Back to Journals » Clinical Epidemiology » Volume 6
Systemic AA amyloidosis: epidemiology, diagnosis, and management
Authors de Asúa D, Costa R, Galván JM, Filigheddu MT, Trujillo D, Cadiñanos J
Received 1 May 2014
Accepted for publication 5 June 2014
Published 29 October 2014 Volume 2014:6 Pages 369—377
DOI https://doi.org/10.2147/CLEP.S39981
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Diego Real de Asúa, Ramón Costa, Jose María Galván, María Teresa Filigheddu, Davinia Trujillo, Julen Cadiñanos
Department of Internal Medicine, Fundación de Investigación Biomédica, Hospital Universitario de La Princesa, Madrid, Spain
Abstract: The term "amyloidosis" encompasses the heterogeneous group of diseases caused by the extracellular deposition of autologous fibrillar proteins. The global incidence of amyloidosis is estimated at five to nine cases per million patient-years. While amyloid light-chain (AL) amyloidosis is more frequent in developed countries, amyloid A (AA) amyloidosis is more common in some European regions and in developing countries. The spectrum of AA amyloidosis has changed in recent decades owing to: an increase in the median age at diagnosis; a percent increase in the frequency of primary AL amyloidosis with respect to the AA type; and a substantial change in the epidemiology of the underlying diseases. Diagnosis of amyloidosis is based on clinical organ involvement and histological evidence of amyloid deposits. Among the many tinctorial characteristics of amyloid deposits, avidity for Congo red and metachromatic birefringence under unidirectional polarized light remain the gold standard. Once the initial diagnosis has been made, the amyloid subtype must be identified and systemic organ involvement evaluated. In this sense, the 123I-labeled serum amyloid P component scintigraphy is a safe and noninvasive technique that has revolutionized the diagnosis and monitoring of treatment in systemic amyloidosis. It can successfully identify anatomical patterns of amyloid deposition throughout the body and enables not only an initial estimation of prognosis, but also the monitoring of the course of the disease and the response to treatment. Given the etiologic diversity of AA amyloidosis, common therapeutic strategies are scarce. All treatment options should be based upon a greater control of the underlying disease, adequate organ support, and treatment of symptoms. Nevertheless, novel therapeutic strategies targeting the formation of amyloid fibrils and amyloid deposition may generate new expectations for patients with AA amyloidosis.
Keywords: amyloidosis, epidemiology, nephrotic syndrome, rheumatoid arthritis, Congo red, eprodisate
Introduction
The term “amyloidosis” encompasses a heterogeneous group of diseases caused by the extracellular deposition of autologous fibrillar proteins, which aggregate into a three-dimensional ß-lamina disposition that impairs normal organ function.1 The term “amyloid” was first used in 1853 by Rudolf Virchow, who observed the close similarity to starch after dyeing these deposits in iodine and sulfuric acid.2 Among the many other tinctorial characteristics of amyloid deposits, the avidity for Congo red and metachromatic birefringence under unidirectional polarized light remain the best known. These entities can be classified according to their constitutive proteins, of which up to 25 have been identified to date, or according to the classification of amyloid deposits into localized or systemic forms. The main subtypes of systemic amyloidosis are primary AL amyloidosis, secondary amyloid A (AA) amyloidosis, familial amyloidosis, and ß2-microglobulin-related amyloidosis (Table 1).3
 | Table 1 Classification of the main types of systemic amyloidosis |
AA amyloidosis is probably the most common type of amyloidosis worldwide, given that most reported cases from developing countries are associated with underlying infections. Systemic AL amyloidosis, on the other hand, which was previously known as primary amyloidosis, is the most prevalent type in developed countries.4 Amyloid deposits in AA amyloidosis are composed mainly of the serum amyloid A (SAA) protein, an apolipoprotein of high-density lipoproteins that serves as a dynamic acute phase reactant.5 It is synthesized as a precursor by hepatocytes in response to transcriptional stimuli from various proinflammatory cytokines, such as interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF) alpha.6 Two different isoforms of SAA have been isolated, and SAA1 is responsible for most of its serum elevation during the acute phase response. Sustained abnormally high levels of SAA, which is usually present at low levels in serum, are essential for the development of amyloidosis, although only a small number of patients with inflammatory conditions will eventually develop amyloidosis. Amyloid fibrils are not only composed of the N-terminal segment of SAA, but also of the serum amyloid P component (SAP), which is derived from a normal circulating plasma protein of the pentraxin family. The SAP binds to all types of amyloid precursors in a calcium-dependent manner and stabilizes their tertiary structure. Heparan sulfate and glycosaminoglycan (GAG) chains from the extracellular matrix are also crucial for amyloid fibrillogenesis.5
