Back to Journals » Drug Design, Development and Therapy » Volume 13
Synthesis, comparative docking, and pharmacological activity of naproxen amino acid derivatives as possible anti-inflammatory and analgesic agents
Authors Elhenawy AA ![]() , Al-Harbi LM
, Al-Harbi LM ![]() , Moustafa GO
, Moustafa GO ![]() , El-Gazzar MA, Abdel-Rahman RF
, El-Gazzar MA, Abdel-Rahman RF ![]() , Salim AE
, Salim AE
Received 28 November 2018
Accepted for publication 4 April 2019
Published 24 May 2019 Volume 2019:13 Pages 1773—1790
DOI https://doi.org/10.2147/DDDT.S196276
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Tuo Deng
Ahmed A Elhenawy,1 LM Al-Harbi,2 Gaber O Moustafa,3 MA El-Gazzar,1 Rehab F Abdel-Rahman,4 Abd Elhamid Salim1
1Chemistry Department, Faculty of Science, Al-Azhar University (Boys’ Branch), Nasr City, Cairo, Egypt; 2King Abdulaziz University, Jeddah 21589, Saudi Arabia; 3Peptide Chemistry Department, Chemical Industries Research Division, National Research Centre, Dokki, Giza 12622, Egypt; 4Department of Pharmacology, National Research Centre, Giza, Egypt
Background and aim: Naproxen is a member of the Nonsteroidal anti-inflammatory drugs (NSAIDs). This work aimed to synthesize a safe NSAID agent based on a peptide derivative.
Methods: The structure of compounds 5–20 was established on the basis of spectral data. Frontier molecular orbitals and chemical reactivity were discussed to clarify inter- and intramolecular interactions among tested compounds. We applied competitive molecular docking using polynomial logarithms to identify the most accurate algorithm for pharmacological activity prediction for the tested compounds. The docking protocol with the lowest RMSD was selected for analyzing binding affinity.
Results: Docking results illustrated that the binding interaction increased after introduction of an acidic fragment to the parent compound. These compounds were selected for additional study against adsorption, distribution, metabolism, excretion, and toxicity (ADMET) in silico. The compounds tested had good oral bioavailability without any carcinogenesis effect; no marked health effects were observed via rodent toxicity. Compounds passed through docking and ADMET profiles for them (5–16) were examined as anti-inflammatory and analgesic agents. Compounds 8 and 16 showed higher anti-inflammatory potency than the reference drug and tested compounds. Compounds 8, 10, and 14 exhibited the highest analgesic potency compared to the other tested compounds.
Conclusion: The tested compounds have shown negligible ulcerogenic effects, and may be considered safer drugs than naproxen for treating inflammatory conditions.
Keywords: naproxen, isothiocyanate, peptide candidates, anti-inflammatory and analgesic agents, docking
Introduction
Naproxen is a propionic acid derivative widely used as an analgesic and anti-inflammatory agent. Its action is thought to be related to its inhibition of cyclooxygnase (COX), which leads to a reduction in the concentration of prostaglandin in different tissue types and fluids.1,2 Naproxen and other aroyl propionic acids have been reported to cause gastrointestinal problems due to the presence of a free carboxylic group in the parent compound.3 Therefore, masking of this acidic group may be a promising means to decrease or abolish gastrointestinal toxicity.4–6 Syntheses of ester prodrugs of naproxen as promoieties have been reported to improve the therapeutic index for oral delivery of NSAIDs.7 Naproxen amide compounds synthesized with methyl esters of amino-acid derivatives exhibit reliable anti-inflammatory activity.8 In addition, the oral anti-inflammatory activity of naproxen glycolamide nitrate was found to cause less gastric damage9 Also, 2-(1-2(2(2-methoxynaphthalen-6-yl)propanoyl)1H-indol-2-yl) acetic acid and m-aminobenzoic acid (an analogue of naproxen) have been reported to increase anti-inflammatory potency, with lower ulcerogenic activity10 An amide prodrug of naproxen derivatives has shown significant anti-inflammatory activity compared to the standard drug naproxen.11 Propane-amide derivatives of naproxen show good antibacterial activity against the Gram-positive bacteria Staphylococcus aureus and Bacillus subtilis and the Gram-negative bacteria Escherichia coli and Pseudomonas aeruginosa, comparable to standard drugs: ampicillin for Gram-positive and ciprofloxacin for Gram-negative bacteria.12 Additionally, the rate of tumor growth in tumor-bearing rats was reduced by 58% when treated with naproxen13 A hydroxamic acid derivative of naproxen has shown potent histone deacetylase inhibition.14 Propanamide derivatives and urea derivatives of naproxen have been found to have promising inhibitory effects against the colon cancer cell line HCT–116.15 Moreover, naproxen is expected to be a promising leader for novel compounds with antiviral activity against influenza A virus. Naproxen esters have been synthesized with tocopherol, and exhibited good antioxidant activity and inflammatory and antioxidant properties.16 The aim of this research in our laboratory was to study the effect of a combination of amino acids with different aromatic and heterocyclic compounds on their biological and pharmacological activities.17–20 Peptides being used as therapeutic agents include Lupron, Sandostatin, and Zoladex.21,22 Peptides are intrinsically able to interact with biological systems, and are thus potent therapeutics.23–25 Due to the importance of naproxen derivatives, amino acids and peptides act as bioactive compounds. The objective of this study was to synthesize novel anti-inflammatory and analgesic agents based on peptide compounds containing 2-(6-methoxynaphthalen-2-yl)propanoyl isothiocyanate.
Designing synthesized compounds as anti-inflammatory agents
In order to identify the structural basis of an anti-inflammatory candidate drug (naproxen), we applied an electrostatic potential map and lipophilicity map, using the PM3 semiempirical method in the MOPAC16 package.26
From molecular electrostatic potential analysis, it was observed that a high-electron-density region at the carbonyl group (red) was responsible for the activity of naproxen. The lipophilicity map demonstrated that the hydrophilic part on the carboxy group (yellow) and the lipophilic part on the naphthalene scaffold (blue were necessary for the activity (Chart 1). In addition, highest occupied molecular orbital (HOMO)–lowest unoccupied MO (LUMO) electron-density maps showed HOMO and LUMO overlapped over the naphthalene ring (Chart 1). This clearly suggests that the naphthalene ring is able to include charge-transfer mechanisms or π–π interaction, which demonstrates the importance of the naphthalene scaffold in the molecular structure for enhancement of efficacy. Therefore, firstly (Chart 2), we increased electron density in the hydrophilic region as an initial hit region by replacing COOH naproxen with a thiourea linker linked with hydrophilic or lipophilic amino-acid residues (5–8) to take into consideration whether this was better or not (Chart 2). Finally, the carboxy group of different amino-acid derivatives (5–8) was converted to both methoxy (9–12) and hydrazide (13–16) derivatives, then elongated with a bulker phthalyl group (17–20), to judge which types of fragments were preferable as new active molecules.
Results and discussion
Chemistry
This study aimed to conjugate amino acids and naproxen. Some of the novel derivatives (5–20) were synthesized based on naproxol isothiocyanate (4), which may be expected to possess various anti-inflammatory and analgesic agents. The compound naproxol isothiocyanate (4) was synthesized according to a reported method8 (Scheme 1).
Condensation of naproxolisothiocyanate (4) with amino acids was done by stirring in ethanol using pyridine as an organic base. This reaction produced substituted naproxol thioureido amino acids (5–8;Scheme 2). Infrared (IR) spectra showed absorption bands at 3,444 and 3,207 cm–1 due to OH overlapping NH, in addition to absorptions of carbonyl groups at bands 1,709 and 1,718 cm–1 for compounds 5 and 8, respectively.1H nuclear magnetic resonance (NMR) spectra of 5–8 showed the characteristic signal of OH for the carboxylic group of newly coupled amino acids in the range δ=9.9–11.70 ppm.
On the other hand, compound 4 was reacted with free amino-acid methyl esters (L-alanine, β-alanine, L-serine and L-tyrosine) in dry tetrahydrofuran (THF) and drops of triethylamine (TEA) as a catalyst, which produced naproxol thioureido–amino acid methyl esters (9–12), respectively (Scheme 3).
IR spectra showed absorption bands at 3,445 and 3,450 cm–1 due to the presence of NH for compounds 9 and 10, respectively. Meanwhile, NH + OH for compounds 11 and 12 exhibited bands at 3,451 and 3,449 cm–1, respectively. Carbonyl groups appeared in the range 1,735–1,732 cm–1 for compounds 9–12, respectively. 1H-NMR spectra of 9–12 showed the characteristic signal of OCH3 for the new ester groups of formed amino-acid esters in the range δ=3.88–3.72ppm. The mass spectrum of compound (12) showed a molecular ion peak equal to its molecular weight at m/z=466 (M+) and revealed a base peak at m/z=185.
Compounds 9–12 were reacted with hydrazine hydrate in ethanol to give naproxol thioureido–amino acid hydrazides 13–16, respectively; Scheme 3). IR spectra showed absorption bands in the range 3,453–3,360 cm–1 due to (NH2, NH) for compounds 13–16. The carbonyl groups for these compounds exhibited absorption bands in the range ν=1,736–1,734 cm–1. 1H-NMR spectra of 13–16 showed characteristic signals of NHNH2 in the range δ=10.3–9.99 ppm, in addition to the characteristic signal of NHNH2 in the range δ=7.01–6.99 ppm. The mass spectrum of compound 14 showed a peak equal to its molecular weight at m/z=374 (20.52%) and base peak at m/z=188 (100%).
On the other hand, compounds 13–16 were reacted with phthalic anhydride to give the corresponding N-phthalimido derivatives (17–20; Scheme 3). IR spectra showed absorption bands in the range 3,451–3,443 cm–1 due to OHSer and OHTyr for compounds 19 and 20, respectively. 1H-NMR spectra of 17–20 showed disappearance of the characteristic signal of OH-Tyr at δ=12.5 for compound 20. The characteristic signals (6.0 and 6.3 ppm) of amino protons NH2for the hydrazide moiety were diapered in 1H-NMR spectra of 17–20, which confirmed the occurrence of the reaction for 13–16 with phthalic anhydride. The mass spectrum of compounds 19 and 20 showed a peak equal to its molecular weight at m/z=522 (46.52%) and 596 (32.71%), respectively, and revealed a base peak at m/z=185 (100%).
