in mice and rabbits was 350 and 120 MBq/kg (9.5 and 3.2 mCi/kg), respectively. Because of the low molecular weight of PAI2, radiation nephropathy was the dose limiting effect. However, the renal uptake can be decreased ×3 if lysine is injected prior to the TAT.18
Th-227 AIC
The anti-CD20 antibody rituximab labeled with the alpha-particle-emitting radionuclide 227Th is of interest for treatment of lymphoma. The MTD was determined for late toxicity of 227Th-rituximab.19 BALB/c mice were injected with saline, cold rituximab or 0.05, 0.2 or 1.0 MBq/kg (and followed for up to 1 year). Toxicity was evaluated by measurements of mouse body weight, white blood cell and platelet counts, serum clinical chemistry parameters, and histological examination of tissues. Only the 1.0 MBq/kg (27 μCi/kg) dose resulted in decreased mouse body weight. A short-term decrease in white blood cell and platelet count was observed at 0.4 and 1.0 MBq/kg. The MTD was found to be between 600 and 1,000 kBq/kg. The MTD to bone marrow was between 2.1 and 3.5 Gy. The MTD is very much less than Bi-213 because of the five decay alpha emissions. Therapeutically relevant dose levels of 227Th-rituximab were well tolerated in mice. Bone marrow suppression, as indicated by a decrease in white blood cell count, was the dose-limiting radiotoxicity.
Microdosimetry
TAT has the advantage of giving therapeutic doses to individual cancer cells and low dose radiation to distant normal cells. However, because of the very short range of alpha radiation, tumor or organ dose is difficult to estimate. Moreover, because of heterogeneous antigen expression among cancer cells and the random nature of short-range, high-LET alpha radiation, dosimetry is challenging. It is therefore inappropriate to investigate the therapeutic efficacy of TAT by conventional dosimetry. Microdosimetry of TAT is a function of the cell geometry, source-target configuration, cell sensitivity, and biological factors. A detailed knowledge of each of these parameters is required for accurate microdosimetric calculations.
As such, the dosimetry of TAT is different from that of beta immunotherapy or external beam radiotherapy as follows:
- Short path length of alpha particles.
The energy loss of alpha particles in cell nuclei is a stochastic process, with some cell nuclei receiving multiple alpha particle hits, while others receive no hits. The energy deposited varies greatly from target to target, leading to a broad frequency distribution for energy loss. - Small target volume.
Given the high LET delivered along an alpha-particle track and its potential cytotoxicity, the concept of mean absorbed dose may not always yield physically or biologically meaningful information. - Heterogeneous distribution of RIs.
Heterogeneous antigen expression and tumor uptake leads to variable spatial microdosimetric distributions of the AIC. Spatial and temporal changes of the source activity in the target can also occur. When the distribution of the AIC is non-uniform, dose averaging over volumes greater in size than the individual target volumes will be inadequate predictors of the biological effect. - Specific energy.
Specific energy is the energy deposited (J) in the target mass (kg) and has the same units as absorbed dose (Gy). Although microdosimetry is concerned with the same concept of energy deposition per unit mass as dosimetry, the difference in the range of the alpha particle and the small size of the target volume introduces stochastic effects, which are negligible in conventional dosimetry. The stochastic quantity of specific energy can be used to investigate biological effects as it can be used to directly calculate the actual cancer cell survival rate. - Monte Carlo models.
Metastatic melanoma lesions experienced marked regression after systemic TAT in a Phase I clinical trial.9 This unexpected response was ascribed to TAVAT,8 in which effective tumor regression is achieved by killing ECs in tumor capillaries and thus depriving cancer cells of nutrition and oxygen. Quantitative analysis of the therapeutic efficacy and safety of TAVAT required building up Monte Carlo microdosimetric models.20
The intraluminal model was designed to simulate the background dose to normal tissue capillary ECs from the non-targeted activity in the blood. The Perivascular model calculates the EC dose from the activity bound to the perivascular cancer cells. The key parameters are the probability of an alpha particle traversing an EC nucleus, the energy deposition, the lineal energy transfer and the specific energy. These results were then applied to interpret the clinical trial. Using the maximum activity of 25 mCi in the melanoma clinical trial (Huang, unpublished data, 2014) the specific energy to the tumor EC nucleus was found to be 3.2 Gy and to a normal capillary EC nucleus to be 1.8 cGy. These data give a maximum therapeutic gain of about 180 and support the TAVAT hypothesis to deliver a cytotoxic dose to tumor capillaries without toxicity to normal tissue capillaries. - Alpha versus beta radiation.
Monte Carlo calculations (Huang, unpublished data, 2014)20 show that alpha emitters can deposit much more energy than beta emitters into the target cell nucleus for the same number of decays. This means that to achieve the same specific energy to cancer cells, a much higher activity is required for the beta emitter which may cause serious normal tissue radiation damage. Alpha emitting RIs with multiple alpha decays give much higher specific energies than those for single decays. For example, 223Ra or 225Ac emit four alphas, giving a much higher specific energy than 211At or 213Bi. However, the recoil of daughters can free the RI from the chelator and increase normal tissue toxicity, especially in the kidneys.
Because of their short range, alpha emitters deliver from 100–1,000 times the specific energy of beta emitters to the nuclei of ECs in the perivascular model. Of the beta emitters, the high energy betas from 90Y and 32P give the lowest specific energies. Even if all antigenic sites are saturated out to 25 cell layers from the capillary, beta radiation spares some 50% of EC cells (for D0 =1.4 Gy), whereas all ECs are killed by alphas (Huang, unpublished data, 2014). The tumor control probability (TCP) is the probability of destroying every clonogen in a tumor as a result of a radiation therapy treatment. Assuming absorbed dose homogeneity throughout the tumor volume, TCP can be derived from a cell survival model. If the absorbed dose is non-homogeneous, its distribution has to be taken into account because survival fractions depend on dose.
The tumor control probability was found to be TCP=1 for all alpha emitters, compared with TCP=0 for beta emitters.
These results are consistent with experimental data, which show comparable or superior efficacy with reduced toxicity for Bi-213 compared with Lu-177 in mouse models for radio-immunotherapy for peritoneal carcinomatosis21 and for radio-peptide therapy for prostate cancer.22
Biological dosimetry
Real time dosimetry of alpha therapy is problematical. Characteristic gamma rays could give an estimate of the macroscopic alpha dose but this is not the dose of interest. Rather, the microscopic alpha dose is required and direct measurement of alpha emission by MOSFET detectors requires implanted detectors in the blood or key organs. An alternative approach is to use radiation induced micronuclei in lymphocytes as a direct measure of the biologically effective dose in the blood.23,24 This is not real time dosimetry but would be of value in fractionated therapy to establish the biological dose after the first fraction.
