")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Tanshinone IIA Ameliorates Progression of CAD Through Regulating Cardiac H9c2 Cells Proliferation and Apoptosis by miR-133a-3p/EGFR Axis
Authors Xu H, Li H, Zhu P, Liu Y, Zhou M, Chen A
Received 14 January 2020
Accepted for publication 16 June 2020
Published 20 July 2020 Volume 2020:14 Pages 2853—2863
DOI https://doi.org/10.2147/DDDT.S245970
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Hong Xu,* Haiqing Li,* Pengxiong Zhu, Yun Liu, Mi Zhou, Anqing Chen
Department of Cardiac Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200020, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Anqing Chen
Department of Cardiac Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200020, People’s Republic of China
Email [email protected]
Background: Coronary artery disease (CAD) leads to the highest mortality worldwide, seriously threatening human health. Tanshinone IIA (Tan IIA), which could be extracted from Danshen, is applied in the treatment of cardiovascular and cerebrovascular diseases. MicroRNAs (miRNAs, miRs) play pivotal roles in cell proliferation and cell apoptosis of the cardiovascular system. The aim of the present study was to explore the role of Tan IIA in CAD in vitro and the underlying molecular mechanism.
Methods: Real-time polymerase chain reaction (RT-PCR) and Western blot were used for the detection of miRNA/mRNA and protein, respectively. Target genes of miR-133a-3p were searched in TargetScan, and the targeting relationship was verified by dual-luciferase reporter assay. Cell proliferation was determined using a Cell Counting Kit-8 (CCK-8) and EdU labeling. Cell apoptosis was detected by flow cytometry and TUNEL staining.
Results: In the present study, lower miR-133a-3p level and higher epidermal growth factor receptor (EGFR; the target of miR-133a-3p) level were found in H2O2-induced H9c2 cells. In addition, Tan IIA upregulated miR-133a-3p and downregulated EGFR expression. Moreover, Tan IIA promoted cell proliferation and suppressed apoptosis and enhanced G0/G1, which was reversed by miR-133a-3p inhibitor, while siRNA-EGFR abolished the effects induced by miR-133a-3p in H2O2-induced H9c2 cells.
Conclusion: Tan IIA reversed H2O2-induced cell proliferation reduction, cell apoptosis induction, and G0/G1 arrest reduction in H9c2 cells by miR-133a-3p/EGFR axis. The findings suggested a potential molecular basis of Tan IIA in treating patients with CAD.
Keywords: tanshinone IIA, coronary artery disease, miR-133a-3p, EGFR
Introduction
Coronary artery disease (CAD) leads to the highest mortality globally, which seriously threatens human health.1 Meanwhile, CAD remains the leading cause of cardiovascular deaths (CVD) worldwide.2 The mortality of CAD has been mounted in the past decades, which is estimated to cause >80% new cases in underdevelopment and developing countries,3 and accounts for 11.1 million deaths in 2020 globally.4 The onset of CAD is correlated with age, tobacco/meat consumption and obesity.5 Myocardial cell apoptosis is the common characteristic during the pathological processes of cardiovascular diseases, with the underlying molecular mechanisms remaining to be expounded.6 Consequently, medical therapies aiming to preserve heart function by mitigating myocardial cell apoptosis are needed to be further intensively investigated.
Tanshinone IIA (Tan IIA; C19H18O3) is a compound that isolated from a Traditional Chinese Medicine (TCM) – Danshen (the root of Salviae miltiorrhizae).7,8 In East Asia, Tan IIA is widely applied in treating cardiovascular and cerebrovascular diseases, such as myocardial infarction, hypertension, acute ischemic stroke, etc.9,11 Moreover, Tan IIA exerts numerous biological activities in the cardiovascular system, possessing anti-apoptotic, anti-oxidant and anti-coagulant properties.12,13 Despite the cardioprotective roles of Tan IIA have been explored for multiple years, the underlying molecular mechanisms remain to be complicated.
miRNAs (miRs) are a group of small non-coding RNAs (20–23 nucleotides), which are highly conserved and widely expressed in eukaryotes.14 miRs degrade target mRNAs or inhibit the translation of target mRNAs by binding to there 3ʹUTR.15 In the cardiovascular system, miRs regulate the biological and cellular processes including cell proliferation and cell apoptosis.16 Tan IIA was reported to protect H9c2 cells from apoptosis by elevating miR-133 level.17,18 However, the molecular mechanism underlying miR-133 is still unclear.
In the present study, we aimed to investigate whether there are molecules that can be targeted by miR-133 and regulated by Tan IIA in CAD. We found Tan IIA will attenuate CAD through miR-133a-3p/EGFR axis in vitro experiment. The findings may provide a theoretical basic for Tan IIA treatment of CAD.
Materials and Methods
RT-qPCR
Total RNA was extracted from H9c2 cells by TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA concentration was detected by NanoDrop2000 spectrophotometer. Afterwards, RNA was subjected to reverse transcribed to obtain complementary deoxyribose nucleic acids (cDNAs) by Primescript RT Reagent (TaKaRa, Otsu, Shiga, Japan). cDNA was amplified by qPCR with SYBR®Premix Ex Taq™ (TaKaRa) following the conditions as listed: 94°C for 30 s, 55°C for 30 s and 72°C for 90 s, for 40 cycles. Relative levels of genes were quantified by 2−ΔΔCt method.19 Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were used as internal references for mRNA and miRNA, respectively.
