Back to Journals » Drug Design, Development and Therapy » Volume 15
Synthesis of Michael Adducts as Key Building Blocks for Potential Analgesic Drugs: In vitro, in vivo and in silico Explorations
Authors Ahmad S, Mahnashi MH, Alyami BA, Alqahtani YS ![]() , Ullah F, Ayaz M
, Ullah F, Ayaz M ![]() , Tariq M
, Tariq M ![]() , Sadiq A, Rashid U
, Sadiq A, Rashid U
Received 25 November 2020
Accepted for publication 19 February 2021
Published 23 March 2021 Volume 2021:15 Pages 1299—1313
DOI https://doi.org/10.2147/DDDT.S292826
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Sajjad Ahmad,1 Mater H Mahnashi,2 Bandar A Alyami,2 Yahya S Alqahtani,2 Farhat Ullah,1 Muhammad Ayaz,1 Muhammad Tariq,3 Abdul Sadiq,1 Umer Rashid4
1Department of Pharmacy, Faculty of Biological Sciences, University of Malakand, Khyber Pakhtunkhwa, Pakistan; 2Department of Pharmaceutical Chemistry, College of Pharmacy, Najran University, Najran, Saudi Arabia; 3Department PCB, Rokhan University, Jalalabad, Nangrahar, Afghanistan; 4Department of Chemistry, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, 22060, Pakistan
Correspondence: Abdul Sadiq
Department of Pharmacy, Faculty of Biological Sciences, University of Malakand, Chakdara, 18000 Dir (L), Khyber Pakhtunkhwa, Pakistan
Tel +92 301 2297 102
Email [email protected]
Umer Rashid
Department of Chemistry, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, 22060, Pakistan
Tel +92-334 5171999
Email [email protected]
Background: Organocatalytic asymmetric Michael addition is a strong approach for C-C bond formation. The objective of the study is to design molecules by exploiting the efficiency of Michael Adducts. We proceeded with the synthesis of Michael adducts by tailoring the substitution pattern on maleimide and trans-β-nitro styrene as Michael acceptors. The synthesized compounds were evaluated for dual cyclooxygenases (COX) and lipoxygenase (LOX) inhibition.
Methods: The compounds (4, 9– 11) were synthesized through Michael additions. The cyclooxygenases (COX-1 and 2) and lipoxygenase (5-LOX) assays were used for in vitro evaluations of compounds. After the acute toxicity studies, the in vivo analgesic potential was determined with acetic acid induced writhing, tail immersion, and formalin tests. Furthermore, the possible roles of adrenergic and dopaminergic receptors were also studied. Extensive computational studies were performed to get a better understanding regarding the binding of this compound with protein target.
Results: Four Michael adducts (4, 9– 11) were synthesized. Compound 4 was obtained in enantio- and diastereopure form. The stereopure compound 4 showed encouraging COX-1 and-2 inhibitions with IC50 values of 128 and 65 μM with SI of 1.94. Benzyl derivative 11 showed excellent COX-2 inhibition with the IC50 value of 5.79 μM and SI value 7.96. Compounds 4 and 11 showed good results in in vivo models of analgesia like acetic acid test, tail immersion, and formalin tests. Our compounds were not active in dopaminergic and adrenergic pathways and so were acting centrally. Through extensive computational studies, we computed binding energies, and pharmacokinetic predictions.
Conclusion: Our findings conclude that our synthesized Michael products (pyrrolidinedione 4 and nitroalkane 11) can be potent centrally acting analgesics. Our in silico predictions suggested that the compounds have excellent pharmacokinetic properties. It is concluded here that dual inhibition of COX/LOX pathways provides a convincing step towards the discovery of safe lead analgesic molecules.
Keywords: Michael products, succinimides, cyclooxygenase, lipoxygenase, analgesic, molecular docking, adrenergic and dopaminergic
Introduction
Pain is an unpleasant multidimensional experience that is necessary for the maintenance of healthy life and informs us about the potential harm after trauma and disease.1 It is an explicit interceptive sensation that arises from a specific part of the body.2 Temporal properties of pain mediated by peripheral and central neurons include stinging, throbbing, pricking, aching, and burning sensations.3 The most frequent and uncomfortable perceptions of pain include fatigue in muscles, muscular spasms, toothache, migraine, and many more.4
The discovery of aspirin was subsequently followed by various discoveries for the management of analgesia and inflammation. One of such major discoveries was the well-known NSAID (Nonsteroidal Anti-inflammatory Drugs) group of analgesics.5,6 In the living cells. Arachidonic acid (AA, as an ester) is covalently bonded in the cell membrane. The cyclo and lipoxygenase metabolize the AA and lead to the formation of eicosanoids and lead to inflammation. The COX and LOX pathways leads to the formation of prostaglandins and leukotrienes respectively, which are correlated with inflammatory properties.7 Among the two isoforms, COX 1 is mainly responsible for various soft tissues of kidney, GIT, and platelets, while COX 2 is involved for inflammation. Therefore, the discovery of selective COX 2 provided patients’ relief from analgesia and inflammation, leaving the gastrointestinal tract protected.8–10 There are various approaches to treat analgesia and inflammation with gastrointestinal protection. It is obvious that COX 1/2 NSAIDs leads to lower accretion of leukotriene via the LOX pathway. Therefore, an effective approach is the dual inhibition of cyclo and lipoxygenase pathways which produce their effect without gastrointestinal toxicity.11
The Michael addition are vital reactions in the complex C-C bond formation. The Michael addition to nitro-olefins and maleimide lead to diverse structures of nitroalkane and succinimide types of products. These diverse structures are either itself vital drug analogs or can be important building blocks in drug and bioactive moieties. In Michael and other vital reactions, chiral compounds have a vital importance in the drug design and discovery.12–14 The exploration of chiral molecules in drugs and natural products can help in launching and marketing of new drugs.15 Optically active molecules are considered to be more suited for pharmacological uses.16 Similarly, the legend based molecular design is also a powerful approach in the discovery of new molecules with diverse pharmacological properties.17–20 Previously, we have explored different Michael products for biological targets. In maleimide to succinimide Michael products, we evaluated its aldehyde,21–24 ketone,2 cyanoacetate,25 and ketoester derivatives.26,27 The nitrogenous synthetic compounds have a variety of pharmacological activities.28–31 Besides synthetic compounds, our group have also explored various natural products for analgesic antinociceptive potentials.32–34 We have recently also converted one of the marketed drugs aceclofenac to a nano-formulation for enhanced analgesic activity.35 Based on our experience with analgesic drugs and the available literature, we feel an opportunity for new, safe, and effective analgesic drugs. Therefore, we have designed this project to synthesize a stereopure organic compound for the possible management of analgesia. Besides in vitro and in vivo studies, we also have performed extensive computational studies for all possible parameters. Through computational studies, we have performed the binding interactions, pharmacokinetics and site of metabolism. Overall, our designed study provides a convincing step towards drug discovery.
Materials and Methods
Chemicals and Drugs
The chemicals and drugs used in this study are 2-methylpentanal 98% (Cat. No 258563, Sigma, Germany), N-phenylmaleimide 97% (Cat. No P27100, Sigma, Germany), trans-β-nitrostyrene (Cat. No N26806, Sigma, Germany), cyclohexanone (Cat. No 398241, Sigma, Germany), acetone (Cat. No 650501, Sigma Germany), 1-benzyl-4-piperidone (Cat No B29806, Sigma, Germany), O-tert-butyl-L-threonine 98% (Cat. No 20644, Sigma, Germany), potassium hydroxide (Sigma, Germany), Acetic acid (Sigma, Germany), yohimbine (Sigma, Germany), formalin, local supplier of Merk, morphine as morphine sulfate (P.D.H Laboratories, Pakistan), indomethacin (Siza International, Pakistan), naloxone hydrochloride injection (RotexMedica, Triitau, Germany), haloperidol decanoate injection (Jhonson and Jhonson, Pakistan), and diclofenac sodium (Novartis Pharma, Pakistan).