Various genetic factors regulate susceptibility to the deposition of SAA, and the ulterior development of amyloidosis. At least five allelic variants of SAA have been identified, and these differ in individual amino acids at codons 52 and 57.7 The SAA genotype is an important determinant of amyloidogenesis in patients with rheumatoid arthritis (RA), or with familial Mediterranean fever (FMF), although substantial ethnic differences have been observed. For instance, in Caucasian patients with RA, the presence of the SAA1.1 allele indicated a higher risk of developing AA amyloidosis;8 whereas, among Japanese patients, homozygosity for the SAA1.3 allele was related to a more pronounced increase in serum SAA levels, a shorter latency period before disease onset, more severe systemic damage, and shorter survival than other allelic variants.9
Epidemiology
Until recently, and despite probably being underdiagnosed, amyloidosis had only been reported through retrospective case series.10 The most comprehensive epidemiological study in recent decades was conducted by Kyle et al at the Mayo Clinic with data collected from 1950–1990 from the general population residing in Olmsted County, MN, USA.11 These authors reported an incidence of AL amyloidosis in nine cases per million person-years (95% confidence interval; 5.1–12.8 cases per million person-years). The authors inferred that approximately 2,200 new cases of AL amyloidosis could occur annually in the USA. Recently, similar studies have been carried out in two European regions. Pinney et al from the National Health Service National Amyloidosis Centre in the UK, estimated a global incidence of amyloidosis in England of five cases per million person-years.4 Of these, close to three cases per million person-years would have the AL type, and one case, the AA type. Using similar methods, Hemminki et al calculated an incidence of eight patients per million person-years in Sweden; three cases per million person-years were ascribed to the AL type and two to the AA type.12
The spectrum of AA amyloidosis has changed in recent decades owing to an increase in median age at diagnosis, a percent increase in primary AL amyloidosis with respect to the AA type, and a substantial change in the epidemiology of the underlying diseases leading to AA amyloidosis (Table 2).13 In the previously cited population-based studies, the incidence of amyloidosis was highest among adults aged 60–80 years; whereas, prior series showed a substantially younger median age at a diagnosis of around 45–55 years.4,13,18
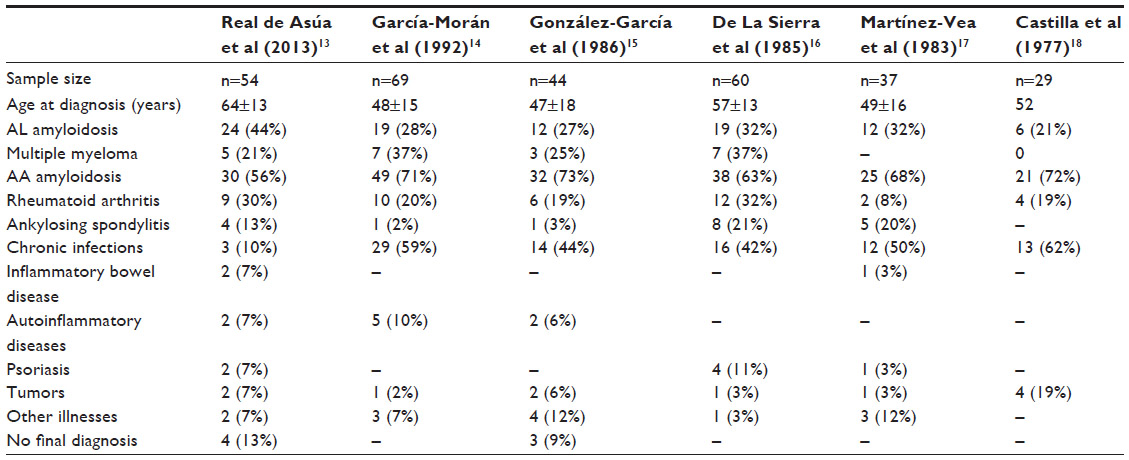 | Table 2 Distribution of underlying diseases causing amyloidosis in six Spanish series |
The percent increase in the frequency of AL amyloidosis with respect to the AA type can be explained by earlier diagnosis of many predisposing conditions for AA amyloidosis and widespread access to more effective treatments that better control many disease processes. Moreover, these factors would also explain the gradual reduction in the frequency of multiple myeloma-related AL amyloidosis. Better access to antimicrobial agents and a stricter control of tuberculosis have led to a significant decrease in the frequency of chronic infectious diseases in developed countries, where autoimmune diseases such as RA, ankylosing spondylitis, chronic juvenile arthritis, and inflammatory bowel disease, or hereditary inflammatory diseases, such as FMF, TNF receptor-associated periodic syndrome, or Muckle–Wells syndrome, can account for up to 90% of recent cases of amyloidosis.13 Nevertheless, tuberculosis and other chronic infections are still relevant causes of AA amyloidosis in other European countries, as well as in developing countries.19,20
It is difficult to establish the approximate prevalence of AA amyloidosis among patients with inflammatory diseases, since these estimations vary substantially with the diagnostic method used (autopsy, affected organ biopsy, or indirect biopsy, such as abdominal fat, rectal mucosa, or minor salivary glands), clinical status (preclinical or symptomatic amyloidosis), or study type (case series or population-based). The prevalence of AA amyloidosis in patients with RA has been estimated to be 5%–78%;21,22 whereas, it is 10%–13% in patients with FMF.23 However, most of these studies were conducted before the generalized use of biologic therapies and current, more intensive, treatment protocols, that enable prompt control of the inflammatory process. The use of biologic therapies and of more intensive treatment protocols have essentially modified the natural history of inflammatory joint disease in developed countries, and it is expected that they may exert a significant influence on the incidence of amyloidosis in the coming decades.24 Finally, new cases of amyloidosis have been detected in groups that were not traditionally associated with amyloidosis. Thus, AA amyloidosis was the most frequent cause of kidney disease among intravenous drug users evaluated at two different reference centers in Frankfurt, Germany.25 Amyloidosis accounted for 50% of the cases, which is much more than any other type of kidney disease. Patients more frequently had chronic HIV infection and a previous medical history of repeated skin and soft tissue infections. The authors postulate that amyloidosis could have been caused both by direct action of HIV and by its associated immunosuppression, which would increase the frequency and duration of the acquired infections. However, the hypothesis for a direct pathogenic effect of HIV remains to be elucidated.