Lymphocytes in the peripheral blood are exposed to background alpha and beta radiation on systemic administration of the AIC. The AIC is designed to target the cancer cells, not the lymphocytes, and as such background dose levels are expected to be low. However, there will be random hits of lymphocytes by untargeted alpha and beta decays of the RI in the blood.
Pre- and post-irradiation peripheral blood samples can be assessed for chromosome breaks and micronuclei. During proliferation assays, cells that have suffered double strand breaks in their DNA will exhibit limited fragmentation, which can form discrete entities of bilayer phospholipid enclosed DNA fragments, called micronuclei, within the lymphocyte prior to division.23 The degree of chromosome fragmentation can be determined by staining and counting under the microscope.
The biological dose measured by the micronuclei and fragmentation assay is not the whole body dose. It is the dose to the blood volume and to the vascular walls, as defined by the 80 μm range of alpha particles from the AIC in the lumen. As such, the biological dose to the bone marrow can be determined. This biological dosimeter provides a direct measure of the stochastic radiation damage without the need to implant a detector to measure absorbed dose and determine relative biological effectiveness factors.
Equivalent dose
Equivalent dose is defined to take into account the International Commission on Radiological Protection radiation weighting factor of 20, because alpha particles are 20 times more likely to cause cancer and other stochastic effects for the same absorbed dose of electrons or photons. The effective dose is a weighted average of individual organ equivalent doses. Both equivalent and effective dose values are associated with the unit Sievert.25
Toxicity and efficacy are the relevant end points for treatment evaluation. These are deterministic quantities for which the end points for alpha radiation have three to seven times more toxicity or efficacy per unit absorbed dose than electrons or photons. The factor used to weight the absorbed dose for deterministic end points is the relative biological effectiveness (RBE), which is defined for specific end points. As such, the unit for biological dose is RBE.Gy, not the Sv. It is recommended that the absorbed dose (in Gy) should be listed separately for (electron and photon) emissions and for alpha-particle emissions.25
Mutagenesis
The mutagenic potential of 213Bi conjugated to a human melanoma antigen-specific antibody (9.2.27) has been investigated.26 The lacZ transgenic mouse model was used, which contains multiple copies of a lacZ target gene in every cell, allowing the quantification and comparison of mutagenesis in different organs. Mice received an intraperitoneal injection of 16.65 MBq (0.45 mCi) of 213Bi-cDTPA-9.2.27, and were sacrificed at 24 hours, 1 week, and 4 weeks post-injection. Pharmacokinetic studies gave the absorbed and effective doses for each organ.
The mutant frequency and mutant spectra were analyzed for the brain, spleen, and kidneys. The brain and spleen did not show significant increases in induced mutation frequencies compared to spontaneous background levels or changes in mutant spectra; these results being independent of p53 status. However, elevated but not significant mutation frequencies and persistent size change mutations were observed in the kidneys. These effects were time dependent and levels returned to those of the controls at 4 weeks post-irradiation due to DNA repair.
These effects were observed at an activity of 830 MBq/kg (22.5 mCi/kg) which is very much higher than that expected for the therapy of human patients of ~37 MBq/kg (1 mCi/kg). As such, the risk of secondary mutations arising from 213Bi-cDTPA-conjugates is not expected to be a significant problem in the clinic.
In vivo models
Nude mice and transgenic mice are used for specific cancer studies. Human cancers can grow in nude mice because of their incompetent immune systems. Transgenic mice have gene changes for specific purposes, such as spontaneous cancer.
The short range of alpha particles, and the short half-life of useful alpha emitting RIs argue against TAT with Bi-213 for regression of solid tumors.27 Consequently, our studies relate to the killing of isolated cancer cells and metastatic cell clusters and the inhibition of tumor growth through anti-vascular effects. A great deal of preclinical work in TAT paved the way for the advance to clinical trials in recent years.28 The Sloan Kettering Memorial Cancer Center led the way, first with the application of Bi-213 immunotherapy7 and later with Ac-225.29 Bi-213 is eluted from an Ac-225 generator.30 Stable alpha-conjugates were synthesized by labeling chelated MAb with Bi-213 to form the AIC. These were tested in vitro and in vivo, inter alia, for melanoma,31,32 leukemia,33 carcinomatosis,21 colorectal,34 prostate,22,35–37 glioma,38 ovarian,14 lymphoma,39 and pancreatic cancers.40–43
Targeting vectors
The following MAbs are at the clinical trial stage:
- Humanized HuM195 targets acute myelogenous leukemia (AML) (see “Leukemia” section)
- Murine 9.2.27 targets the MCSP antigen on melanoma and glioblastoma multiforme (GBM) cells and vascular pericytes (see “Intralesional TAT for metastatic melanoma” and “Systemic TAT for metastatic melanoma” sections)
- Anti-CD20 for lymphoma (see “Glioma” section)
- MX35 F(ab’)2 for ovarian cancer44
- Human-mouse chimeric anti-tenascin 81C6 for GBM (see “Glioma” section)
- RaCl2 for bone metastases (see “233RaCl2 (Alpharadin® or Xofigo®)” section)
Preclinical results for some other targeting vectors not yet in clinical trial for TAT are reviewed below.
Bevacizumab (BZ) targets the vascular endothelial growth factor and is now part of the standard treatment for advanced colorectal cancer. Castrate resistant prostate cancers also express vascular endothelial growth factor but are only marginally responsive to BZ alone or in combination with chemotherapy. The efficacy of orthotopic administration of 213Bi-BZ was studied in the nude mouse, prostate cancer xenograft model.45 The control mice received a saline injection in equal volume to each BZ administration. 213Bi-BZ was significantly more efficacious in inhibiting xenograft progression in the prostate gland compared with BZ antibody alone (P=0.009) and when compared to the controls (P<0.0001). Orthotopic administration of 213Bi-BZ greatly improved the early control of organ confined prostate cancer compared to BZ alone (P<0.01).
