Back to Journals » Drug Design, Development and Therapy » Volume 14
Synthesis, Cytotoxic Analysis, and Molecular Docking Studies of Tetrazole Derivatives via N-Mannich Base Condensation as Potential Antimicrobials
Authors Atef Hatamleh A, Al Farraj D, Salah Al-Saif S, Chidambaram S ![]() , Radhakrishnan S
, Radhakrishnan S ![]() , Akbar I
, Akbar I ![]()
Received 6 July 2020
Accepted for publication 21 September 2020
Published 23 October 2020 Volume 2020:14 Pages 4477—4492
DOI https://doi.org/10.2147/DDDT.S270896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Ashraf Atef Hatamleh,1 Dunia Al Farraj,1 Sarah Salah Al-Saif,1 SathishKumar Chidambaram,2 Surendrakumar Radhakrishnan,2 Idhayadhulla Akbar2
1Botany and Microbiology Department, College of Science, King Saud University, Riyadh, Kingdom of Saudi Arabia; 2Research Department of Chemistry, Nehru Memorial College (Affiliated with the Bharathidasan University), Puthanampatti, Tiruchirappalli District, Tamil Nadu, South India
Correspondence: Idhayadhulla Akbar Email [email protected]
Purpose: A new series of tetrazole derivatives, which are renowned antimicrobials possessing a five-membered aromatic heterocyclic group, are synthesized herein and subjected to antimicrobial and cytotoxicity screening.
Methods: The tetrazole derivatives were synthesized via ultrasonication using Mannich base condensation. Structural verification of the products was performed using IR, 1H NMR, and 13C NMR spectroscopy, as well as mass spectroscopic and elemental analyses. The compounds were then screened for antimicrobial and cytotoxic activity against HepG2 (liver), MCF-7 (breast), and HeLa (cervical) cell lines. Inter- and intra-molecular binding interactions were determined using molecular docking studies. The exact binding mode between the most active tetrazole derivatives (ie, 1b, 2a, and 2b) and the proteins (ie, 4OR7, 1AI9, and 4FM9) was established using Autodock Vina 1.1.2 software and compared to the binding mode of the reference compounds (ie, cefazolin, clotrimazole, and fluorouracil).
Results: Compound 1b was extremely active against Enterococcus faecalis relative to the positive control cefazolin. Compounds 1b and 1e were active against Candida albicans and Microsporum audouinii compared to the positive control clotrimazole in antifungal screening. The HepG2 (liver) and MCF-7 (breast) cancer cell lines were particularly susceptible to the synthesized compounds. Compared to the control compound fluorouracil, 2a and 2b were extremely active against all three cancer cell lines. Molecular docking studies showed that 2b exhibited higher binding affinity (− 7.8 kcal/mol) to the 4OR7 protein than the control cefazolin (− 7.2 kcal/mol).
Conclusion: Generally, 1b, 2a, and 2b exhibited impressive inhibitory capabilities in antibacterial, antifungal, and cytotoxic screenings relative to the reference compounds. The results of the molecular docking studies and both the microbial and anticancer screenings indicate that these novel derivatives could be developed into potential therapeutic agents for medical applications.
Keywords: antibacterial, antifungal, cytotoxic, molecular docking, N-Mannich base, ultrasonication
Introduction
As promising molecular structures in new drug design concepts, heterocyclic compounds, in particular azoles such as imidazoles, triazoles, and tetrazoles, are renowned for their antimicrobial, antifungal, and anticancer capabilities and are widely used for treating topical mycotic pathologies associated with AIDS.1 Tetrazole derivatives are reputed to have antinociceptive,2,3 antimycobacterial,4 antibacterial, and antifungal properties.5,6 Many important tetrazole analogs (Figure 1) have been reported, including the angiotensin II receptor blocker losartan (commonly used for treating hypertension) and the oral agonist of the human growth hormone secretagogue (GHS) receptor BMS-317,180.7 The tetrazole derivative, cefazolin, possesses antibacterial activity.8 Antifungal capabilities are well-documented in TAK-456 and other tetrazole-based derivatives.9
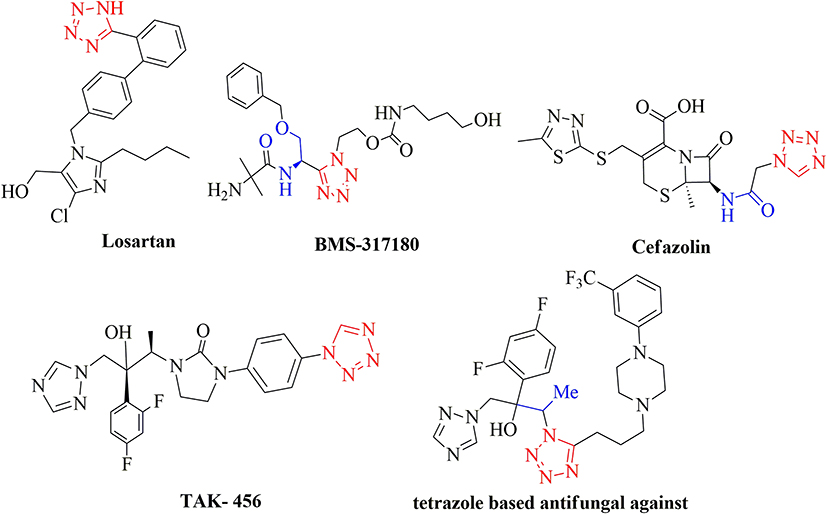 |
Figure 1 Tetrazole bioactive compounds. |
Ultrasonication is a good protocol for organic synthesis compared to traditional methods because more high-yielding organic reactions can be conducted in relatively shorter reaction times or under milder reaction conditions.10–12 This is best exemplified when synthesizing tetrazole derivatives via an ultrasound-promoted, one-pot cyclization reaction.13 The Mannich reaction has become the cornerstone in multidrug synthesis, leading to the simplified synthesis of numerous antimicrobial,14–18 cytotoxin,19 and anticonvulsant20 agents. Inspired by the widespread applicability of the Mannich reaction and the high product yield obtained under ultrasonication, we synthesized a new series of tetrazole derivatives using a combination of these techniques. Herein, the antibacterial, antifungal, and cytotoxic activities of the products were evaluated against three major cancer cell lines, namely, HepG2 (liver), MCF-7 (breast), and HeLa (cervical), and various microbes, including Candida albicans, Microsporum audouinii, and Enterococcus faecalis. Additionally, molecular docking studies were performed to gain a deeper insight into the binding modes of the most active derivatives using the proteins 4OR7, 1AI9, and 4FM9.
Experimental
Synthesis
All chemicals were purchased from Merck and Sigma–Aldrich, and were used without further purification. Pre-coated silica gel plates impregnated with a fluorescent indicator were used for analytical TLC. An ethyl acetate/hexane solvent mixture was used as the eluent for both TLC and column chromatography. The FT–IR spectra were recorded using KBr pellets on a Shimadzu 8201pc spectrometer operating between 4000 and 400 cm−1. The 1H and 13C NMR spectra were obtained on Bruker DRX-300 MHz and Bruker DRX-75 MHz Avance spectrometers, respectively, and the resulting chemical shifts were expressed in ppm using tetramethylsilane as the internal standard. JEOL JMS D-300 spectrometer was used to analysis the mass spectra (EI) all compounds. Elemental analysis (C, H, N, and S) was conducted using a Varian EL III elemental analyzer.
Synthesis of Compounds 1a – e
A mixture of 1H-tetrazole (0.01 mol), thiosemicarbazone (0.01 mol, 0.9 g), and the respective aromatic aldehyde (0.01 mol) in ethanol was subjected to ultrasonication for 3 min. The final products were identified using TLC (hexane/EtOAc, 4:1, v/v). After all starting materials were consumed, the reaction was quenched with water (10 mL) before being extracted with ethyl acetate (10 mL × 3). The solvent was removed under vacuum and purification was conducted via column chromatography on silica gel using the above-mentioned 4:1 hexane/ethyl acetate solvent mixture to give the desired products (1a – e).
Synthesis of Compounds 2a – e
A mixture of 1H-tetrazole (0.01 mol), 1.1-dimethylurea (0.01 mol), and the respective aromatic aldehyde (0.01 mol) in ethanol was subjected to ultrasonication for 6 min. The endpoint of the reaction was monitored via TLC using a mixed hexane/EtOAc (4:1, v/v) solution. Once all starting materials were consumed, the reaction was quenched using water (10 mL) before being extracted using ethyl acetate (10 mL × 3). The solvent was removed under vacuum and purification was conducted via column chromatography on silica gel using the above-mentioned 4:1 hexane/ethyl acetate solvent mixture to give the desired products (2a – e).