Diagnosis
Clinical suspicion and initial aspects of diagnosis
Proteinuria leading to nephrotic syndrome and renal insufficiency is the earliest and most frequent clinical manifestation that should raise suspicion of AA amyloidosis in patients with chronic inflammatory conditions. Proteinuria may be present in up to 95% of patients and determines prognosis.26 Although amyloid deposits are common in the liver and spleen, their clinical significance is relatively minor in the early stages of the disease. Nevertheless, hepatosplenomegaly and adrenal insufficiency may complicate the disease course in the advanced stages. The gastrointestinal tract may also be affected, causing malabsorption, intestinal pseudo-obstruction, diarrhea, or bleeding. Peripheral polyneuropathy, restrictive myocardiopathy leading to heart failure, and skin and soft tissue involvement, such as macroglossia, are extremely uncommon, especially when compared with other types of systemic amyloidosis.26
Diagnosis is confirmed based on clinical organ involvement and histological demonstration of amyloid deposits. Diagnosis cannot be confirmed based on the finding of amyloid deposits in indirect biopsy in the absence of clinical organ involvement or in the presence of a predisposing condition, even with highly elevated amyloidogenic proteins in serum and no histological evidence of organ damage.3 Once the initial diagnosis has been made, the amyloid subtype must be identified, and systemic organ involvement evaluated. Since there are no specific reference criteria to define organ damage in AA amyloidosis, those accepted for the AL type can generally be applied, albeit with caution (Figure 1 and Table 3).27
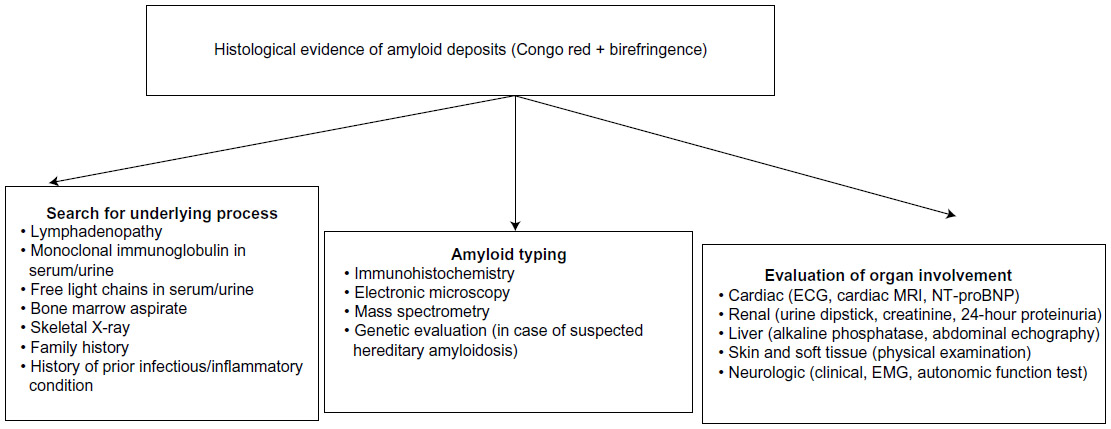 | Figure 1 Diagnostic algorithm in patients with suspected amyloidosis. |
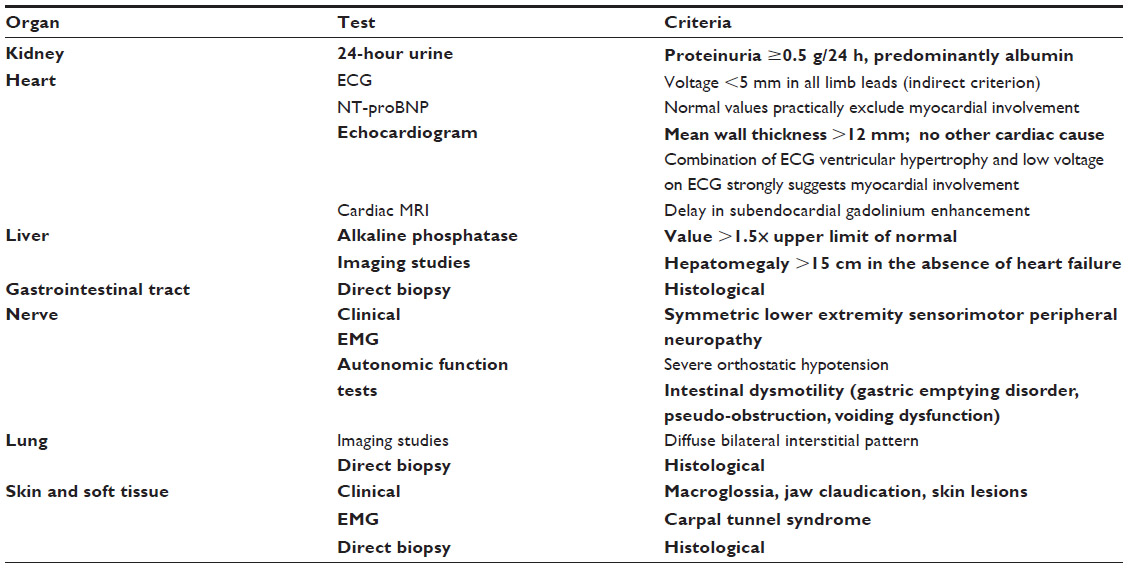 | Table 3 Criteria for organ involvement in AL amyloidosis |
Selecting biopsy site
In the 1960s, rectal and gingival mucosa biopsies were considered useful indirect alternatives to deep organ biopsies for the diagnosis of amyloidosis. In 1973, Westermark and Stenkvist described subcutaneous abdominal fat tissue aspiration as a new, noninvasive technique, and this has now become the preferred initial option in most centers throughout the world.28 It should be noted that a negative result in any of these indirect biopsies is insufficient to rule out a diagnosis of amyloidosis, especially with the appropriate clinical suspicion, and a biopsy of a potentially affected deep organ should be performed.
- Subcutaneous abdominal fat tissue aspiration: this simple procedure can be performed in the outpatient setting by aspirating two to five samples of abdominal fat with an 18–23 G needle connected to a 10 mL syringe. Several case series show that, while its specificity for the diagnosis of amyloidosis is high (93%–100%), its sensitivity varies between 57%–82%, which makes it equally useful as the rectal biopsy.29 However, these findings can vary depending on the study population, disease course and severity, and the collecting, processing, and visualization technique. While some authors have shown sensitivity as high as 90% with three samples evaluated by two expert pathologists, others have reported sensitivity as low as 0%–19% in the early stages of the disease.29,30