Synthesis of (S)-2-((S)-2,5-Dioxo-1-Phenylpyrrolidin-3-yl)-2-Methylpentanal (4)
To a small reaction vial was added 2-methylpentanal (1.2 mmol, 1.2 equiv, 149 µL), O-tert-butyl-L-threonine (5 mol%, 8.75 mg), and KOH (5 mol%, 2.80 mg) in dichloromethane (2.0 M, 0.5 mL). This mixture was stirred for 2–3 minutes to form the enamine. Afterwards, N-phenylmaleimide (1.0 mmol, 1.0 equiv, 173.17 mg) was added to it and stirring was continued. The reaction was monitored with thin layer chromatography. The reaction was completed in 16 hours. The reaction was stopped physically and quenched by adding 15 mL of water to it. The organic layer was separated with 15 mL of dichloromethane. The extraction was repeated three times to get the maximum possible compound out of the mixture. The organic layers were combined and dried with anhydrous sodium sulfate. The dried solvent mixture was filtered and was put on a rotary evaporator to get the dried crude product. The crude product was purified with column chromatography and the structure was confirmed.21
Crystallization of Compound 4
Crystallization of the purified product was performed to enrich the diastereomeric ratio. Using 15 mL of methanol and pet ether (1:1), the diastereomeric mixture (3:1) was dissolved at 70°C. Then it was allowed to cool slowly which resulted in the crystallization of the major diastereomer.
Stereochemistry of Compound 4
The absolute and relative stereochemistry of the compound was determined by comparing with the reported data.21 The initial diastereoselectivity of the compound was determined by the crude 1H NMR and chiral HPLC analysis. The final diastereoselectivity was confirmed with 1H NMR of the final product after crystallization. The enantioselectivity of the final single diastereomer was determined by comparing the HPLC spectra of with achiral 2-(2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal using chiral HPLC. The HPLC parameters were Chiralcel OD-H column, i-propanol/n-heptane 20/80, flow rate 1 mL/min and λ was 210 nm. The retention times of the stereoisomers (2-(2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal) were T1 = 43.2 minutes, T2 = 63.2 minutes, T3 = 75.0 minutes, and T4 = 95.0 minutes. T1 and T2 represent the minor diastereomer in which T1 is the major enantiomer and T2 is the minor one. Similarly, T3 and T4 are the major and minor enantiomers of the major diastereomer, respectively.
Synthesis of Nitroalkane Michael Products (9–11)
Compounds 9–11 were synthesized by Michael additions reactions. In a small vial was added two equivalents of ketone (acetone for compound 9, cyclohexanone for compound 10, 1-benzyl-4-piperidone for compound 11) to Ot-Bu-L-threonine 0.1 equiv and potassium hydroxide 0.1 equiv and stirred in 1 M of dichloromethane for 2 or 3 minutes. Afterwards, 1 equiv of trans-β-nitrostyrene was added as Michael acceptor to complete the reaction. The time of reaction was monitored by TLC analysis. When we observed that the limiting reagent trans-β-nitrostyrene was not visible on TLC, the reaction was quenched in-situ with water. The reaction was extracted three times in dichloromethane and combined. The combined organic layer was dried and was purified by column chromatography using n-hexane and an ethyl acetate solvent system.12
Cyclooxygenase (COX-1 and 2) Inhibition Assays
Both the cyclooxygenase 1 and 2 inhibitory assays were carried out according to the standard reported procedure.36 The dimethyl sulfoxide (DMSO) solvent was used in the blank. The reaction was initiated via the addition of arachidonic acid and the solutions were incubated for exactly 8–10 minutes in a water bath at 37°C. After that, the reaction was terminated with 10 μL 2N HCl. In COX 1 and 2 experiments, 5 μM and 200 μM of standard indomethacin were used, respectively. Test compounds were tested in concentrations ranging from 31.25–250 μg/mL. The cyclooxygenase enzyme inhibition was calculated as per the given standard formula;
Percent cyclooxygenase inhibition=[1−(DSNtest sample−DSNbackground/DSNsolvent blank−DSNbackground)]×100
5-Lipoxygenase (5-LOX) Inhibition Assay
The 5-LOX potentials of the compounds (4, 9–11) was determined by the previously established reported method.2 Different dilutions (500–31.25 μg/mL) of the synthesized compound were prepared. The lipoxygenase enzyme solution was also prepared 10,000 U/mL. The substrate linoleic acid of 80 mM was also prepared. The phosphate buffer solution of 50 mM having pH 6.3 was also prepared. A mixture of all the solutions (2 mL) having equivalent volumes of all were prepared and mixed thoroughly. The dilutions made of the compounds and also that of the standard drug were also added to the mixture. The rate of reaction was found for the compounds and control. The enzyme activity was measured at 234 nm by using a double beam spectrophotometer. The percentage activity/inhibition was determined by raising the absorbance of the compounds compared to the negative control. The zileuton was used as a positive control.
Experimental Animals
Balb C mice of either sex were obtained from Pakistan Council of Scientific and Industrial Research, KPK, Pakistan. They were kept at standard laboratory conditions with the provision of standard food and water ad libitum in the animal house, University of Malakand, Pakistan. The experimental mice were used in in vivo studies according to the approval of the Ethical Committee, Department of Pharmacy, Faculty of Biological Sciences, University of Malakand, Pakistan via letter No. DREC/22 under the guidelines of Scientific Procedures Issue-1 of Animal Bylaws – 2008. After the experimental procedures, the animals were euthanized properly as per the standard procedure using AVMA Guidelines for the Euthanasia of Animals.37 Halothane vapors were slowly given to the animals to induce anesthesia. The overdose and prolonged time euthanized the animals.
Acute Toxicity Study
Before the analgesic studies on experimental animals, the acute toxicity profiling of the selected compounds (4 and 11) was performed as per the standard protocols.38 Six groups of experimental mice were made, having six mice in each group. Test compounds were administered to the mice in different doses, ie, 25, 50, 75, 100, 125 and 150 mg/kg body weight through I/P route. The animals were initially monitored for any unwanted symptoms and then observed for any mortality in the next 24 hours.
Acetic Acid Induced Writhing Test
The peripheral nociception was determined with an acetic acid induced writhing model. In this protocol, two groups of mice, having six mice in each were made. Sixty minutes prior to 1% acetic acid injection (10 mL/kg, intraperitoneally), both groups were administered the compounds (4 and 11) in 25 and 50 mg/kg body weight. Tween 80 (1%, 10 mL/kg) solution was used as negative control, while diclofenac sodium (50 mg/kg, i.p.) was used as positive control. The numbers of writhing/stretching were observed, and the percent result was calculated.32
Tail Immersion Test
The tail immersion test was used for determination of central antinociceptive response. The compounds (4 and 11) were administered at doses of 25 and 50 mg/kg i.p., morphine (5 mg/kg) and vehicle 2% half an hour prior to tail immersion. The mice tail was dipped up to 3 cm in hot water of temperature 55±0.5°C. After the administration of the compounds, the reactions of mice were observed with a gap of 15 minutes up to 90 minutes.33
Formalin-Induced Paw-Licking Test
In formalin method, the selected compounds (4 and 11) at concentrations of 25 and 50 mg/kg were administered to the labeled mice. After some time (30 minutes), 20 μL of 2.5% of formalin was injected into the hind paw of mice. In early and late phase (ie, start to 5 minutes and 15–30 minutes), the mice were observed for their reaction in licking their paw. Subcutaneous naloxone (2 mg/kg), morphine (5 mg/kg) and indomethacin (10 mg/kg) were also used.34
Possible Mechanism of Action
Adrenergic Receptors
The mice were given a 1 mg/kg dose of yohimbine i.p. to check the involvement of the adrenergic system. The animals were given compounds (4 and 11) at 25 and 50 mg/kg after 15 minutes of yohimbine administration. After a the gapof 30 minutes, administration of acetic acid was done to the mice. The amount of stretching–writhing were observed, and the percent result was calculated.33
Dopaminergic Receptors
The mice were given haloperidol i.p. to check the involvement of dopaminergic receptors. The animals were given selected compounds (4 and 11) at 25 and 50 mg/kg after 15 minutes of haloperidol administration. After 30 minutes, administration of acetic acid was done to the mice. The amount of stretching–writhing was observed, and the percent result was calculated.33
Statistical Analysis
The experiments were repeated three times and the values were expressed as mean±SEM. The statistical analysis was performed as per the standard procedure by using one-way ANOVA method.