Western Blot
Total protein was extracted from H9c2 cells by radioimmunoprecipitation assay (RIPA, Roche, Shanghai, China). Protein concentration was determined by the method of bicinchoninic acid (BCA, Beyotime, Shanghai, China). Then, protein (20 μg) was separated by 8% electrophoresis, followed by transferring onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5% skimmed milk at room temperature for 2 h, the PVDF membranes were incubated with primary antibodies (Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight and secondary antibody at room temperature for 1 h, successively. Protein bands were exposed by enhanced chemiluminescence (ECL) and quantified by Image Software (NIH, Bethesda, MD, USA). The information of primary antibodies were showed as follows: anti-EGFR (#4267; 1:1000); anti-p21 (#2947; 1:1000); anti-p53 (#9282; 1:1000); anti-cleaved caspase-3 (#9661; 1:1000); anti-caspase-3 (#9662; 1:1000); anti-Bax (#2774; 1:1000); anti-Bcl-2 (#4223; 1:1000); anti-cyclin D1 (#2922; 1:1000); anti-CDK4 (#12790; 1:1000) and anti-β-actin (#8457; 1:1000). The secondary antibody is anti-rabbit IgG, HRP-linked antibody (#7074; 1:2000; Cell Signaling Technology).
Cell Culture
293T cells were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). Cardiac H9c2 cells are obtained and authenticated by the American Type Culture Collection (ATCC). 293T and H9c2 cells were incubated in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Sigma-Aldrich) in a humidified incubator at 37°C with 5% CO2.
H9c2 cells were starved of serum in DMEM supplemented with 1% FBS for 24 h at 37°C, followed by treatment with H2O2 (400 μM), Tan IIA (3 μM), or in combination for 24 h at 37°C prior to the subsequent experiments.
Cell Transfection
H9c2 cells (107cells/mL) in the logarithmic growth phase at 70–80% confluence were seeded into 6-well plates and subjected to transfection. miR-NC inhibitor, miR-133a-3p inhibitor, siRNA-NC or siRNA‐EGFR (100 pM, GenePharma, Shanghai, China) and LipofectamineTM 2000 (5 µL, Invitrogen, Carlsbad, CA, USA) were added into Opti-MEM (250 µL, Gibco). After incubation for 20 min, the mixture was added into each well and incubated for another 24 h at 37°C prior to the subsequent experiments. The transfection efficiency was determined by using RT-qPCR.
Target mRNA Prediction
The target mRNAs of miR-133a-3p were searched and selected in TargetScan (https://www.targetscan.org) for the further functional and mechanistic verification in the present study.
Dual-Luciferase Reporter Assay
The EGFR 3ʹUTR (forward, 5ʹ-CCgTCTAgATCTTTTgCTCTCTCTTgCTCTC-3ʹ; reverse, 5ʹ-AgCTCTAgAACggATAAACAgTAgCACCAA-3ʹ) was cloned into the XbaI site of the luciferase reporter pGL3 plasmid (Promega, Madison, WI, USA). The EGFR mutant (MUT) was obtained by the QuikChange Site-Directed Mutagenesis Kit (Stratagene, California, USA). As for the reporter assay, miR-133a-3p mimics or miR-NC mimics and reporter plasmids (pGL3-EGFR-WT or pGL3-EGFR-MUT) were co-transfected into 293T cells by lipofectamine 2000 (Life Technologies, Darmstadt, Germany). The relative activity was measured by the Dual-Luciferase Report Assay System (Promega). Luciferase activity was normalized to Rellina luciferase activity.
Cell Proliferation Assay
H9c2 cell proliferation was detected by Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan). In brief, cells were cultured in 96-well plates for 24 h. Afterward, CCK-8 reagent (10 µL) was added to each well and incubated for another 2 h. Thereafter, the cell proliferation rate was detected by a VICTOR X multi-label reader (PerkinElmer, Inc., Waltham, MA, USA) with a wavelength of 450 nm.
EdU Labeling
H9c2 cells were plated into a 12-well plate and recovered overnight in a humidified incubator at 37°C with 5% CO2, followed by serum starvation for 24 h in the same incubation condition. Afterward, cells were exposed to serum for 24 h, during which cells were treated with EdU (20 uM) from the Click-iT EdU Alexa Fluor 488 Imaging Kit (Life technologies, Carlsbad, CA) at the last 3 h. After then, cells were fixed and permeabilized by 4% PFA and 0.5% Triton-X, followed by incubation with Click-iT reaction cocktail (including CuSO4 and Alexa Fluor Azide), and nuclear staining by DAPI. Images were captured by a fluorescence microscope (Magnification: ×40; Leica, Leica Microsystems GmbH), and data were analyzed by ImageJ software (NIH).
Flow Cytometry
At 48 h after transfection, H9c2 cells were harvested, re-suspended in 500 μL binding buffer (1×106/mL), and incubated with Annexin V-FITC (5 μL, fluorescein isothiocyanate) and Propidium Iodide (5 μL, PI) from a FITC Annexin-V Apoptosis Detection Kit I (Becton-Dickinson Biosciences, San Jose, CA, USA) for 30 min in the dark at room temperature. For cell cycle, Cycletest PLUS DNA Reagent Kit (Becton-Dickinson Biosciences) was used. Cell apoptosis and cyclin were determined by FACScan flow cytometer (Becton-Dickinson Biosciences) and analyzed by Cell Quest software (Becton-Dickinson Biosciences).
TUNEL Staining
The H9c2 cell apoptosis was detected by TUNEL staining. In brief, at 24 h after transfection, cells were harvested and fixed with 4% PFA for 30 min at room temperature. Afterward, cells were incubated by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay kit (Roche, Switzerland). Nuclei were stained by DAPI at 37°C for 15 min. Next, the apoptotic cells were captured by a confocal microscope (Magnification: ×40; Leica) and digitized by LAS AF Lite software (versions 2.0; Leica). The apoptotic cells were exerted to be green color, while the nuclei were blue.
Statistics
Results were obtained from at least three independent experiments. Data were analyzed by GraphPad Prism 6 and expressed as mean ± standard deviation (SD). The Student’s t-test was used for the analysis of the difference between 2 groups. One-way analysis of variance (ANOVA) followed by Turkey’s multiple comparisons test was used for the analysis of differences among 3 or more groups. P<0.05 was considered a statistically significant difference.