- Rectal mucosa biopsy: numerous studies support the use of a biopsy of the rectal mucosa and submucosa, with a sensitivity of ~75%–85% for the detection of amyloid deposits.30 However, some series have shown lower sensitivity than abdominal fat aspiration. Considering that the involvement of the gastrointestinal tract is segmentary, other locations along the gastrointestinal tract (eg, stomach or colon) may also be useful for diagnosis.
- Minor salivary gland biopsy: This technique offers sensitivity of ~83%–100% for the diagnosis of both AL amyloidosis and AA amyloidosis.31 It is also useful for the detection of mutated transthyretin, although it has lower sensitivity in ß2-microglobulin-associated amyloidosis. Biopsy of the gingival mucosa may also be of help, but apparently its sensitivity is lower.
Other biopsy-susceptible organs include the skin, tongue, peripheral nerves, endocardium, and bone marrow, for which sensitivity varies, depending on the type of amyloidosis and the extent of systemic involvement. The diagnostic yield of biopsy of these organs is generally inferior to that of the techniques described previously. For instance, the sensitivity of bone marrow biopsy for the detection of AL amyloidosis has been estimated at ~50%–60%.30
Histological identification of amyloid deposits
Staining with hematoxylin-eosin reveals amyloid deposits to be homogeneous and eosinophilic. Amyloid material is identified based on its metachromatic properties with aniline dyes, such as gentian violet, thioflavin T, or Congo red. This optical effect is caused by the spatial orientation of the dye molecules between amyloid fibrils, which determines their capacity to transmit and to absorb light.32 Congo red is still considered the gold standard dye owing to its higher sensitivity and specificity when differentiating amyloid from other protein deposits.33 The classic apple-green birefringence under polarized light can only be seen under ideal circumstances, and other chromatic combinations such as green–yellow, blue–yellow, and red–green are equally characteristic.34 Avidity for Congo red or any other aniline dye alone without this optical effect is not sufficient to differentiate amyloid from other protein deposits and should be considered inappropriate for diagnosis. The sensitivity and the specificity of the technique depend to a large extent on the experience of the pathologist, the amount of amyloid, and the quality of the dye process, and both false-positive and false-negative results are frequent.35 Adding phenol to the classic Congo red dye and using fluorescence microscopy or electronic microscopy may help improve the sensitivity of this technique for the detection of amyloid deposits.36–38
Amyloid typing
Amyloid precursors are heterogeneous. However, after deposition, their microscopic morphology and histochemical properties are very similar, and they cannot be differentiated using classic dyes. Consequently, even though the clinical context may suggest a particular amyloid type, it is essential to characterize and confirm the type.