Computational Studies
Docking Studies Using a Molecular Operating Environment
Molecular Operating Environment (MOE) docking program version 2016.08 was used for docking studies. Crystal structure of ovine COX-1 (PDB code 1EQG) complexed with ibuprofen and crystal structure of COX-2 (PDB code 1CX2) with co-crystallized inhibitor SC-558 were used for the study. The possible water molecules were eliminated from the target protein structure. Afterwards, H-atoms were added instead, and optimization of energy was calculated out via default force field. The three-dimensional structures (3D) of compounds were patterned through the MOE builder program. Geometrical parameters for three dimensional structures of (S)-2-((S)-2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal were optimized. Before molecular docking, partial charges were also calculated. The protonation (3D of both isozymes) was done. Using default parameters of MOE energy minimization algorithm, ie, gradient = 0.05, Force Field = MMFF94X, energy minimization of target protein molecule was carried out. The model obtained was put forward to systematic conformational search at default parameters. The active site of the prepared enzymes was defined as the residues within 10 Å of the reference ligands. The residue and backbone remained fixed and energy minimization was performed. For onward analysis, the lowest energy minimized pose was used. Ligand-interaction module of MOE was used to calculate ligand–enzyme interactions. Docking observations and investigations of their surface with graphical representations were done using MOE and a discovery studio visualizer.39
Results and Discussion
Design Rational
Selectivity for COX-2 inhibition depends on its structural template, ie, the presence of two vicinal aryl rings or 1.3-aryl groups attached with central five or six-membered heterocyclic or carbocyclic motif. We are constantly searching for the new templates for the inhibition of both isoforms of cyclooxygenase. Previously, we discovered a flexible methylene linker containing aryl carbonyl derivatives that showed excellent COX-2 inhibition. In the current research, we considered it worthwhile to design molecules by exploiting the efficiency of Michael Adducts. We proceeded with the synthesis of Michael adducts by tailoring the substitution pattern on maleimide and trans-β-nitro styrene as Michael acceptors. Although we planned to use aldehyde as a nucleophile for the synthesis of succinimide derivative, however, we were interested to explore the effects of the small nitro group instead of traditional sulfonamide. Due to a difference in size of active site of COX isoforms, the structure activity relationship was designed to investigate the effect of addition of acetone, cyclohexanone, and 1-benzylpiperidin-4-one to trans-β-nitro styrene.
Chemistry
The target molecule was obtained in a single step Michael addition followed by crystallization, as shown in Scheme 1. Using the asymmetric organocatalytic approach, we synthesize the target compound 4, ie, ‘(S)-2-((S)-2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal’ with 90% isolated yield, 99% ee and 3:1 dr as depicted from initial 1H NMR analysis. To enrich the diastereomeric ratio of 3:1 to a single diastereomer, we tested several different solvent systems. In methanol/pet ether (1:1), we obtained a pure single diastereomer (major diastereomer), ie, dr of >99:1 as shown in 1H NMR (Figure S1, Supporting Information) and HPLC spectra (Figure S2 and S3, Supporting Information). The final yield of the compound after crystallization was 45%.
 |
Scheme 1 Organocatalytic synthetic approach to synthesize (S)-2-((S)-2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal (4). |
The compounds 9–11 were synthesized by Michael additions from the respective ketones as shown in Scheme 2. The 1H NMR spectra of compound 9 and 10 are shown in Figure S4 and S5 of the supporting Information respectively. Compound 9 was synthesized from the addition of acetone to trans-β-nitrostyrene. The purified product was obtained as Off-White powder with an isolated yield of 78%. The observed and calculated Rf value of the compound 5-nitro-4-phenylpentan-2-one was 0.41 (n-hexane: ethyl acetate; 80:20). Similarly, compound 10 was obtained as White powder with an 86% yield, while compound 11 was obtained as a brown semisolid with isolated yields of 89%. The NMRs spectra of the compounds are provided in the supporting information.
 |
Scheme 2 Synthesis of compounds 9–11 through Michael additions. |
In vitro Inhibition Studies Against COX-1, COX-2, and 5-LOX
The COX isozyme inhibitory activity of compounds 4, 9–11 were determined by using COX-1 and COX-2 enzymes. Indomethacin was used as a positive control for COX-1 and 2 assays. The results are presented as IC50 values in μM concentrations, as shown in Table 1. Selectivity index (SI) was also calculated by using formula IC50 of COX-1/IC50 of COX-2. The results presented in Table 1 showthat benzyl derivative 11 showed good COX-2 inhibition with the IC50 value of 5.79 μM and SI value 7.96. The observed IC50 values for COX-1 of compounds 4, 9, and 10 were found to be 128.1, 67.59, and 57.40 μM, respectively. Similarly, in COX-2 assay, compounds 4, 9, and 10 exhibited IC50 values of 65.91, 73.53, and 49.68 μM, respectively.
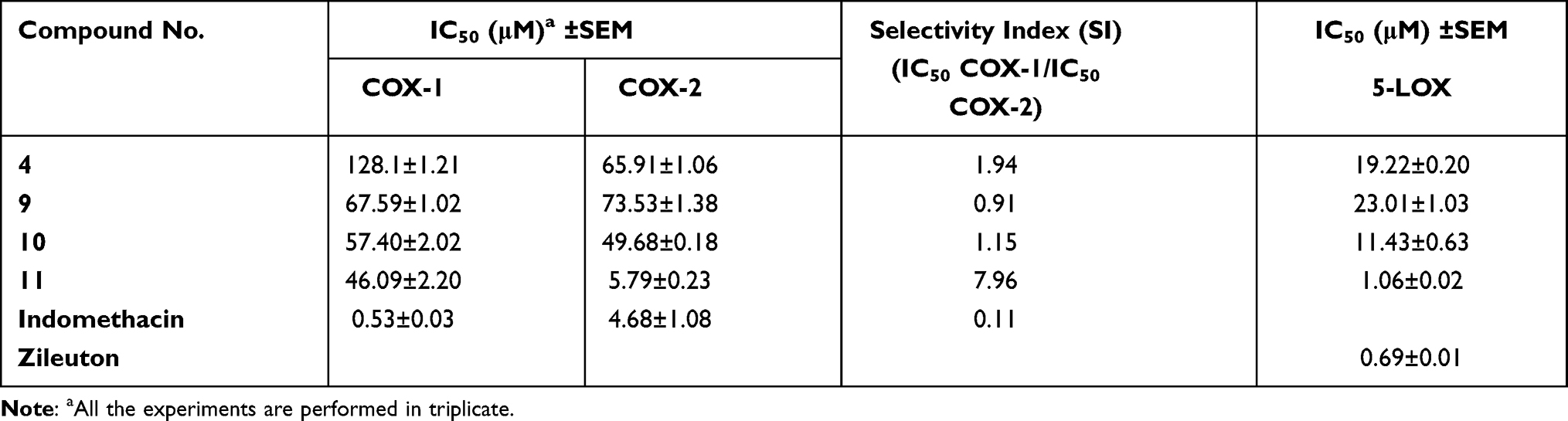 |
Table 1 In vitro Cyclooxygenase-1/2 and 5-LOX Inhibitions of the Michael Products |
Moreover, the 5-lipoxygenase inhibitions values are also summarized in Table 1. Compound 11 emerged as the most potent compound of the series, with an IC50 value of 1.06 μM. Standard drug zileuton exhibited the IC50 value 0.69 μM.
In vivo Studies
Acute Toxicity
As per the standard procedure, all the samples subjected to experimental animals should be tested for tests necessary for safety before administration. To test a novel drug or sample in humans, the acute toxicity test is a simple and vital test which can be carried out in experimental animals. The major purpose behind this test is to find out the minimum concentration which can cause adverse side-effects and lethality in animals.
Based on our in vitro experiments, we selected pyrrolidine derivative 4 and nitroalkane derivative 11 for in vivo studies. The Michael products 4 and 11 at highest tested dose, ie, 150 mg/kg for body weight proved to be safe, with no adverse effect, change in body weight, or mortality observed (as shown in Table 2). Consequently, the dose of compound 25 and 50 mg/kg was taken as safe for the in vivo anti-nociceptive studies.
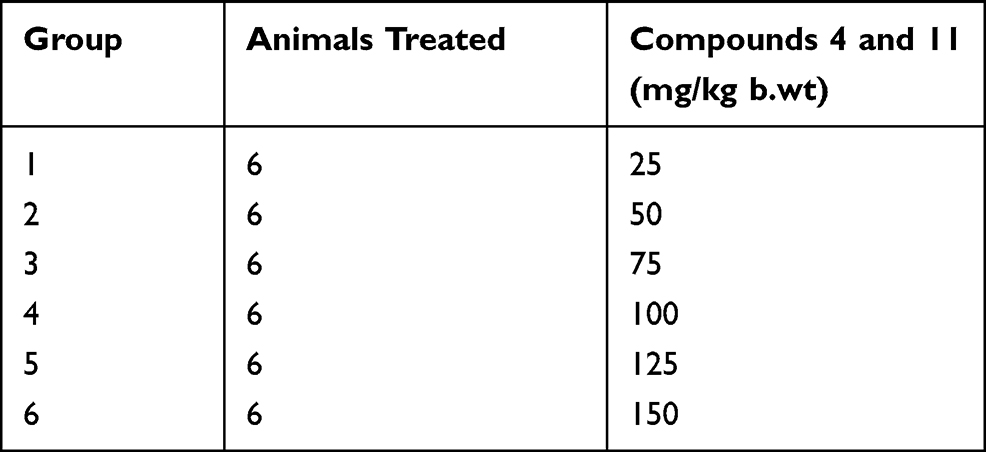 |
Table 2 Animal Group Specification and Quantity of Drug Administered for Acute Toxicity Studies with Synthesized Michael Products |
Acetic Acid Induced Writhing Test
In the current analgesic investigations using acetic acid induced writhing essay, our compounds 4 and 11 exhibited encouraging in vivo results. At 50 mg/kg, compound 4 reached to maximum inhibition value of 52.2%, P<0.01, n=6 as shown in Figure 1. In comparison, the standard medicine diclofenac sodium at concentration of 50 mg/kg reached to 85% inhibition. Compound 11 inhibited the writhing up to 48.71% (P<0.01, n=6, 50 mg/kg, Figure 1). Comparatively, diclofenac (50 mg/kg) exhibited a dominant inhibition of 84.96%.