Synthesis of Compounds 3a – e
A mixture of 1H-tetrazole (0.01 mol), benzylidenehydrazine (0.01 mol), and the respective aromatic aldehyde (0.01 mol) in ethanol was subjected to ultrasonication for 7 min. The endpoint of the reaction was monitored via TLC in a mixed hexane/EtOAc (4:1, v/v) solvent. Once all starting materials were consumed, the reaction was quenched using water (10 mL) before being extracted using ethyl acetate (10 mL × 3). The solvent was removed under vacuum and purification was conducted via column chromatography on silica gel using the aforementioned 4:1 hexane/ethyl acetate solvent mixture to give the pure products (3a – e).
Synthesis of Compounds 4a – e
A mixture of 1H-tetrazole (0.01 mol), (furan-2-ylmethylene) hydrazine (0.01 mol), and the respective aromatic aldehyde (0.01 mol) in ethanol was subjected to ultrasonication for 8 min, and the reaction endpoint was monitored via TLC in a hexane/EtOAc (4:1, v/v) solvent mixture. After all starting materials were consumed, the reaction was quenched in water (10 mL) and extracted with ethyl acetate (10 mL × 3). The solvent was then removed under vacuum and purification of the crude mixture was conducted via column chromatography on silica gel using the aforementioned 4:1 hexane/ethyl acetate solvent mixture to give the desired products (4a – e).
Biological Screening
In vitro Antibacterial Screening
Compounds 1a – e, 2a – e, 3a – e, and 4a – e were subjected to in vitro antibacterial analysis against Staphylococcus aureus (MTCC-96), Escherichia coli (MTCC-739), Enterococcus faecalis (MTCC-439), Pseudomonas aeruginosa (MTCC-2488), and Klebsiella pneumonia (recultured) via the disc diffusion method22,23 using the Mueller–Hinton agar (Hi-Media) medium. Each compound was tested at a concentration of 100 μg/mL in DMSO. The zone of inhibition was measured after an incubation period of 24 h at 37 ºC.
In vitro Antifungal Screening
Compounds 1a – e, 2a – e, 3a – e, and 4a – e were subjected to in vitro antifungal analysis using Aspergillus niger (MTCC-872), Candia albicans (MTCC-227), Cryptococcus neoformans (recultured), and Microsporum audouinii (MTCC-8197) via the disc diffusion method24,25 with Sabouraud’s dextrose agar (SDA; Hi-Media). Each compound was tested at a concentration of 100 μg/mL in DMSO. The zone of inhibition (mm) was measured at 37 °C.
Determining the Minimal Inhibitory Concentration (MIC)
The compounds were dissolved in dimethyl sulfoxide at a concentration of 64 µg/mL. Next, 2-fold dilutions of the respective solutions were prepared at 64, 32, 16, 8, 4, 2, 1, 0.5, and 0.25 µg/mL. Suspensions of the respective microorganisms at a concentration of 106 CFU/mL (colony-forming unit/mL) were incubated in the corresponding wells at 37 ºC for 24 h. The minimum inhibitory concentration (MIC) was noted by observing the lowest concentration of the drug at which there was no visible growth.
Cytotoxic Activity
Compounds 1a – e, 2a – e, 3a – e, and 4a – e were subjected to cytotoxic screening per the procedure detailed in previously reported studies.26
Molecular Docking Analysis
Molecular docking studies were conducted to determine the binding mode and interactions between the most active compounds of the tetrazole series (ie, 1b, 2a, and 2b) and the proteins 4OR7, 1AI9, and 4FM9 using Autodock Vina 1.1.2.27 The results obtained were compared to the molecular docking models of the reference compounds, namely, cefazolin, clotrimazole, and fluorouracil. Here, the crystal structure of Klebsiella pneumonia dihydrofolate reductase complexed with 4OR7 protein,28 dihydrofolate reductase from C. albicans (PDB ID: 1AI9),29 and human topoisomerase II alpha bound to DNA (PDB ID: 4FM9)30 were downloaded from the Protein Data Bank, and used for antibacterial, antifungal, and cytotoxic screening, respectively. The 3D structures of 1b, 2a, 2b, cefazolin, clotrimazole, and fluorouracil were obtained using ChemDraw Ultra 12.0 software. The search grid of the 4OR7 protein was identified as center_x = 47.231, center_y = 28.209, and center_z = 11.012 with the dimensions of size_x = 24, size_y = 24, and size_z = 30 at a spacing of 1.0 Å. The search grid of the 1AI9 protein was identified as center_x = 27.873, center_y = −10.945, and center_z = 12.224 with the dimensions of size_x = 24, size_y = 24, and size_z = 28 at a spacing of 1.0 Å. The search grid of the 4FM9 protein was identified as center_x = 14.41, center_y = 40.808, and center_z = 26.199 with the dimensions of size_x = 14, size_y = 16, and size_z = 16 at a spacing of 1.0 Å. The exhaustiveness value was set to 8. All other parameters were set to the default values for the Vina docking program and are not mentioned herein. The compound with the least binding affinity value was the best-scoring compound. Discovery Studio 2019 software was used to analysis all results via visually.
Results and Discussion
Chemistry
Compounds 1a – e, 2a – e, 3a – e, and 4a – e were synthesized via a condensation reaction method using ultrasonication. The general synthetic procedure used in this study is illustrated in Scheme 1. Here, 2-[phenyl(1H-tetrazol-1-yl)methyl]hydrazinecarbothioamides (1a – e) were prepared from tetrazole reacted with benzaldehyde and thiosemicarbazone in the presence of ethanol. The reactions performed via condensation only afforded low product yields, whereas the reactions conducted under ultrasonication were high-yielding reactions. Optimization studies were undertaken using various solvents (ie, H2O, MeOH, H2O/EtOH, and EtOH) and adjusting the reaction times. An ethanol medium was used to maximize the product yield while minimizing the required reaction time relative to the other solvents or conventional synthesis (Table 1). Additionally, the ethanol medium was ideal for ultrasonication, as evidenced by the findings presented in (Table 2). Generally, 1a, 2a, 3a, and 4a were obtained from high-yielding reactions with percentage yields of 96, 78, 88, and 81% in EtOH with a 3-min ultrasonication cycle. Conversely, the product yields obtained via the conventional method of synthesis were low at 66%. The product yields for 1b – e, 2b – e, 3b – e, and 4b – e were 76–89%, 89–79%, 80–89%, and 79–88%, respectively, and were obtained in EtOH with a 3- to 5-min reaction time under ultrasonication; the corresponding products obtained via the conventional reaction methods were low.