Lachmann et al39 studied 350 patients with presumptive AL amyloidosis based on clinical and laboratory findings, with no definitive histological diagnosis. Genetic testing made it possible to confirm a diagnosis of hereditary amyloidosis in 34 patients (9.7% of the sample), eight of whom (8/34; 23.5%) had concomitant low-grade monoclonal gammopathy of undetermined significance.39
These findings highlight the importance of reaching a definitive histological diagnosis, since critical treatment options may depend on it. However, none of the currently available typing techniques has established itself as the gold standard. Evaluation is generally based on immunohistochemical testing, biochemistry (serum and urine), imaging studies, and/or genetic testing (Figure 1).40 New techniques, such as the proteomic analysis by mass spectrometry, are emerging as powerful complementary tools to improve amyloid typing.41
Scintigraphic evaluation of global amyloid burden
SAP accounts for ~15% of amyloid deposit. Following the intravenous injection of the 123I-labeled SAP, the tracer distributes between the free and amyloid-bound SAP pools in proportion to their size and can then be imaged and quantified.3 Although SAP scintigraphy should not substitute histological evaluation as the cornerstone of diagnosis, this safe and noninvasive technique has revolutionized diagnosis and monitoring of treatment in systemic amyloidosis.
First, its sensitivity and specificity for detection of amyloid deposits are estimated at ~90% and 93%, respectively.42 Second, it can identify anatomical patterns of amyloid deposition throughout the body, some of which may be pathognomonic (eg, bone marrow deposits in AL amyloidosis), even in the presymptomatic phase.37 Finally, the intensity of the scintigraphic signal is proportional to the underlying amyloid burden. This quantitative information helps not only to enable initial prognostic estimations (since patients with different amounts of amyloid may benefit from different risk–benefit balances when they receive cytotoxic chemotherapy), but also to monitor disease course and response to treatment.26
Unfortunately, scintigraphy has several major limitations. Given the short half-life of the isotope, it is not useful for assessing cardiac involvement, nor can it be applied to evaluate intracerebral or peripheral nerve amyloidosis, owing to the slow penetration of SAP in the nervous system.3,26 Furthermore, given its cost and technical complexity, the technique is only available in highly specialized centers.
Treatment
Owing to the etiologic diversity of AA amyloidosis, common therapeutic strategies are scarce. Therefore, all treatment options should be based upon greater control of the underlying disease, adequate organ support treatment, and symptomatic relief (Table 4).
 | Table 4 Useful treatments for systemic AA amyloidosis |
Treatment of underlying predisposing disease
Controlling the underlying disease, which leads to a subsequent reduction in acute phase reactant levels, including circulating serum SAA levels, is the most effective strategy for stabilization or even regression of amyloid deposition. Surgical excision of highly inflammatory neoplasm, such as localized Castleman’s disease, has enabled drastic reductions in serum acute phase reactant levels and in amyloid deposits detected by scintigraphy in many cases.43 Remission of amyloidosis-associated nephrotic syndrome has also been achieved with tuberculostatic treatment.44 High-dose colchicine (1.5–2 mg/day) is effective in controlling systemic inflammation in autoinflammatory syndromes, such as FMF, and has been able to induce remission of associated AA amyloidosis.45 Moreover, anti-IL-1 antibodies, such as anakinra or canakinumab, which have already proven effective as first-line therapies in several autoinflammatory syndromes, may also prove effective in patients with refractory FMF, in whom clinical remission with high-dose colchicine has not been achieved, patients in whom significantly elevated serum SAA levels are still detectable despite treatment with colchicine, patients with colchicine intolerance, and patients in whom the clinical picture is suggestive of an overlap between FMF and vasculitis (predominantly with concomitant polyarteritis nodosa, or Henoch–Schönlein purpura).46,47 Finally, immunomodulatory drugs have also proven extremely useful in controlling the progression of amyloidosis-associated proteinuria and improving long-term survival in several inflammatory joint diseases and in inflammatory bowel disease. Although an in-depth analysis of each of these drugs is well beyond the scope of this review, they include, but are not limited to, chlorambucil, cyclophosphamide, tacrolimus, and anti-TNF-alpha or anti-IL-6 antibodies, such as infliximab, etanercept, or tocilizumab.48–50
Treatment targeting amyloid deposits