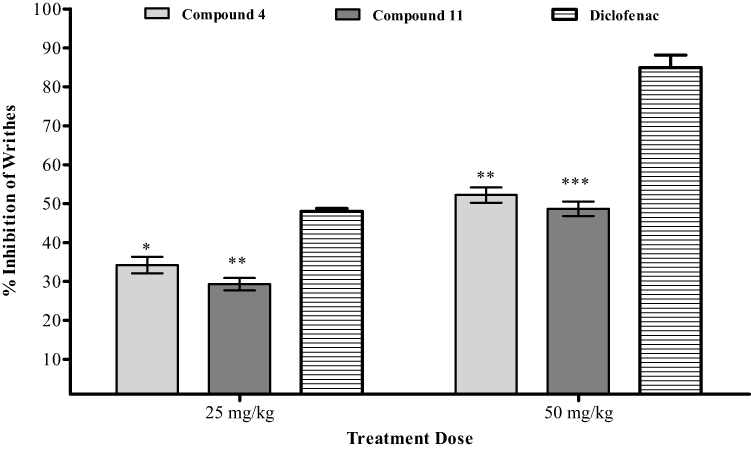 |
Figure 1 Acetic acid induced writhing results of compounds 4 and 11. The observed values are represented as ±SEM (*P<0.05, **P<0.01, and ***P<0.001 in comparison to the standard). |
Tail Immersion Test
Furthermore, in tail flick model, compound 4 exhibited a 29.5% response at 25 mg/kg body weight as shown in Table 3. The latency time was a maximum at 75 minutes and reduced back to 19.8% at 90 minutes. (S)-2-((S)-2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal (4) at a dose of 50 mg/kg showed a 53.4% response at 75 minutes interval. The analgesic response was again reduced at 90 minute interval to 46.2%. Compound 11 also showed an overwhelming response in tail flick method compared to compound 4. At 25 mg/kg, the response reached 42.2% in 75 minutes. Similarly, for compound 11 at 50 mg/kg, the response was maximum at 75 minutes (63.5%) which reduced back to 58.7% at 90 minutes. Centrally acting opioid analgesic, displayed 80.2% response at a dose of 5 mg/kg after 75 minutes and 78.4% response at 90 minutes. The level of significance was P<0.001 at 90% confidence interval in comparison to positive control (morphine).
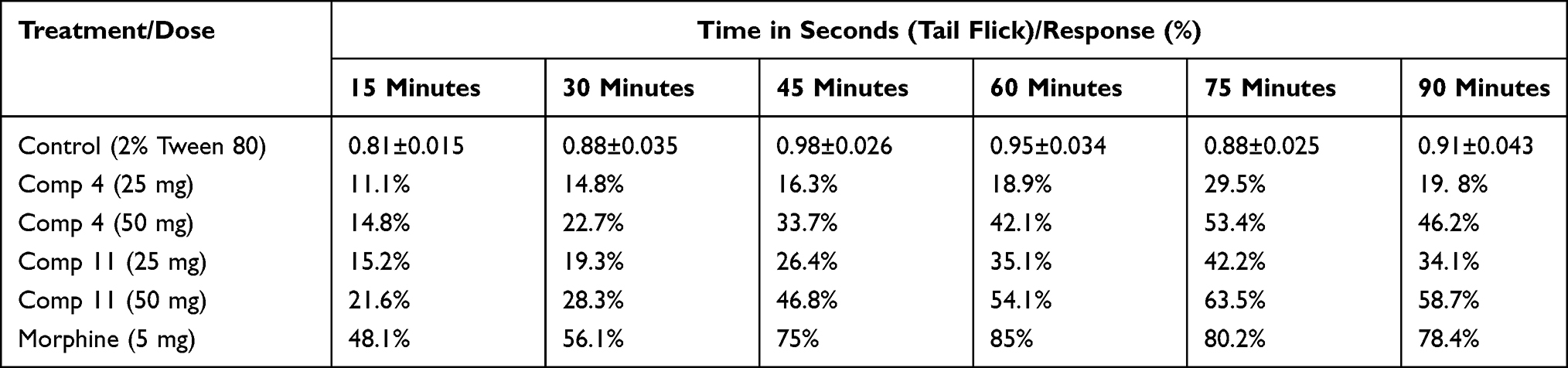 |
Table 3 Tail Flick Method Results of the Michael Products 4 and 11 |
Formalin Test
To check the involvement of central analgesic pathway, animals previously treated with compounds (4 and 11) were administered formalin, and a dose dependent response was observed. The compound 4 significantly inhibited the two phases, ie, 36.16±4.47% in 1st phase and 50.66±3.84% in 2nd phase at 50 mg/kg. In comparison, mice treated with positive control (morphine 5 mg) inhibited 87.16±3.32% of phase 1 and 96.34±0.66% for phase 2 (Table 4). Similarly, in formalin assay, the inhibitions of compound 11 were 33.66% (**P<0.01, n=6) in the phase first while 47.78% (**P<0.01, n=6) in the second at 50 mg/kg. In mice pre-treated with naloxone, no obvious reversal inhibition was observed. We observed that, due to naloxone, there was no turnaround in analgesic effect of morphine in both the phases. Similarly, administration of 10 mg/kg of standard indomethacin also inhibited the two phases significantly.
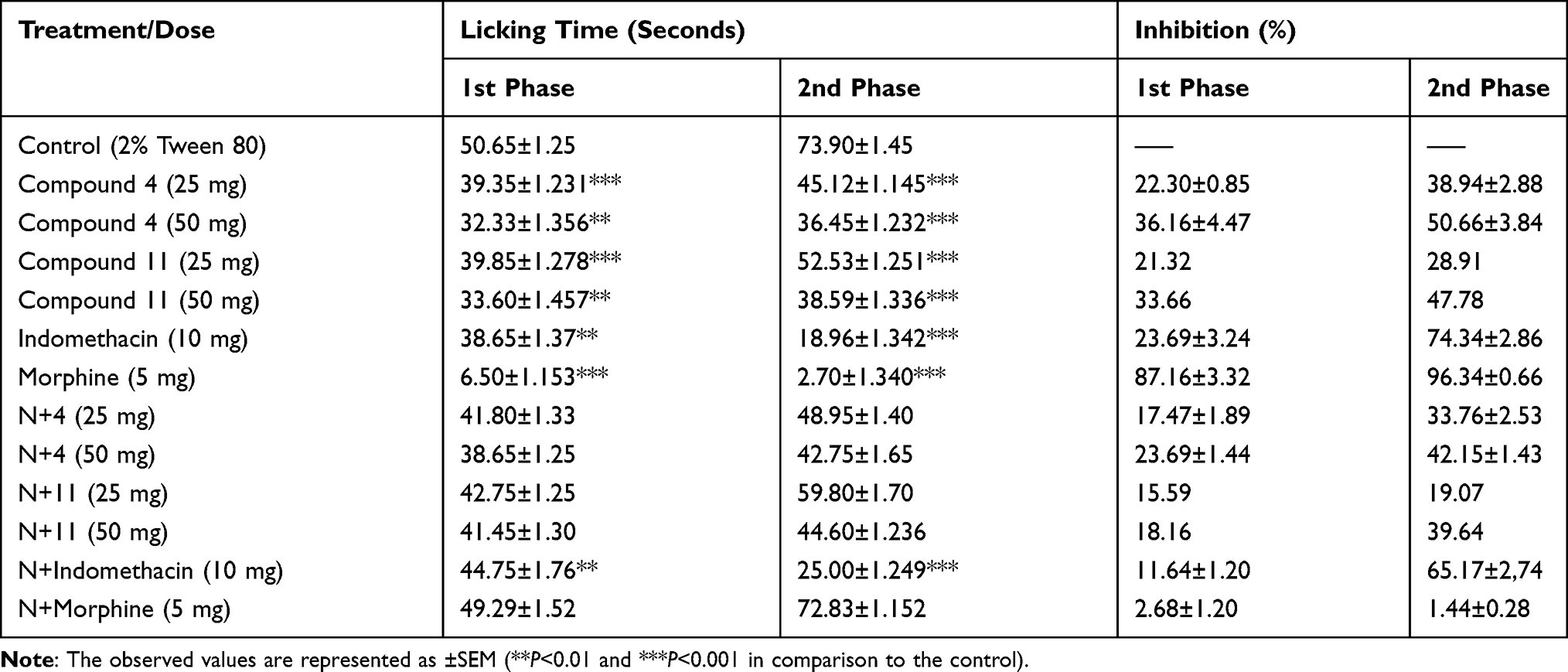 |
Table 4 Formalin-Induced Paw-Licking Response for Elucidation of Central Analgesic Pathway of Compounds 4 and 11 |
Possible Role of Adrenergic and Dopaminergic Receptors
Data on the possible involvement of the compounds on adrenergic receptor is given in Table 5. The yohimbine and haloperidol were used for confirmation/involvement of adrenergic and dopaminergic receptors, respectively. Both yohimbine and haloperidol are antagonists of adrenergic (alpha 2) and dopaminergic (D1/D2) receptors, respectively. Experimental mice administered with yohimbine give almost a similar response with our compounds, so it is obvious that adrenergic receptors are not involved in the mechanism for the compounds (Table 5). Similarly, pretreatment with haloperidol (as shown in Table 6) also does not affect the anti-nociceptive action of the compound. So, it is obviously observed that the dopaminergic pathway is also not involved in the mechanism.