Enantiomeric purity was evaluated for compounds 5, 7–9, 11–13, 15–17, 19, and 20 through determination of values of the specific rotation, enantiomeric excess (ee) and diastereoisomeric excess (de); these values were unchanged after repeated crystallization for several times. Thin-layer chromatography (TLC) analysis and optical purity for the obtained compounds were >97%. Therefore, as we expected, stereochemical configuration at the α-carbon atom of the acid was practically unaffected without undergoing any significant loss of optical activity.
Molecular modeling studies
Stability of inter- and intramolecular interaction of synthesized compounds based on global chemical reactivity
Optimization geometry and conformational analysis for compounds 5–20 were performed using the PM3 semiempirical method in MOPAC16 package 27, as implemented in MOE.2015.,27 All calculated energies are summarized in
The binding interaction takes place between the HOMO drug and the LUMO receptor and vice versa. This interaction is stabilized inversely with an energy gap. Increasing HOMO energy for a receptor and decreasing LUMO energy of the drug molecule leads to enhanced stabilization of the drug–receptor interaction29 Frontier-MO energy is directly related to soft nucleophiles and hard electrophiles. As such, we can describe the electrophiles and nucleophiles as “soft“ and “hard“. ΔG is a measure of stability. All compounds (5–20) displayed nearly the same of ΔG values (~–7.41 to −7.96 eV), softness (~0.25–0.27 eV) and hardness (~3.70–3.8 eV; (Table 1). Molecules with small ΔG values have high softness and chemical reactivity, as well as increased nucleophilicity (offer electrons easily to an acceptor). Therefore, all compounds had nearly the same reactivity against the biological environment.
 | Table 1 Pharmacokinetic parameters for ligands 5–20 |
The presence of both carboxyl (5–8) and phthalyl (17–20) groups in the parent molecule led to maximum electroacceptance. All compounds were almost equal for the donating power of the electron ω−. Compounds bearing alanyl residues (5, 9, 13, and 17) had the highest electron acceptance. All methoxy (9–12) and hydrazide (13–16) members showed more electrophilicity (ω±) and electronegativity (χ) than other members. From these data, the hydrazide derivative was the best nuclophile, with high donating power for the electronic charge. As such, we think these compounds may attack the hydrophilic part of the receptor.
Docking studies
To identify a suitable anti-inflammatory agent from the synthesized compounds, the biological data were clarified on a structural basis using a docking study. The docking experiment was performed using three search algorithms. These algorithms generated different scores (dG, MolDock, and Plants). We obtained an X-ray of the crystal structure of COX2 (ID 1PXX)30 complexed with naproxen as reference inhibitor.31
Analysis of COX2-active site has shown that Tyr385 and Ser530 are vital chelation sites against ligand32 Arachidonic acid (by the carboxyl group) was coordinated with Tyr385 and Ser530 through a tetrahedral intermediate, which was stabilized in a binding pocket via a negative charge.32,33 NSAIDs inhibit COX2 in the same manner as arachidonic acid.30 The tested compounds (5–20) redocked into the COX2-active site after elimination of the reference inhibitor. The docking result was analyzed based on a scoring function. A docking protocol, scored by lowest RMSD was selected for the binding affinity of the inhibitor–COX2 complex. These complexes were energetically minimized with an MMFF94 force field,34 then visualized with Discovery Studio 2017 software35
Scatterplots of RMSD vs docking scores for different docking protocols are given in Figure 1. The MOE protocol exhibited a correlation coefficient of 0.472 P<0.002), with an RMSD range of 0.594–0.94 Å for all tested compounds. We used the MOE technique (with the lowest RMSD) to perform docking visualization against COX2 for all members (
 | Table 2 Anti-inflammatory effect of synthesized compounds on carrageenan-induced paw edema in rats after 1, 2, 3, and 4 hours of test-drug administration |
 | Figure 1 Scatteplots of docking scores and RMSD for 5–20. |
Interaction strength was free acids (5–8) > methyl esters (9–12) > hydrazide derivatives (13–16). Compounds 5, 8, 12, and 15 formed important H-bond interactions (four, one, three, and one, respectively) with binding pockets (Phe205, Ala527, Ser530, Gly536, Tyr348, Tyr355, and Tyr385; Figures 2 and 3). These compounds were trapped in the amino-acid backbone of the binding pocket through adjustment of naphthalene rings in perpendicular mode with Tyr385 (Figure 3). Furthermore, these compounds stabilized with caped binding pockets through arranged naphthalene rings in orthogonal position with amino-acid fragments (Figure 3). The binding interaction increased with increasing hydrophobicity of ligands (Figure 3). The results obtained clearly revealed that the amino-acid residues close to the reference molecules were mostly the same as observed in the tested compounds. The high docking scores and strong binding process indicated that the compounds tested may be suitable anti-inflammatory agents as COX2 inhibitors.
 | Figure 2 2D view of interactions of the highest-binding interaction compounds (5, 8, 12, and 15) into the active site of COX2 with the lowest RMSD, using the MOE tool. |
 | Figure 3 3-D view of interactions and distance for compounds (4, 6, 7, 8, 10,and 11) into the hydrophobic surface of the active site of COX2. |
ADMET profile
To be an ideal drug, a bioactive molecule should have low toxicity, as well as good pharmacokinetic properties. Oral bioavailability plays a vital role in enhancement of any therapeutic bioactive molecule. Therefore, it is useful to know the adsorption, distribution, metabolism, excretion and toxicity (ADMET) profile before beginning experimental assessment, which can be expensive and labor-intensive.
ADMET descriptors were elucidated using both MOE,27 and ADMET SAR36 tools (Table 1). Compounds 5–20 obeyed Lipinski rules (Figure 4),37 Lipophilicity was >5.0,38 absorption ~50.64%–71.48.%,39 and topological polar surface area <140 (molecules passively absorbed if >140)38 These data suggested that these compounds may have good absorption (Figure 4). Also, carcinogen behaviors were investigated through comparison of our compounds with 981 carcinogenic molecules from the Carcinogenic Potency Database. No compounds possessed a carcinogenic effect (~0.6–0.9 mg/kg body weight/day) or any acute oral toxicity (~0.47–0.63 mg/kg). The compounds tested exhibited low LD50 values (~2.43–2.66 mol/kg).
 | Figure 4 3-D principal component–analysis plot for compounds 5–20.Notes: x-axis, structure number; y-axis, TPSA; z-axis, ball sizes represent log P-values. Tumorigenicity shown above each ball. |
No compounds acted as inhibitors or substrates against Pgp. Therefore, these compounds can be retained safely without any effect.40 All compounds were able to pass through the blood–brain barrier. Permeability of the compounds through the BBB indicated that they may be effective in treating inflammation. The compounds displayed weak inhibition against the hERG gene. These features can conduct to long QT syndrome.41 As such, we have concluded that, these compounds may be have a good oral bioavailability without observed any marked health effects via rodent toxicity profiles.
Pharmacological activities
Anti-inflammatory activity
In vivo anti-inflammatory screening was performed for the compounds (5–16) using a carrageenan-induced rat-paw-edema assay.42 Naproxen is one of the most potent NSAIDs, and was used as a reference drug in this study. Reduction in tested compounds for edema in comparison with the control and naproxen (reference drug) was calculated (Table 2). Generally, compared to naproxen, all tested compounds exhibited lower potency, except compounds 8 and 16, which showed about 1.4 times the potency of the reference drug. After 1 hour of induced inflammation, compounds 5, 8, 15, and 16 showed the highest anti-inflammatory activity compared to the reference drug and other members, while 6 7, and 11 exhibited moderate potency among the tested compounds. The other compounds showed lower potency. For the entire experiment, compounds bearing tyrosine (8) and tyrosin hydrazide (16) fragments showed the highest anti-inflammatory activity (131.2% and 133.9%, respectively, after 4 hours) of all synthesized compounds. These compounds exhibited activities 1.3 times better than naproxen, which may be explained by the increasing hydrophilicity of the parent compound. Ester (9–12) and β-Ala hydrazide derivatives (14) exhibited a lowest anti-inflammatory potency throughout the experiment. Also, most carboxylic acid members (5–7) showed moderate potency throughout (60.3%–85.2%, after 4 hours). After 2, 3, and 4 hours of induced inflammation, the presence of alanyl-hydrazide (13) and serinyl-hydrazide (15) in the parent compound showed moderate potency compared to the reference drug (Table 2).
Analgesic activity (tail-flick latency test)
The analgesic activity of tested compounds was evaluated using the tail-flick test in rats.43 Tail-withdrawal latency for the tested drugs is given in (Table 3). All tested compounds showed a significant delay in latency. Latency showed a significant increase in most treated groups, analgesic potencies arranged in descending order for compounds (8, 10, 14, 6, 7, 16, 9, 5, 12, 13) with activity percentages (50.0%–84.3% after 120 minutes), compared with the reference drug (Table 3). At 30 minutes postinjection, only compound (14) showed increased tail-withdrawal latency value (6.1±0.4) over naproxen. After 120 minutes, compounds 8, 10, and 14 exhibited the ; highest potency (84.3%, 81.4%, and 81.4%, respectively; significant) compared to synthesized members (5–16). Compounds bearing free Tyr (8) fragments showed the highest potency, in addition to elongation alkyl chain was increased activity as β-AlaO-Me (10) and β-Ala hydrazide (14) derivatives. -The remaining compounds showed moderate analgesic activity at all time points (Table 3).
 | Table 3 Analgesic activity using tail-flick test for tested compounds |
Acute ulcerogenesis
Compounds were screened for gastric irritation activity. The ulcerogenic effect of naproxen and newly synthesized compounds was studied. Naproxen showed moderate gastric ulceration. No synthesized compounds showed any lesions in the stomach on macroscopic examination. These compounds showed negligible ulcerogenic effect, and may be considered safer drugs for treating inflammatory conditions.