Greater understanding of the pathophysiological mechanisms underlying amyloid deposition has enabled the development of new treatment strategies, specifically those targeting formation of amyloid proteins. In this sense, tocilizumab, a humanized anti-IL-6 monoclonal antibody, proved to be extremely effective in reducing circulating SAA levels and controlling the progression of amyloidosis in several autoimmune joint diseases. Its effect was independent of the underlying disease and of that attained with the aforementioned immunomodulators or with abatacept and rituximab. Tocilizumab has not only been successfully tested in RA or chronic juvenile arthritis, but also in Behçet’s disease and even in a patient with tuberculosis, in whom rapidly progressive amyloidosis-induced nephrotic syndrome was controlled with a combination of tuberculostatic drugs and tocilizumab.51,52
Dimethyl sulfoxide is a derivative molecule of intracellular low-density lipoprotein, which disrupts hydrogen bonding. It has been tested in patients with gastrointestinal and renal amyloidosis and can lower acute phase reactant levels and improve gastrointestinal complaints while reducing local amyloid deposits. However, improved renal function was only tangentially achieved in patients with mild proteinuria at the onset of treatment.53
Eprodisate is a low molecular-weight molecule that is similar to heparan sulfate. By binding competitively to GAG union sites, it inhibits polymerization of amyloid fibrils and prevents the stabilization of amyloid deposits. Phase II trials showed stabilization of renal function in 42% of cases, although the drug was unable to modify serum SAA levels and had no significant effect on proteinuria or overall survival.54 A Phase III randomized controlled trial is under way to establish the efficacy and safety of eprodisate in preventing decline in renal function in patients with AA amyloidosis.55
Heparins and statins also have beneficial effects on the outcome of AA amyloidosis. The former can slow progression by breaking the stabilizing bonds between GAG and SAA in the deposits, in a manner analogous to that of eprodisate;56 the latter seem to exert their effect through inhibition of the isoprenoid pathway by specifically blocking farnesyltransferase. This mechanism is shared by some autoinflammatory diseases, such as hyperimmunoglobulinemia D with recurrent fever syndrome, in which amyloidosis is rare despite vigorous, recurrent inflammation.57
Supportive treatment of specific symptoms
Although symptoms of dysautonomia are rare in AA amyloidosis, severe orthostatic hypotension may lead to recurrent syncope. In these cases, fludrocortisone or midodrine may prove useful. Furthermore, diarrhea or protein-wasting enteropathy may be ameliorated by the use of antibiotics to diminish bacterial overgrowth, prednisone pulses, or the combination of prednisolone and octreotide.58 In the case of progression of organ damage, transplantation may be considered, especially if the underlying inflammatory illness has been controlled.59
Novel therapeutic targets
Most novel research targets focus on controlling and reducing amyloid deposition in tissue. The new anti-SAP antibody R-1-[6-[R-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC) is expected to improve the clearance of serum SAP. The molecule, tested on 31 patients with systemic amyloidosis (any type), achieved a partial response in kidney function, with no significant side effects.60 Kluve-Beckerman et al used murine models to block SAA-transcribing messenger RNA with two complementary antisense oligonucleotides.61 They achieved a reduction >50% in circulating SAA levels, as well as a significantly lower tissue amyloid burden.61 Finally, murine models have also been used to explore the use of clodronate in phagocytic depletion, which may also serve as a potential target for preventing and treating amyloidosis.62 It is hoped that these novel targets will generate further therapeutic options for patients with AA amyloidosis.
Acknowledgments
The authors would like to thank Dr S Castañeda, Dr R García-Vicuña, Dr E Tomero, and Mr T O’Boyle for their kind review of the final draft of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583–596. | |
Campistol JM, Amiloidosis, En Rozman C, Cardellach F, editors. Farreras/Rozman, Medicina Interna,16ª ed. Barcelona: Elsevier España; 2009:1147–1150. | |
Pepys MB. Amyloidosis. Annu Rev Med. 2006;57:223–241. | |
Pinney JH, Smith CJ, Taube JB, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013;161(4):525–532. | |
Obici L, Perfetti V, Palladini G, Moratti R, Merlini G. Clinical aspects of systemic amyloid diseases. Biochim Biophys Acta. 2005;1753(1):11–22. | |
Urieli-Shoval S, Linke RP, Matzner Y. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr Opin Hematol. 2000;7(1):64–69. | |
Obici L, Raimondi S, Lavatelli F, Bellotti V, Merlini G. Susceptibility to AA amyloidosis in rheumatic diseases: a critical overview. Arthritis Rheum. 2009;61(10):1435–1440. | |
van der Hilst JC. Recent insights into the pathogenesis of type AA amyloidosis. ScientificWorldJournal. 2011;11:641–650. | |