Results
miR-133a-3p Targeted EGFR 3ʹ-UTR
To explore the underlying molecular mechanisms, we carried out bioinformatics analysis and dual-luciferase reporter assay. Binding sites for miR-133a-3p were found at EGFR 3ʹ-UTR (Figure 1A). Moreover, compared with miR-133a-3p mimics NC, miR-133a-3p mimics significantly decreased the luciferase activity of reporter plasmid for EGFR-WT 3ʹ-UTR but not EGFR-MUT 3ʹ-UTR (Figure 1B, p<0.01).
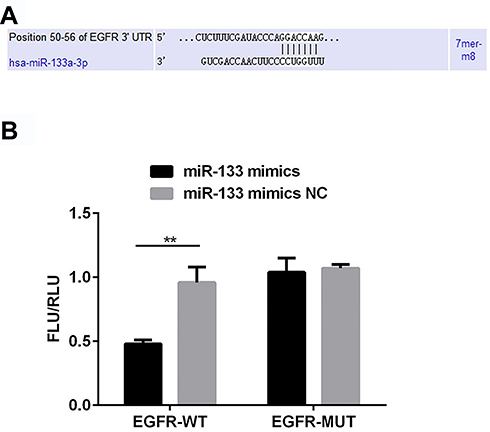 |
Figure 1 miR-133a-3p targets EGFR 3ʹ-UTR. There were binding sites for miR-133a-3p on EGFR 3ʹ-UTR (A). miR-133a-3p mimics significantly decreased the luciferase activity when co-transfected with pGL3-EGFR-WT (B). **p<0.01. |
miR-133a-3p Was Decreased and EGFR Was Increased in H2O2-Induced H9c2 Cells
Compared with the control group, miR-133a-3p level was significantly decreased by H2O2, which was reversed by Tan IIA in H9c2 cells (Figure 2A, p<0.001). Inversely, compared with the control group, the EGFR level was significantly increased by H2O2, which was reduced by Tan IIA in H9c2 cells (Figure 2B, p<0.001).
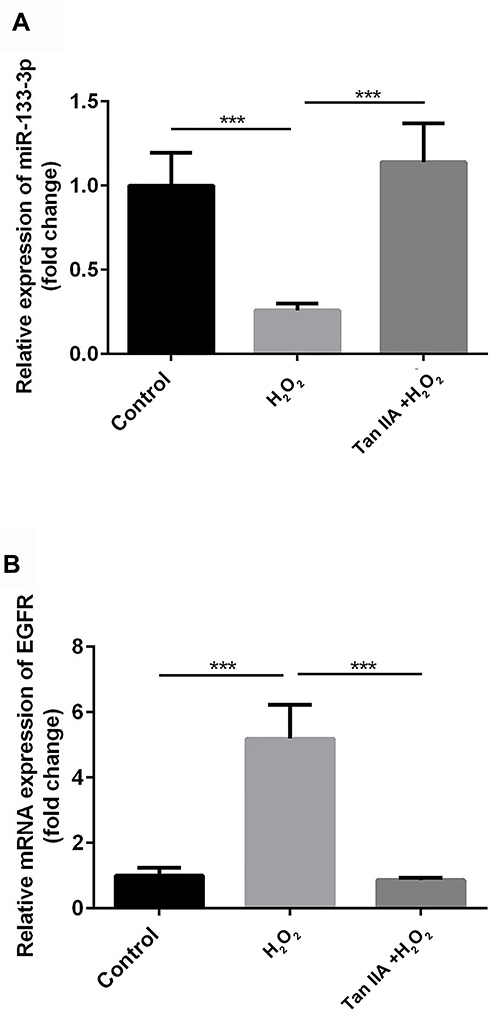 |
Figure 2 miR-133a-3p was decreased and EGFR was increased in H2O2-induced H9c2 cells. Compared with control group, miR-133a-3p level was significantly lower in H2O2 group, which was reversed by Tan IIA (A). Compared with control group, EGFR level was significantly increased by H2O2, which was reduced by Tan IIA in H9c2 cells (B). ***p<0.001. |
miR-133a-3p Inhibitor Increased the Expression of EGFR, and Rescued the Downregulation of EGFR Level Caused by Transfection of siRNA-EGFR in H9c2 Cells
Compared with the control group, there was no significant difference in miR-133a-3p level in the miR-NC inhibitor group, which was significantly inhibited by miR-133a-3p inhibitor (Figure 3A, p<0.001). Additionally, compared with the control group, the expression of EGFR was significantly reduced by transfection of siRNA-EGFR-1 and siRNA-EGFR-2 at mRNA or protein level, especially in siRNA-EGFR-2 group. Therefore, siRNA-EGFR-2 was used in the subsequent experiments (Figure 3B, p<0.001). Furthermore, compared with miR-NC inhibitor group, there was a significant increase expression of EGFR due to inhibition of miR-133a-3p at both mRNA (Figure 4A, p<0.01) and protein (Figure 4B, p<0.01) levels. However, EGFR expression was deceased in miR-133a-3p inhibitor +siRNA-EGFR group, compared with miR-133a-3p inhibitor group (Figure 4 A and B, mRNA: p<0.01; protein: p<0.05). As compared to miR-133a-3p inhibitor +siRNA-EGFR group, the downregulation of EGFR expression was found by the transfection of siRNA-EGFR (Figure 4 A and B, p<0.01).