 |
Scheme 1 Synthesis of tetrazole derivatives 1a–e, 2a–e, 3a–e, and 4a–e via ultrasonication. |
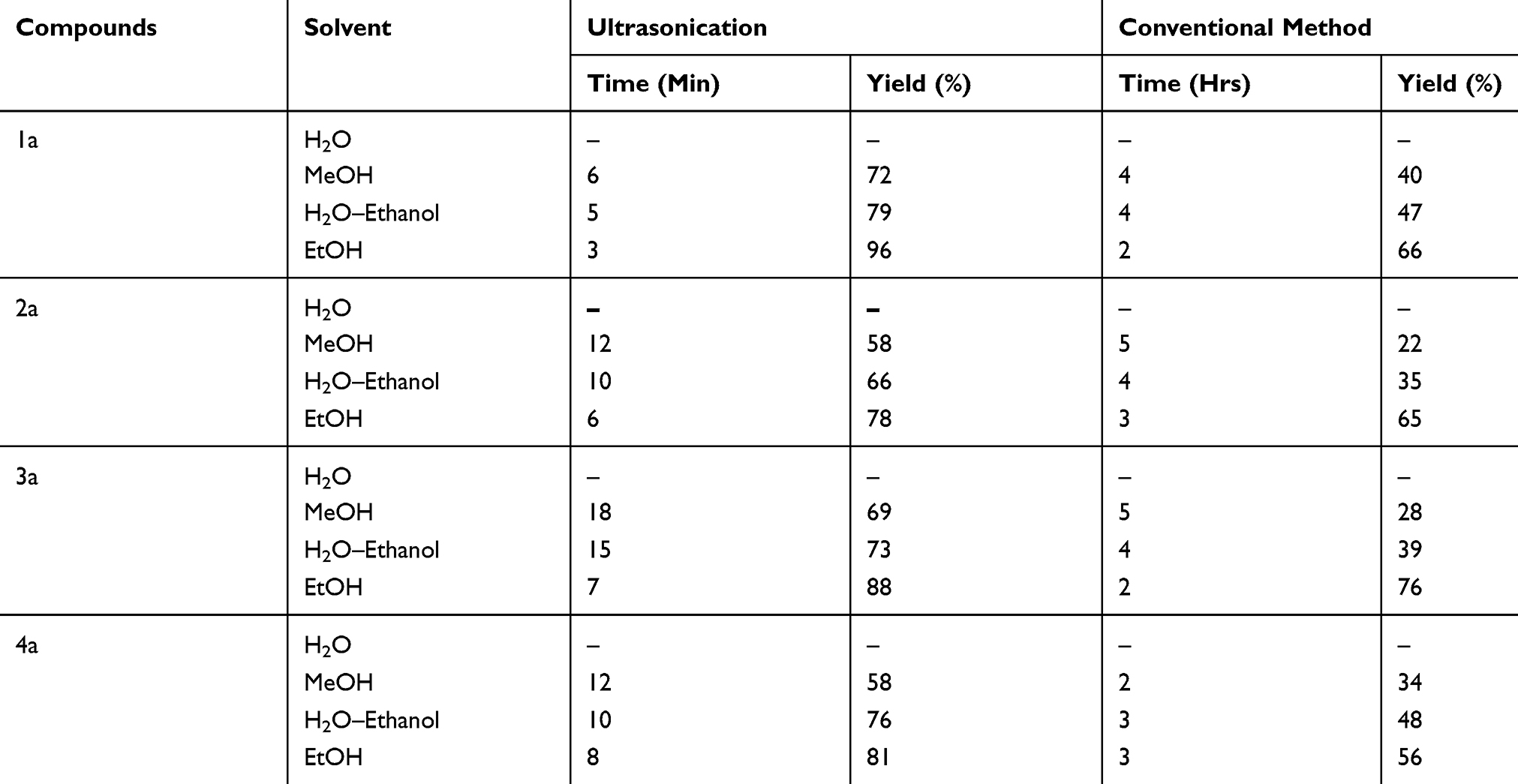 |
Table 1 Effect of Various Solvents on the Product Yield of 1a, 2a, 3a, and 4a |
 |
Table 2 Effect of Using Ethanol as the Solvent on the Product Yield |
Characterization of the synthesized derivatives was conducted using mass, IR, 1H NMR, and 13C NMR spectroscopic techniques. The characteristic IR bands assigned to 1a – e, 2a – e, 3a – e, and 4a – e revealed strong absorption bands at 2932–2909 cm−1, 2916–2936 cm−1, 2908–2919 cm−1, and 2912–2978 cm−1, respectively, which corresponded to the CH bonds in each derivative. 1H NMR spectra showed that the peak doublets within the ranges of 6.12–6.43 ppm, 6.15–6.21 ppm, 6.12–6.24 ppm, and 6.14–6.28 ppm corresponded to the –CH proton. The 13CNMR spectra contained signals within the range of 72.1–74.9 ppm, 66.1–69.1 ppm, 72.1–73.9 ppm, and 72.1–73.2 ppm that corresponded to the carbon of –CH– for the respective derivatives. Additionally, the mass spectroscopic analysis revealed that the molecular ion signals were consonant with the projected molecular weights of all synthesized compounds.
Biological Activities
Antibacterial Activity
Compounds 1a – e, 2a – e, 3a – e, and 4a – e were screened for antibacterial activity. Here, we noted that 1b was most active against Enterococcus faecalis (Minimum inhibitory concentration (MIC) = 8 µg/mL at 22 mm), P. aeruginosa (MIC = 32 µg/mL at 16 mm), and K. pneumoniae (MIC = 4 µg/mL at 18 mm) when compared to the results obtained using the reference compound, cefazolin. The lowest activity for this compound was noted against S. aureus (MIC = 64 µg/mL at 12 mm), whereas moderate activity was observed against E. coli (MIC = 8 µg/mL at 19 mm). Compound 1c was very active against E. faecalis (MIC = 16 µg/mL at 17 mm). Compound 2a was very active (MIC = 8 µg/mL at 16 mm) against K. pneumoniae, whereas 2b exhibited equipotency against E. coli (MIC = 8 µg/mL at 22 mm) and high activity against K. pneumoniae (MIC = 4 µg/mL at 18 mm). Relative to the reference standard, namely, cefazolin, compounds 3c (MIC = 16 µg/mL at 13 mm) and 4e (MIC =16 µg/mL at 13 mm) were very active against E. faecalis. The antibacterial screening results are summarized in Table 3.
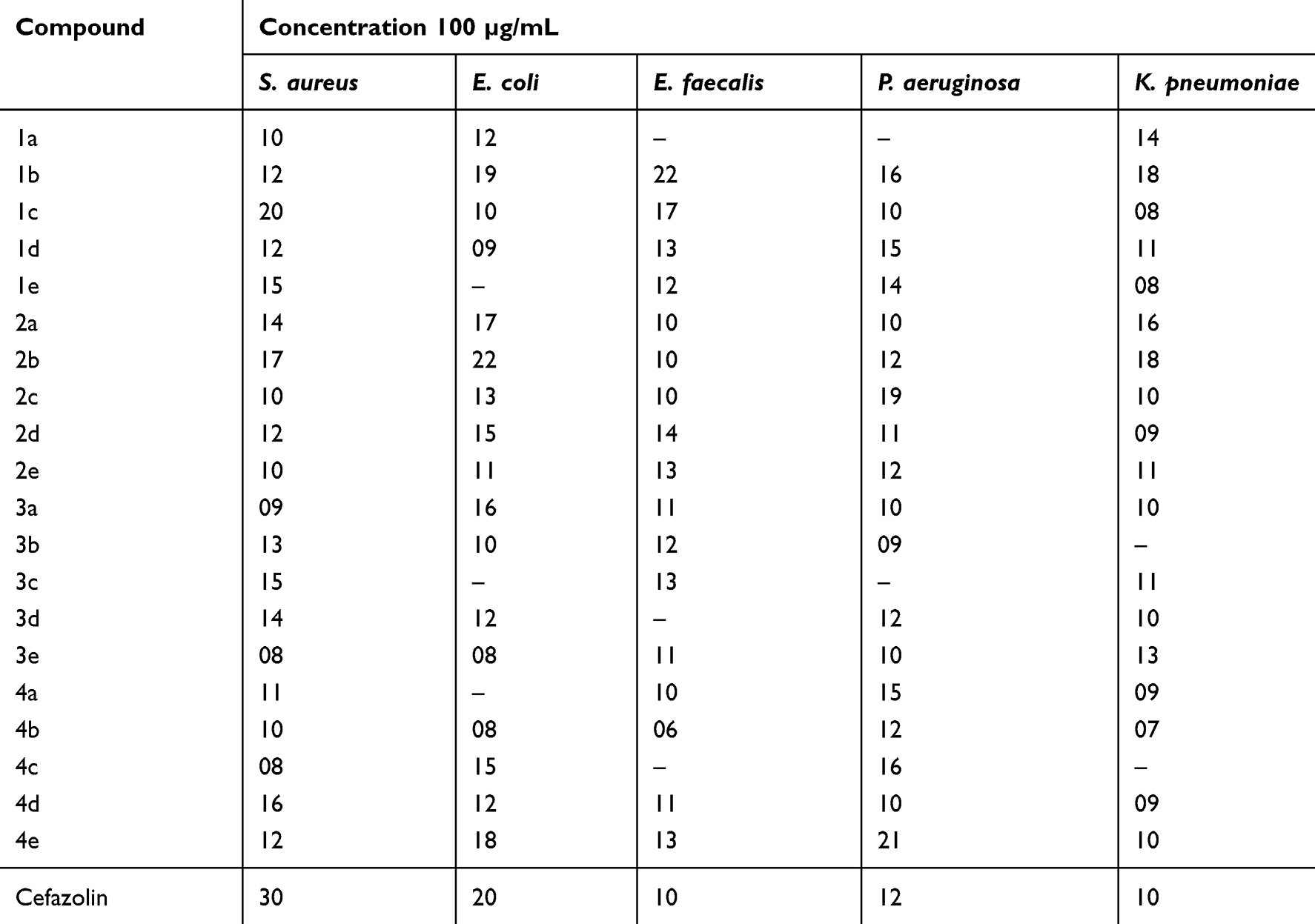 |
Table 3 Antibacterial Effects Measured by the Zone of Inhibition (Mm) |
Antifungal Activity
Compounds 1a – e, 2a – e, 3a – e, and 4a – e were subjected to antifungal screening. Here, we observed that 1b was very active against Candia albicans (MIC = 0.25 µg/mL at 26 mm) and 1e was very active against Microsporum audouinii (MIC = 1 µg/mL at 28 mm) when compared to clotrimazole. The antifungal screening results are summarized in Table 4, whereas the MIC values against various bacteria and fungi are summarized in Table 5.