Nakamura T, Higashi S, Tomoda K, Tsukano M, Baba S, Shono M. Significance of SAA1.3 allele genotype in Japanese patients with amyloidosis secondary to rheumatoid arthritis. Rheumatology (Oxford). 2006;45(1):43–49. | |
Simms RW, Prout MN, Cohen AS. The epidemiology of AL and AA amyloidosis. Baillieres Clin Rheumatol. 1994;8(3):627–634. | |
Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79(7):1817–1822. | |
Hemminki K, Li X, Försti A, Sundquist J, Sundquist K. Incidence and survival in non-hereditary amyloidosis in Sweden. BMC Public Health. 2012;12:974. | |
Real de Asúa D, Costa R, Contreras MM, Gutiérrez Á, Filigghedu MT, Armas M. Clinical characteristics of the patients with systemic amyloidosis in 2000–2010. Rev Clin Esp. 2013;213(4):186–193. | |
García Moran JI, Barat Cascante A, Oliva Aldámiz H. Amiloidosis sistémica: Estudio clínico-patológico de 69 casos [Systemic amyloidosis: clinico-pathologic study of 69 cases]. Rev Clin Esp. 1992;191:181–186. | |
González-García JJ, García-Alegría J, Peña JM, Vicandi B, Picazo ML, Fernández-Capitán MC, et al. Espectro y correlación clinicopatológica en la amiloidosis renal [Spectrum and clinico-pathological correlations in renal amyloidosis. Study of 44 cases]. Estudio de 44 casos. Rev Clin Esp. 1986;178:421–427. | |
De la Sierra A, Cardellach F, Ingelmo M, Roselló R, Balcells A. Amiloidosis: aspectos clínicos y biológicos de 60 casos [Amyloidosis: clinical and biological aspects of 60 cases]. Med Clin (Barc). 1985;84:297–301. | |
Martínez-Vea A, Torras A, Darnell A, Carrera M, Revert L. Amiloidosis primaria y secundaria renal: manifestaciones clínicas, evolución y supervivencia. Estudio comparativo de 37 pacientes [Primary and secondary renal amyloidosis: clinical manifestations, development and survival. Comparative study of 37 patients]. Med Clin (Barc). 1983;80:191–195. | |
Castilla J, Montes R, Quereda C, Marcen R, Rodríguez G, Pereira P, et al. Amiloidosis renal: estudio clínico en 29 pacientes [Renal amyloidosis: clinical study in 29 patients]. Rev Clin Esp. 1977;147:349–354. | |
Bergesio F, Ciciani AM, Santostefano M, et al; Immunopathology Group, Italian Society of Nephrology. Renal involvement in systemic amyloidosis – an Italian retrospective study on epidemiological and clinical data at diagnosis. Nephrol Dial Transplant. 2007;22(6):1608–1618. | |
Dixit R, Gupta R, Dave L, Prasad N, Sharma S. Clinical profile of patients having pulmonary tuberculosis and renal amyloidosis. Lung India. 2009;26(2):41–45. | |
Barile L, Ariza R, Muci H, et al. Tru-cut needle biopsy of subcutaneous fat in the diagnosis of secondary amyloidosis in rheumatoid arthritis. Arch Med Res. 1993;24(2):189–192. | |
Kobayashi H, Tada S, Fuchigami T, et al. Secondary amyloidosis in patients with rheumatoid arthritis: diagnostic and prognostic value of gastroduodenal biopsy. Br J Rheumatol. 1996;35(1):44–49. | |
Tunca M, Akar S, Onen F, et al; Turkish FMF Study Group. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine (Baltimore). 2005;84(1):1–11. | |
Immonen K, Finne P, Grönhagen-Riska C, et al. A marked decline in the incidence of renal replacement therapy for amyloidosis associated with inflammatory rheumatic diseases – data from nationwide registries in Finland. Amyloid. 2011;18(1):25–28. | |
Jung O, Haack HS, Buettner M, et al. Renal AA-amyloidosis in intravenous drug users – a role for HIV-infection? BMC Nephrol. 2012;13:151. | |
Lachmann HJ, Goodman HJ, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361–2371. | |
Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, April 18–22, 2004. Am J Hematol. 2005;79(4):319–328. | |
Westermark P, Stenkvist B. A new method for the diagnosis of systemic amyloidosis. Arch Intern Med. 1973;132(4):522–523. | |
van Gameren II, Hazenberg BP, Bijzet J, Van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum. 2006;54(6):2015–2021. | |
Hachulla E, Grateau G. Diagnostic tools for amyloidosis. Joint Bone Spine. 2002;69(6):538–545. | |
Caporali R, Bonacci E, Epis O, Bobbio-Pallavicini F, Morbini P, Montecucco C. Safety and usefulness of minor salivary gland biopsy: retrospective analysis of 502 procedures performed at a single center. Arthritis Rheum. 2008;59(5):714–720. | |
Howie AJ, Brewer DB. Optical properties of amyloid stained by Congo red: history and mechanisms. Micron. 2009;40(3):285–301. | |
Cooper JH. An evaluation of current methods for the diagnostic histochemistry of amyloid. J Clin Pathol. 1969;22(4):410–413. | |
Howie AJ, Owen-Casey MP. Discrepancies between descriptions and illustrations of colours in Congo red-stained amyloid, and explanation of discrepant colours. Amyloid. 2010;17(3–4):109–117. | |
Linke RP. On typing amyloidosis using immunohistochemistry. Detailled illustrations, review and a note on mass spectrometry. Prog Histochem Cytochem. 2012;47(2):61–132. | |
Ishii W, Matsuda M, Nakamura N, et al. Phenol Congo red staining enhances the diagnostic value of abdominal fat aspiration biopsy in reactive AA amyloidosis secondary to rheumatoid arthritis. Intern Med. 2003;42(5):400–405. | |