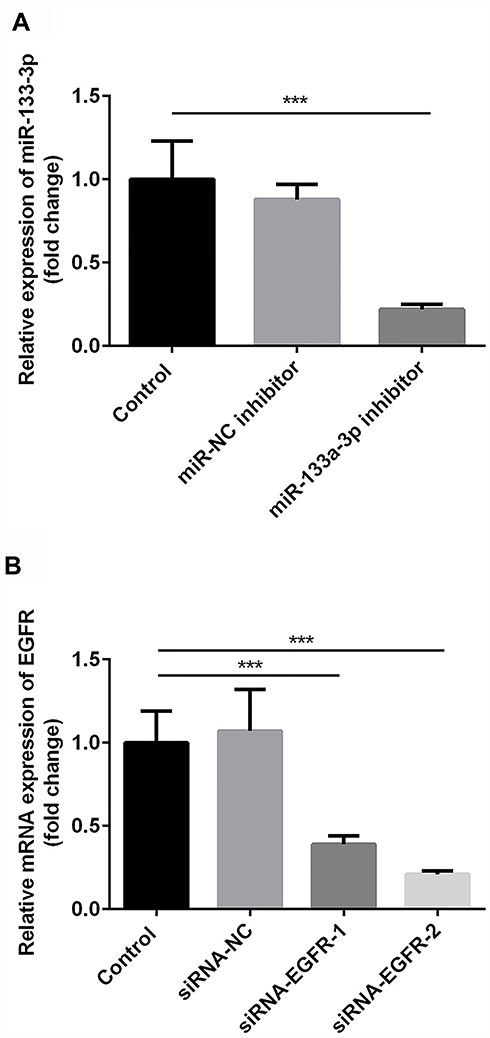 |
Figure 3 The successful transfection of miR-133a-3p inhibitor and siRNA-EGFR. Compared with control group, miR-133a-3p level was significantly inhibited by miR-133a-3p inhibitor but not miR-NC inhibitor (A). Compared with control group, EGFR level was significantly reduced by siRNA-EGFR-1and siRNA-EGFR-2 but not siRNA-NC (B). ***p<0.001. |
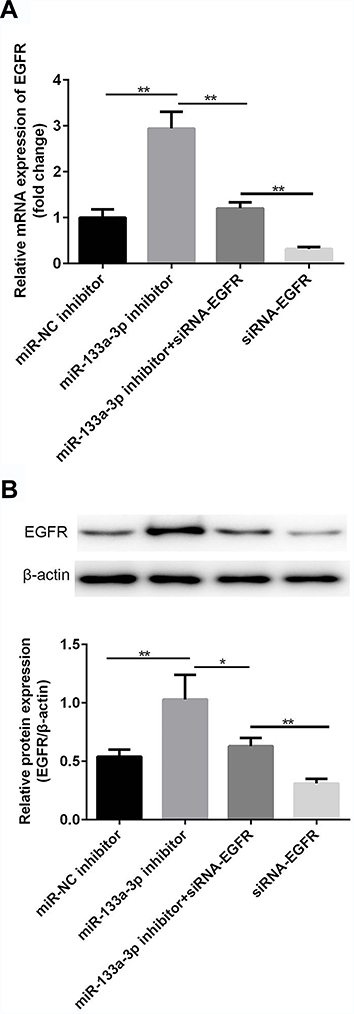 |
Figure 4 miR-133a-3p inhibitor negatively regulated and siRNA-EGFR positively regulated the expression of EGFR. Compared with miR-NC inhibitor group, a significant increase of mRNA level of EGFR was found in miR-133a-3p inhibitor group, which was reversed by siRNA-EGFR (A). Compared with miR-NC inhibitor group, protein level of EGFR was upregulated by inhibition of miR-133a-3p, which was reversed by siRNA-EGFR (B). **p<0.01, *p<0.05. |
Tan IIA Improved Cell Viability in H9c2 Cells Subjected to H2O2-Induced Injury by Regulating miR-133a-3p/EGFR Axis
Next, cells were divided into 5 different groups: A, Control group; B, H2O2; C, Tan IIA+ H2O2; D, Tan IIA+miR-133a-3p inhibitor+ H2O2; E, Tan IIA+miR-133a-3p inhibitor +siRNA-EGFR + H2O2. Results of CCK-8 (Figure 5A) and EdU (Figure 5B and C) staining exhibited that, compared with the control group, H9c2 cell proliferation was significantly decreased by H2O2 (p<0.01), which was rescued by Tan IIA (p<0.05). In addition, the effect of Tan IIA on proliferation was partially attenuated by miR-133a-3p inhibitor (p<0.05), while knockdown of EGFR abolished the effects induced by inhibiting miR-133a-3p (p<0.05). Consistently, the expressions of p21 and p53 showed the opposite trends to cell proliferation (Figure 5D and E, p<0.01 or p<0.001).