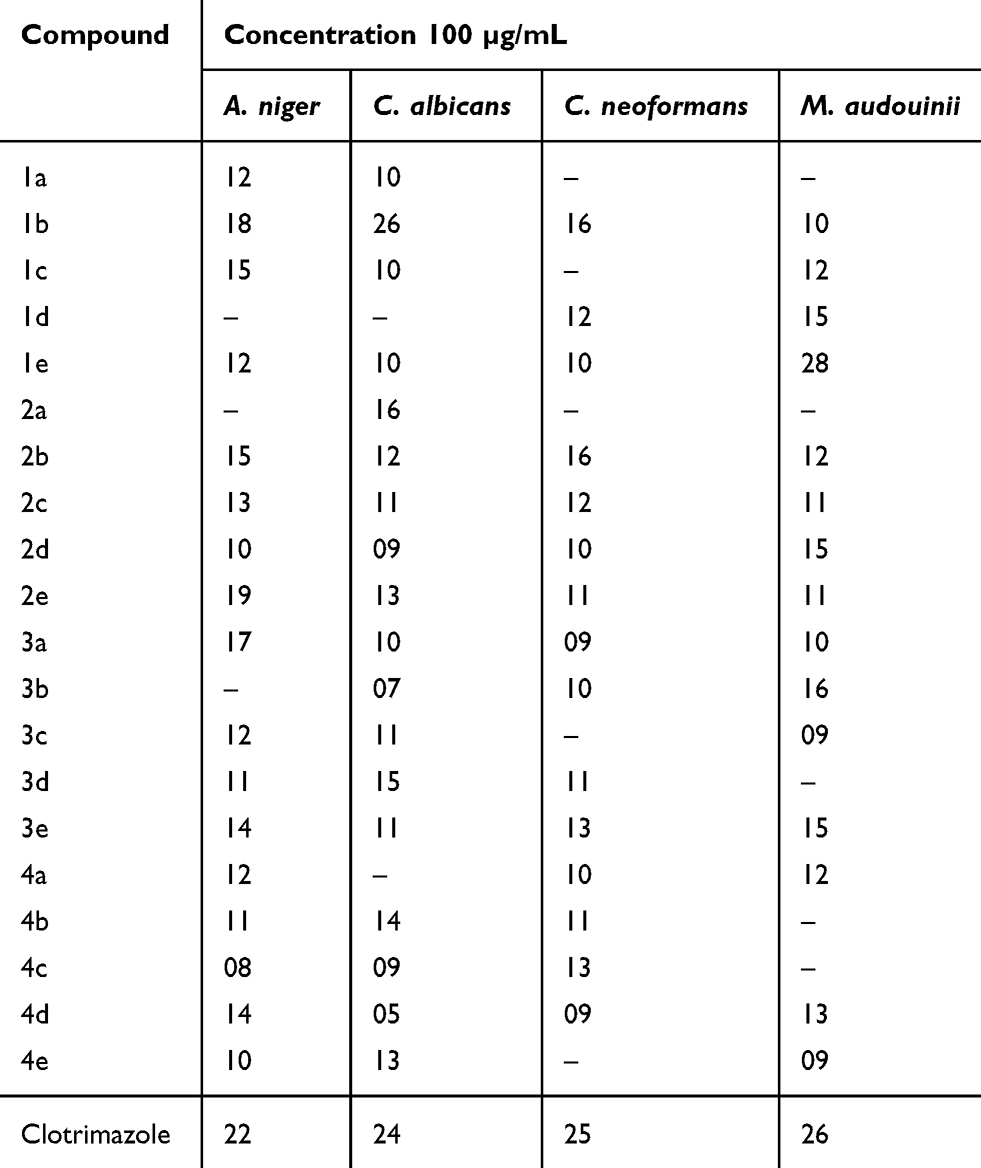 |
Table 4 Antifungal Effects Measured by Zone of Inhibition (Mm) |
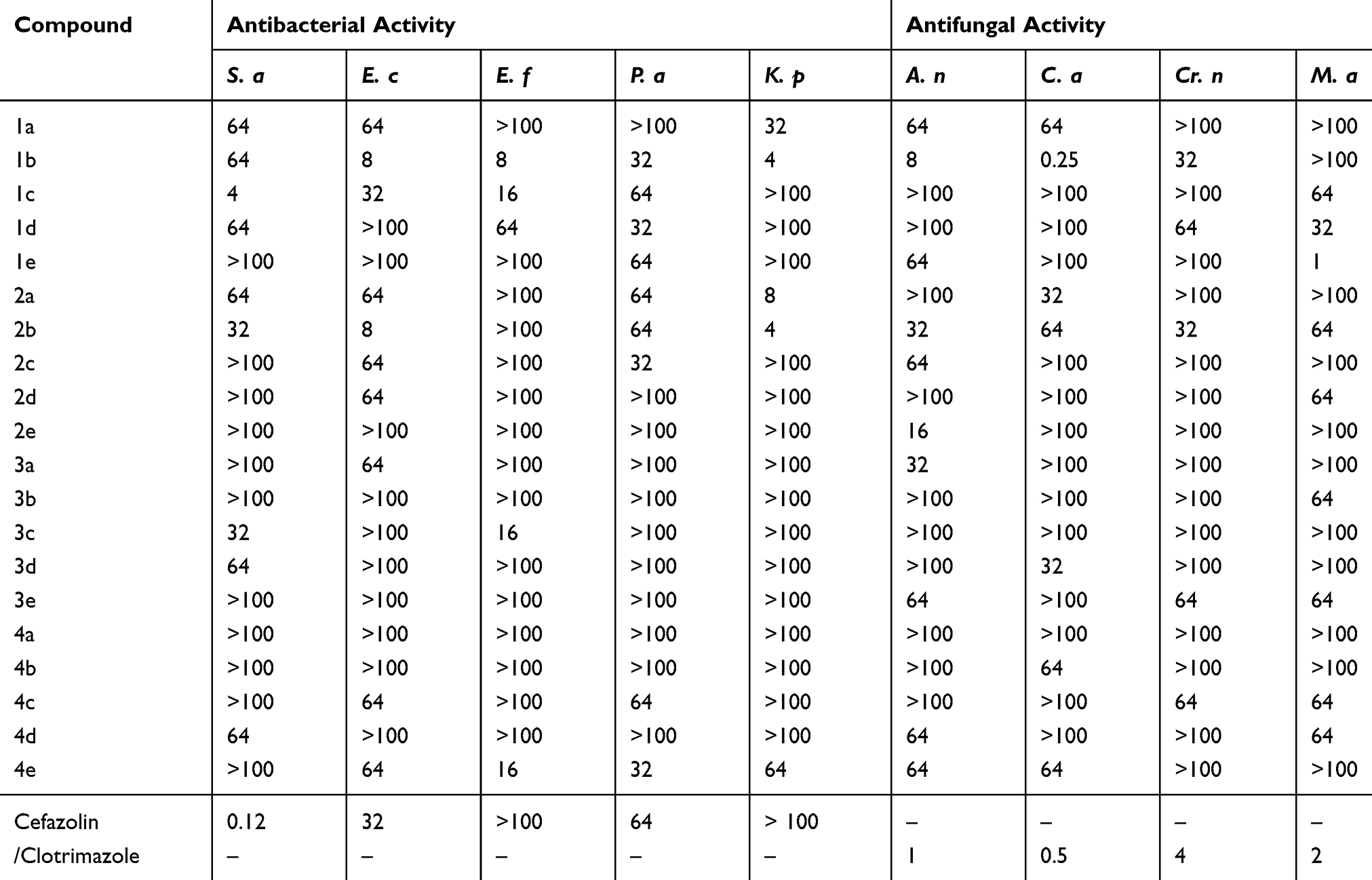 |
Table 5 The Minimal Inhibitory Concentrations (MIC; µg/mL) of 1a – e, 2a – e, 3a – e, and 4a –e |
Cytotoxic Activity
Compounds 1a – e, 2a – e, 3a – e, and 4a – e were also screened for their anticancer potency against three cancer cell lines, namely, liver, cervical, and breast cell lines. The GI50, TGI, and LC50 values were determined for the respective derivatives, and the results are expressed in terms of GI50 growth inhibitor concentration. We noted that 2a was extremely active against MCF-7 (GI50 = 5.2 μm) and HeLa cancer cell lines (GI50 = 8.1 μm) compared to the other compounds and the reference standard. Compounds 2a and 2b were very active against HepG2 cancer cell lines, with GI50 values of 6.3 and 5.4 μm, respectively, when compared to the other compounds and the standard fluorouracil. When compared with the standard fluorouracil, the other compounds exhibited either moderate activity or were equipotent against the HepG2 cancer cell lines, but generally showed low activity against MCF-7 and HeLa cancer cell lines. The results of the cytotoxic screening are summarized in Table 6.