Materials and methods
Animals
Mature Wistar rats (weighing 130–150 g) were purchased from the Animal House Colony at the National Research Centre, Egypt. Standard conditions were a 12:12-hour light:dark cycle and well-ventilated rooms for housing of the animals. Animals were kept in hygienic cages and given free access to clean standard pellet-diet food and water. One week before experimentation, all animals were shifted to adapt to the laboratory environment. This study was done according to standards of the ethics committee of the National Research Centre (approval MREC–17–141), which is in accordance with the national regulations on animal welfare and Institutional Animal Ethics Committee.
Chemistry
Solvents, chemicals, and thin-layer chromatography used in this work were obtained from international chemical companies: Sigma-Aldrich ( St Louis, MO, USA), Honeywell (Charlotte, NC, USA), and Merck (Kenilworth, NJ, USA). Carrageenan was obtained from Sigma-Aldrich. Melting points were determined using a digital electrothermal melting-point apparatus in opened glass capillary tubes and were uncorrected. Elemental microanalyses for carbon, nitrogen, and hydrogen (at the Microanalytical Unit, Cairo University, Cairo, Egypt) were obtained within good limits of theoretical values. IR spectra were obtained using KBr disks using Fourier-transform IR spectrophotometry (IRAffinity 1S; Shimadzu, Kyoto, Japan) at the Microanalytical Unit. Measurements of mass were taken using gas chromatography–mass spectrometry (QP2010 Ultra;Shimadzu) at the Microanalytical Unit. 1H-NMR spectra were run on 500 MHz instruments (JEOL, Tokyo, Japan) in DMSO-d6.
Synthesis of naproxen (2), naproxol chloride (3), and naproxol isothiocyanate (4)
These compounds were prepared according to previously reported methods.8
Synthesis of naproxol thioureido amino acids (5–8)
These compounds were synthesized by stirring free amino acids with naproxolisothiocynate (4) in THF and a few drops of pyridine. The progress of the reaction was monitored by TLC. After neutralization by 1N HCl, the crude materials purified by recrystallization from ethanol, yields (68–80%). All the synthesized compounds (5–8) were chromatographically homogeneous when developed with iodine solution, benzidine, and gave negative ninhydrin tests. Structures of the synthesized compounds (5–8) were confirmed from elemental analysis, chromatographic studies, spot reactions, IR spectra, and 1H NMR spectra. Complete acid hydrolysis of the synthesized compounds (5–8) using 6NHCl at 110°C for 24 hours gave positive ninhydrin spots of L-alanine, β-alanine, L-serine and L-tyrosine.
Yield: 75%; melting point: 118–120 °C, Rf: 0.55 (S), 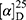 =−22.1 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3444 broad band (OH overlapping NH), 3075(CHArom), 2959 (CHali), 1709(CO), 1389 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=9.9(s, 1H, OHCOOH), 7.83 (d, J=9.3 Hz, 2H, NHCSNH), 7.40 (t, J=1.5 Hz, 2H-ArH), 7.30 (dd, J=7.5, 1.6 Hz, 2H-ArH), 7.40(m, J=7.6, 2H-ArH), 3.89 (m, J=7.6 Hz, 1H-CHCH3-Nap.), 3.82 (m, J=6.8 Hz, 1H-CHCH3 Ala-)]), 3.78(s, 3H-OCH3-Nap), 1.43 (m,J=6.8 Hz,3H-CHCH3−Nap.),1.41 (d, J=6.8 Hz, 3H-CHCH3Ala). Molecular formula (molecular weight): C18H20N2O4S (360.4). Calculated analysis: C, 59.98; H, 5.59; N, 7.77; S, 8.90; Found: C, 59.92; H, 5.56; N, 7.78 S, 8.88.
=−22.1 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3444 broad band (OH overlapping NH), 3075(CHArom), 2959 (CHali), 1709(CO), 1389 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=9.9(s, 1H, OHCOOH), 7.83 (d, J=9.3 Hz, 2H, NHCSNH), 7.40 (t, J=1.5 Hz, 2H-ArH), 7.30 (dd, J=7.5, 1.6 Hz, 2H-ArH), 7.40(m, J=7.6, 2H-ArH), 3.89 (m, J=7.6 Hz, 1H-CHCH3-Nap.), 3.82 (m, J=6.8 Hz, 1H-CHCH3 Ala-)]), 3.78(s, 3H-OCH3-Nap), 1.43 (m,J=6.8 Hz,3H-CHCH3−Nap.),1.41 (d, J=6.8 Hz, 3H-CHCH3Ala). Molecular formula (molecular weight): C18H20N2O4S (360.4). Calculated analysis: C, 59.98; H, 5.59; N, 7.77; S, 8.90; Found: C, 59.92; H, 5.56; N, 7.78 S, 8.88.
Yield: 73%; melting point: 114–116 °C, Rf: 0.56 (S), 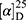 =−25.2 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3448, 3236 broad band (overlapping of OH NH and CHarom.), 2975 (CHali.), 1725(CO), 1615 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=11.70 (s, 1H, OH), 9.12(s, 1H, NHCS), 7.81(m, 1H, J=5.4, 5.4, CSNH), 7.66 (dd, J=7.5, 1.5, 2H, ArHNap), 7.30 (td, J=1.6, 1.6, 0.7, 2H, ArHNap), 7.14 (dd, J=7.5, 1.5, 2H, ArHNap), 3.9 (q, J=6.8, 6.8, 6.8, 1HNap), 3.80(s, 3H,CH3Nap), 2.67–2.58 (4H, CH2CH2 B-ala), 1.46 (d, J=6.8, 3H, CH3 Nap), Molecular formula (molecular weight): C18H20N2O4S (360.4). Calculated analysis: C, 59.98; H, 5.59; N, 7.77; S, 8.90; Found: C, 59.94; H, 5.57; N, 7.75 S, 8.86.
=−25.2 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3448, 3236 broad band (overlapping of OH NH and CHarom.), 2975 (CHali.), 1725(CO), 1615 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=11.70 (s, 1H, OH), 9.12(s, 1H, NHCS), 7.81(m, 1H, J=5.4, 5.4, CSNH), 7.66 (dd, J=7.5, 1.5, 2H, ArHNap), 7.30 (td, J=1.6, 1.6, 0.7, 2H, ArHNap), 7.14 (dd, J=7.5, 1.5, 2H, ArHNap), 3.9 (q, J=6.8, 6.8, 6.8, 1HNap), 3.80(s, 3H,CH3Nap), 2.67–2.58 (4H, CH2CH2 B-ala), 1.46 (d, J=6.8, 3H, CH3 Nap), Molecular formula (molecular weight): C18H20N2O4S (360.4). Calculated analysis: C, 59.98; H, 5.59; N, 7.77; S, 8.90; Found: C, 59.94; H, 5.57; N, 7.75 S, 8.86.
3-hydroxy-2-(3-(2-(6-methoxynaphthalen-2-yl)propanoyl)thioureido)propanoic acid (7)
Yield: 80%; melting point: 108–110 °C, Rf: 0.65 (S), 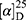 =−30.6 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3452, 3213 broad band (overlapping of OH NH and CHarom.), 2971 (CHali), 1731(CO), 1608 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=11.18 (s, 1H-OH−COOH), 9.12 (d, J=11.6, 2H, NHCS), 7.81 (s,1H, CSNH), 7.69 (td, J=1.6, 1.6, 0.5, 2H, ArHNap), 7.31 (dd, J=7.5, 1.5 2H, ArHNap), 7.14 (td, J=1.6, 1.6, 0.7, 2H,), 6.31(s,1H,OHSer), 3.90 (dt, J=11.6, 7.0, 7.0, 2H (1H, CHSer +1H, CHNap), 3.88 (s, 3H, OCH3), 3.33–3.69 (3H,(m, 2H, CH2Ser),1.47 (d, J=6.8, 3H), Molecular formula (molecular weight): C18H20N2O5S (376.4). Calculated analysis: C, 57.43; H, 5.36; N, 7.44; S, 8.52; Found: C, 57.45; H, 5.32; N, 7.45 S, 8.50.
=−30.6 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3452, 3213 broad band (overlapping of OH NH and CHarom.), 2971 (CHali), 1731(CO), 1608 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=11.18 (s, 1H-OH−COOH), 9.12 (d, J=11.6, 2H, NHCS), 7.81 (s,1H, CSNH), 7.69 (td, J=1.6, 1.6, 0.5, 2H, ArHNap), 7.31 (dd, J=7.5, 1.5 2H, ArHNap), 7.14 (td, J=1.6, 1.6, 0.7, 2H,), 6.31(s,1H,OHSer), 3.90 (dt, J=11.6, 7.0, 7.0, 2H (1H, CHSer +1H, CHNap), 3.88 (s, 3H, OCH3), 3.33–3.69 (3H,(m, 2H, CH2Ser),1.47 (d, J=6.8, 3H), Molecular formula (molecular weight): C18H20N2O5S (376.4). Calculated analysis: C, 57.43; H, 5.36; N, 7.44; S, 8.52; Found: C, 57.45; H, 5.32; N, 7.45 S, 8.50.
Yield: 68%; melting point: 122–124 °C, Rf: 0.67 (S), [α]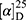 =−19.3 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3207 broad band (overlapping of OH NH and CHarom.), 1718(CO), 1616 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6 δ=12.5(s, 1H, NHCS), 10.12(s, 1H, OH-COOH), 7.80 (d, J=10.8 Hz, 1H, CSNH), 7.77(s,1H,OHTyr), 7.69 (dd, J=7.5, 1.5 Hz, 2H,ArH), 7.68–7.63 (m, 2H, ArH), 7.31 (d, J=7.5 Hz, 2H-Ar-H), 7.13 (d, J=1.4 Hz, 1H-Ar-HTyr), 7.20 (m, J=7.5 Hz, 1H, Ar-HTyr), 7.11 (m, J=7.5, 2H, ArHTyr), 3.80(m, J=6.85 Hz, 1H-CH-Tyr), 3.80(m, J=6.85 Hz, 1H-CH-Nap),3.80(m, J=6.85 Hz, 3H-OCH3-Nap), 3.3 (m, J=6.9 Hz, 2H-CH2-Tyr), 1.4 (d, 6.8 Hz, 3H-CH3−Nap). Molecular formula (molecular weight): C24H24N2O5S (452.5). Calculated analysis: C, 63.70; H, 5.35; N, 6.19; S, 7.09; Found: C, 63.72; H, 5.31; N, 6.19 S, 7.05.