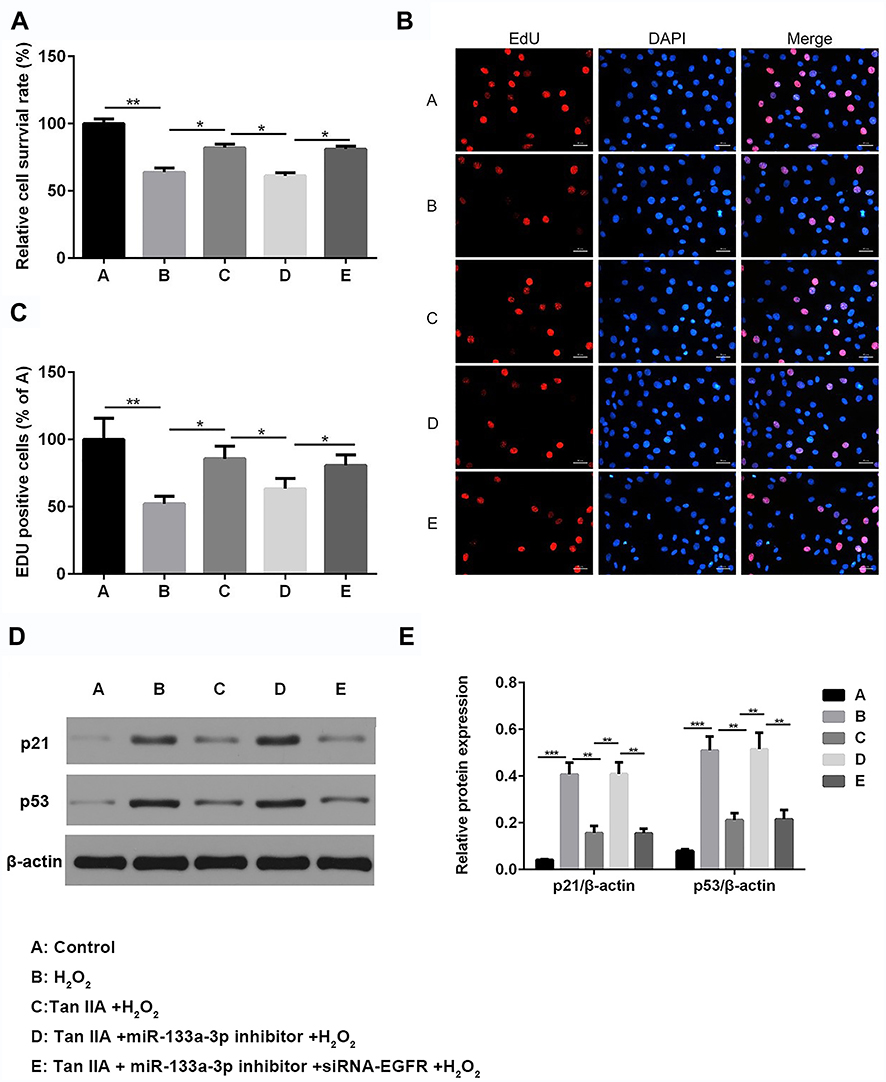 |
Figure 5 Tan IIA reversed H2O2-induced H9c2 cell proliferation reduction by miR-133a-3p/EGFR axis. Results of CCK-8 (A) analysis showed Tan IIA reversed H2O2-induced H9c2 cell proliferation reduction, which was partially attenuated by miR-133a-3p inhibitor but not miR-133a-3p inhibitor+siRNA-EGFR (A). *p<0.05 and **p<0.01. Results of EdU staining showed that Tan IIA reversed cell proliferation decreased in H2O2-induced H9c2 cells, which was partially rescued by miR-133a-3p inhibitor but not in miR-133a-3p inhibitor+siRNA-EGFR group (B). Quantified results of B (C). *p<0.05 and **p<0.01. Western blot analysis indicated that the expressions of p21 and p53 showed the opposite trends (D). Quantified results of D (E). **p<0.01 and ***p<0.001. |
Tan IIA Protected H9c2 Cells from H2O2-Induced Apoptosis and G0/G1 Phase Arrest Reduction by Regulating miR-133a-3p/EGFR Axis
Results of flow cytometry (Figure 6) and TUNEL staining (Figure 7) exhibited that, compared with the control group, H9c2 cells apoptosis was significantly increased by H2O2, which was reversed by Tan IIA. In addition, cell apoptosis was attenuated by miR-133a-3p inhibitor in Tan IIA treated cells, compared with Tan IIA alone treatment group, which was rescued by EGFR knockdown. The expression of cleaved caspase-3 and Bax was similar to that of cell apoptosis, while Bcl-2 showed the opposite trends to cell apoptosis (Figure 8). However, the expression level of caspase-3 was not affected by Tan IIA treatment(Figure 8). Moreover, for the cell cycle, compared to the control group, H2O2 significantly decreased the cellular population at the G0/G1 phase, which was reversed by Tan IIA. Moreover, the effect of Tan IIA on the cell cycle was partially attenuated by miR-133a-3p inhibitor but not miR-133a-3p inhibitor+siRNA-EGFR (Figure 9). Meanwhile, the expression of CyclinD1 and CDK4 exhibited the similar trends with that of the cell cycle (Figure 8).
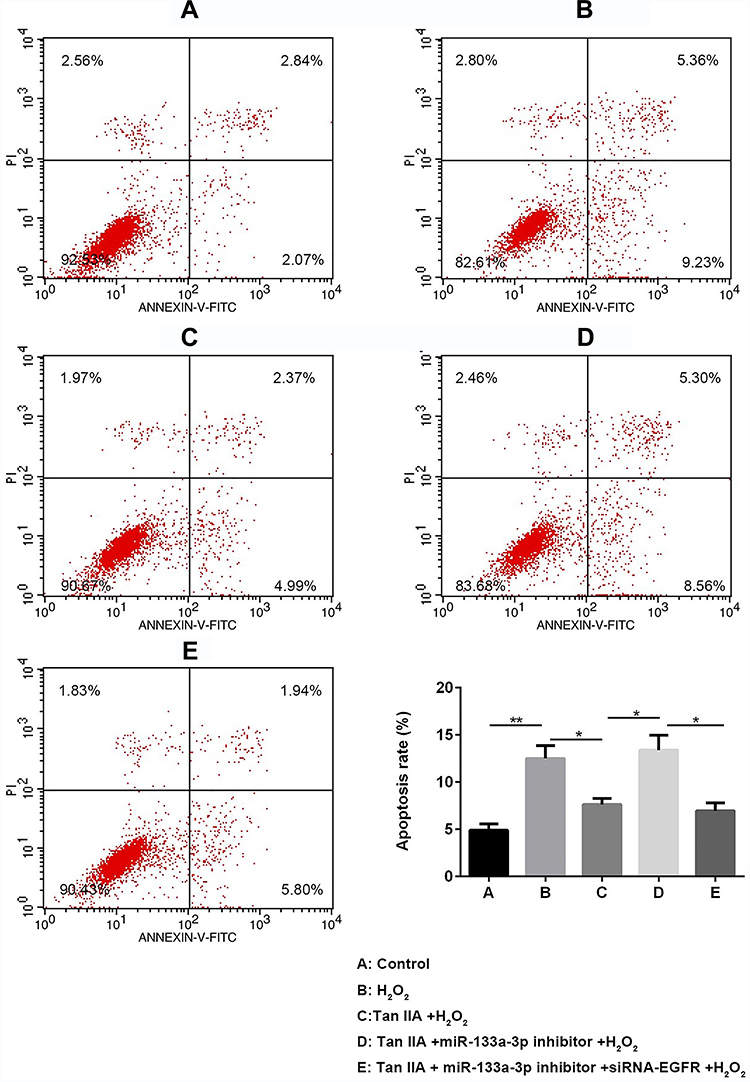 |
Figure 6 Flow cytometry results showed Tan IIA protected H2O2-induced H9c2 cells from apoptosis by miR-133a-3p/EGFR axis. Results of FCM analysis showed Tan IIA protected H2O2-induced H9c2 cells from apoptosis, which was partially attenuated by miR-133a-3p inhibitor but not miR-133a-3p inhibitor+siRNA-EGFR. *p<0.05, **p<0.01. |