 |
Table 6 Cytotoxic Effects of 1a – e, 2a – e, 3a – e, and 4a – e |
Structure–Activity Relationship
The structure–activity relationship (SAR) can be used to determine the relationship between an active molecule’s chemical structure and its biological activity in a specific assay system. This aids in identifying the chemical group/atom pivotal to modulating the biological potency of the compound. Figure 2 shows that the para substituted phenyl ring acts as a lipophilic domain, the C=S and C=O moieties as electron donors, the NH group as the hydrogen bonding domain, and the tetrazole moiety is an essential pharmacophore for biological activity. We noted several key points while conducting SAR studies:
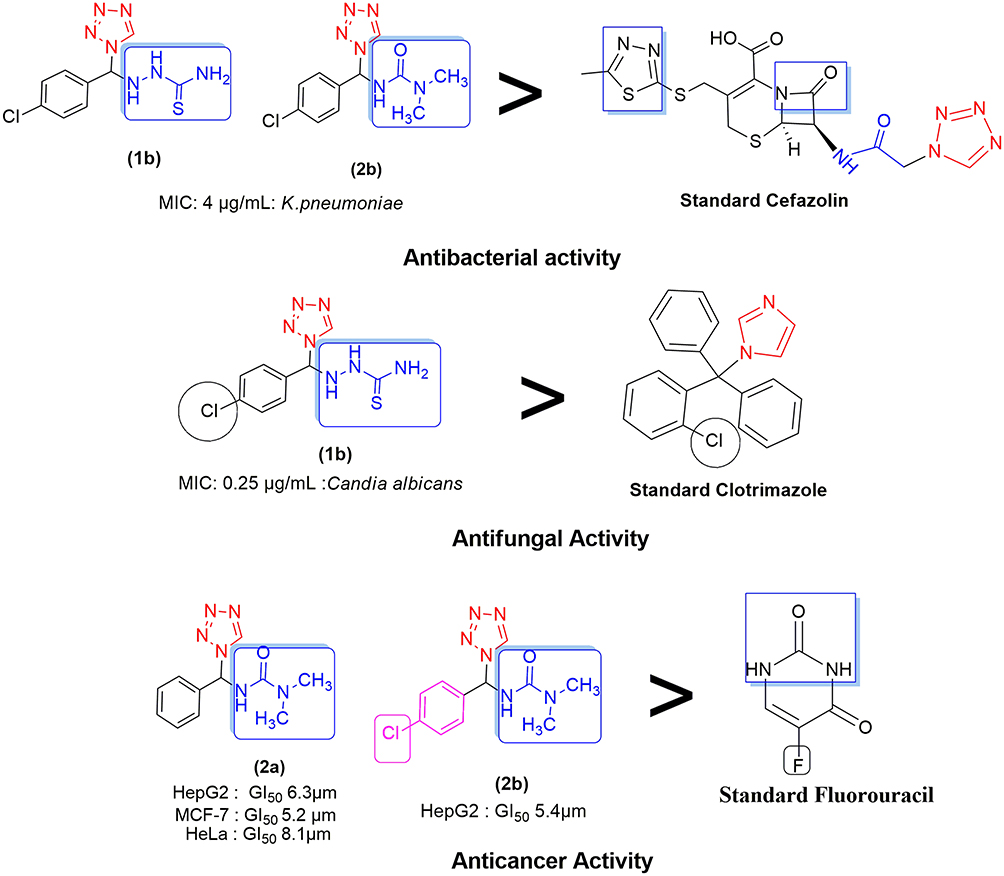 |
Figure 2 Structure–activity relationship. |
- Compound 1a, which contains a thiosemicarbazone group attached to the tetrazole, was moderately active against all bacterial and fungal species tested and exhibited anticancer activity.
- Compound 1b was very active against E. coli, (MIC = 8 µg/mL), E. faecalis, (MIC = 8 µg/mL), and P. aeruginosa (MIC = 32 µg/mL) in antibacterial screenings. K. pneumoniae was particularly susceptible to 1b (MIC = 4 µg/mL) when compared to the cefazolin reference standard due to the presence of a 4-Cl phenyl group attached to the thiosemicarbazonein the tetrazole moiety. Figure 2 showed that the sulfur groups in the thiourea moiety exhibited a similar binding mode as that seen for the sulfur-containing thiadiazole functional group in cefazolin. Additionally, 1b was more potent against C. albicans (MIC = 0.5 µg/mL) than clotrimazole, but exhibited notably weaker activity against all other fungal species. Compound 1b showed only moderate activity against the HepG2 cancer cell line, but exhibited abysmal activity against all other cell lines tested, especially when compared to the fluorouracil standard.
- The 4-OH-substituted phenyl group of thiosemicarbazone in the tetrazole moiety boosted the potency of 1c against E. faecalis (MIC = 16 µg/mL) and resulted in equipotent activity against E. coli (MIC = 32 µg/mL), especially when compared to cefazolin; conversely, S. aureus was the most susceptible microbe (MIC = 4 µg/mL) to the antibacterial effects of 1c. The antifungal activity of 1c was notably lower than that of clotrimazole. Generally, 1c was only moderately active against the HepG2 and MCF-7 cancer cell lines and exhibited inferior activity against the HeLa cancer cell line when compared to fluorouracil.
- The 4-NO2-substituted phenyl moiety attached to thiosemicarbazone in the tetrazole group imparted abysmal antifungal and antibacterial activity to 1d. For the cancer cell lines, 1d exhibited equipotent activity against HepG2 and very low activity against the MCF-7 and HeLa cancer cell lines when compared to fluorouracil.
- Despite the low potency of 1e against all bacterial species, this compound was extremely active against Microsporum audouinii (MIC = 1 µg/mL) due to the 4-OCH3-substituted phenyl group attached to the thiosemicarbazone moiety. Conversely, no other fungi were susceptible to the antifungal influence of this compound. The anticancer activity of 1d was on par with the reference standard for the HepG2 cell line.
- Compound 2a, which contained a diamide substituent in the tetrazole functional group, was extremely active against K. pneumoniae (MIC = 8 µg/mL), but showed very low activity against the other bacterial species and notably poor activity against all the fungi tested herein. Note, however, that 2a was remarkably potent against the MCF-7, HeLa, and HepG2 cancer cell lines, with GI50 values of 5.2, 8.1, and 6.3 µm, respectively. Figure 2 shows that the diamide in 2a is comparable to the diamide moiety present in fluorouracil (Malani et al, 2014).
- The 4-Cl-substituted phenyl group combined with the diamide moiety in tetrazole imparted significant antimicrobial properties to 2b, as evidenced by the MIC value (4 µg/mL) obtained when screening was conducted against K. pneumoniae. This was decidedly much better than the results seen with cefazolin. Figure 2 shows the presence of the NH–CO–N– moiety that serves a comparable function as its NH–CO–C– counterpart in cefazolin. Relative to the respective reference standards, 2b exhibited equipotent activity against E. coli and low activity against the other remaining bacterial and all fungal species. Compound 2b showed good activity against the HepG2 cancer cell line (GI50 = 5.4 µm), but low activity against the other cell lines tested compared to fluorouracil.
- Compound 2c, which contained a 4-OH-substituted phenyl group combined with a diamide functional group in the tetrazole moiety, was very active only against P. aeruginosa compared to the cefazolin standard. Additionally, 2c was shown to be a poor antifungal agent. Compound 2c was equipotent against HepG2, but exhibited poor activity against the MCF-7 and HeLa cancer cell lines compared to fluorouracil.
- Generally, 2d and 2e exhibited inferior antifungal and antibacterial properties. Additionally, these compounds were shown to be equipotent against HepG2, but were not potent against the MCF-7 and HeLa cancer cell lines, especially compared to the results obtained for fluorouracil.
- Compounds 3a – e and 4a – e, which contained a hydrazine group, exhibited inferior antifungal and anticancer activity. Additionally, the potency of 3a and 3b was abysmal against all bacterial and fungal species tested, and their anticancer properties were generally much lower than the reference compound fluorouracil.
- For 3c, the 4-OH-substituted phenyl group combined with the hydrazine substituent in the tetrazole moiety imparted excellent antibacterial properties only against E. faecalis (MIC = 16 µg/mL), but had little effect on the derivative’s antifungal activity, as evidenced by its poor activity relative to the reference standard cefazolin. Of the three cell lines tested, the HepG2 cancer cell line was the only one that was susceptible to 3c (GI50 = 18.6 µm) compared to fluorouracil. Here, low activity was noted for the MCF-7 and HeLa cancer cell lines.
- Compounds 3d, 3e, 4a, 4b, 4c, and 4d were all very poor antibacterial and antifungal agents. The anticancer activity of 3d, 3e, 4a, 4b, and 4d was equipotent against the HepG2 cell line compared to fluorouracil, and exhibited poor activity against the MCF-7 and HeLa cancer cell lines. Compounds 4b, 4c, and 4e exhibited very low activity against all three cell lines.
- For 4e, the 4-OCH3-substituted phenyl group combined with a hydrazine moiety imparted excellent activity against E. faecalis and P. aeruginosa only. Otherwise, 4e was shown to be a poor antibacterial and antifungal agent.