C595, a murine monoclonal antibody against mucin (MUC1), is expressed by prostate, breast, ovarian, and pancreatic cancers. C595 is an IgG3 antibody raised against the protein core of human urinary epithelial MUC1. MUC1 is found to be frequently up-regulated and abnormally glycosylated in a number of common malignancies, including breast, bladder, colon, ovarian, prostate, and gastric cancer. Cancer-associated MUC1 is structurally different to normal MUC1 in that the former has shorter and less dense O-glycan chains, which exposes novel regions of the protein core. Preclinical results show inhibition of tumor growth and regression of cell clusters. Over 90% of primary prostate, pancreatic and ovarian tumors expressed MUC1 while 95% of normal tissues did not. Further, MUC1 expression was found on the surface of cancer cell lines. The lethal pathway in all in vitro studies after TAT was found to be predominantly by apoptosis.10,36,42,46–48
PAI2 is a human recombinant protein that targets uPA, which is expressed by many cancers at their most malignant stage. The PAI2-uPAR targeting system has several important advantages over monoclonal approaches to TAT. First, PAI2 is a human protein, rather than a murine antibody, so overcoming problems of immune response. Secondly, it is a much smaller targeting molecule so can penetrate tissue more efficiently leading to faster targeting, which is important considering the short half-life of the alpha conjugate (47 minutes). Finally, pre-clinical studies of over-expression show that uPA is highly expressed in around 75% of pancreatic adenocarcinomas, while expression of uPA mRNA in a normal pancreas is only 6% of that for pancreatic adenocarcinoma.40,41 The PAI-2 protein was successfully tested in breast,49 ovarian,14,18 prostate,35 and pancreatic41 cancers. These conjugates are highly selective of and cytotoxic to targeted cancer cells. In vitro cytotoxicity of alpha-conjugates is very much greater than beta conjugates, non-specific alpha-conjugates, and free alpha isotope. The lethal pathway for alpha therapy is predominantly apoptosis.36
J591 targets the prostate specific membrane antigen expressed by most prostate cancer cells. The first preclinical studies for TAT with 213Bi-J591 in 2002 were promising in their efficacy and safety.50 A recent Phase II trial51 of the efficacy of a single infusion of J591 labeled with Lu-177 has been completed. Two cohorts with progressive metastatic castration-resistant prostate cancer received one dose of 177Lu-J591 (15 patients at 65 mCi/m2, 17 at 70 mCi/m2) with radionuclide imaging. An additional cohort (n=15) received 70 mCi/m2 to verify response rate and examine biomarkers.
In terms of prostate specific antigen (PSA) expression, 11% of patients experienced ≥50% decline in PSA, 36% experienced ≥30% decline, and 60% experienced a significant PSA decline. All experienced reversible hematologic toxicity, with grade 4 thrombocytopenia occurring in 47% without significant hemorrhage; 26% experienced grade 4 neutropenia, with one episode of febrile neutropenia. The Phase I maximum tolerated dose of 70 mCi/m2 resulted in more PSA declines of 30% (46.9% versus 13.3%, P=0.048) and longer survival (21.8 versus 11.9 months, P=0.03), but also more grade 4 hematologic toxicity and platelet transfusions.
A single dose of 177Lu-J591 was well tolerated with reversible myelosuppression. Accurate tumor targeting and PSA responses were seen with evidence of dose response. The results for one patient, with a rapidly rising PSA pre-treatment, with a PSA doubling time of 3.9 months, showed a PSA decline of 90% immediately following a single TAT injection.51
Clinical trials
RIs
Alpha therapy is following the well-worn trail of radio-immunotherapy with beta emitters. While I-131 was the therapeutic mainstay for many years, Sr-89, Sm-153, Lu-177 and Re-188/186 have proven effective, especially for bone cancer treatment.
The radiopharmaceuticals 90Y-Zevalin® and 131I-Bexxar® have been found to be effective against non-Hodgkin’s lymphoma and have been registered for the treatment of this disease. Somatostatin receptor peptides labeled with Y-90 or Lu-177 showed effectiveness for the treatment of neuroendocrine tumors in ongoing clinical trials, though they are not yet approved for clinical usage. More recently, radio-immunotherapy with 177Lu-J591 has been effective in extending life time and reducing PSA in post-hormonal therapy of castrate resistant prostate cancer.51
Some laboratories have concentrated on At-211, which, with a 7.2 hour half-life, needs to be used at or near the production site. The advantages of the Bi RIs are that they can be generated from long-lived parents, Ac-225 (T1/2=10 days) and Th-228 (T1/2=1.91 years), which can be transported over long distances. The 225Ac-213Bi generator has an additional advantage in that it decays in house and does not need long-term waste disposal. While the half-lives of Bi-213 (47 minutes) and Bi-212 (61 minutes) are rather short, there is sufficient time for synthesis of the AIC and for vascular distribution throughout the body. However, there is inadequate time for infusion into tumors, which can take 24–48 hours. On the other hand, Ac-225 with a 10 day half-life allows plenty of time for infusion of the AIC through the target tumors. The short ranges of the alpha products require a high degree of homogeneity if all cancer cells are to be lysed. Each cancer may require a different vector to target over-expressed membrane bound antigens. However, generic vectors do exist that target many different cancers. As such, a number of vectors are being used or are yet to be introduced into the clinic.
Phase I and II clinical trials
Seven clinical trials were reported at the Berlin TAT symposium in 2011. Bi-213 was used for studies in AML, melanoma, and lymphoma; Ac-225 for AML; Ra-223 for bone cancer, and At-211 for glioblastoma and ovarian cancer. The Phase I Bi-213 trial for AML has been completed and the current trial is Phase II with chemotherapy pre-treatment. The intralesional melanoma trial with Bi-213 has also been completed, being followed by an incomplete systemic trial with the same alpha conjugate.
The following sections review the results of past and current clinical trials, and the objectives of proposed trials. Current and completed clinical trials are as follows:
- Completed Phase I study for AML.7
- Ongoing Phase II study for post-chemotherapy of AML.52 Ongoing Phase I study with Ac-225.52
- Completed Phase I trial for intralesional melanoma.15
- Phase I trial of systemic melanoma.9,53,54
- Completed Phase I trial of glioblastoma.55
- Completed pilot trial of glioma.56
- Completed trial of Ra-223 for bone metastases.57,58
- Phase I for lymphoma.59
- Phase I trial in neuroendocrine tumors.61
- Phase I trial of intraperitoneal TAT for ovarian cancer.44 While solid tumors have never been envisaged as suitable targets for TAT, in contrast with liquid cancers and micrometastases,27 stage 4 advanced cancer patients are used in Phase I trials for toxicity studies.