=−19.3 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3207 broad band (overlapping of OH NH and CHarom.), 1718(CO), 1616 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6 δ=12.5(s, 1H, NHCS), 10.12(s, 1H, OH-COOH), 7.80 (d, J=10.8 Hz, 1H, CSNH), 7.77(s,1H,OHTyr), 7.69 (dd, J=7.5, 1.5 Hz, 2H,ArH), 7.68–7.63 (m, 2H, ArH), 7.31 (d, J=7.5 Hz, 2H-Ar-H), 7.13 (d, J=1.4 Hz, 1H-Ar-HTyr), 7.20 (m, J=7.5 Hz, 1H, Ar-HTyr), 7.11 (m, J=7.5, 2H, ArHTyr), 3.80(m, J=6.85 Hz, 1H-CH-Tyr), 3.80(m, J=6.85 Hz, 1H-CH-Nap),3.80(m, J=6.85 Hz, 3H-OCH3-Nap), 3.3 (m, J=6.9 Hz, 2H-CH2-Tyr), 1.4 (d, 6.8 Hz, 3H-CH3−Nap). Molecular formula (molecular weight): C24H24N2O5S (452.5). Calculated analysis: C, 63.70; H, 5.35; N, 6.19; S, 7.09; Found: C, 63.72; H, 5.31; N, 6.19 S, 7.05.
Synthesis of naproxool thioureido–amino acid methyl esters (9–12)
These compounds were synthesized by stirring amino-acid methyl ester hydrochlorides (L-alanine, β-alanine, L-serine, and L-tyrosine) with naproxol isothiocyanate (4) in THF and a few drops of TEA for 3 hours. Progress of the reaction was monitored by TLC. After TEA-HCl had been filtered off, the solvent was removed in vacuum and crude materials purified by recrystallization from ethanol (yields 65%–80%). All synthesized compounds (9–12) were chromatographically homogeneous when developed with iodine solution and benzidine, gave positive hydroxamate reactions, and gave negative ninhydrin tests. Structures of the synthesized compounds (9–12) were confirmed by elemental analysis, chromatographic studies, spot reactions, IR spectra, 1H NMR spectra, and mass spectra. Complete acid hydrolysis of the synthesized compounds (9–12) using 6 N HCl at 110°C for 24 hours gave positive ninhydrin spots of L-alanine, β-alanine, L-serine, and L-tyrosine.
Yield: 79%; melting point: 90–92 °C, Rf: 0.58 (S), 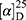 =−28.1 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3445 broad band (NH + CHarm.), 2970 (CHAli), 1732(CO), 1607 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=δ 8.85 (s, 1H, NHCS), 7.80 (d, J=8.6 Hz, 1H,CSNH), 7.69 (dd, J=7.5, 1.5 Hz, 2H,ArH), 7.59 (dd, J=7.6, 1.6 Hz, 2H,ArH), 7.31(t, J=1.6 Hz, 1H, ArH), 7.13 (q, J=1.2, 0.8 Hz, 1H,ArH), 3.9 (s,1H-CH-Ala(, 3.8 (m, J=6.9 Hz, 3H-OCH3Nap), 3.72 (s,3H-OCH3Ala(, 3.70 (q, J=6.8 Hz, 1H-CH-Nap), 1.47 (d, J=6.8 Hz, 3H-CH3Nap), 1.36 (d, J=6.8 Hz, 3H- CHCH3−Ala). Molecular formula (molecular weight): C19H22N2O4S (374.5). Calculated analysis: C, 60.94; H, 5.92; N, 7.48; S, 8.56; Found: C, 60.96; H, 5.88; N, 7.49 S, 8.54.
=−28.1 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3445 broad band (NH + CHarm.), 2970 (CHAli), 1732(CO), 1607 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=δ 8.85 (s, 1H, NHCS), 7.80 (d, J=8.6 Hz, 1H,CSNH), 7.69 (dd, J=7.5, 1.5 Hz, 2H,ArH), 7.59 (dd, J=7.6, 1.6 Hz, 2H,ArH), 7.31(t, J=1.6 Hz, 1H, ArH), 7.13 (q, J=1.2, 0.8 Hz, 1H,ArH), 3.9 (s,1H-CH-Ala(, 3.8 (m, J=6.9 Hz, 3H-OCH3Nap), 3.72 (s,3H-OCH3Ala(, 3.70 (q, J=6.8 Hz, 1H-CH-Nap), 1.47 (d, J=6.8 Hz, 3H-CH3Nap), 1.36 (d, J=6.8 Hz, 3H- CHCH3−Ala). Molecular formula (molecular weight): C19H22N2O4S (374.5). Calculated analysis: C, 60.94; H, 5.92; N, 7.48; S, 8.56; Found: C, 60.96; H, 5.88; N, 7.49 S, 8.54.
Yield: 75%; melting point: 82–84 °C, Rf: 0.67 (S), 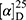 =−35.7 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3450 (NH + CHarm.), 2973 (CHali), 1735(CO), 1612 (CONH) cm–1.; δ=9.18 (s, 1H, NHCS), 9.09 (t, J=5.0, 5.0 Hz, 1H, CSNH), 7.72 (m, J=7.5, 1.5 Hz, - 7.14 (m, J=1.5, Hz, 5H, ArH), 4.24–4.16 (m, 1H, CH Nap.), 3.88 (s, 3H, OCH3), 3.73–3.56 (m, 2H, CH2-β-Ala), 3.63 (s, 3H, CH3Nap), 2.77–2.65 (m, 2H, CH2β-Ala); MS (EI, 70 eV): m/z (%)=374 (32.38%) which is corresponding to the molecular formula and revealed a base peak at m/z 247(100%). Molecular formula (molecular weight) C19H22N2O4S (374.5). Calculated analysis: C, 60.94; H, 5.92; N, 7.48; S, 8.56; Found: C, 60.96; H, 5.88; N, 7.49 S, 8.54.
=−35.7 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3450 (NH + CHarm.), 2973 (CHali), 1735(CO), 1612 (CONH) cm–1.; δ=9.18 (s, 1H, NHCS), 9.09 (t, J=5.0, 5.0 Hz, 1H, CSNH), 7.72 (m, J=7.5, 1.5 Hz, - 7.14 (m, J=1.5, Hz, 5H, ArH), 4.24–4.16 (m, 1H, CH Nap.), 3.88 (s, 3H, OCH3), 3.73–3.56 (m, 2H, CH2-β-Ala), 3.63 (s, 3H, CH3Nap), 2.77–2.65 (m, 2H, CH2β-Ala); MS (EI, 70 eV): m/z (%)=374 (32.38%) which is corresponding to the molecular formula and revealed a base peak at m/z 247(100%). Molecular formula (molecular weight) C19H22N2O4S (374.5). Calculated analysis: C, 60.94; H, 5.92; N, 7.48; S, 8.56; Found: C, 60.96; H, 5.88; N, 7.49 S, 8.54.
Yield: 80%; melting point: 94–96 °C, Rf: 0.66 (S), 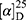 =−20.5 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3451 broad band (NH + OH), 2972 (CHali), 1735(CO), 1610 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=8.50 (d, J=10.9, 2H, NHCS), 7.81(s,1H, CSNH), 7.75(dd, J=7.6, 1.6, 2H, ArH), 7.69 (dd J=7.5, 1.5, 0.5, 2H, ArH), 7.13 (tq, J=1.6, 1.6, 0.5, 0.5, 0.5, 2H, ArH), 4.54 (dt, J=10.9, 7.0, 7.0, 1H, CHSer), 4.44 (t, J=6.7, 6.7, 1H,OH), 3.88 (s, 3H,OCH3Ser), 3.69 (s, 3H,OCH3-Nap), 3.3 (d, J=7.5, 2H-CH2Ser), 1.47 (d, J=6.8, 3H, CH3), Molecular formula (molecular weight): C19H22N2O5S (390.5). Calculated analysis: C, 58.45; H, 5.68; N, 7.17; S, 8.21; Found: C, 58.46; H, 5.64; N, 7.18; S, 8.22.
=−20.5 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3451 broad band (NH + OH), 2972 (CHali), 1735(CO), 1610 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=8.50 (d, J=10.9, 2H, NHCS), 7.81(s,1H, CSNH), 7.75(dd, J=7.6, 1.6, 2H, ArH), 7.69 (dd J=7.5, 1.5, 0.5, 2H, ArH), 7.13 (tq, J=1.6, 1.6, 0.5, 0.5, 0.5, 2H, ArH), 4.54 (dt, J=10.9, 7.0, 7.0, 1H, CHSer), 4.44 (t, J=6.7, 6.7, 1H,OH), 3.88 (s, 3H,OCH3Ser), 3.69 (s, 3H,OCH3-Nap), 3.3 (d, J=7.5, 2H-CH2Ser), 1.47 (d, J=6.8, 3H, CH3), Molecular formula (molecular weight): C19H22N2O5S (390.5). Calculated analysis: C, 58.45; H, 5.68; N, 7.17; S, 8.21; Found: C, 58.46; H, 5.64; N, 7.18; S, 8.22.