From these preliminary SAR investigations, we concluded that the position of the tetrazole substituent bearing the secondary amine was important for biological activity. Additionally, the presence of a substituent on the phenyl ring enhanced the bioactivity of this series.
Docking Studies
The docking behavior of the most potent synthesized compounds (ie, 1b, 2a, and 2b) and the controls (ie, cefazolin, clotrimazole, and fluorouracil) were examined using the proteins 4OR7, 1AI9, and 4FM9 via Autodock Vina program. Here, 2b showed better binding affinity (−7.8 kcal/mol) than the control cefazolin (−7.2 kcal/mol) for 4OR7. Indeed, hydrogen bonding seemed to be pivotal in stabilizing the protein–ligand bonding interactions, thereby ensuring a favorable bond distance of less than 3.5 Å between the H-donor and the H-acceptor atoms.21 The distances of the hydrogen-bonding interactions of the synthesized compounds (ie, 1b, 2a, and 2b) and the controls (ie, cefazolin, clotrimazole, and fluorouracil) were less than 3.5 Å in the respective proteins, signifying that these interactions were very strong. Compound 2b formed two hydrogen-bonding interactions with the receptor 4OR7. Here, the residue Thr46, with bond lengths of 2.13 and 2.95, was involved in the aforementioned interactions. Also, residues Ile14, Met20, Leu45, and Arg98 were involved in hydrophobic interactions. For comparison, the control cefazolin formed seven hydrogen-bonding interactions with 4OR7. Here, residues Arg44 (bond lengths = 2.01 and 2.56), Leu45 (bond length = 2.21), Gly96 (bond length = 1.88), Thr46 (bond lengths = 1.94 and 2.96), and Val99 (bond length = 2.98) were complex in hydrogen-bonding interactions, whereas residues Gly43 and Arg98 were complex in hydrophobic interactions. The overall bonding connections of the respective amino acid residues in 4OR7 protein, when analyzed using 2b and cefazolin, are shown in Figures 3 and 4. Compound 1b showed moderate binding affinity (−6.5 kcal/mol) to 4OR7 compared to clotrimazole (−6.8 kcal/mol), and formed five hydrogen-bonding interactions with 1AI9. The residues Ala111 (bond lengths = 2.63 and 2.97), Tyr118 (bond lengths = 1.91 and 2.84), and Ile112 (bond length = 2.41) were involved in the aforementioned hydrogen-bonding interactions, whereas residues Ile9, Met25, Phe36, and Leu69 were complex in hydrophobic connections. Clotrimazole, the reference compound, did not form any hydrogen bonding interactions with 1AI9. The amino acid residues Met25, Ile33, Phe36, Lys37, Ile62, and Leu69 were involved in hydrophobic interactions. The hydrogen-bonding and hydrophobic interactions of the amino acid residues in 1AI9 with 1b and clotrimazole are shown in Figures 5 and 6, respectively. Compound 2a shows a higher binding affinity (−7.1 kcal/mol) than fluorouracil (−5.4 