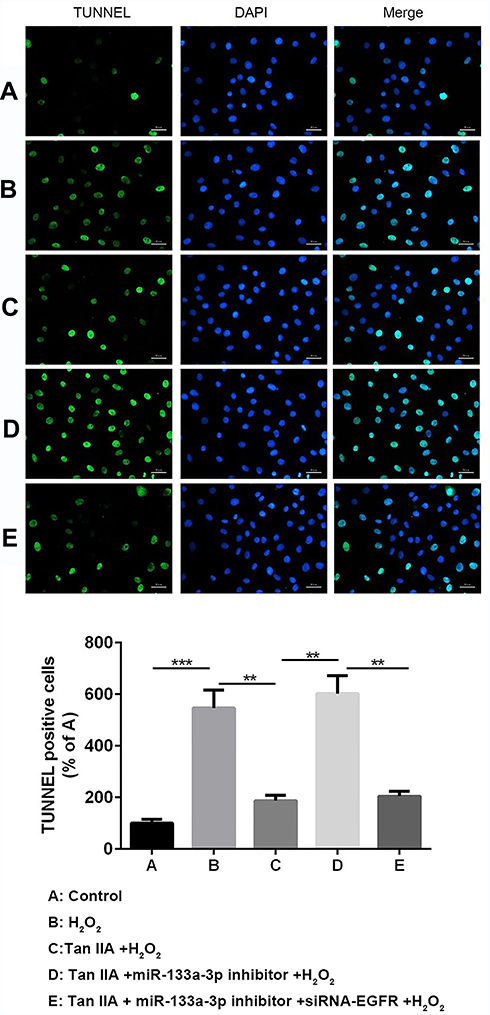 |
Figure 7 Tunnel staining results showed Tan IIA protected H2O2-induced H9c2 cells from apoptosis by miR-133a-3p/EGFR axis. Results of TUNNEL analysis showed Tan IIA protected H2O2-induced H9c2 cells from apoptosis, which was partially attenuated by miR-133a-3p inhibitor but not miR-133a-3p inhibitor+siRNA-EGFR. **p<0.01, ***p<0.001. |
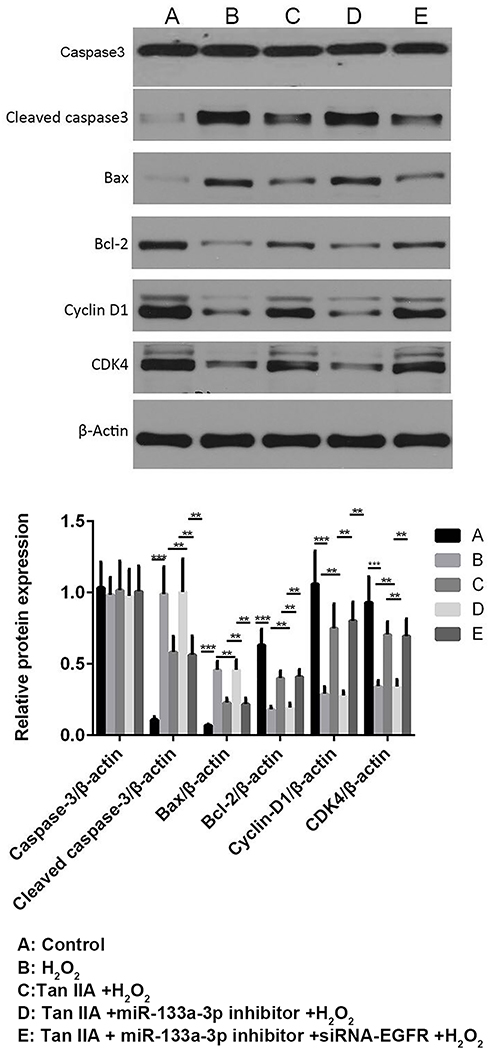 |
Figure 8 Tan IIA reversed H2O2-induced changes in the expressions of apoptosis and cell cycle-related proteins of H9c2 cells by miR-133a-3p/EGFR axis. The expression profile of cleaved caspase-3 and Bax showed the similar trends to cell apoptosis; while Bcl-2 showed the opposite trends. The expression profile of CyclinD1 and CDK4 exhibited the similar trends with that of cell cycle. **p<0.01, ***p<0.001. |
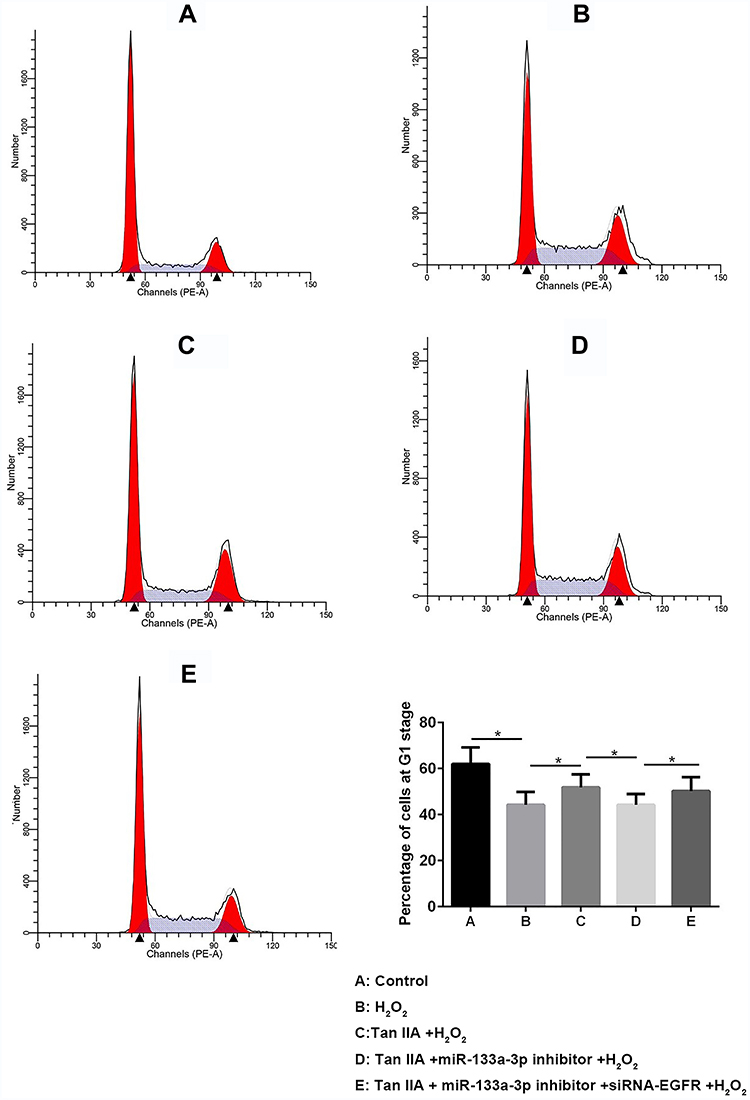 |
Figure 9 Tan IIA reversed H2O2-induced G0/G1 phase reduction of H9c2 cells by miR-133a-3p/EGFR axis. Tan IIA reversed H2O2-induced G0/G1 phase reduction of H9c2 cells, which was partially attenuated by miR-133 inhibitor but not miR-133a-3p inhibitor +siRNA-EGFR. *p<0.05. |
Discussion
Tan IIA was reported to ameliorate cell apoptosis and induced cell growth of myocardiocytes by upregulating miR-133 level.17 In addition, Tan IIA was discovered to protect H9c2 cells from oxidative stress-induced cell death by upregulating miR-133 level.18 Consistently, in the present study, the lower miR-133a-3p level was found in H2O2-induced H9c2 cells, which was rescued by Tan IIA.
Thereafter, the higher EGFR level was found in H2O2-induced H9c2 cells, in line with the reports about the central role of EGFR in atherosclerosis.20,22 In addition, EGFR was identified as a target of miR-133a-3p, which expression was negatively regulated by miR-133a-3p in H2O2-induced H9c2 cells.