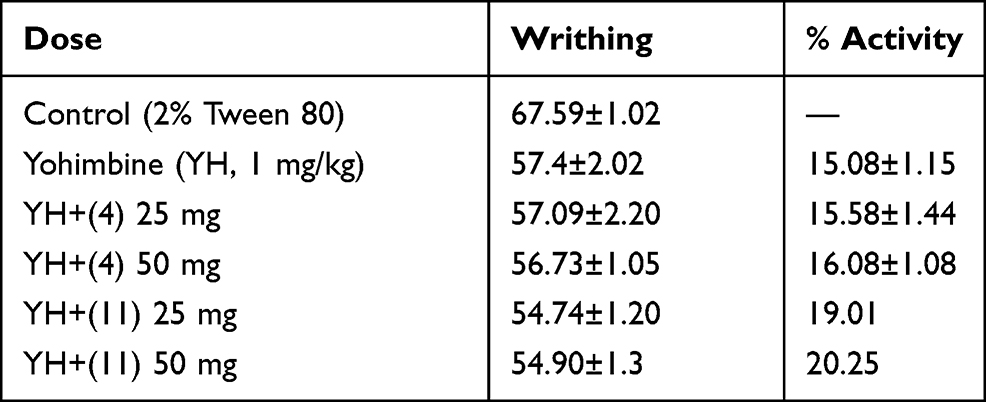 |
Table 5 Possible Involvement of Alpha-2 Adrenergic Receptor in the Analgesic Response |
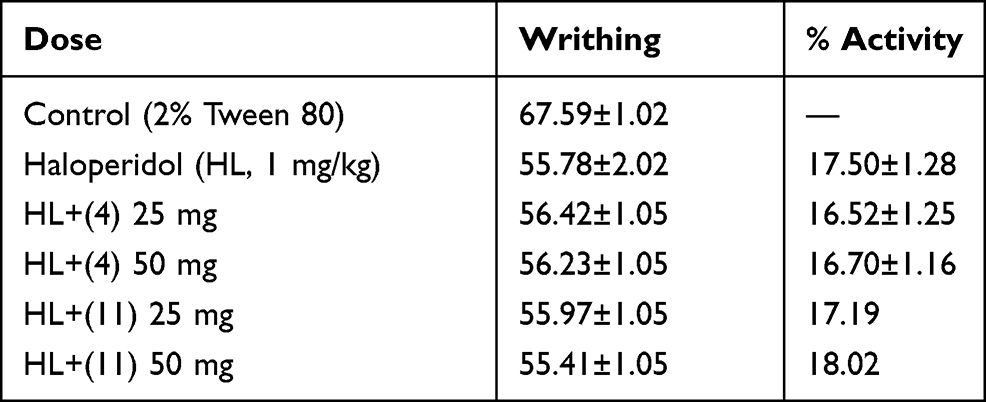 |
Table 6 Possible Involvement of Dopaminergic Receptor in the Analgesic Response |
Pain is a vital sensory feeling that makes us aware regarding life threatening infections and disease conditions.40 Similar to hunger and thirst, pain is a motivational somatic situation that drives suitable behavioral responses. However, chronic pathological pain can entirely overlook attention, consciousness, and lead to intolerable suffering.41 In such conditions, the use of analgesic drugs is important to relieve patient suffering besides treating the basic cause of disease. The origin of pain including spinal cord, brain and peripheral nervous system is greatly understood. Several neurochemicals are implicated in the interaction between different systems like thermoregulatory, immune, and sympathetic systems and make pain an integrated physiological phenomenon.42 The clinical management of pain is greatly improved with the development of local, spinal analgesia and the discovery of novel therapeutic tools like adrenergic, serotonergic agonists.43 Still the development of novel and cost-effective drugs is necessary for mitigation of chronic pain.
By inhibition of prostaglandin synthesis mediated by cyclooxygenase enzymes, non-steroidal anti-inflammatory drugs (NSAIDs) relieve pain and inflammation. Two important enzymes COX-1 and COX-2 have been recognized, one predominating at sites of inflammation (COX-2), whereas COX-2 is predominantly expressed in the gastrointestinal tract. COX-2 inhibition is known to play an important role in the development of highly effective yet safe analgesic drugs. Whereas, COX-1 inhibitors are associated with mucosal damage, and ulceration in the gastrointestinal tract. In the current study, our test compound displayed a non-selective inhibition of the COX-1 and COX-2 enzymes.
The peripheral anti-nociceptive response can be sensibly determined by using the acetic acid animal model of analgesia.32 The acetic acid model relies on several pathways/mechanisms which include the release of biogenic amines (sympathetic pathway), cyclooxygenase enzymes, and opioid pathways.33 On the other hand, the formalin method is a well-known test for the central analgesic activity.44 In this research, the synthesized compound inhibited both phases of formalin pathway. Furthermore, the naloxone (an antagonist of opioid) did not alter the analgesic activity. So, this suggests that the main pathway is involved, and opioid receptor is not involved.
The international regulations related to the health of human beings are at the opinion that any new molecule must be initially checked for the safety profile prior to the pharmacological activity. The safety test must be conducted in experimental animals. So, one of such tests is acute toxicity studies which is used to identify what can cause adverse effects and also the minimum dose of the compound which cause mortality. The acute toxicity test is an approved method for the safety profile before the in vivo experiments.45 The synthesized compound ((S)-2-((S)-2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal) was observed to be safe for a concentration of up to 150 mg/kg. The experimental animals were checked for all types of adverse reaction as per the standard guidelines and hence a dose of 25 and 50 mg/kg was safe for various activities in animal models.
Succinimides, group of biologically active compounds with known anticonvulsant potential. The commonly employed of them are phensuximide, methsuximide, and ethosuximide. The basic structure of all succinimides contain a cyclic five membered ring with two amide carbonyls connected by an amide nitrogen for both.2,12,21 Various researchers have substituted the basic nucleus of succinimide and have formed a diverse array of substituted derivatives of it.46 Being a vital class of medicinally important compounds, succinimide have shown various pharmacological potentials.47 In this article, we have shown the synthetic approach to enantiopure and diastereo-pure derivative of succinimide, ie (S)-2-((S)-2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal.
In silico Studies
We have performed docking studies to explore the mechanism of binding and selectivity for two isozymes, ie, COX-1 and COX-2. Furthermore, in silico pharmacokinetic prediction of the synthesized derivatives was also computed by using an online computational tool.
Docking Studies
Docking studies of the synthesized compound were carried out to explore the binding affinities and its selectivity against COX-1/COX-2. These docking studies were carried out using Molecular Operating Environment (MOE).