Yield: 65%; melting point: 103–105 °C, Rf: 0.68 (S), 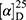 =−23.1 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3449broad band (NH + OH), 2971 (CHali), 1735(CO), 1609 (CONH) cm–1; 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=δ 10.29 (s, 1H, NHCS), 8.30 (d, J=10.2 Hz, 1H, OH-COOH), 7.73–7.11–7.03 (m, 10H,Ar-H), 6.68–6.62 (m, 2H), 4.57 (d, J=10.2, 7.0 Hz, 1H-CH-Tyr), 4.24 (m, 1H, CH-Nap), 3.86 (s, 3H), 3.67 (s, 3H, OCH3Nap.), (s, 3H, OCH3-Tyr), 3.67 (s, 3H), 3.12–3.00 (m, 2H, CH2-Tyr), 1.47 (d, J=6.8 Hz, 3H, CH3−Nap).; MS (EI, 70 eV): m/z (%)=466 (22.3%) which is corresponding to the molecular formula and revealed a base peak at m/z 185 (100%). Molecular formula (molecular weight): C25H26N2O5S (466.5). Calculated analysis: C, 64.36; H, 5.62; N, 6.00; S, 6.87; Found: C, 64.38; H, 5.58; N, 6.01; S, 7.00.
=−23.1 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3449broad band (NH + OH), 2971 (CHali), 1735(CO), 1609 (CONH) cm–1; 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=δ 10.29 (s, 1H, NHCS), 8.30 (d, J=10.2 Hz, 1H, OH-COOH), 7.73–7.11–7.03 (m, 10H,Ar-H), 6.68–6.62 (m, 2H), 4.57 (d, J=10.2, 7.0 Hz, 1H-CH-Tyr), 4.24 (m, 1H, CH-Nap), 3.86 (s, 3H), 3.67 (s, 3H, OCH3Nap.), (s, 3H, OCH3-Tyr), 3.67 (s, 3H), 3.12–3.00 (m, 2H, CH2-Tyr), 1.47 (d, J=6.8 Hz, 3H, CH3−Nap).; MS (EI, 70 eV): m/z (%)=466 (22.3%) which is corresponding to the molecular formula and revealed a base peak at m/z 185 (100%). Molecular formula (molecular weight): C25H26N2O5S (466.5). Calculated analysis: C, 64.36; H, 5.62; N, 6.00; S, 6.87; Found: C, 64.38; H, 5.58; N, 6.01; S, 7.00.
Synthesis of naproxol thioureido–amino acid hydrazides (13–16)
These compounds were synthesized by heating naproxol thioureido–amino acid methyl esters (9–11) with ethanolic hydrazine hydrate solution for 30 minutes. Progress of the reaction was monitored by TLC. The desired materials were filtered off and recrystallized from ethanol (yields 67%–76%). All synthesized compounds (13–16) were chromatographically homogeneous when developed with benzidine and iodine solution, gave positive silver nitrate reactions, and gave negative hydroxamate and ninhydrin reactions. Structures of the synthesized compounds (13–16) were confirmed by elemental analysis, chromatographic studies, spot reactions, IR spectra, 1H NMR spectra, and mass spectra. Complete acid hydrolysis of the synthesized compounds (13–16) using 6 N HCl at 110°C for 24 hours, gave positive ninhydrin spots of L-alanine, β-alanine, L-serine, and L-tyrosine.
Yield: 67%; melting point: 98–100 °C, Rf: 0.71 (S), 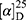 =−28.3 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3360 (NH2, NH), 2970 (CHali), 1734 (CO), 1605 (CONH) cm1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=10.1 (m, J=4.5 Hz, 2H, NHCS), 9.99 (s, 1H, NHNH2),7.78(s, 1H,CSNH), 7.57 (dd, J=7.5, 1.5 Hz, 2H, ArHNap), 7.31 (dd, J=7.6, 1.6 Hz, 2H, ArHNap), 7.31–7.28 (m, 2H, ArHNap), 6.99 (d, J=4.6 Hz, 2H, NH2), 3.95–3.88 (m, 2H,CHNap + Ala), 3.78 (d, J=6.8 Hz, 3H,OCH3), 1.47 (d, J=6.8 Hz, 3H,CH3Nap), 1.31 (d, J=6.8 Hz, 3H, CH3Ala), Molecular formula (molecular weight): C18H22N4O3S (374.5). Calculated analysis: C, 57.73; H, 5.92; N, 14.96; S, 8.56; Found: C, 57.75; H, 5.88; N, 14.97; S, 8.55.
=−28.3 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3360 (NH2, NH), 2970 (CHali), 1734 (CO), 1605 (CONH) cm1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=10.1 (m, J=4.5 Hz, 2H, NHCS), 9.99 (s, 1H, NHNH2),7.78(s, 1H,CSNH), 7.57 (dd, J=7.5, 1.5 Hz, 2H, ArHNap), 7.31 (dd, J=7.6, 1.6 Hz, 2H, ArHNap), 7.31–7.28 (m, 2H, ArHNap), 6.99 (d, J=4.6 Hz, 2H, NH2), 3.95–3.88 (m, 2H,CHNap + Ala), 3.78 (d, J=6.8 Hz, 3H,OCH3), 1.47 (d, J=6.8 Hz, 3H,CH3Nap), 1.31 (d, J=6.8 Hz, 3H, CH3Ala), Molecular formula (molecular weight): C18H22N4O3S (374.5). Calculated analysis: C, 57.73; H, 5.92; N, 14.96; S, 8.56; Found: C, 57.75; H, 5.88; N, 14.97; S, 8.55.
Yield: 72%; melting point: 88–90 °C, Rf: 0.69 (S), 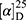 =−26.4 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3430 (NH2, NH), 2970 (CHali), 1734(CO), 1605 (CONH) cm–1. . 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=9.24 (t, J=5.2 Hz, 1H, NHCS), 9.18 (s, 1H, NHNH2), 8.96 (t, J=5.1 Hz, 1H, CSNH), 7.72–7.07 (m,, 7H, 5Ar-H+2H-NH2), 4.24–4.16 (m, 1H,CH-Nap), 4.06 (d, J=5.1 Hz, 2H-CH2 β-Ala), 3.88 (s, 3H- OCH3), 3.63–3.46 (m, 2H,CH2 β-Ala)MS (EI, 70 eV): m/z (%)=374 (20.52%) which is corresponding to the molecular formula and revealed a base peak at m/z 188 (100%),
=−26.4 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3430 (NH2, NH), 2970 (CHali), 1734(CO), 1605 (CONH) cm–1. . 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=9.24 (t, J=5.2 Hz, 1H, NHCS), 9.18 (s, 1H, NHNH2), 8.96 (t, J=5.1 Hz, 1H, CSNH), 7.72–7.07 (m,, 7H, 5Ar-H+2H-NH2), 4.24–4.16 (m, 1H,CH-Nap), 4.06 (d, J=5.1 Hz, 2H-CH2 β-Ala), 3.88 (s, 3H- OCH3), 3.63–3.46 (m, 2H,CH2 β-Ala)MS (EI, 70 eV): m/z (%)=374 (20.52%) which is corresponding to the molecular formula and revealed a base peak at m/z 188 (100%),
Yield: 75%; melting point: 100–102 °C, Rf: 0.75 (S), 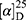 =−34.3 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3453(NH2, NH), 2970 (CHali), 1736 (CO), 1607 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=10.15 (s, 1H, NHCS), 9.93 (s, 1H, NHNH2), 7.78 (s, 1H,CSNH), 7.66–7.56 (m, 2H,ArHNap), 7.34–7.28 (m, 4H, ArHNap), 7.01 (d, J=5.1, 1H, NH2), 4.15(s,1H,OHser) 3.88 (s, 1H,CH-Ser), 3.78 (dd, J=6.8, 0.1, 3H, OCH3), 3.67–3.50 (m, 3H (1H,CHNap) +(2H,CH2Ser), 1.49 (d, J=6.8 Hz, 3H,CH3Nap), Molecular formula (molecular weight): C28H22N4O4S (390.5). Calculated analysis: C, 55.37; H, 5.68; N, 14.35; S, 8.21; Found: C, 55.38; H, 5.64; N, 14.36; S, 8.25.
=−34.3 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3453(NH2, NH), 2970 (CHali), 1736 (CO), 1607 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=10.15 (s, 1H, NHCS), 9.93 (s, 1H, NHNH2), 7.78 (s, 1H,CSNH), 7.66–7.56 (m, 2H,ArHNap), 7.34–7.28 (m, 4H, ArHNap), 7.01 (d, J=5.1, 1H, NH2), 4.15(s,1H,OHser) 3.88 (s, 1H,CH-Ser), 3.78 (dd, J=6.8, 0.1, 3H, OCH3), 3.67–3.50 (m, 3H (1H,CHNap) +(2H,CH2Ser), 1.49 (d, J=6.8 Hz, 3H,CH3Nap), Molecular formula (molecular weight): C28H22N4O4S (390.5). Calculated analysis: C, 55.37; H, 5.68; N, 14.35; S, 8.21; Found: C, 55.38; H, 5.64; N, 14.36; S, 8.25.
Yield: 76%; melting point: 111–113 °C, Rf: 0.81 (S), 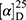 =−17.2 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3448 (NH2, NH), 2970 (CHali), 1734 (CO), 1605 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=12.5 (s, 2H, NHCS), 10.3 (d, J=8.8 Hz, 1H, NHNH2), 7.85(d, J=9.5, 1H, CSNH), 7.76 (d, J=10.2 Hz, 0H), 7.71 (m, 2H-ArHNap), 7.31 (dd, J=7.5, 1.5 Hz, 2H,ArHNap), 7.25–7.20 (m, 2H, ArHNap), 7.13–7.09 (m, 5H, Ar-HTyr), 6.99 (s, 2H, NH2), 4.08 (d, J=5.1 Hz, 1H,CHTyr), 3.93–3.90 (m,1H,CHCH3-Nap (3.86 (s, 3H,OCH3), 3.30 (ddt, J=12.4, 7.0, 1.0, 1.0 Hz, 2H,CH2Tyr), 1.47–1.44 (m, d, J=6.8 Hz, 3H,CH3Nap)., Molecular formula (molecular weight): C24H26N4O4S (466.6). Calculated analysis: C, 61.78; H, 5.62; N, 12.01; S, 6.87; Found: C, 61.80; H, 5.58; N, 12.02; S, 6.80.