kcal/mol) in 4FM9. Compound 2a did not form any hydrogen bonds with the receptor 4FM9. The residues Pro593 and Leu705 were complex in hydrophobic connections. The control fluorouracil formed three hydrogen bonds with the receptor 4FM9. The residue Gln542 (bond length = 2.81), Tyr590 (bond length = 2.72), and Leu685 (bond length = 2.24) were complex in hydrogen-bonding interactions, whereas Tyr686 was involved in hydrophobic interactions. The hydrogen-bonding and hydrophobic interactions of the residues in 4FM9 with 2a and fluorouracil are shown in Figures 7 and 8, respectively. In general, the results show that 1b, 2a, and 2b exhibited better antifungal, antibacterial, and anticancer properties than the reference compounds (Table 7).
 |
Table 7 Molecular Docking Interactions of 2b, 1b, and 2a with 4OR7, 1AI9, and 4FM9 |
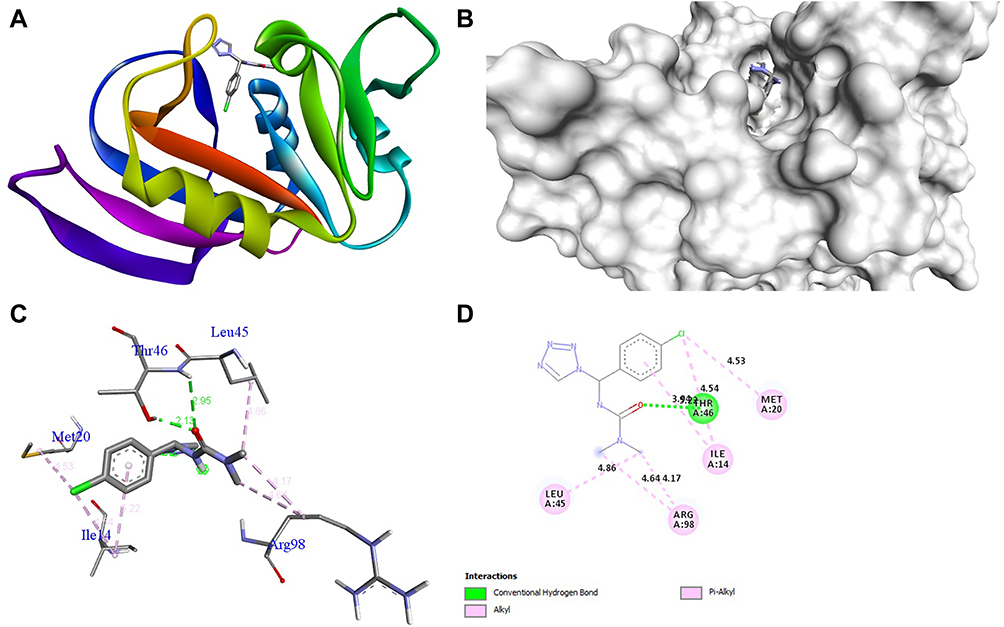 |
Figure 3 Molecular docking of compound 2b binding site of 4OR7. (A) Docked complex. (B) Molecular surface. (C) 3D. (D) 2D. |
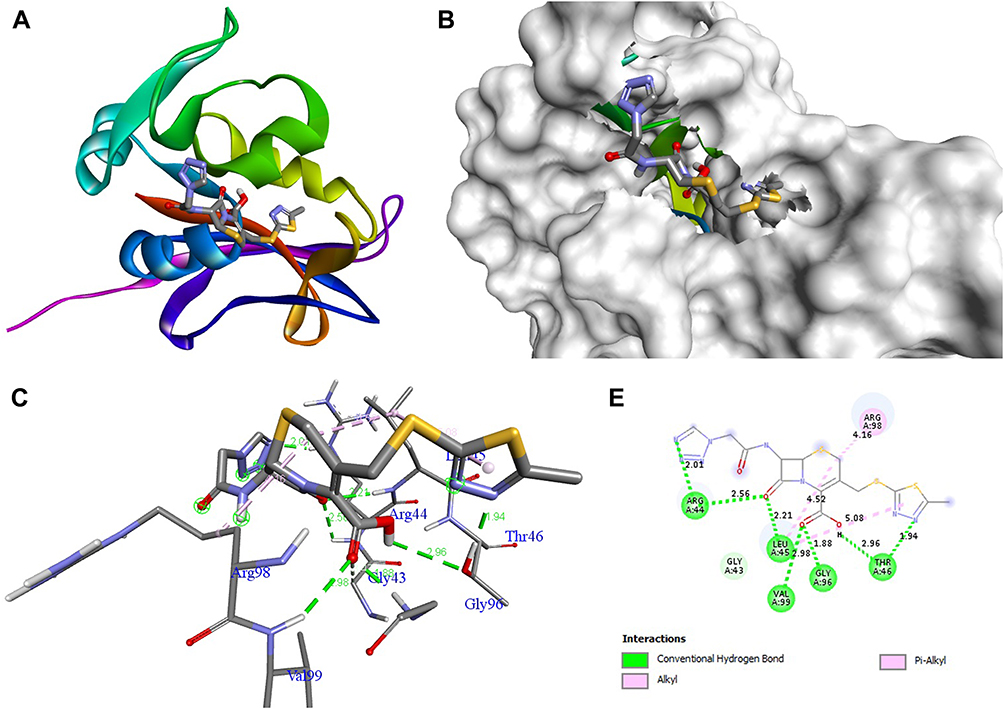 |
Figure 4 Molecular docking of cefazolin binding site of 4OR7. (A) Docked complex. (B) Molecular surface. (C) 3D. (D) 2D. |
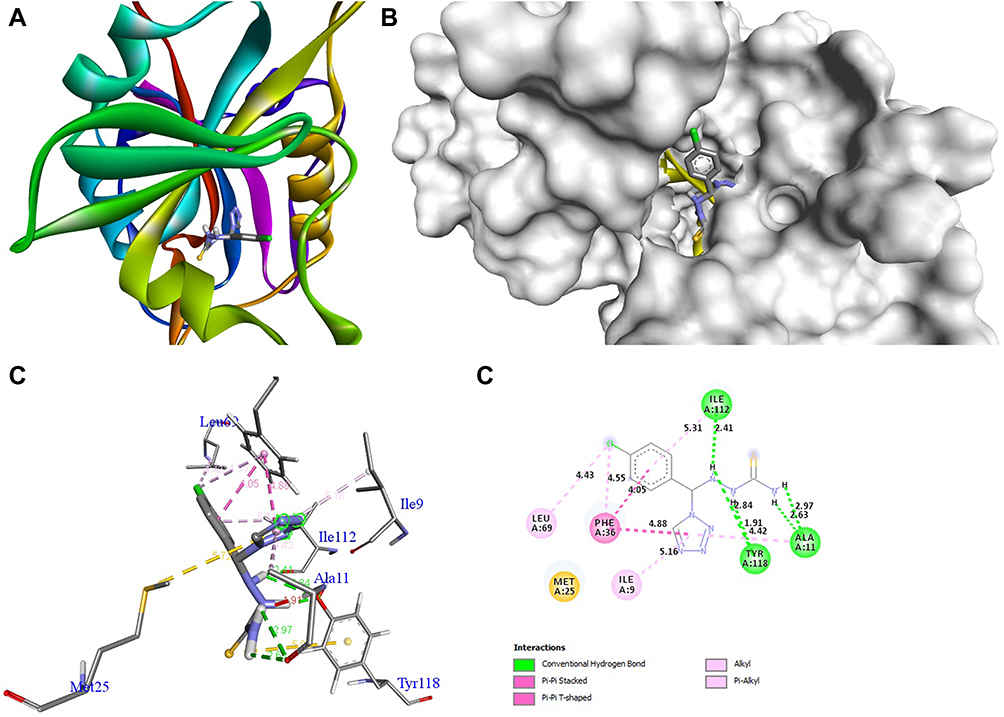 |
Figure 5 Molecular docking of compound 1b binding site of 1AI9. (A) Docked complex. (B) Molecular surface. (C) 3D. (D) 2D. |
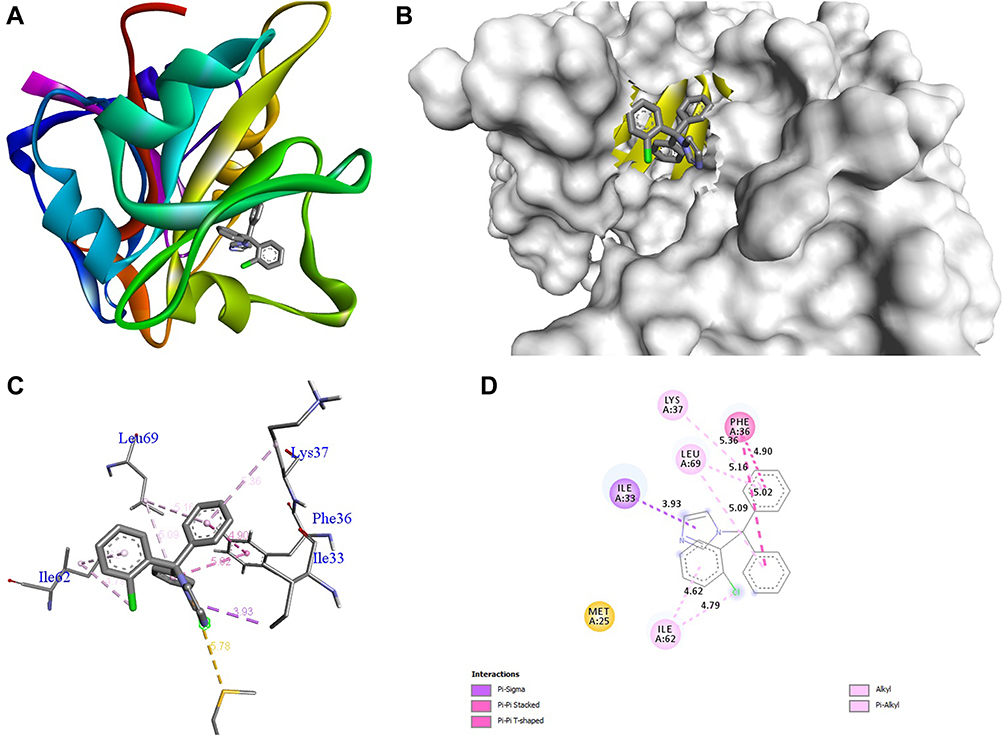 |
Figure 6 Molecular docking of clotrimazole binding site of 1AI9. (A) Docked complex. (B) Molecular surface. (C) 3D. (D) 2D. |
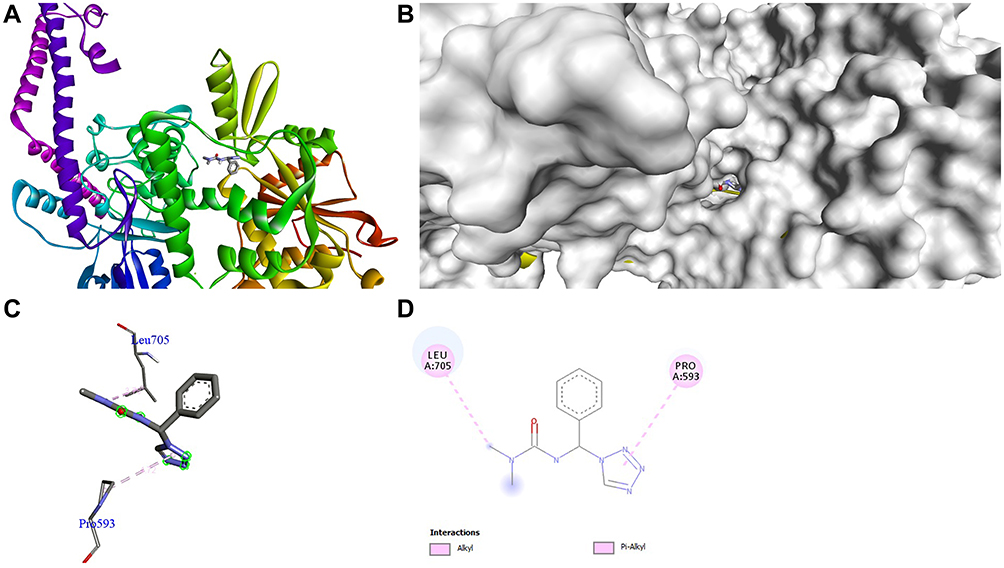 |
Figure 7 Molecular docking of compound 2a binding site of 4FM9. (A) Docked complex. (B) Molecular surface. (C) 3D. (D) 2D. |
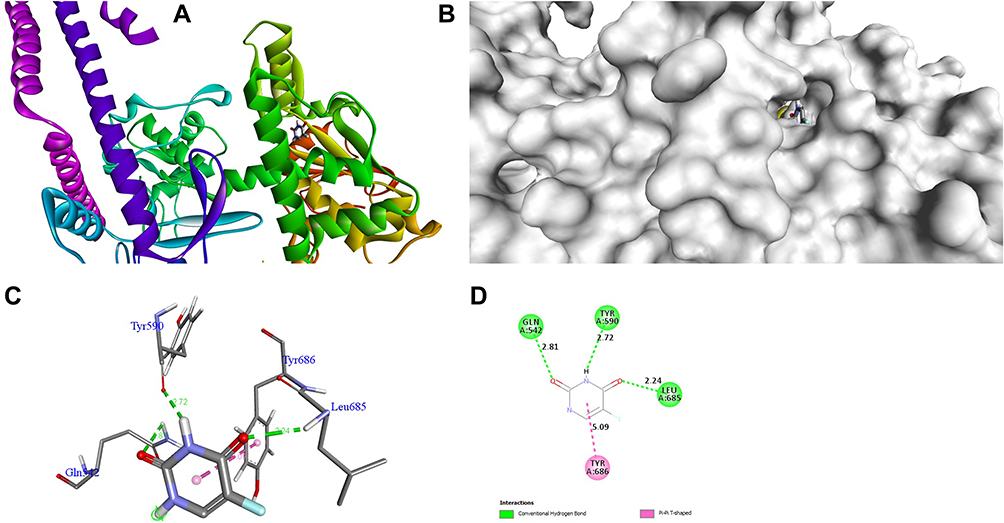 |
Figure 8 Molecular docking of fluorouracil binding site of 4FM9. (A) Docked complex. (B) Molecular surface. (C) 3D. (D) 2D. |
Conclusion
Herein, an efficient and practical synthesis of bioactive tetrazole derivatives, namely, 1a – e, 2a – e, 3a – e, and 4a – e, via ultrasonication is described. The results showed that 1b was highly active in antibacterial screening against Enterococcus faecalis (MIC = 8 µg/mL) then cefazolin drug. Compounds 1b and 1e were very active against Candia albicans (MIC = 0.25 µg/mL) and Microsporum audouinii (MIC = 8 µg/mL), respectively, when compared to the standard clotrimazole. Compound 2a was very active against MCF-7 (GI50 = 5.2 µm) and HeLa (GI50 = 8.1µm) cancer cell lines when compared to the other compounds. Compound 2b was extremely active against the HepG2 (GI50 = 5.4 µm) cancer cell line when compared to fluorouracil, and exhibited better binding affinity (−7.8 kcal/mol) than cefazolin (−7.2 kcal/mol) in 4OR7. Given these results, we believe that 1b, 1e, 2a, and 2b could function of novel classes of clinically useful antimicrobial and anticancer agents.