Neuroendocrine tumors
Neuroendocrine cancer affects cells that translate neuronal information into hormonal information. Hormones and neuroendocrine nerve cells control a range of physiological processes, including efficiency of digestion, cellular metabolism, blood flow, and the reproductive cycle. Standard therapy for neuroendocrine cancer is surgery, chemotherapy, and radiotherapy.
This first alpha-immunotherapy dose escalation study involved the injection of a specific tumor-targeted peptide tagged with an alpha-emitter, using 213Bi-DOTATOC peptide receptor alpha-therapy.61 DOTATOC is a tumor-targeting probe that mimics the endocrine-system regulating hormone somatostatin. Fourteen patients with neuroendocrine liver metastases resistant to previous treatment with beta-particle peptide therapy were treated. The therapy was found to be highly effective for targeting neuroendocrine tumors and inducing remission of metastases without dangerous toxicity to healthy tissues. Additional studies are scheduled to escalate dosage further for even greater cancer-killing power for metastatic neuroendocrine cancer patients.
Leukemia
The first clinical trial with 213Bi was conducted at Memorial Sloan Kettering Cancer Center in New York in collaboration with the National Institutes of Health and the Institute for Transuranium Elements in Karlsruhe.7 Eighteen patients with relapsed and refractory AML or chronic myelomonocytic leukemia were treated with 10.4 to 37.0 MBq/kg 213Bi-HuM195. The humanized antibody HuM195 targets the CD33 antigen, a 67 kDa glycoprotein expressed on most myeloid leukemias and clonogenic leukemia progenitors. Fourteen (93%) of 15 evaluable patients had reductions in circulating blasts and 14 (78%) of 18 patients had reductions in the percentage of bone marrow blasts. Extra-medullary toxicity was not observed. The safety, feasibility, and efficacy of 213Bi-HuM195 provided the first proof of concept of systemic targeted α particle immunotherapy in humans.
Based on the encouraging results of this pioneering study, a follow-up Phase I/II study was conducted to determine the maximum tolerated dose and efficacy of 213Bi-HuM195 after partially cyto-reductive chemotherapy with cytarabine.62 Thirty-one patients with newly diagnosed (n=13) or relapsed/refractory (n=18) AML were treated. Significant reductions in marrow blasts were seen at all dose levels and complete remissions and partial responses were seen in six (24%) of the 25 patients who received doses of ≥37 MBq/kg.
Myelosuppression was tolerable and no significant extra-medullary toxicity was observed.
Intralesional TAT for metastatic melanoma
The 9.2.27 MAb was used to target the MCSP receptor in an intralesional melanoma trial.15 MCSP is expressed by the lesions of more than 90% of melanoma patients. This antigen is the same as the heavy molecular weight melanoma associated antigen and is thought to be identical to the NG2 murine antigen. The antibody is covalently coupled to the cDTPA chelator and labeled with the Bi-213 alpha emitting RI to form the AIC 213Bi-cDTPA-9.2.27, turning the benign targeting vector 9.2.27 into a highly cytotoxic agent for targeted melanoma cells.
The intra-lesional TAT was the first part of a program to establish a new systemic therapy for metastatic melanoma. The safety and efficacy of intra-lesional AIC in patients with metastatic skin melanoma was investigated in 16 melanoma patients, all with melanomas that were positive to the monoclonal antibody 9.2.27.15 AIC doses from 50 to 450 μCi were injected into lesions of different sizes, causing massive tumor cell death as observed by the presence of tumor debris. The AIC was very effective in delivering a high dose to the tumor while sparing other tissues. There were no significant changes in blood proteins and electrolytes. There was no evidence of a human anti-mouse antibody reaction. Evidence of significant decline in serum marker melanoma inhibitory activity protein at 2 weeks post-TAT was observed. Intra-lesional TAT was found to be safe and efficacious to 1,350 μCi. Tumors were resected at 8 weeks post-TAT, to show massive cell debris in the injected volume, but no effect in the untreated tumor or in the antibody-only treated tumor in the same patient. Tumor to kidney activity ratios were ~3,000. Reductions in MIA and the Ki67 proliferation marker and increases in apoptosis tests all indicated that TAT is a promising therapy for the control of inoperable secondary melanoma or primary ocular melanoma. As such, intra-lesional TAT is indicated for uveal melanoma and brain metastases.
Systemic TAT for metastatic melanoma
The aim of this study was to assess toxicity and response of systemic alpha therapy for metastatic melanoma using the AIC 213Bi-cDTPA-9.2.27.9 Tools used to investigate the responses were physical examination, imaging of tumors, pathology comparisons over 12 weeks, glomerular filtration rate, computed tomography comparisons, and changes in tumor markers over 8 weeks. Responses are based on the Response Evaluation Criteria in Solid Tumors (RECIST). Forty patients with stage IV melanoma/in-transit metastasis were treated with activities of 55-947 MBq. Using RECIST criteria, 50% of subjects experienced stable disease and 12% showed partial response. One patient showed near complete response after a 5 mCi intravenous injection of the AIC. Twenty/21 lesions completely disappeared with no evidence of residual melanoma in tumor beds. Another patient showed response in a mandible tumor and reduction in lung lesions. The melanoma inhibitory activity protein tumor marker reduced over 8 weeks in most patients. However, there was a disparity of dose with responders.
Toxicity was not observed over the range of administered activities. The observation of responses without any toxicity indicates that TAT has the potential to be a safe and effective therapeutic approach for metastatic melanoma. The observation of both safety and efficacy at quite low doses showed that this trial, while adequate as a Phase I, was inadequate to investigate the underlying factors that were contributing to the unexpected efficacy. As such, the trial was terminated in June 2007 without reaching the MTD and the results were published.8,9,53,54 A new trial was designed to provide more detailed information but was not funded.
Non-Hodgkin’s lymphoma
The first clinical trial of TAT on non-Hodgkin’s lymphoma was conducted in collaboration between the German Cancer Research Center, the University Hospital Düsseldorf, and the Institute for Transuranium Elements.59 In this Phase I dose-escalation trial, 12 patients were treated at dose levels ranging from 555 to 2,165 MBq of 213Bi-antiCD20 MAb. The maximum tolerated dose was not reached; acute or extra-medullary toxicity was not observed at any dose level.