=−17.2 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3448 (NH2, NH), 2970 (CHali), 1734 (CO), 1605 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=12.5 (s, 2H, NHCS), 10.3 (d, J=8.8 Hz, 1H, NHNH2), 7.85(d, J=9.5, 1H, CSNH), 7.76 (d, J=10.2 Hz, 0H), 7.71 (m, 2H-ArHNap), 7.31 (dd, J=7.5, 1.5 Hz, 2H,ArHNap), 7.25–7.20 (m, 2H, ArHNap), 7.13–7.09 (m, 5H, Ar-HTyr), 6.99 (s, 2H, NH2), 4.08 (d, J=5.1 Hz, 1H,CHTyr), 3.93–3.90 (m,1H,CHCH3-Nap (3.86 (s, 3H,OCH3), 3.30 (ddt, J=12.4, 7.0, 1.0, 1.0 Hz, 2H,CH2Tyr), 1.47–1.44 (m, d, J=6.8 Hz, 3H,CH3Nap)., Molecular formula (molecular weight): C24H26N4O4S (466.6). Calculated analysis: C, 61.78; H, 5.62; N, 12.01; S, 6.87; Found: C, 61.80; H, 5.58; N, 12.02; S, 6.80.
Synthesis of naproxol thioureido–N-phthalimido amino-acid hydrazides (17–20)
These compounds were synthesized by fusing naproxool thioureido–amino acid hydrazides (13–16) with phthalic anhydride in an oil bath for 15 minutes. The solid obtained was recrystallized from ethanol–water to give the desired compounds (yields 62%–72%). All synthesized compounds (17–20) were chromatographically homogeneous when developed with benzidine and iodine solution, gave positive silver nitrate reactions, and gave negative hydroxamate and ninhydrin reactions. Structures of the synthesized compounds (17–20) were confirmed by elemental analysis, chromatographic studies, spot reactions, IR spectra, 1H NMR spectra, and mass spectra. Complete acid hydrolysis of the synthesized compounds (17–20) using 6 N HCl at 110°C for 24 hours gave positive ninhydrin spots of L-alanine, β-alanine, L-serine, and L-tyrosine.
Yield: 62%; melting point: 110–112 °C, Rf: 0.81 (S), 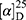 =−17.3 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3455 (NH), 3323(CHArm.), 2971 (CHali), 1735 (CO), 1607 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=10.06 (s, 1H, NH-NPht), 8.60 (d, 2H, J=7.6 Hz, 2H, NHCSNH-Ala), 7.83(d, J=0.8, 4H, ArHPht.), 7.61 (dd, J=7.5, 1.5 Hz, 2H, ArHNap), 7.31–7.12 (m, 4H, ArHNap), 4.39 (m, 1H, q, J=6.9, CH-Ala), 3.90 (dd, J=6.8, 6.8, 1H,CHNap), 3.85 (s, 3H-OCH3-Nap), 1.4)m, J=6.8 Hz, 3H-CH3−Nap),1.33 (q, J=7.5 Hz, 3H-CH3-Ala). Molecular formula (molecular weight): C26H24N4O5S (504.6). Calculated analysis: C, 61.89; H, 4.79; N, 11.10; S, 6.36; Found: C, 61.90; H, 4.76; N, 11.11; S, 6.34.
=−17.3 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3455 (NH), 3323(CHArm.), 2971 (CHali), 1735 (CO), 1607 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=10.06 (s, 1H, NH-NPht), 8.60 (d, 2H, J=7.6 Hz, 2H, NHCSNH-Ala), 7.83(d, J=0.8, 4H, ArHPht.), 7.61 (dd, J=7.5, 1.5 Hz, 2H, ArHNap), 7.31–7.12 (m, 4H, ArHNap), 4.39 (m, 1H, q, J=6.9, CH-Ala), 3.90 (dd, J=6.8, 6.8, 1H,CHNap), 3.85 (s, 3H-OCH3-Nap), 1.4)m, J=6.8 Hz, 3H-CH3−Nap),1.33 (q, J=7.5 Hz, 3H-CH3-Ala). Molecular formula (molecular weight): C26H24N4O5S (504.6). Calculated analysis: C, 61.89; H, 4.79; N, 11.10; S, 6.36; Found: C, 61.90; H, 4.76; N, 11.11; S, 6.34.
Yield: 68%; melting point: 115–117 °C, Rf: 0.85 (S), 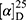 =−19.9 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3619 (NH), 3452(CHArm.), 2971 (CHali), 1734(CO), 1607 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=10.29 (s, 1H, NH-NPht), 8.63 (d, 2H, J=9.5 Hz, 2H, NHCSNH- β-Ala), 7.81–7.71 (m, 4H-ArHPht.), 7.46 (dd, J=7.5, 1.5 Hz, 2H, ArHNap), 7.33–7.13 (m, 4H, ArHNap), 4.34–4.26 (m, 1H,CHNap), 3.83 (s, 3H,OCH3), 3.29–3.18 (4H, CH2CH2B-Ala), 1.48–1.43 (m, 3H, CH3Nap), Molecular formula (molecular weight): C26H24N4O5S (504.6). Calculated analysis: C, 61.89; H, 4.79; N, 11.10; S, 6.36; Found: C, 61.90; H, 4.76; N, 11.11; S, 6.35.
=−19.9 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3619 (NH), 3452(CHArm.), 2971 (CHali), 1734(CO), 1607 (CONH) cm–1. 1H-NMR (500 MHz, δ, ppm, DMSO-d6): δ=10.29 (s, 1H, NH-NPht), 8.63 (d, 2H, J=9.5 Hz, 2H, NHCSNH- β-Ala), 7.81–7.71 (m, 4H-ArHPht.), 7.46 (dd, J=7.5, 1.5 Hz, 2H, ArHNap), 7.33–7.13 (m, 4H, ArHNap), 4.34–4.26 (m, 1H,CHNap), 3.83 (s, 3H,OCH3), 3.29–3.18 (4H, CH2CH2B-Ala), 1.48–1.43 (m, 3H, CH3Nap), Molecular formula (molecular weight): C26H24N4O5S (504.6). Calculated analysis: C, 61.89; H, 4.79; N, 11.10; S, 6.36; Found: C, 61.90; H, 4.76; N, 11.11; S, 6.35.
Yield: 72%; melting point: 107–109 °C, Rf: 0.88 (S), 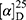 =−22.7 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3451 (OH, NH), 3200(CH arom.), 2970 (CHali), 1735 (CO), 1607 (CONH) cm–1, 1H NMR (500 MHz, Chloroform-d) δ=10.94 (s, 1H, NHNPht), 10.29 (s, 1H, NHCS), 8.72 (d, J=9.5 Hz, 1H, CSNHSer), 7.92–7.84 (m, 4H-ArHPht.), 7.33 (dd, J=7.5, 1.5 Hz, 2H, ArHNap), 7.16–7.13 (m, 4H, ArHNap) 4.83–4.76 (m, 1H,CHSer), 3.83 (s, 3H,OCH3), 3.69–3.61 (m, 1H, CHNap.), 1.48–1.43 (m, 3H, CH3Nap), MS (EI, 70 eV): m/z (%)=522 (46.52%) which is corresponding to the molecular formula and revealed a base peak at m/z 185 (100%). Molecular formula (molecular weight): C26H24N4O6S (520.6). Calculated analysis: C, 59.99; H, 4.65; N, 10.76; S, 6.16; Found: C, 60.00; H, 4.63; N, 10.77; S, 6.18.
=−22.7 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3451 (OH, NH), 3200(CH arom.), 2970 (CHali), 1735 (CO), 1607 (CONH) cm–1, 1H NMR (500 MHz, Chloroform-d) δ=10.94 (s, 1H, NHNPht), 10.29 (s, 1H, NHCS), 8.72 (d, J=9.5 Hz, 1H, CSNHSer), 7.92–7.84 (m, 4H-ArHPht.), 7.33 (dd, J=7.5, 1.5 Hz, 2H, ArHNap), 7.16–7.13 (m, 4H, ArHNap) 4.83–4.76 (m, 1H,CHSer), 3.83 (s, 3H,OCH3), 3.69–3.61 (m, 1H, CHNap.), 1.48–1.43 (m, 3H, CH3Nap), MS (EI, 70 eV): m/z (%)=522 (46.52%) which is corresponding to the molecular formula and revealed a base peak at m/z 185 (100%). Molecular formula (molecular weight): C26H24N4O6S (520.6). Calculated analysis: C, 59.99; H, 4.65; N, 10.76; S, 6.16; Found: C, 60.00; H, 4.63; N, 10.77; S, 6.18.
Yield: 69%; melting point: 120–122 °C, Rf: 0.89 (S), 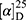 =−22.1 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3443 (OH, NH), 2970 (CHali), 1732 (CO), 1606 (CONH) cm–1, 1H NMR (500 MHz, Chloroform-d) δ=10.29 (s, 1H, NHNPht), 9.99 (s, 1H, NHCS), 8.63 (d, J=8.8 Hz, 1H, CSNH), 7.92–7.83 (m, 4H, ArHPth), 7.74–7.66 (m, 2H, ArHNap), 7.61 (m, J=0.8 Hz, 1H,ArHNap), 7.56 (s, 1H, OH-Tyr, which disappeared by D2O), 7.33 (dd, J=7.4, 1.5 Hz, 2H, ArHNap), 7.14 (q, J=1.1 Hz, 2H, ArHNap), 7.09 (d, J=7.6, 7.5 Hz, 4H- ArHTyr), 4.52 (t, J=7.0 Hz, 1H, CHTyr), 4.20 (q, J=6.9 Hz, 1H, CHNap), 3.83 (s, 3H,OCH3), 3.03 (d, J=7.0, 1.2 Hz, 2H, CH2Tyr), 1.45 (d, J=6.9 Hz, 3H-Naproxine),MS (EI, 70 eV): m/z (%)=596 (32.71%) which is corresponding to the molecular formula and revealed a base peak at m/z 185 (100%). Molecular formula (molecular weight): C32H28N4O6S (596.7). Calculated analysis: C, 64.42; H, 4.73; N, 9.39; S, 5.37; Found: C, 65.08; H, 4.75; N, 9.49 S, 5.31.