Previous studies revealed that activated EGFR was related to numerous cellular processes, including cell proliferation and cell apoptosis.23,24 Consequently, we investigated whether Tan IIA regulated cell proliferation, cell apoptosis and cell cycle of H9c2 cells by regulating miR-133/EGFR axis as well. The results indicated that Tan IIA reversed H2O2-induced cell proliferation reduction by regulating miR-133a-3p/EGFR axis in H9c2 cells. We also tested the molecules, p53 and p21, which were correlated with cell proliferation. p53, a tumor suppressor, could be stabilized and activated by hypoxia stimulation, thus resulting in cessation of cell proliferation.25 And p21 is a downstream target for p53.26 Consistently, our findings exerted that p53 and p21 expression levels showed the opposite profile to cell proliferation.
Afterward, Tan IIA was found to reverse H2O2-induced cell apoptosis induction by regulating miR-133a-3p/EGFR axis in H9c2 cells. Next, we also explored the molecules that were correlated with cell apoptosis, for instance, cleaved caspase-3, Bax and Bcl-2. Bax is a pro-apoptotic protein, while Bcl-2 is an anti-apoptotic protein. When the ratio of Bax/Bcl-2 is elevated, the expression of cleaved caspase-3 might also be elevated, thus suggesting an elevation in cell apoptosis.27 Consistently, our findings showed that the expression of cleaved caspase-3 and Bax was similar to that of cell apoptosis, while Bcl-2 showed the opposite trends.
Thereafter, Tan IIA was found to reverse H2O2-induced G0/G1 arrest reduction by regulating miR-133a-3p/EGFR axis in H9c2 cells. We also tested the molecules that were correlated with cell cycle, for instance, CyclinD1 and CDK4. Cyclin D1 is an important promoter for cell cycle, which could activate CDK4, thus initiating DNA replication.28 Consistently, our findings demonstrated that the expression of CyclinD1 and CDK4 exhibited similar trends with that of the cell cycle.
Taken together, the present study manifested that, Tan IIA attenuated the progression of CAD in vitro through promoting cell proliferation, suppressing apoptosis and increasing G0/G1 arrest by targeting miR-133a-3p/EGFR axis. These findings provide potential molecular basis for the application of Tan IIA in the treatment of patients with CAD.
Data Sharing Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Disclosure
The authors declare that they have no competing interests.