Glioma
Targeted 213Bi therapy of glioma was first clinically tested at the University Hospital in Basel in collaboration with the Institute for Transuranium Elements in 1999. Two patients received intracavitary injections of 213Bi-[Thi8,Met(O2)11]-substance P (213Bi-SP) after surgical removal of the main tumor mass.38 Substance P (SP) is a neuropeptide, a substance that functions as a neurotransmitter and as a neuromodulator, and is the physiological ligand of the neurokinin-1 receptor, which is over expressed in the World Health Organization classification grades II–IV gliomas. It has also been detected on tumor cells infiltrating the intra- and peri-tumoral vasculature. It diffuses rapidly and localizes in remote satellite lesions. Treatment with 213Bi-SP was well tolerated in both patients. One patient with progressive glioblastoma, had an overall survival of 26 months. The other patient, with a post-central low-grade oligodendroglioma World Health Organization II, had a resection at 33 months post-therapy, which disclosed radiation necrosis and absence of vital tumor cells. The further course has been excellent to date (>11 years after therapy) without any signs of tumor recurrence.
Based on these encouraging results, a Phase I study in patients diagnosed with functionally critically located gliomas was conducted in 2007/2008.56 In this study, five patients received intratumoral injections ranging from 1.1 to 7.4 GBq of 213Bi-SP in one to four therapy cycles. Excellent retention of the radiolabeled peptide at the tumor site was observed by single photon emission computed tomography imaging monitoring the 440 keV photon emission of 213Bi and was confirmed by low values (<1.5%) of injected dose found in the blood pool.
The treatment was well tolerated by all patients and adverse effects were not observed up to 7.4 GBq of 213Bi-SP. Extensive radiation necrosis was observed in the tumor tissue in four/five patients who underwent tumor resection after 213Bi therapy. In two patients receiving activities ≥100 MBq/cm3, tumor resection was significantly facilitated by pseudo-encapsulation. Tumor resection disclosed extensive radiation necrosis and absence of vital tumor cells. This study demonstrated that targeted local radiotherapy using 213Bi-SP is feasible, safe, and therapeutically effective. Inoperable gliomas may become resectable with this treatment, potentially improving prognosis.
233RaCl2 (Alpharadin® or Xofigo®)
Radium-223 has been approved by the Food and Drug Administration for palliative therapy of prostate cancer bone metastases following a Phase III trail that showed improvements in the overall survival and delayed symptomatic progression. Radium is a calcium analogue and is taken up by osteoblasts in the repair of bone damaged by the cancer cells. Clinical studies of radium-223 are listed in Table 1 and have not shown significant daughter redistribution.
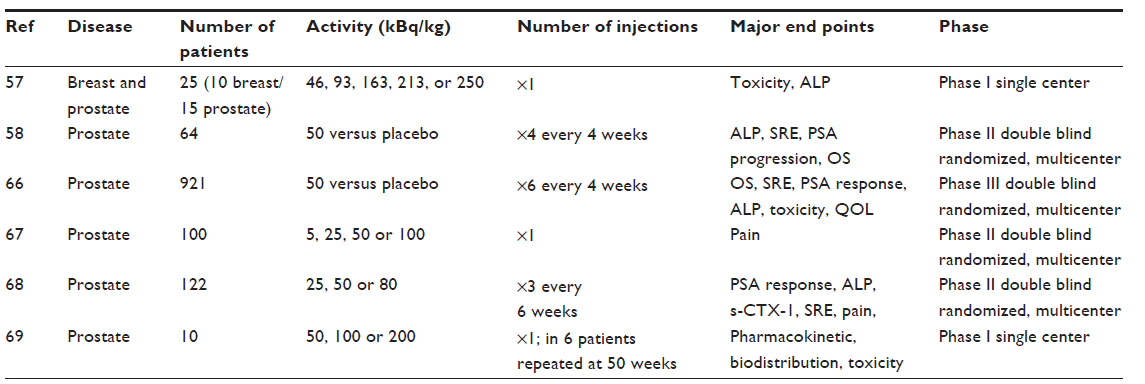 | Table 1 Ra-223 dichloride for the treatment of metastatic disease to bone |
Recommendations and future directions
The following recommendations are based, inter alia, on selected recommendations arising from the International Atomic Energy Agency Technical Meeting on Alpha Emitting Radionuclides and Radiopharmaceuticals for Therapy, Vienna, 2013:63
- A new paradigm is needed to move away from single values of mean absorbed dose to take into account the short path length of alpha particles and stochastic effects. Biodosimetry could be that paradigm.
- Preclinical studies are required to identify new targets and disease settings such as intra-vesicle delivery, vascular targeting, and pre-targeting.
- More clinical studies are urgently required to establish the clinical utility of alpha therapy, including combination studies to investigate the potential for synergistic activity with other agents and to encourage clinical research into use of alpha-therapeutics earlier in disease history.
- New applications of alpha-emitters would be highly useful for setting up new clinical trials.
There are a number of approaches that need to be tested to enhance the efficacy and reduce the potential toxicity of TAT.
Chelator
The melanoma trial used the cDTPA chelator to link the antibody and RI, as this was the only commercially available chelator at that time. As delayed radiation nephrosis is the main concern, the more stable CHX-A” is expected to reduce the renal uptake of free Bi-213 and so increase the MTD for the kidneys. Further, commercial production of CHX-A” is now available (Macrocyclics, Inc., Dallas, TX, USA). Recoils in Ac-252 decay require more stable chelation such as DOTA. A complete review on the chelating chemistry was recently given on all the alpha emitters.64
Specific activity
MAbs target the membrane expressed antigens and if expression is low, antigens can be more readily saturated and blocked by unlabeled antibody, thus limiting efficacy. One way around this problem is to increase the specific activity of the AIC to >20 μCi/mg. The AIC is usually prepared by minimizing the free RI in the labeling process. However, the objective should be to minimize the unlabeled antibody fraction. This then leads to a higher specific activity, less blocking of target antigens, and more effective therapy. Further, the higher specific activity will reduce the amount of antibody injected, even at higher activities, therefore reducing the potential human anti-mouse antibody effect.
Fractionation
A fractionated dose regime will be more practical at high doses and may give improved efficacy as tumor capillaries may be damaged, resulting in increased extravascular permeability for the AIC. Daily fractionation over 4–5 days would not be of concern for immune response (human anti-mouse antibody) as the time period is too short for the generation of an immune response (7–14 days).