The binding site of these two isozymes is elongated and a narrow hydrophobic channel that is extending from membrane-binding domain to catalytic domain. The structural difference between the active site of the two isoforms is the Val523 in COX-2 (Ile523 in COX-1). Three-dimensional X-ray crystallographic structures of the enzymes were downloaded from the Protein Data Bank (PDB). Crystal structure of ovine COX-1 (PDB code 1EQG) complexes with ibuprofen and crystal structure of COX-2 (PDB code 1CX2) with co-crystallized inhibitor SC-558 were used for the study.
First, the docking procedure was validated by re-docking of the native ligands; ibuprofen (COX-1) and SC-558 (COX-2). Docking was carried out using Triangle matcher algorithm (placement stage) and scored by London dG scoring function. The results of the re-docking experiments were evaluated using root-mean square deviation (RMSD) for higher ranked pose. The reasonable performance protocol with RMSD value of 1.14 Å for COX-1 and 0.74 Å for COX-2 (threshold RMSD < 2.0 Å) was adopted for the docking of all the isolated compounds.10,48–50
In the best possible binding mode, compound 4 showed the same type of interactions with the active site residues of both isozymes. Arg120 and Tyr355 showed hydrogen bond donor interactions with the aldehydic carbonyl oxygen atom in the binding site of COX-1 (Figure 2A) and COX-2 (Figure 2B). The MOE binding energy of ligand-COX-1 complex is −6.4259 Kcal/mol and for ligand-COX-2 complex is −6.8730 Kcal/mol (Table 7). The details related to extensive docking studies of compound 4 are provided in supporting information (Table S1–S6).
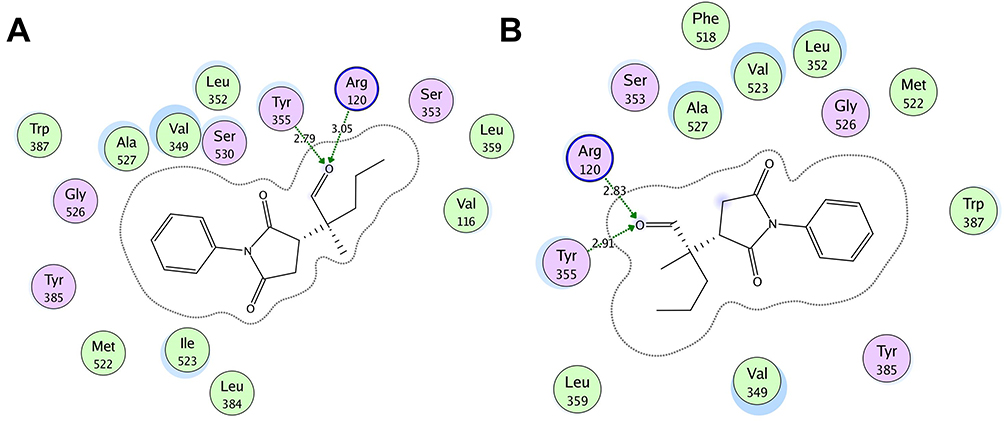 |
Figure 2 Two-dimensional (2D) interaction plot of compound 4 into the binding site of COX 1 (A) and COX-2 (B). The diagram is generated from MOE software. |
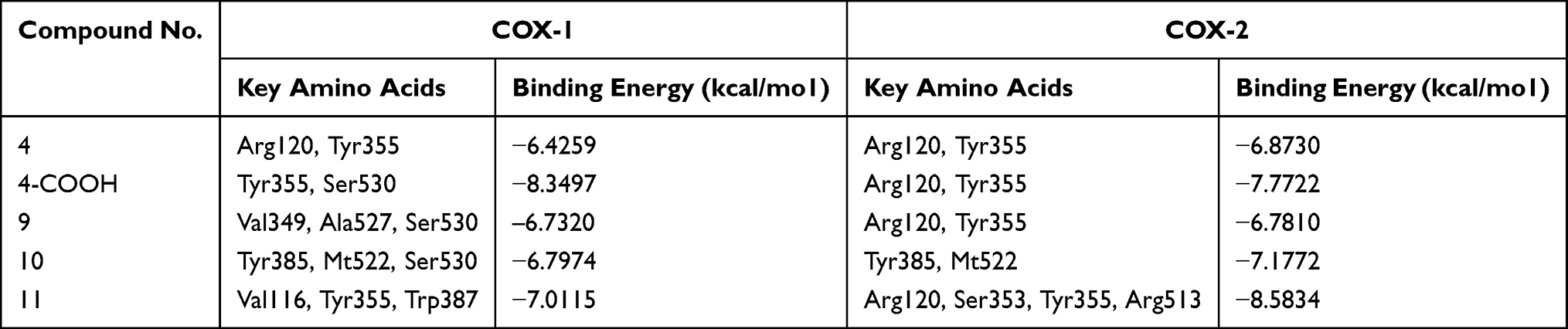 |
Table 7 Interaction with the Amino Acid Residues and Binding Energy Value of the Compounds |
We extended our study and carried out docking studies on the oxidized form of our synthesized aldehyde. These studies were performed on corresponding carboxylic acid by using MOE. The interactions of the compound on both isoforms are shown in Figure 3. It can be seen from Figure 3A that carboxylic acid derivative forms four hydrogen bond (HB) interactions with the cavity amino acid residues. Arg120 forms a bifurcated HB with carbonyl oxygen and hydroxyl oxygen. Another hydrogen bond was found between Tyr355 and hydroxyl oxygen. Ser530 establishes another HB with carbonyl oxygen of pyrrolidine-2,5-dione moiety. For COX-2, carbonyl oxygen forms two interactions with Arg120 and Tyr355 (Figure 3B). The binding energy comparison of the carboxylic acid form with its parent compound (aldehydic) shows that the carboxylic acid derivative is more tightly bound into the binding site of the enzymes. The binding energy for COX-1 is −8.3497 kcal/mol and for COX-2 is −7.7722 kcal/mol (Table 7). Therefore, it may be predicted here that the carboxylic acid form will be more potent than its parent aldehydic form.
 |
Figure 3 COX 1 and 2 molecular docking on the corresponding carboxylic acid form of compound 4. |
Next, we docked trans-β-nitro styrene derivatives 9–11 into the binding site of both COX isozymes. The binding orientation of derivatives 9–11 into the binding site of COX-1 (PDB code 1EQG) is shown in Figure 4A. Compound 9 forms one hydrogen bond interaction and two π–σ types of interactions (Figure 4B). Compound 10 establishes two hydrogen bond interactions with Ty385 and Ser530, while phenyl ring forms π-sulfur interaction with Met522 (Figure 4C). Phenyl rings of compound 11 form two π–π interactions with Tyr355 and Trp387. Val116 forms a π–σ interaction with phenyl ring (Figure 4D). The MOE binding energy values of ligands-COX-1 complex of compounds 9–11 are –6.7320, −6.7974, and −7.0115 Kcal/mol, respectively.
 |
Figure 4 (A) Superposed binding orientation of compounds 9–11 on native ibuprofen (yellow) into the binding site of COX 1. (B–D) Three-dimensional interaction plot of compounds 9–11, respectively, into the binding site of COX-1. |
All the three trans-β-nitro styrene derivatives (9–11) were then docked into the binding site of COX-2 (PDB ID=1CX2). Binding orientation of all the compounds superposed on the native SC-558 is shown in Figure 5A. Three-dimensional interaction plot of the compounds (Figure 5B–D) revealed that only compound 11 interacts with the conserved COX-2 residues. The nitro group forms a bifurcated hydrogen bond interaction with deeply located COX-2 pocket residue Arg513, while Ser353 forms π–σ types of interactions (Figure 5D). Cyclohexanone carbonyl oxygen oriented itself towards the Arg120/Tyr355 present below the COX-2 selectivity pocket. The MOE binding energy values of ligands-COX-2 complex of compounds 9–11 are −6.7810, −7.1772, and −7.9834 Kcal/mol, respectively.
 |
Figure 5 (A) Superposed binding orientation of compounds 9–11 on native SC-558 (yellow) into the binding site of COX 2. (B–D) Three-dimensional interaction plot of compounds 9–11, respectively, into the binding site of COX-2. |
Preliminary in silico Pharmacokinetic Studies
In silico pharmacokinetic studies were carried out using online AdmetSAR predictor. Through computational studies, several molecular descriptors were studied to get an idea about the pharmacokinetics of the compound (Table 8). The computed molecular descriptors include the Lipinski Rule of Five (Lipinski Ro5), human intestinal absorption, blood–brain barrier (BBB) penetration, bioavailability, carcinogenicity, hepatotoxicity, and plasma protein binding. The oral absorption permeability and in agreement with the were also assessed. The parameters in the rule of five include formation of H-bond, size of the compound, lipophilic nature, and the flexibility of the compound. All the compounds are predicted to have excellent human intestinal absorption, BBB penetration, and bioavailability. From the positive value of BBB penetration, it can be predicted that all the synthesized compounds can be used to treat the neuroinflammation.