=−22.1 (C=0.04, MeOH); IR (cm–1): (KBr):ν=3443 (OH, NH), 2970 (CHali), 1732 (CO), 1606 (CONH) cm–1, 1H NMR (500 MHz, Chloroform-d) δ=10.29 (s, 1H, NHNPht), 9.99 (s, 1H, NHCS), 8.63 (d, J=8.8 Hz, 1H, CSNH), 7.92–7.83 (m, 4H, ArHPth), 7.74–7.66 (m, 2H, ArHNap), 7.61 (m, J=0.8 Hz, 1H,ArHNap), 7.56 (s, 1H, OH-Tyr, which disappeared by D2O), 7.33 (dd, J=7.4, 1.5 Hz, 2H, ArHNap), 7.14 (q, J=1.1 Hz, 2H, ArHNap), 7.09 (d, J=7.6, 7.5 Hz, 4H- ArHTyr), 4.52 (t, J=7.0 Hz, 1H, CHTyr), 4.20 (q, J=6.9 Hz, 1H, CHNap), 3.83 (s, 3H,OCH3), 3.03 (d, J=7.0, 1.2 Hz, 2H, CH2Tyr), 1.45 (d, J=6.9 Hz, 3H-Naproxine),MS (EI, 70 eV): m/z (%)=596 (32.71%) which is corresponding to the molecular formula and revealed a base peak at m/z 185 (100%). Molecular formula (molecular weight): C32H28N4O6S (596.7). Calculated analysis: C, 64.42; H, 4.73; N, 9.39; S, 5.37; Found: C, 65.08; H, 4.75; N, 9.49 S, 5.31.
Computational model
All quantum chemical computations were performed using the PM3 semiempirical Hamiltonian MO calculation MOPAC16 package 27, as implemented in the MOE 2015 package. Optimization geometry for molecular structures was carried out to improve knowledge of chemical structures. Global chemical reactivity was computed for molecules (Table 1): S, softness (measures stability of molecule, which is directly proportional to chemical reactivity); η, hardness (reciprocal of softness); μ, chemical potential; χ, electronegativity (catching electron strength); μ−, electron-donating potency; μ+; electroacceptance potency; ω−, electron-donating capacity; ω+; electron-acceptance capacity; ω±, net electrophilicity (measuring relative potential powers between electron acceptance and electron donation); ωi, electrophilicity index in ground state (determines decreasing energy obtained from maximal movement of electrons current between donor and acceptor); ωiVS, electrophilicity index in valence state; ωi, and second electrophile energy (if molecule saturated with electrons).44 These parameters are represented in ionization-potential and electron-affinity terms: I, ionization potential is total energy variance, when an electron is lost (k−1) from parent molecule (k) electrons; and A, electron affinity determining acceptance of the electron (k+1) in the same conditions. υ(r) is external potential of an N-electron system,45 and the other terms are represented in equations
Docking study
Docking studies were carried out for the target compounds into EGFR using MOE 2015, AutoDock Vina, MVD,46 and PLANTS.47 Crystal structures of COX2 complexes with naproxen (1PXX33) were obtained prepared using MOE 2015. Water and inhibitor molecules were removed and hydrogen atoms added. Parameters and charges were assigned with an MMFF94× force field. We defined active sites based on the original ligand in the crystal using the site-finder module of MOE. Optimized 3-D structures of molecules were subjected to generate different poses of ligands using triangular matcher placement, which generates poses by aligning ligand triplets of atoms on triplets of α-spheres represented in receptor site points. A random triplet of α-sphere centers was used to determine the pose during each iteration. The pose generated was rescored using the London dG scoring function. Docked poses were clustered using two methods. First, analysis of protein–lipid interaction by fluorescence was carried out with MOE and four bits extracted. Cluster analysis was performed with the k-means algorithm.18 Second, MOE was used for cluster analysis of ligand poses on the basis of the RMSDs of the compounds‘ atomic coordinates. Then, representative structures were selected by comparison of the docking scores (generalized Born volume integral/weighted surface area ΔG) of the poses in each cluster. The structures selected by this process were used as initial structures in the subsequent MD simulations. The poses generated were refined with the MMFF94× force field and solvation effects treated. The Born solvation model (generalized Born/volume integral) was used to calculate final energy, and finally assigned poses were assigned a score based on free energy (kcal/mol). The results were analyzed with Discovery Studio 2017 software.35
ADMET predictions
We used ADMET profiles for in silico identification of ADMET. MOE and ADMET SAR tools were used for prediction of pharmacokinetic parameters.
Pharmacological activities
Pharmacological activities for the newly synthesized compounds were estimated in rats (130–150 g). In a preliminary test to choose the dose for biological testing, animals in groups of six rats for each group received 0.2 mMol/100 g orally in DMSO for the tested compounds and 0.1 mMol/100 g orally in DMSO for naproxen. Animals were observed for 24 hours for signs of toxicity and number of deaths. No deaths were recorded, and there were no observed signs of distress, dyspnea, impaired movement, seizures, or any other abnormal clinical signs.
Anti-inflammatory activity
Anti-inflammatory effects of the newly synthesized compounds were evaluated using a carrageenan-induced rat-paw-edema assay Edema was induced in the left hind paw of all rats by subcutaneous injection of 0.1 mL 1% (w:v) carrageenan in distilled water into their footpads. Rats were divided into 18 groups of six rats each. The first group was assigned as the control and orally given a matching volumes of solvent (1% DMSO). The other groups were orally administered the tested drugs in doses of 5 mg/kg and 2.5 mg/kg naproxen dissolved in 1% DMSO 1 hour before carragennan injection. Paw volumes were measured using plethysmometry (Ugo Basile, Italy) 1, 2, 3 and 4 hours after injection of 1% carragennan. Edema rate and inhibition rate were measured at the sameintervals, in accordance with Khalifa et al:42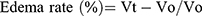
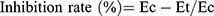
where Vo is the volume before carragennan injection (mL), Vt the volume at t hours after carrageenin injection (mL), Ec the edema rate of the control group, and Et the edema rate of the treated groupv
Analgesic activity
Analgesic validation of the tested compounds was evaluated using tail-flick test.3 Latency of the tail-withdrawal reflex was measured 60 minutes following drug administration. Rats were divided into groups of six animals each. Group 1 was kept as control and received vehicle only, while group 2 received pure naproxen (2.5 mg/kg orally) in DMSO. Groups 3–18 received 5 mg/kg orally of the tested compounds in DMSO. Rats were gently held with the tail put on the tail-flick apparatus (Ugo Basile) and the tail-flick response elicited by applying a radiant-heat stimulus to the ventral surface of the tail about 3–4 cm from the tip. The time in seconds from initial heat-source activation until tail withdrawal was recorded. The mean of two measures was used for each experimental animal as tail-withdrawal latency. In order to avoid excessive suffering of animals, a cutoff was set at 30 seconds.
Acute ulcerogenesis
This study was carried out on healthy albino rats. Animals were divided into groups of six each group 1 served as control and received vehicle only, group 2 received pure naproxen 2.5 mg/kg orally in DMSO, and the other 16 groups received 5 mg/g orally of the tested compounds in DMSO. Food but not water was removed 24 hours prior to administration of the tested compounds. Rats were fed a normal diet for 17 hours and then killed after drug treatment. Stomachs were removed and opened along the greater curvature, washed with distilled water, and cleaned gently by dipping in saline. Mucosal damage was examined by magnifying glass. For each stomach, mucosal damage was assessed macroscopically.
Statistical analysis
Results were compared to untreated and standard groups and analyzed using one-way ANOVA followed by Dunnett’s multiple comparisons using SPSS 17.0 and expressed as means ± SE.
Conclusion
The present work aimed to synthesi some novel naproxl thiourea nucle. Synthesized compounds were characterized by different spectral data. Chemical reactivity analysis wasperformed, which introduced a possible explanation for reactivity of ligands against receptors. Data obtained indicated that the carboxyl (5–8) and phthalyl (17–20) derivatives possessed lower electrophilicity than methoxy (9–12) and hydrazide (13–16) members. Th, these compounds may attack the hydrophilic part the receptor. Molecular docking showed that the presence of tyrosine residues (8,12, and 16) resulted in the highest MOE scores, while introduction of β-amino acid to the parent compound resulted in the lowest binding energies. In addition, he presence of serine and alanine residues resulted in moderate binding potency. Binding-interaction strength was free acids (5–8) > methyl esters (9–12) > hydrazide derivatives (13–16). Also, binding interactions increased with increasing hydrophobicity of ligands. The ADMET profile in silico showed that these compounds probably have good oral bioavailability without any carcinogenesis effect or marked health effects via rodent toxicity profiles. Compounds (5–16) passed through docking and ADMET profiles were examined as anti-inflammatory and analgesic agents. Compounds 8 and 16 showed higher anti-inflammatory potency than reference drug and tested compounds. Compounds 8, 10,and 14 exhibited the highest analgesic potency compared to other tested compounds. All compounds showed negligible ulcerogenic effect, and may be considered safer drugs than naproxen for treating inflammatory conditions.
 | Chart 1 Design of initial hit molecules (5–8) compared with naproxen structure. |
 | Chart 2 Design of synthesized compounds (5–20) derived from naproxen. |
 | Scheme 1 Synthetic route for naproxoylisothiocyanate (4). |
 | Scheme 2 Synthetic routes for compounds 5–8. |
 | Scheme 3 Synthetic routes for compounds 9–20. |
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tripathi K. Drugs for cough and bronchial asthma. In: Tripathi KD Essential of Medical Pharmacology.