Dose normalization
Body surface area with a dose cutoff is used for dose normalization in chemotherapy whereas body weight is used for radio-immunotherapy. The volume of interaction would be the preferred quantity as it determines drug concentration. The lean body mass can be readily measured and may be a more practical parameter to determine the injected dose for each patient.
MTD
Alpha-emitting drugs are in human clinical development for several different solid cancer types and leukemia. However, MTDs need to be established in most cases. This has been determined with 213Bi-AIC for acute AML at ~1 mCi/kg.7 For intra-cavity therapy of GBM, it is ~0.14 mCi/kg for 211At-IC.55 The MTD has not yet been established for metastatic melanomas, but the efficacious dose for some melanomas is certainly less than 0.3 mCi/kg.54
Palliation
Only one alpha drug, Xofigo® (Alpharadon, radium-223 chloride), has been registered so far in the US, demonstrating the clinical feasibility of TAT. Xofigo shows a favourable efficacy profile and good tolerance but is not a cancer targeting drug. Rather, radium is a chemical analogue and is taken up by osteoblasts as they repair bone damage by cancer cells.
Therapeutic efficacy
The next stage is to determine efficacy in Phase II trials at the MTD or the effective dose. A current study of combined modalities for AML is ongoing at Memorial Sloan Kettering Cancer Center using chemotherapy to reduce the cancer load followed by 213Bi or 225Ac-AIC to further reduce the cancer. The melanoma trial showed significant efficacy in the Phase I trial achieving 10% near complete or partial response, 40% stable disease, and 13% long-term survival of 2–5 years, without any evidence of adverse events. The Duke study achieved a median survival of 52 weeks for GBM.55
It is now appropriate TAT be applied to high risk subjects in remission, for whom recurrence is likely to arise from subclinical disease, as well as end-stage patients for palliative therapy. Perhaps the most appropriate application could be for post-hormonal therapy in prostate cancer, when the PSA is at its nadir. TAT could target the surviving hormone insensitive cells that lead to fatal outcomes. Evidence for this application is already available with 177Lu-HuJ591, which achieved this result with low energy beta emission.51,65
TAVAT
The metastatic melanoma clinical trial showed surprising tumor regressions at doses very much below the MTD and if further research is successful, TAVAT could change the prognosis for many end-stage cancers. The demonstration of the synergy between tumor vascular disruption agents and TAVAT could bring about a sea change in cancer therapy.
Systemic dosimetry
This ongoing problem could be resolved with biological dosimetry using micronuclei technology. Evaluation of systemic therapeutic response requires measurement of cancer cells in transit, yet to be established.
RI supply
The limited supplies of Ac-225 available at present from separation from Th-229 are adequate for clinical trials. However, should TAT become a clinical procedure, then new supplies must be found. Accelerator production centers could ensure adequate supply of Ac-225 for international distribution and At-211 for local distribution.
Future prospects
These have been described previously60 and were recently reviewed at the International Atomic Energy Agency Technical Meeting on Alpha Emitting Radionuclides and Radiopharmaceuticals for Therapy, Vienna, 2013. The Meeting63 concluded that:
[…] TAT will play an important role in the treatment of disseminated, chemoresistant and radioresistant metastatic disease, against which there are no efficacious treatment options. Combined with other therapies, including chemotherapy, signalling pathway inhibitors and even targeted beta emitting radionuclides, TAT is likely to improve survival.63
Alpha therapy is still a work in progress with translation of preclinical studies to clinical trials. Ideally suited to leukemia, alpha therapy is demonstrating efficacy, but at the MTD. However, efficacy for TAT of GBM and metastatic melanoma has been found well below the MTD.
Disclosure
The authors report that there are no conflicts of interest in this work.
References
Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–497. | |
Goldenberg DM. Targeted therapy of cancer with radiolabeled antibodies. J. Nucl Med. 2002;43(5):693–713. | |
Brons S, Jakob B, Taucher-Scholz G, Kraft G. Heavy ion production of single- and double-strand breaks in plasmid DNA in aqueous solution. 2001. Phys Med. 17 Suppl 1:217–218. | |
Kerr JF. A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J. Pathol. Bacteriol. 1965;90(2):419–435. | |
Elgqvist J. Targeted Alpha Therapy. Current Radiopharmaceuticals. 2011;4(3):176. | |
Sgouros G. Radioimmunotherapy of micrometastases: sidestepping the solid-tumor hurdle. J Nucl Med. 1995;36(10):1910–1912. | |
Jurcic GJ, Larson SM, Sgouros G, et al. Targeted αparticle immunotherapy for myeloid leukemia. Blood. 2002;100(4):1233–1239. | |
Allen BJ, Raja C, Rizvi S, Song EY, Graham P. Tumour anti-vascular alpha therapy: a mechanism for the regression of solid tumours in metastatic cancer. Phys Med Biol. 2007;52(13):L15–L19. | |
Raja C, Graham P, Rizvi SM, et al. Interim analysis of toxicity and response in phase 1 trial of systemic targeted alpha therapy for metastatic melanoma. Cancer Biol Ther. 2007;6(6):846–852. | |
Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999; 59(13):3192–3198. | |
Pandit-Taskar N, Lason SM, Carrasquillo JA. Bone-seeking radiopharmaceuticals for treatment of osseous metastases, Part 1: α therapy with 223Ra-dichloride. J Nucl Med. 2014;55(2):268–274. | |