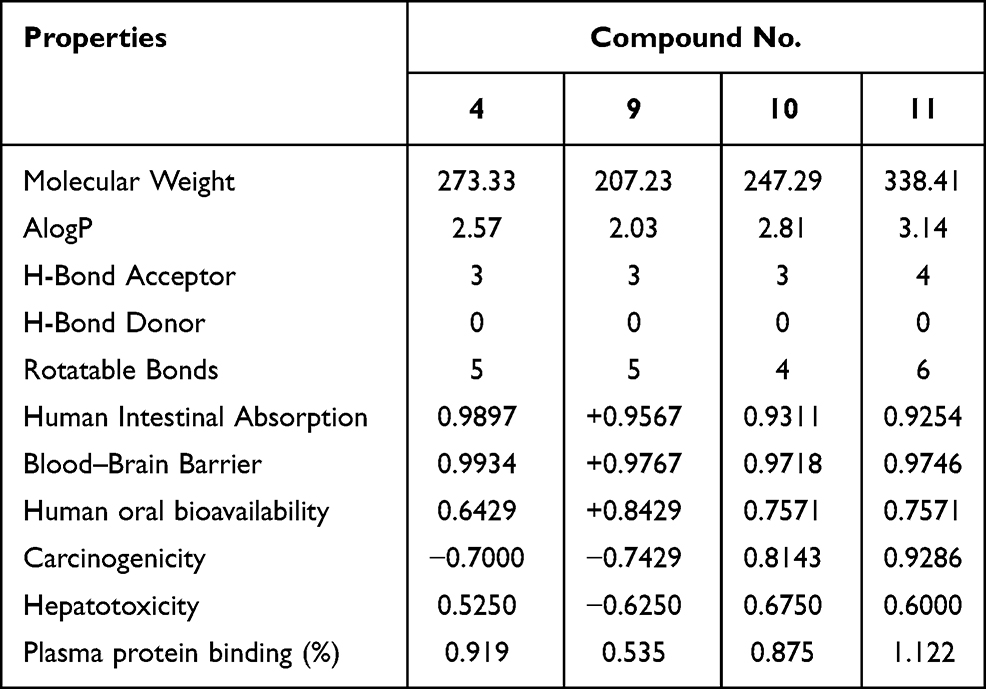 |
Table 8 Predicted Properties of the Synthesized Compounds (4, 9–11) |
Conclusions
Dual inhibition of the enzymatic pathways of cyclooxygenases (COX-1/COX-2) and lipoxygenase (LOX) is a rational approach for developing more efficient and safe analgesic/anti-inflammatory agents. In the current research, we have efficiently synthesized the Michael products (4, 9–11). Compound 4, ie, (S)-2-((S)-2,5-dioxo-1-phenylpyrrolidin-3-yl)-2-methylpentanal was obtained in stereopure form. Moreover, compound 11 has been synthesized for the first time. All of our compounds showed encouraging results against in vitro COX-1, COX-2, and 5-LOX enzymes. We selected one pyrrolidinedione (4) and one nitroalkane (11) Michael products for in vivo activities. Both of our compounds were active in animals’ studies following acetic acid, tail flick, and formalin methods. We also explored that the central pathway blockade is the possible analgesic mechanism of our compounds. Moreover, we also confirmed that the adrenergic and dopaminergic receptors are not amplified in the analgesic pathway. Extensive molecular docking simulations provided us insight into the binding modes of the compounds (4 and 11). We also find the pharmacokinetic and site of metabolism in compound 4 through in silico studies. Furthermore, with molecular docking approach, we also find that the corresponding carboxylic acid of compound 4 can be a potent analgesic lead compound. Our designed studies provide scientific basis for the use of Michael products (pyrrolidinedione and nitroalkane) as lead analgesic molecules.
Acknowledgment
The authors are thankful to the Najran University, Ministry of Education, Kingdom of Saudi Arabia for extending their support in completion of the project (Project No. NU/MID/17/046). We are thankful to the Higher Education Commission of Pakistan for financial support (10562/KPK/NRPU/R&D/2017). Dr. Umer Rashid is thankful to the Higher Education Commission for financial support for the purchase of MOE license under HEC-NRPU project 5291/Federal/NRPU/R&D/HEC/2016.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interest.
References
1. Craig AD, Sorkin LS Pain and analgesia. e LS.
2. Jan MS, Ahmad S, Hussain F, et al. Design, synthesis, in-vitro, in-vivo and in-silico studies of pyrrolidine-2, 5-dione derivatives as multitarget anti-inflammatory agents. Eur J Med Chem. 2020;186:111863. doi:10.1016/j.ejmech.2019.111863
3. Crombez G, Eccleston C, Baeyens F, Eelen P. The disruptive nature of pain: an experimental investigation. Behav Res Ther. 1996;34(11–12):911–918. doi:10.1016/S0005-7967(96)00058-7
4. Fields HL. Analgesic drugs. Pain. 1987;1:272.
5. Altman R, Bosch B, Brune K, Patrignani P, Young C. Advances in NSAID development: evolution of diclofenac products using pharmaceutical technology. Drugs. 2015;75(8):859–877. doi:10.1007/s40265-015-0392-z
6. Petrovska BB. Historical review of medicinal plants’ usage. Pharmacogn Rev. 2012;6(11):1–5. doi:10.4103/0973-7847.95849
7. Tallima H, El Ridi R. Arachidonic acid: physiological roles and potential health benefits–a review. J Adv Res. 2018;11:33–41. doi:10.1016/j.jare.2017.11.004
8. Yadav DK, Kumar S, Misra S, et al. Molecular insights into the interaction of RONS and Thieno [3, 2-c] pyran analogs with SIRT6/COX-2: a molecular dynamics study. Sci Rep. 2018;8(1):1–6. doi:10.1038/s41598-018-22972-9
9. Meena A, Yadav DK, Srivastava A, Khan F, Chanda D, Chattopadhyay SK. In silico exploration of anti-inflammatory activity of natural coumarinolignoids. Chem Biol Drug Des. 2011;78(4):567–579. doi:10.1111/j.1747-0285.2011.01173.x
10. Yadav DK, Sharma P, Misra S, et al. Studies of the benzopyran class of selective COX-2 inhibitors using 3D-QSAR and molecular docking. Arch Pharm Res. 2018;41(12):1178–1189. doi:10.1007/s12272-017-0945-7
11. Martel-Pelletier J, Lajeunesse D, Reboul P, Pelletier JP. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann Rheum Dis. 2003;62(6):501–509. doi:10.1136/ard.62.6.501
12. Sadiq A, Nugent TC. Catalytic access to succinimide products containing stereogenic quaternary carbons. ChemistrySelect. 2020;5(38):11934–11938. doi:10.1002/slct.202003664
13. Nugent TC, Negru DE, El-Shazly M, et al. Sequential reductive amination-hydrogenolysis: a one-pot synthesis of challenging chiral primary amines. Adv Synth Catal. 2011;353(11–12):2085–2092. doi:10.1002/adsc.201100250
14. Nugent TC, Bibi A, Sadiq A, Shoaib M, Umar MN, Tehrani FN. Chiral picolylamines for Michael and aldol reactions: probing substrate boundaries. Org Biomol Chem. 2012;10(46):9287–9294. doi:10.1039/c2ob26382c
15. Agranat I, Caner H, Caldwell J. Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov. 2002;1(10):753–768. doi:10.1038/nrd915
16. Ariens EJ. Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology. Eur J Clin Pharmacol. 1984;26(6):663–668. doi:10.1007/BF00541922
17. Yadav DK, Kumar S, Saloni HS, et al. Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Des Devel Ther. 2017;11:1859. doi:10.2147/DDDT.S130601
18. Kumar Yadav D, Dhawan S, Chauhan A, et al. QSAR and docking based semi-synthesis and in vivo evaluation of artemisinin derivatives for antimalarial activity. Curr Drug Targets. 2014;15(8):753–761. doi:10.2174/1389450115666140630102711
19. Yadav DK, Khan F, Negi AS. Pharmacophore modeling, molecular docking, QSAR, and in silico ADMET studies of gallic acid derivatives for immunomodulatory activity. J Mol Model. 2012;18(6):2513–2525. doi:10.1007/s00894-011-1265-3
20. Yadav DK, Khan F. QSAR, docking and ADMET studies of camptothecin derivatives as inhibitors of DNA topoisomerase‐I. J Chemom. 2013;27(1–2):21–33. doi:10.1002/cem.2488
21. Nugent TC, Sadiq A, Bibi A, et al. Noncovalent bifunctional organocatalysts: powerful tools for contiguous quaternary‐tertiary stereogenic carbon formation, scope, and origin of enantioselectivity. Chem Eur J. 2012;18(13):4088–4098. doi:10.1002/chem.201103005
22. Ahmad A, Ullah F, Sadiq A, et al. Comparative cholinesterase, α-glucosidase inhibitory, antioxidant, molecular docking, and kinetic studies on potent succinimide derivatives. Drug Des Devel Ther. 2020;14:2165. doi:10.2147/DDDT.S237420
23. Bibi A, Shah T, Sadiq A, Khalid N, Ullah F, Iqbal A. L-isoleucine-catalyzed michael synthesis of N-alkylsuccinimide derivatives and their antioxidant activity assessment. Russian J Organic Chem. 2019;55(11):1749–1754. doi:10.1134/S1070428019110174
24. Ahmad A, Ullah F, Sadiq A, et al. Pharmacological evaluation of aldehydic-pyrrolidinedione against HCT-116, MDA-MB231, NIH/3T3, MCF-7 cancer cell lines, antioxidant and enzyme inhibition studies. Drug Des Devel Ther. 2019;13:4185. doi:10.2147/DDDT.S226080
25. Hussain F, Khan Z, Jan MS, et al. Synthesis, in-vitro α-glucosidase inhibition, antioxidant, in-vivo antidiabetic and molecular docking studies of pyrrolidine-2, 5-dione and thiazolidine-2, 4-dione derivatives. Bioorg Chem. 2019;91:103128. doi:10.1016/j.bioorg.2019.103128
26. Jan MS, Shahid M, Ahmad S, et al. Synthesis of pyrrolidine-2, 5-dione based anti-inflammatory drug: in vitro COX-2, 5-LOX inhibition and in vivo anti-inflammatory studies. Latin Am J Pharm. 2019;38(11):2287–2294.