Giorgadze TA, Shiina N, Baloch ZW, Tomaszewski JE, Gupta PK. Improved detection of amyloid in fat pad aspiration: an evaluation of Congo red stain by fluorescent microscopy. Diagn Cytopathol. 2004;31(5):300–306. | |
Devata S, Hari P, Markelova N, Li R, Komorowski R, Shidham VB. Detection of amyloid in abdominal fat pad aspirates in early amyloidosis: Role of electron microscopy and Congo red stained cell block sections. Cytojournal. 2011;8:11. | |
Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346(23):1786–1791. | |
Kebbel A, Röcken C. Immunohistochemical classification of amyloid in surgical pathology revisited. Am J Surg Pathol. 2006;30(6):673–683. | |
Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114(24):4957–4959. | |
Hazenberg BP, van Rijswijk MH, Piers DA, et al. Diagnostic performance of 123I-labeled serum amyloid P component scintigraphy in patients with amyloidosis. Am J Med. 2006;119(4):355.e15–355.e24. | |
Lachmann HJ, Gilbertson JA, Gillmore JD, Hawkins PN, Pepys MB. Unicentric Castleman’s disease complicated by systemic AA amyloidosis: a curable disease. QJM. 2002;95(4):211–218. | |
Castellano I, Gómez-Martino JR, Hernández MT, Novillo R, Covarsí A. [Remission of nephrotic syndrome caused by renal amyloidosis secondary to pulmonary tuberculosis after tuberculostatic treatment]. Nefrologia. 2001;21(1):88–91. Spanish. | |
Cerquaglia C, Diaco M, Nucera G, La Regina M, Montalto M, Manna R. Pharmacological and clinical basis of treatment of Familial Mediterranean Fever (FMF) with colchicine or analogues: an update. Curr Drug Targets Inflamm Allergy. 2005;4(1):117–124. | |
Meinzer U, Quartier P, Alexandra JF, Hentgen V, Retornaz F, Koné-Paut I. Interleukin-1 targeting drugs in familial Mediterranean fever: a case series and a review of the literature. Semin Arthritis Rheum. 2011;41(2):265–271. | |
Ter Haar N, Lachmann H, Özen S, et al; Paediatric Rheumatology International Trials Organisation (PRINTO) and the Eurofever/Eurotraps Projects. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis. 2013;72(5):678–685. | |
Chevrel G, Jenvrin C, McGregor B, Miossec P. Renal type AA amyloidosis associated with rheumatoid arthritis: a cohort study showing improved survival on treatment with pulse cyclophosphamide. Rheumatology (Oxford). 2001;40(7):821–825. | |
Fernández-Nebro A, Tomero E, Ortiz-Santamaría V, et al. Treatment of rheumatic inflammatory disease in 25 patients with secondary amyloidosis using tumor necrosis factor alpha antagonists. Am J Med. 2005;118(5):552–556. | |
Inoue D, Arima H, Kawanami C, et al. Excellent therapeutic effect of tocilizumab on intestinal amyloid a deposition secondary to active rheumatoid arthritis. Clin Rheumatol. 2010;29(10):1195–1197. | |
Magro-Checa C, Navas-Parejo Casado A, et al. Successful use of tocilizumab in a patient with nephrotic syndrome due to a rapidly progressing AA amyloidosis secondary to latent tuberculosis. Amyloid. 2011;18(4):235–239. | |
Redondo-Pachón MD, Enríquez R, Sirvent AE, et al. Tocilizumab treatment for nephrotic syndrome due to amyloidosis in Behcet’s disease. Ren Fail. 2013;35(4):547–550. | |
Amemori S, Iwakiri R, Endo H, et al. Oral dimethyl sulfoxide for systemic amyloid A amyloidosis complication in chronic inflammatory disease: a retrospective patient chart review. J Gastroenterol. 2006;41(5):444–449. | |
Dember LM, Hawkins PN, Hazenberg BP, et al; Eprodisate for AA Amyloidosis Trial Group. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. 2007;356(23):2349–2360. | |
CT Development America, Inc. Efficacy and Safety Study of KIACTA in Preventing Renal Function Decline in AA Amyloidosis. Available from: http://clinicaltrials.gov/ct2/show/NCT01215747?term=eprodisate&rank=1. NLM identifier: NCT01215747. Accessed August 19, 2014. | |
Zhu H, Yu J, Kindy MS. Inhibition of amyloidosis using low-molecular-weight heparins. Mol Med. 2001;7(8):517–522. | |
van der Hilst JC, Kluve-Beckerman B, Bodar EJ, van der Meer JW, Drenth JP, Simon A. Lovastatin inhibits formation of AA amyloid. J Leukoc Biol. 2008;83(5):1295–1299. | |
Fushimi T, Takahashi Y, Kashima Y, et al. Severe protein losing enteropathy with intractable diarrhea due to systemic AA amyloidosis, successfully treated with corticosteroid and octreotide. Amyloid. 2005;12(1):48–53. | |
Ozdemir BH, Ozdemir FN, Sezer S, Sar A, Haberal M. Among therapy modalities of end-stage renal disease, renal transplantation improves survival in patients with amyloidosis. Transplant Proc. 2006;38(2):432–434. | |
Gillmore JD, Tennent GA, Hutchinson WL, et al. Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. Br J Haematol. 2010;148(5):760–767. | |
Kluve-Beckerman B, Hardwick J, Du L, et al. Antisense oligonucleotide suppression of serum amyloid A reduces amyloid deposition in mice with AA amyloidosis. Amyloid. 2011;18(3):136–146. | |
Kennel SJ, Macy S, Wooliver C, et al. Phagocyte depletion inhibits AA amyloid accumulation in AEF-induced huIL-6 transgenic mice. Amyloid. 2014;21(1):45–53. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.