Martin VM, Siewert C, Scharl A, et al. Immunomagnetic enrichment of disseminated epithelial tumour cells from peripheral blood by MACs. Exp Hematol. 1998;26(3):252–264. | |
Wang J, Abbas Rizvi SM, Madigan MC, et al. Control of prostate cancer spheroid growth using 213Bi-labeled multiple targeted alpha immunoconjugates. Prostate. 2006;66(16):1753–1767. | |
Song YJ, Qu CF, Rizvi SMA, et al. Cytotoxicity of PAI2, C595 and Herceptin vectors labeled with the alpha-emitting radioisotope Bismuth-213 for ovarian cancer cell monolayers and clusters. Cancer Lett. 2006;234(2):176–183. | |
Allen BJ, Raja C, Rizvi S, et al. Intralesional targeted alpha therapy for metastatic melanoma. Cancer Biol Ther. 2005;4(12):1318–1324. | |
Azzam EI, Little JB. The radiation-induced bystander effect: evidence and significance. Hum Exp Toxicol. 2004;23(2):61–65. | |
Allen, BJ. Internal high linear energy transfer (LET) targeted radiotherapy for cancer. Phys Med Biol. 2006;51(13):R327–R341. | |
Song EY, Rizvi SMA, Qu CF, et al. Pharmacokinetics and toxicity of 213Bi-labelled PAI2 in preclinical targeted alpha therapy for cancer. Cancer Biol Therapy. 2007;6(6):898–904. | |
Dahle J, Jonasdottir TJ, Heyerdahl H, et al. Assessment of long-term radiotoxicity after treatment with the low-dose-rate alpha-particle-emitting radioimmunoconjugate (227)Th-rituximab. Eur J Nucl Med Mol Imaging. 2010;37(1):93–102. | |
Huang CY, Oborn BM, Guatelli S, Allen BJ. Monte Carlo calculation of the maximum therapeutic gain of tumor antivascular alpha therapy. Med Phys. 2012;39(3):1282–1288. | |
Seidl C, Zöckler C, Beck R, Quintanilla-Martinez L, Bruchertseifer FR. 177Lu-immunotherapy of experimental peritoneal carcinomatosis shows comparable effectiveness to 213Bi-immunotherapy, but causes toxicity not observed with 213Bi. Eur J Nucl Med Mol Imaging. 2011;38(2):312–322. | |
Wild D, Frischnknect M, Zhang H, et al. Alpha- versus beta-particle radiopeptide therapy in a human prostate cancer model (213Bi-DOTA-PESIN and 213Bi-AMBA versus 177Lu-DOTA-PESIN). Cancer Res. 2011;71(3):1009–1018. | |
Song EY, Rizvi SM, Qu CF, et al. The cytokinesis–block micronucleus assay as a biological dosimeter for targeted alpha therapy. Phys Med Biol. 2008;53(2):319–328. | |
Huang CY, Guatelli S, Oborn B, Allen BJ. Background dose for systemic targeted alpha therapy. Prog Nucl Sci Tech. 2011;2:187–190. | |
Sgouros G, Roeske JC, McDevitt MR, et al. MIRD Pamphlet No 22 (abridged): radiobiology and dosimetry of alpha-particle emitters for targeted radionuclide therapy. J Nucl Med. 2010;51(2):311–328. | |
Allen BJ, So T, Rizvi SMA, Song EY, Fernandez HR, Lutz-Mann L. Mutagenesis induced by targeted alpha therapy using 213Bi-cDTPA-9.2.27 in lacZ transgenic mice. Cancer Biol Therapy. 2009;8(9):777–781. | |
Allen BJ. Can alpha-immunotherapy succeed where other systemic modalities have failed? Nucl Med Commun. 1999;20(3):205–207. | |
Allen BJ. Clinical trials of targeted alpha therapy for cancer. Rev Recent Clin Trials. 2008;3(3):185–191. | |
McDevitt MR, Ma D, Lai LT, et al. Tumor therapy with targeted atomic nanogenerators. Science. 2001;294(5546):1537–1540. | |
Finn RD, McDevitt MR, Scheinberg DA, et al. Refinements and improvements for Bi-213 production and use as a targeted therapeutic radiopharmaceutical. J Labelled Compds Radiopharm. 1997;40:293. | |
Abbas Rizvi SM, Sarkar S, Goozee G, Allen BJ. Radioimmunoconjugates for targeted alpha therapy of malignant melanoma. Melanoma Res. 2000;10(3):281–289. | |
Allen BJ, Rizvi SM, Tian, Z. Preclinical targeted alpha therapy for subcutaneous melanoma. Melanoma Res. 2001;11(2):175–182. | |
Abbas Rizvi SMA, Henniker AJ, Goozee G, Allen BJ. In vitro testing of the leukaemia monoclonal antibody WM-53 labeled with alpha and beta emitting radioisotopes. Leuk Res. 2002;26(1):37–43. | |
Rizvi SM, Allen BJ, Tian Z, Goozee G, Sarkar S. In vitro and preclinical studies of targeted alpha therapy (TAT) for colorectal cancer. Colorectal Dis. 2001;3(5):345–353. | |
Li Y, Rizvi SM, Ranson M, Allen BJ. 213Bi-PAI2 conjugate selectively induces apoptosis in PC3 metastatic prostate cancer cell line and shows anti-cancer activity in a xenograft animal model. Br J Cancer. 2002;86(7):1197–1203. | |
Li Y, Rizvi SMA, Brown J, et al. Antigenic expression of human prostate cancer cell lines for in vitro multiple-targeted a-therapy with 213Bi-conjugates. Int J Radiat Oncol Biol Phys. 2004;60(3):896–908. | |
Li Y, Cozzi PJ, Qu CF, et al. Cytotoxicity of human prostate cancer cell lines in vitro and induction of apoptosis using 213Bi-Herceptin alpha-conjugate. Cancer Letters. 2004;205(2):161–171. | |
Kneifel S, Cordier D, Good S, et al. Local targeting of malignant gliomas by the diffusible peptidic vector 1,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid-substance p. Clin Cancer Res. 2006;12(12):3843–3850. | |
Park SI, Shenoi J, Pagel JM, et al. Conventional and pretargeted radioimmunotherapy using bismuth-213 to target and treat non-Hodgkin lymphomas expressing CD20: a preclinical model toward optimal consolidation therapy to eradicate minimal residual disease. Blood. 2010;116(20):4231–4239. | |
Qu CF, Song EY, Li Y, et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by 213Bi-C595 radioimmunoconjugate. Brit J Cancer. 2004:91;2086–2093. | |
Qu CF, Song EY, Li Y, et al. Pre-clinical Study of 213Bi Labeled PAI2 for the Control of Micrometastatic Pancreatic Cancer. Clin Exp Metastasis. 2005;22(7):575–586. | |
Allen BJ, Rizvi SM, Qu CF, Smith RC. Targeted alpha therapy approach to the management of pancreatic cancer. Cancers (Basel). 2011;3(2):1821–1843. | |