27. Sadiq A, Mahmood F, Ullah F, et al. Synthesis, anticholinesterase and antioxidant potentials of ketoesters derivatives of succinimides: a possible role in the management of Alzheimer’s. Chem Cent J. 2015;9(1):31. doi:10.1186/s13065-015-0107-2
28. Munir A, Khushal A, Saeed K, et al. Synthesis, in-vitro, in-vivo anti-inflammatory activities and molecular docking studies of acyl and salicylic acid hydrazide derivatives. Bioorg Chem. 2020;104:104168. doi:10.1016/j.bioorg.2020.104168
29. Ahmad S, Iftikhar F, Ullah F, Sadiq A, Rashid U. Rational design and synthesis of dihydropyrimidine based dual binding site acetylcholinesterase inhibitors. Bioorg Chem. 2016;69:91–101. doi:10.1016/j.bioorg.2016.10.002
30. Sarfraz M, Sultana N, Rashid U, Akram MS, Sadiq A, Tariq MI. Synthesis, biological evaluation and docking studies of 2, 3-dihydroquinazolin-4 (1H)-one derivatives as inhibitors of cholinesterases. Bioorg Chem. 2017;70:237–244. doi:10.1016/j.bioorg.2017.01.004
31. Bibi M, Qureshi NA, Sadiq A, et al. Exploring the ability of dihydropyrimidine-5-carboxamide and 5-benzyl-2, 4-diaminopyrimidine-based analogues for the selective inhibition of L. major Dihydrofolate reductase. Eur J Med Chem. 2020;112986. doi:10.1016/j.ejmech.2020.112986
32. Shah SM, Ullah F, Shah SM, Zahoor M, Sadiq A. Analysis of chemical constituents and antinociceptive potential of essential oil of Teucrium Stocksianum bioss collected from the North West of Pakistan. BMC Complement Altern Med. 2012;12(1):244. doi:10.1186/1472-6882-12-244
33. Sadiq A, Zeb A, Ullah F, et al. Chemical characterization, analgesic, antioxidant, and anticholinesterase potentials of essential oils from Isodon rugosus Wall. Front Pharmacol. 2018;9:623. doi:10.3389/fphar.2018.00623
34. Shah SM, Sadiq A, Shah SM, Ullah F. Antioxidant, total phenolic contents and antinociceptive potential of Teucrium stocksianum methanolic extract in different animal models. BMC Complement Altern Med. 2014;14(1):181. doi:10.1186/1472-6882-14-181
35. Rahim H, Sadiq A, Khan S, et al. Aceclofenac nanocrystals with enhanced in vitro, in vivo performance: formulation optimization, characterization, analgesic and acute toxicity studies. Drug Des Devel Ther. 2017;11:2443. doi:10.2147/DDDT.S140626
36. Zeb A, Ahmad S, Ullah F, Ayaz M, Sadiq A. Anti-nociceptive activity of ethnomedicinally important analgesic plant Isodon rugosus Wall. ex Benth: mechanistic study and identifications of bioactive compounds. Front Pharmacol. 2016;7:200. doi:10.3389/fphar.2016.00200
37. Underwood W, Anthony R. AVMA guidelines for the euthanasia of animals: 2020 edition. Retrieved on March. 2013;30(2020):2020–2021.
38. Ahmad G, Rasool N, Rizwan K, et al. Synthesis, in-vitro cholinesterase inhibition, in-vivo anticonvulsant activity and in-silico exploration of N-(4-methylpyridin-2-yl) thiophene-2-carboxamide analogs. Bioorg Chem. 2019;92:103216. doi:10.1016/j.bioorg.2019.103216
39. Sultana N, Sarfraz M, Tanoli ST, et al. Synthesis, crystal structure determination, biological screening and docking studies of N1-substituted derivatives of 2, 3-dihydroquinazolin-4 (1H)-one as inhibitors of cholinesterases. Bioorg Chem. 2017;72:256–267. doi:10.1016/j.bioorg.2017.04.009
40. Craig AD. A new view of pain as a homeostatic emotion. Trends Neurosci. 2003;26(6):303–307. doi:10.1016/S0166-2236(03)00123-1
41. Miljanich G, Rauck R, Saulino M. Spinal mechanisms of pain and analgesia. Pain Pract. 2013;13(2):114–130. doi:10.1111/j.1533-2500.2012.00564.x
42. Sessle BJ. Acute and chronic craniofacial pain: brainstem mechanisms of nociceptive transmission and neuroplasticity, and their clinical correlates. Crit Rev Oral Biol Med. 2000;11(1):57–91. doi:10.1177/10454411000110010401
43. Moertel CG, Ahmann DL, Taylor WF, Schwartau N. A comparative evaluation of marketed analgesic drugs. N Engl J Med. 1972;286(15):813–815. doi:10.1056/NEJM197204132861504
44. Tjølsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51(1):5–17. doi:10.1016/0304-3959(92)90003-T
45. Sjöberg P, Jones DR. Non-Clinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals: ICH M3 and M3 (R2). In Global Approach in Safety Testing. New York, NY: Springer; 2013:
46. Mahmood F, Jan MS, Ahmad S, et al. Ethyl 3-oxo-2-(2, 5-dioxopyrrolidin-3-yl) butanoate derivatives: anthelmintic and cytotoxic potentials, antimicrobial, and docking studies. Front Chem. 2017;5:119. doi:10.3389/fchem.2017.00119
47. Patil MM, Rajput SS. Succinimides: synthesis, reaction, and biological activity. Int J Pharm Pharm Sci. 2014;6(11):8–14.
48. Yadav DK, Rai R, Kumar N, Singh S, Misra S, Sharma P. “New arylated benzo [h] quinolines induce anti-cancer activity by oxidative stress-mediated DNA damage”. Sci Rep. 2016;6(1):1–13. doi:10.1038/srep38128
49. Gaur R, Yadav DK, Kumar S, Darokar MP, Khan F, Bhakuni RS. Molecular modeling based synthesis and evaluation of in vitro anticancer activity of indolyl chalcones. Curr Top Med Chem. 2015;15(11):1003–1012. doi:10.2174/1568026615666150317222059
50. Sathish Kumar B, Kumar A, Singh J, et al. Synthesis of 2-alkoxy and 2-benzyloxy analogues of estradiol as anti-breast cancer agents through microtubule stabilization. Eur J Med Chem. 2014;86:740–751. doi:10.1016/j.ejmech.2014.09.033
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.