2. Rodrigues MR, Lanzarini CM, Ricci-Junior E. Preparation, in vitro characterization and in vivo release of naproxen loaded in poly-caprolactone nanoparticles. Pharm Dev Technol. 2011;16(1):12–21. doi:10.3109/10837450903460475
3. Husain A. Amide Derivatives of Sulfonamides and Isoniazid: Synthesis and Biological Evaluationü. Acta Poloniae Pharmaceutica-Drug Research. 2009;66(5):513–521.
4. Deshmukh GB, Deshmukh NS, Gaikwad VB, Bhole AD, Patil SV. Synthesis of substituted N-aryl pyrollo-quinolines and study of their antimicrobial activities. Journal of Chemical and Pharmaceutical Research. 2014;6(8):393–399.
5. Katritzky AR, Jishkariani D, Narindoshvili T. Convenient synthesis of ibuprofen and naproxen aminoacyl, dipeptidoyl and ester derivatives. Chem Biol Drug Des. 2009;73(6):618–626. doi:10.1111/j.1747-0285.2009.00811.x
6. Zhao X, Tao X, Wei D, Song Q. Pharmacological activity and hydrolysis behavior of novel ibuprofen glucopyranoside conjugates. Eur J Med Chem. 2006;41(11):1352–1358. doi:10.1016/j.ejmech.2006.05.014
7. Mahfouz NM, Omar FA, Aboul-Fadl T. Cyclic amide derivatives as potential prodrugs II: N-hydroxymethylsuccinimide-/isatin esters of some NSAIDs as prodrugs with an improved therapeutic index. Eur J Med Chem. 1999;34(7–8):551–562.
8. Ammar Y, Salem M, Fayed EA, Helal M, El-Gaby M, Thabet HK. Naproxen derivatives: synthesis, reactions, and biological applications. Synth Commun. 2017;47(15):1341–1367. doi:10.1080/00397911.2017.1328066
9. Ranatunge RR, Augustyniak ME, Dhawan V, et al. Synthesis and anti-inflammatory activity of a series of N-substituted naproxen glycolamides: nitric oxide-donor naproxen prodrugs. Bioorg Med Chem. 2006;14(8):2589–2599. doi:10.1016/j.bmc.2005.11.040
10. Manjunathaiah Raghavendra N, Ramakrishna K, Sirisha V, Divya P, Venkateswara Rao A. Computer aided discovery of potential anti-inflammatory (s)-naproxen analogs as COX-2 inhibitors. Med Chem (Los Angeles). 2013;9(4):553–559.
11. Fernandes J, Kumar A, Kumar P, Singh S, Raju B, Mahaveer J. Synthesis, Anti-Inflammatory and Antimicrobial Activity of Some Novel Carboxamide Derivatives of Naproxen. 2014;3(2):2026–2034, 2014.
12. Helal MH, Abbas SY, Salem MA, Farag AA, Ammar YA. Synthesis and characterization of new types of 2-(6-methoxy-2-naphthyl) propionamide derivatives as potential antibacterial and antifungal agents. Med Chem Res. 2013;22(11):5598–5609. doi:10.1007/s00044-013-0524-5
13. Piffar P, Fernandez R, Tchaikovski O
14. Chen PC, Patil V, Guerrant W, Green P, Oyelere AK. Synthesis and structure–activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorg Med Chem. 2008;16(9):4839–4853. doi:10.1016/j.bmc.2008.03.050
15. Khalifa MM, Ismail MM, Eissa S, Ammar Y. Design and synthesis, of some novel 6-methoxynaphthalene derivatives with potential anti-cancer activity. Der Pharma Chem. 2012;4(4):1552–1566.
16. Spadaro A, Bucolo C, Ronsisvalle G, Pappalardo M. Design, synthesis and antiinflammatory activity of novel γ-tocopherol naproxen ester prodrug. J Pharm Sci Res. 2009;1(4):88–95.
17. Elhenawy AA, El-Gazzar MAE-G, Mohmmed HM. Synthesis, anti-infammatory, analgesic, molecular modeling and ADMET studies of novel diclofenac derivatives containing alanyl moiety. Chem Mater Res. 2014;6(2):69–77.
18. Kadah MS, Abd-Elghany RD, Hassanein MK. Design and synthesis of novel NSAIDs class acting as anticancer agents. Int J Sci. 2016;30(4):32–52.
19. Elhenawy A, Kadh MS, Radwan A, Abd-Elghany RD, Hassanein M. Design, synthesis and discovery potent of novel anticancer agents based on the coumarin scaffold. J Chem Pharm Res. 2015;7(12):1056–1066.
20. Moustafa G, Khalaf H, Naglah A, Al-Wasidi A, Al-Jafshar N, Awad H. Synthesis, molecular docking studies, in vitro antimicrobial and antifungal activities of novel dipeptide derivatives based on N-(2-(2-hydrazinyl-2-oxoethylamino)-2-oxoethyl)-nicotinamide. Molecules. 2018;23(4):761. doi:10.3390/molecules23040761
21. Kimmerlin T, Seebach D. ‘100 years of peptide synthesis’: ligation methods for peptide and protein synthesis with applications to β‐peptide assemblies. J peptide res. 2005;65(2):229–260. doi:10.1111/j.1399-3011.2005.00214.x
22. Monbaliu J-CM, Katritzky AR. Recent trends in Cys-and Ser/Thr-based synthetic strategies for the elaboration of peptide constructs. Chem Commun. 2012;48(95):11601–11622. doi:10.1039/c2cc34434c
23. Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discovery. 2012;11(1):37. doi:10.1038/nrd3591
24. Castanho M, Santos N. Peptide Drug Discovery and Development: Translational Research in Academia and Industry. John Wiley & Sons; 2011.
25. Gademann K, Kimmerlin T, Hoyer D, Seebach D. Peptide folding induces high and selective affinity of a linear and small β-peptide to the human somatostatin receptor 4. J Med Chem. 2001;44(15):2460–2468.
26. Stewart JJ. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J Mol Model. 2013;19(1):1–32. doi:10.1007/s00894-012-1667-x
27. Molecular Operating Environment (MOE) CCGU, 1010 Handbook. St. West, Suite 910, Montreal, QC, Canada, H3A 2R7; 2017.
28. Hofmann P, Rauk A. Orbital interaction theory of organic chemistry. New York: Wiley & Sons; 1994. ISBN 0-471-59389-3. 307 Seiten, mit HMO-Programmdiskette, Preis: $45.50. Berichte der Bunsengesellschaft für physikalische Chemie. 1995;99(8):997–999.
29. Fukui K. Role of frontier orbitals in chemical reactions. Science. 1982;218(4574):747–754. doi:10.1126/science.218.4574.747
30. Hochgesang GP, Rowlinson SW, Marnett LJ. Tyrosine-385 is critical for acetylation of cyclooxygenase-2 by aspirin. J Am Chem Soc. 2000;122(27):6514–6515. doi:10.1021/ja0003932
31. Kiefer JR, Pawlitz JL, Moreland KT, Stegeman RK. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature. 2000;405(6782):97. doi:10.1038/35011103
32. Rowlinson SW, Kiefer JR, Prusakiewicz JJ, et al. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J Biol Chem. 2003;278(46):45763–45769. doi:10.1074/jbc.M305481200
33. Kieffer J, Brémond É, Lienard P, Boccardi G. In silico assessment of drug substances chemical stability. J Mol Struct. 2010;954(1):75–79. doi:10.1016/j.theochem.2010.03.032
34. Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17(5–6):490–519. doi:10.1002/(SICI)1096-987X(199604)17:5/6<>1.0.CO;2-C
35. Biovia DS. Discovery Studio Modeling Environment, Release 2017. San Diego: Dassault Systèmes; 2017.
36. Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105. doi:10.1021/ci300367a
37. Larson ER, Lipinski CA, Sarges R. Medicinal chemistry of aldose reductase inhibitors. Med Res Rev. 1988;8(2):159–186.
38. Clark DE, Pickett SD. Computational methods for the prediction of ‘drug-likeness’. Drug Discov Today. 2000;5(2):49–58.
39. Zhang R, Du B, Sun G, Sun Y. Experimental and theoretical studies on o-, m- and p-chlorobenzylideneaminoantipyrines. Spectrochim Acta A Mol Biomol Spectrosc. 2010;75(3):1115–1124. doi:10.1016/j.saa.2009.12.067
40. Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights. 2013;7:27–34.
41. Witchel HJ, Hancox JC. Familial and acquired long QT syndrome and the cardiac rapid delayed rectifier potassium current. Clin Exp Pharmacol Physiol. 2000;27(10):753–766.
42. Khalifa NM, Al-Omar MA, Amr AE-GE, Baiuomy AR, Abdel-Rahman RF. Synthesis and biological evaluation of some novel fused thiazolo [3, 2-a] pyrimidines as potential analgesic and anti-inflammatory agents. Rus J Biorganic Chem. 2015;41(2):192–200. doi:10.1134/S1068162015020090
43. Lim YJ, Lee JS, Ku YS, Hahm KB. Rescue strategies against non-steroidal anti-inflammatory drug-induced gastroduodenal damage. J Gastroenterol Hepatol. 2009;24(7):1169–1178. doi:10.1111/j.1440-1746.2009.05929.x
44. Ali IO, Salama TM, Bakr MF, El‐Henawy AA, Lateef MA, Guma HA. Synthesis of nano-sized zeolite-Y functionalized with 5-amino-3-thiomethyl 1H-pyrazole-4-carbonitrile for effective Fe (III)-chelating strategy. Res Chem Intermed. 44(9):5193–5222.
45. Lamaka SV, Zheludkevich ML, Yasakau KA, Serra R, Poznyak S, Ferreira M. Nanoporous titania interlayer as reservoir of corrosion inhibitors for coatings with self-healing ability. Prog Org Coat. 2007;58(2):127–135. doi:10.1016/j.porgcoat.2006.08.029
46. Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49(11):3315–3321. doi:10.1021/jm051197e
47. Korb O, Stutzle T, Exner TE. Empirical scoring functions for advanced protein− ligand docking with PLANTS. J Chem Inf Model. 2009;49(1):84–96. doi:10.1021/ci800298z
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.