References
1. Jiang L, Krumholz HM, Li X, Li J, Hu S. Achieving best outcomes for patients with cardiovascular disease in China by enhancing the quality of medical care and establishing a learning health-care system. Lancet. 2015;386(10002):1493–1505. doi:10.1016/S0140-6736(15)00343-8
2. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1–25. doi:10.1016/j.jacc.2017.04.052
3. Bhatnagar P, Wickramasinghe K, Williams J, Rayner M, Townsend N. The epidemiology of cardiovascular disease in the UK 2014. Heart. 2015;101(15):1182–1189. doi:10.1136/heartjnl-2015-307516
4. Mathers CDLD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):442. doi:10.1371/journal.pmed.0030442
5. Singh S, Kullo IJ, Pardi DS, Loftus EV
6. Zhao ZQ, Vinten-Johansen J. Myocardial apoptosis and ischemic preconditioning. Cardiovasc Res. 2002;55(3):438–455. doi:10.1016/S0008-6363(02)00442-X
7. Zhou L. Zuo Z and Chow MS: Danshen: an overview of its chemistry, pharmacology, pharmacokinetics and clinical use. J Clin Pharmacol. 2005;45(12):1345–1359. doi:10.1177/0091270005282630
8. Fish JM, Welchons DR, Kim YS, Lee SH, Ho W-K, Antzelevitch C. Ho WK and Antzelevitch C: dimethyl lithospermate B, an extract of Danshen, suppresses arrhythmogenesis associated with the Brugada syndrome. Circulation. 2006;113(11):1393–1400. doi:10.1161/CIRCULATIONAHA.105.601690
9. Adams JD, Wang R, Yang J, et al. Preclinical and clinical examinations of Salvia miltiorrhiza and its tanshinones in ischemic conditions. Chin Med. 2006;1(1):3. doi:10.1186/1749-8546-1-3
10. Pang H, Han B, Yu T, Peng Z. The complex regulation of tanshinone IIA in rats with hypertension-induced left ventricular hypertrophy. PLoS One. 2014;9(3):e92216. doi:10.1371/journal.pone.0092216
11. Xu S, Liu P. Tanshinone II-A: new perspectives for old remedies. Expert Opin Ther Pat. 2013;23(2):149–153. doi:10.1517/13543776.2013.743995
12. Gao J, Yang G, Pi R, et al. Chen S and Liu P: tanshinone IIA protects neonatal rat cardiomyocytes from adriamycin-induced apoptosis. Transl Res. 2008;151(2):79–87. doi:10.1016/j.trsl.2007.11.005
13. Yang R, Liu A, Ma X, Li L, Su D, Liu J. Su D and Liu J: sodium tanshinone IIA sulfonate protects cardiomyocytes against oxidative stress-mediated apoptosis through inhibiting JNK activation. J Cardiovasc Pharmacol. 2008;51(4):396–401. doi:10.1097/FJC.0b013e3181671439
14. Baehrecke EH. MiRNAs: micro managers of programmed cell death. Curr Biol. 2003;13(12):R473–R475. doi:10.1016/S0960-9822(03)00405-6
15. Mendell JT. MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle. 2005;4(9):1179–1184. doi:10.4161/cc.4.9.2032
16. Landskroner-Eiger S, Moneke I, Sessa WC. miRNAs as Modulators of Angiogenesis. Cold Spring Harb Perspect Med. 2013;3(2):a006643. doi:10.1101/cshperspect.a006643
17. Song T, Yao Y, Wang T, Huang H, Xia H. Tanshinone IIA ameliorates apoptosis of myocardiocytes by up-regulation of miR-133 and suppression of Caspase-9. Eur J Pharmacol. 2017;815:343–350. doi:10.1016/j.ejphar.2017.08.041
18. Gu Y, Liang Z, Wang H, et al. Tanshinone IIA protects H9c2 cells from oxidative stress-induced cell death via microRNA-133upregulation and Akt activation. Exp Ther Med. 2016;12(2):1147–1152. doi:10.3892/etm.2016.3400
19. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
20. Makki N, Thiel KW, Miller F. Miller FJ Jr: the epidermal growth factor receptor and its ligands in cardiovascular disease. Int J Mol Sci. 2013;14(10):20597–20613. doi:10.3390/ijms141020597
21. Schreier B, Gekle M, Grossmann C. Gekle M and Grossmann C: role of epidermal growth factor receptor in vascular structure and function. Curr Opin Nephrol Hypertens. 2014;23(2):113–121. doi:10.1097/01.mnh.0000441152.62943.29
22. Wang S, He W, Wang C. MiR-23a regulates the vasculogenesis of coronary artery disease by targeting epidermal growth factor receptor. Cardiovasc Ther. 2016;34(4):199–208. doi:10.1111/1755-5922.12187
23. McCarthy NJ, Bennett MR. The regulation of vascular smooth muscle cell apoptosis. Cardiovasc Res. 2000;45(3):747–755. doi:10.1016/S0008-6363(99)00275-8
24. Samarakoon R, Higgins PJ. Integration of non-SMAD and SMAD signaling in TGF-beta1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb Haemost. 2008;100(12):976–983. doi:10.1160/TH08-05-0273
25. Leszczynska KB, Foskolou IP, Abraham AG, et al. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J Clin Invest. 2015;125(6):2385–2398. doi:10.1172/JCI80402
26. Valente LJ, Grabow S, Vandenberg CJ, Strasser A, Janic A. Combined loss of PUMA and p21 accelerates c-MYC-driven lymphoma development considerably less than loss of one allele of p53. Oncogene. 2016;35(29):3866–3871. doi:10.1038/onc.2015.457
27. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. doi:10.1038/nrm3722
28. Pozner A, Terooatea TW, Buck-Koehntop BA. Cell-specific kaiso (ZBTB33) regulation of cell cycle through cyclin D1 and cyclin E1. J Biol Chem. 2016;291(47):24538–24550. doi:10.1074/jbc.M116.746370
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.