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project number (RSP-2020/190), King Saud University, Riyadh, Saudi Arabia.
Funding
Financial support came from the Deanship of Scientific Research at King Saud University, Saudi Arabia.
Disclosure
The authors have no conflicts of interest to declare.
References
1. Koltin Y. Targets for antifungal drug discovery. Ann Rep Med Chem. 1990;25:141–148.
2. Cardoso CS, Silva DPB, Silva DM, et al. Mechanisms involved in the antinociceptive and anti-inflammatory effects of a new triazole derivative: 5-[1-(4-fluorophenyl)-1H-1,2,3-triazol-4-yl]-1H-tetrazole (LQFM-096). Inflammopharmacol. 2020;28:877–892. doi:10.1007/s10787-020-00685-8
3. Rajasekaran A, Thampi PP. Synthesis and antinociceptive activity of some substituted-{5-[2-(1, 2, 3, 4-tetrahydrocarbazol-9-yl) ethyl] tetrazol-1-yl} alkanones. Eur J Med Chem. 2005;40:1359–1364. doi:10.1016/j.ejmech.2005.07.013.
4. Mohite PB, Bhaskar VH. In vitro evaluation of tetrazoles as a novel class of Antimycobacterium tuberculosis agents. Adv Pharm Bull. 2012;2(1):31–36. doi:10.5681/apb.2012.005.
5. Feng G, Jiaqi X, Gang H. Current scenario of tetrazole hybrids for antibacterial activity. Eur J Med Chem. 2019;184:111744. doi:10.1016/j.ejmech.2019.111744.
6. Kritchenkov AS, Lipkan NA, Kurliuk AV, et al. Synthesis and antibacterial activity of chitin tetrazole derivatives. Pharm Chem J. 2020;54:138–141. doi:10.1007/s11094-020-02180-4
7. Davulcu AH, McLeod DA, Li JE, et al. Process research and development for a tetrazole-based Growth Hormone Secretagogue (GHS) pharmaceutical development candidate. J Org Chem. 2009;74:4068–4079. doi:10.1021/jo9003508.
8. Afshari NA, Ma JJ, Duncan SM, Pineda R, Starr CE, DeCroos FC. Trends in resistance to ciprofloxacin, cefazolin, and gentamicin in the treatment of bacterial keratitis. J Ocul Pharmacol Ther. 2008;24:217–223. doi:10.1089/jop.2007.0085
9. Upadhyaya RS, Jain S, Sinha N, Kishore N, Chandra R, Rora SK. Synthesis of novel substituted tetrazoles having antifungal activity. Eur J Med Chem. 2004;39:579–592. doi:10.1016/j.ejmech.2004.03.004.
10. Mokhtary M, Torabi M. Nano magnetite (Fe3O4), an efficient and robust catalyst for the one-pot synthesis of 1-(aryl(piperidin-1-yl)methyl)naphthalene-2-ol and 1-(α-amido alkyl)-2-naphthol under ultrasound irradiation. J Saudi Chem Soc. 2017;21:S299- S304. doi:10.1016/j.jscs.2014.03.009
11. Safari J, Zarnegar Z, Ahmadi M, Seyyedi S. An investigation of the catalytic potential of potassium cyanide and imidazolium salts for ultrasound-assisted synthesis of benzoin derivatives. J Saudi Chem Soc. 2015;19:628–633. doi:10.1016/j.jscs.2012.05.005
12. Javanshir S, Ohanian A, Majid Heravi M, Naimi-Jamal MR, Bamoharram FF. Ultrasound-promoted, rapid, green, one-pot synthesis of 2′-aminobenzothiazolomethylnaphthols via a multi-component reaction, catalyzed by heteropolyacid in aqueous media. J Saudi Chem Soc. 2014;18:502–506. doi:10.1016/j.jscs.2011.10.013
13. Petkovsek M, Mlakar M, Levstek M, Strazar M, Sirok B, Dular M. A novel rotation generator of hydrodynamic cavitation for waste-activated sludge disintegration. Ultrason Sono Chem. 2015;26:408–414. doi:10.1016/j.ultsonch.2015.01.006
14. Tramontini M, Angiolini L. Further advances in the chemistry of mannich bases. Tetrahedron. 1990;46:1791. doi:10.1016/S0040-4020(01)89752-0
15. Medic-Saric M, Maysinger D, Movrin M, Dvorzak I. Antibacterial and antifungal activities of nitroxoline Mannich bases. Chemotherapy. 1980;26:263–267. doi:10.1159/000237915
16. Gul HI, Vepsalainen J, Gul M, Erciyas E, Hanninen O. Cytotoxic activities of mono and bis Mannich bases derived from acetophenone against Renca and Jurkat cells. Pharm Acta Helv. 2000;74:393–398. doi:10.1016/S0031-6865(00)00022-4
17. Siatra-Papastaikoudi T, Tsotinis A, Chinou I, Roussakis C. Synthesis and anticancer activity of new phenyl-ring substituted 4-morpholino-1-phenylthio-2-butanones [Mannich bases]. Farmaco. 1994;49:221–223.
18. Idhayadhulla A, Surendra Kumar R, Jamal Abdul Nasser A, Selvin J, Manilal A. Synthesis of some Mannich base derivatives and their antimicrobial activity study. Arabian J Chem. 2014;7:994–999. doi:10.1016/j.arabjc.2010.12.025
19. Atwal MS, Bauer L, Dixit SN, et al. Analgetics. II. Relation between structure and activity of some beta-amino ketones. J Med Chem. 1969;12:994–997. doi:10.1021/jm00306a006
20. Borenstein MR, Doukas PH. Anticonvulsant activity of indanylspirosuccinimide Mannich bases. J Pharm Sci. 1987;76:300–302. doi:10.1002/jps.2600760407
21. Taha M, Ismail NH, Khan A, et al. Synthesis of novel derivatives of oxindole, their urease inhibition and molecular docking studies. Bioorg Med Chem Lett. 2015;25:3285–3289. doi:10.1016/j.bmcl.2015.05.069
22. Bauer AW, Kirby WM, Sherris JC, Turck M. Antibiotic susceptibility testing by a standardized single disk method. Am J Clin Pathol. 1966;45:493–496. doi:10.1093/ajcp/45.4_ts.493
23. Petersdorf RG, Sherris JC. Methods and significance of in vitro testing of bacterial sensitivity to drugs. Am J Med. 1965;39:766–779. doi:10.1016/0002-9343(65)90096-3
24. Collins AH. Microbiological Methods.
25. Verma RS, Khan IK, Singh AP. Antifungal agents: past, present, future prospects. Natl Acad Chem Biol. 1998;55–128.
26. Surendra Kumar R, Moydeen M, Al-Deyab S, Manilal A, Idhayadhulla A. Synthesis of new morpholine-connected pyrazolidine derivatives and their antimicrobial, antioxidant, and cytotoxic activities. Bioorg Med Chem Lett. 2017;27:66–71. doi:10.1016/j.bmcl.2016.11.032
27. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi:10.1002/jcc.21334
28. Lamb KM, Lombardo MN, Alverson J, Priestley ND, Wright DL, Anderson AC. Crystal structures of Klebsiella pneumoniae dihydrofolate reductase bound to propargyl-linked antifolates reveal features for potency and selectivity. Antimicrob Agents Chemother. 2014;58:7484–7491. doi:10.1128/AAC.03555-14
29. Whitlow M, Howard AJ, Stewart D, et al. X-ray crystallographic studies of Candida albicans Dihydrofolate reductase high resolution structures of the holoenzyme and an inhibited ternary complex. J Biol Chem. 1997;272:30289–30298. doi:10.1074/jbc.272.48.30289
30. Wendorff TJ, Schmidt BH, Heslop P, Austin CA, Berger JM. The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J Mol Biol. 2012;424:109–124. doi:10.1016/j.jmb.2012.07.014
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.