Qu CF, Song YJ, Li Y, et al. In Vivo and In Vitro Inhibition of Pancreatic Cancer Growth by Targeted Alpha Therapy using 213Bi-CHX.A”-C595. Cancer Biol Therapy. 2005;4(8):e31–e36. | |
Andersson H, Cederkrantz E, Bäck T, et al. Intraperitoneal alpha-particle radioimmunotheapy of ovarian cancer patients: pharmacokinetics and dosimetry of (211)At-MX35 F(ab’)2 – a phase 1 study. J Nucl Med. 2009;50(7):1153–1160. | |
Rizvi S, Allen B, Lee C, et al. Orthotopic administration of (213)Bi-bevacizumab inhibits progression of PC3 xenografts in the prostate. Immunotherapy. 2012;4(5):549–554. | |
Song EY, Qu CF, Rizvi SM, et al. Bismuth-213 radioimmunotherapy with C595 anti–MUC1 monoclonal antibody in an ovarian cancer ascites model. Cancer Biol Ther. 2008;7(1):76–80. | |
Qu CF, Li Y, Song YJ, et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by (213)Bi-C595 radioimmunoconjugate. Br J Cancer. 2004;91(12):2086–2093. | |
Qu CF, Song YJ, Li Y, Rizvi SM, et al. In vivo and in vitro inhibition of pancreatic cancer growth by targeted alpha therapy using 213Bi-CHX.A”-C595. Cancer Biol Ther. 2005;4(8):848–853. | |
Allen BJ, Tian Z, Rizvi SM, Li Y, Ranson M. Preclinical studies of targeted alpha therapy for breast cancer using 213Bi-labelled-plasminogen activator inhibitor type 2. Brit J Cancer. 2003;88(6):944–950. | |
Li Y, Tian Z, Rizvi SM, Bander NH, Allen BJ, In vitro and preclinical targeted alpha therapy of human prostate cancer with Bi-213 labeled J591 antibody against the prostate specific membrane antigen. Prostate Cancer and Prostatic Dis. 2002;5(1):36–46. | |
Tagawa ST, Milowsky MI, Morris M, et al. Phase II study of Lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for metastatic castration-resistant prostate cancer. Clin Cancer Res. 2013;19(18):5182–5191. | |
Jurcic J, Rosenblat TL, McDevitt MR, et al. Alpha-particle immunotherapy for acute myeloid leukemia with Bismuth-213 and actinium-225. Presented at: 7th Symposium on targeted Alpha Therapy; July 17–19, 2011; Berlin. | |
Allen BJ, Raja C, Rizvi SMA, Graham P, Kearsley JH. New Directions for Clinical Trials of Targeted Alpha Therapy for Metastatic Melanoma. Current Radiopharmaceuticals. 2008;1(3):240–250. | |
Allen BJ, Singla AA, Rizvi SM, et al. Analysis of patient survival in a Phase 1 trial of systemic targeted α-therapy for metastatic melanoma. Immmunotherapy. 2011;3(9):1041–1050. | |
Zalutsky MR, Reardon DA, Akabani G, et al. Clinical experience with the alpha-particle emitting 211At: treatment of recurrent brain tumours patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med. 2008;49(1):30–38. | |
Cordier D, Forrer F, Bruchertseifer F, et al. Targeted alpha-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTA-[Thi8,Met(O2)11]-substance P: a pilot trial. Eur J Nucl Med Mol Imaging. 2010;37(7):1335–1344. | |
Nilsson S, Larsen RH, Fosså SD, et al. First clinical experience with alpha-emitting radium-223 in the treatment of skeletal metastases. Clin Cancer Res. 2005;11(12):4451–4459. | |
Nilsson S, Franzén L, Parker C, et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: a randomised, multicentre, placebo-controlled phase II study. Lancet Oncol. 2007;8(7):587–594. | |
Schmidt D, Neumann F, Antke C, et al. Phase 1 Clinical Study on Alpha-Therapy for Non-Hodgkin Lymphoma. In: 4th Alpha-Immunotherapy Symposium; June 28–29, 2004. Dusseldorf, Germany. pp.12. | |
Allen BJ. Future prospects for targeted alpha therapy. Curr Radiopharm. 2011;4(4):336–342. | |
Kratochwil C, Giesel FL, Bruchertseifer F, et al. Dose escalation study of peptide receptor alpha-therapy with arterially administered 213Bi-DOTATOC in GEP-NET patients refractory to beta-emitters. Presented at: Ann Congress European Assoc Nuclear Medicine; October 15–19, 2011; Birmingham. | |
Rosenblat TL, McDevitt MR, Mulford DA, et al. Sequential cytarabine and alpha-particle immunotherapy with bismuth-213-lintuzumab (HuM195) for acute myeloid leukemia. Clin Cancer Res. 2010;16(21):5303–5311. | |
International Atomic Energy Agency. Alpha Emitting Radionuclides and Radiopharmaceuticals for Therapy. Meeting Report, International Atomic Energy Agency; June 24–28, 2013; Vienna. Available from: http://www-naweb.iaea.org/napc/iachem/working_materials.html. Accessed May 27, 2014. | |
Wilbur DS. Chemical and radiochemical considerations in radiolabeling with α-emitting radionuclides. Curr Radiopharm. 2011;4(3):214–247. | |
Prostate Cancer Foundation; Research News: Moving the Dial to Cure; can Targeted Radiotherapy Actually Eradicate Tumors in Men with Advanced Prostate Cancer? Available from: http://www.pcf.org/site/c.leJRIROrEpH/b.8922409/k.8A41/Moving_the_Dial_to_Cure_Can_Targeted_Radiotherapy_actually_eradicate_tumors_in_men_with_advanced_prostate_cancer.htm?msource=jan14np. Accessed May 27, 2014. | |
Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213–223. | |
Nilsson S, Strang P, Aksnes AK, et al. A randomized, dose–response, multicenter phase II study of radium-223 chloride for the palliation of painful bone metastases in patients with castration-resistant prostate cancer. Eur J Cancer. 2012;48(5):678–686. | |
Parker CC, Pascoe S, Chodacki A, et al. A randomized, double-blind, dose-finding, multicenter, phase 2 study of radium chloride (Ra 223) in patients with bone metastases and castration-resistant prostate cancer. Eur Urol. 2013;63(2):189–197. | |
Carrasquillo JA, O’Donoghue JA, Pandit-Taskar N, et al. Phase I pharmacokinetic and biodistribution study with escalating doses of ßRa-dichloride in men with castration-resistant metastatic prostate cancer. Eur J Nucl Med Mol Imaging. 2013;40(9):1384–1393. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.