Back to Journals » Drug Design, Development and Therapy » Volume 10
Synthesis, anti-inflammatory, analgesic, COX1/2-inhibitory activity, and molecular docking studies of hybrid pyrazole analogues
Authors Alam MJ, Alam O, Khan SA, Naim MJ, Islamuddin M, Deora GS
Received 27 July 2016
Accepted for publication 16 September 2016
Published 31 October 2016 Volume 2016:10 Pages 3529—3543
DOI https://doi.org/10.2147/DDDT.S118297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Md Jahangir Alam,1 Ozair Alam,1 Suroor Ahmad Khan,1 Mohd Javed Naim,1 Mohammad Islamuddin,2 Girdhar Singh Deora3
1Department of Pharmaceutical Chemistry, Faculty of Pharmacy, 2Parasite Immunology Laboratory, Department of Biotechnology, Faculty of Science, Jamia Hamdard, New Delhi, 3Institute of Life Sciences, University of Hyderabad, Hyderabad, India
Abstract: This article reports on the design, synthesis, and pharmacological activity of a new series of hybrid pyrazole analogues: 5a–5u. Among the series 5a–5u, the compounds 5u and 5s exhibited potent anti-inflammatory activity of 80.63% and 78.09% and inhibition of 80.87% and 76.56% compared with the standard drug ibuprofen, which showed 81.32% and 79.23% inhibition after 3 and 4 hours, respectively. On the basis of in vivo studies, 12 compounds were selected for assessment of their in vitro inhibitory action against COX1/2 and TNFα. The compounds 5u and 5s showed high COX2-inhibitory activity, with half-maximal inhibitory concentrations of 1.79 and 2.51 µM and selectivity index values of 72.73 and 65.75, respectively, comparable to celecoxib (selectivity index =78.06). These selected compounds were also tested for TNFα, cytotoxicity, and ulcerogenicity. Docking studies were also carried out to determine possible interactions of the potent compounds (5u and 5s), which also showed high docking scores of -12.907 and -12.24 compared to celecoxib, with a -9.924 docking score. These selective COX2 inhibitors were docked into the active site of COX2, and showed the same orientation and binding mode to that of celecoxib (selective COX2 inhibitor). Docking studies also showed that the SO2NH2 of 5u and 5s is inserted deep inside the selective pocket of the COX2-active site and formed a hydrogen-bond interaction with His90, Arg513, Phe518, Ser353, Gln192, and Ile517, which was further validated by superimposed docked pose with celecoxib.
Keywords: anti-inflammatory activity, analgesic activity, selective COX2 inhibition, TNFα inhibition, molecular docking studies, pyrazole
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the most widely used therapeutic agents for the management of pain and inflammation.1 NSAIDs exert their pharmacological action by inhibiting COX enzymes, which catalyses the conversion of arachidonic acid to prostaglandins, prostacyclins, and thromboxanes.2 Two different isoforms of COX exist (COX1 [constitutive] and COX2 [inducible]), which are assumed to play a vital role in producing pathological and physiological prostaglandins, respectively.3 Currently used NSAIDs employ their anti-inflammatory action mostly by inhibition of both isoforms of COX.4–6 However, absence of selectivity and their long-term clinical use leads to various adverse effects, such as gastric ulceration, gastrointestinal bleeding, cardiovascular toxicity, and nephrotoxicity, which are mainly associated with most of the reported nonselective NSAIDs, due to their inhibition of COX1-mediated physiological prostaglandins.7–9 Similarly, NFκB also plays a key role by activating the inflammatory response and releasing proinflammatory cytokines and enzymes, such as TNFα, IL-1β, COX2, and iNOS.10,11 Improper regulation of NFκB is directly linked to inflammation, together with improper immunodevelopment.12–14
Pyrazole, a key heterocyclic scaffold in medicinal chemistry, possess an array of various biological activities, such as antitumor,15 anticancer,16,17 antiviral,18 anticonvulsant,19 antibacterial,20 and antifungal21 activity. The rationale for evaluating pyrazole analogues (5a–5u) as anti-inflammatory, analgesic, and COX inhibitors is based on successful marketed drugs containing pyrazole as the central core (celecoxib, SC558, deracoxib, SC560, metamizole, antipyrine, SC581, and lonazolac) (Figure 1). The design of NSAIDs with higher therapeutic value and lower risk could be achieved by increasing specificity toward COX2 rather than COX1.22–24 In the present study, the central pyrazole core linked with the phenylsulfonamide group of selective COX2 inhibitors (celecoxib and SC558) was employed, and structural modification led to the design of new pyrazole analogues (Figure 2).
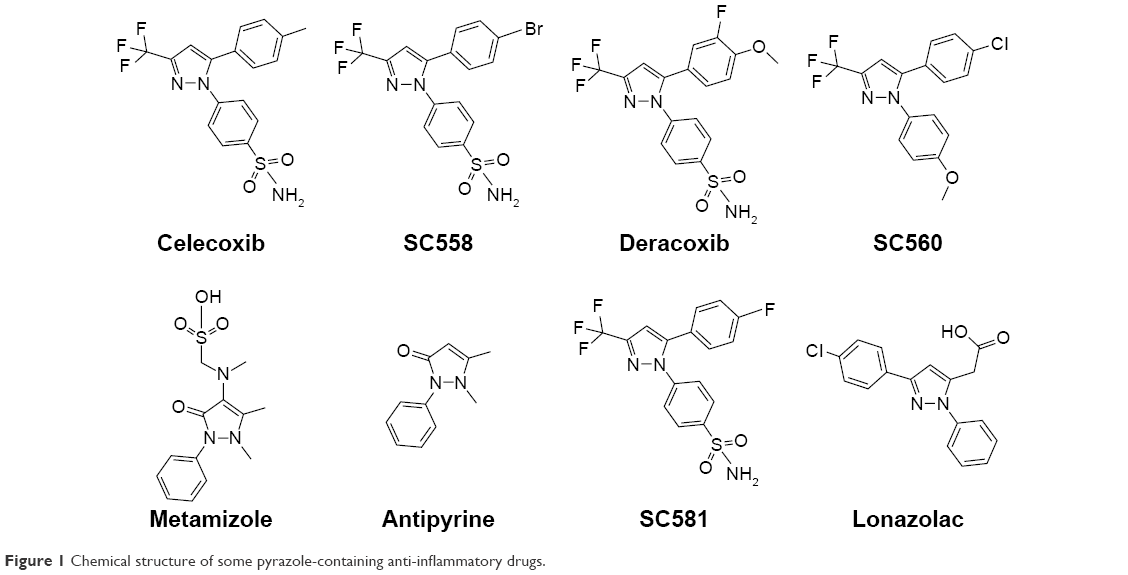 | Figure 1 Chemical structure of some pyrazole-containing anti-inflammatory drugs. |
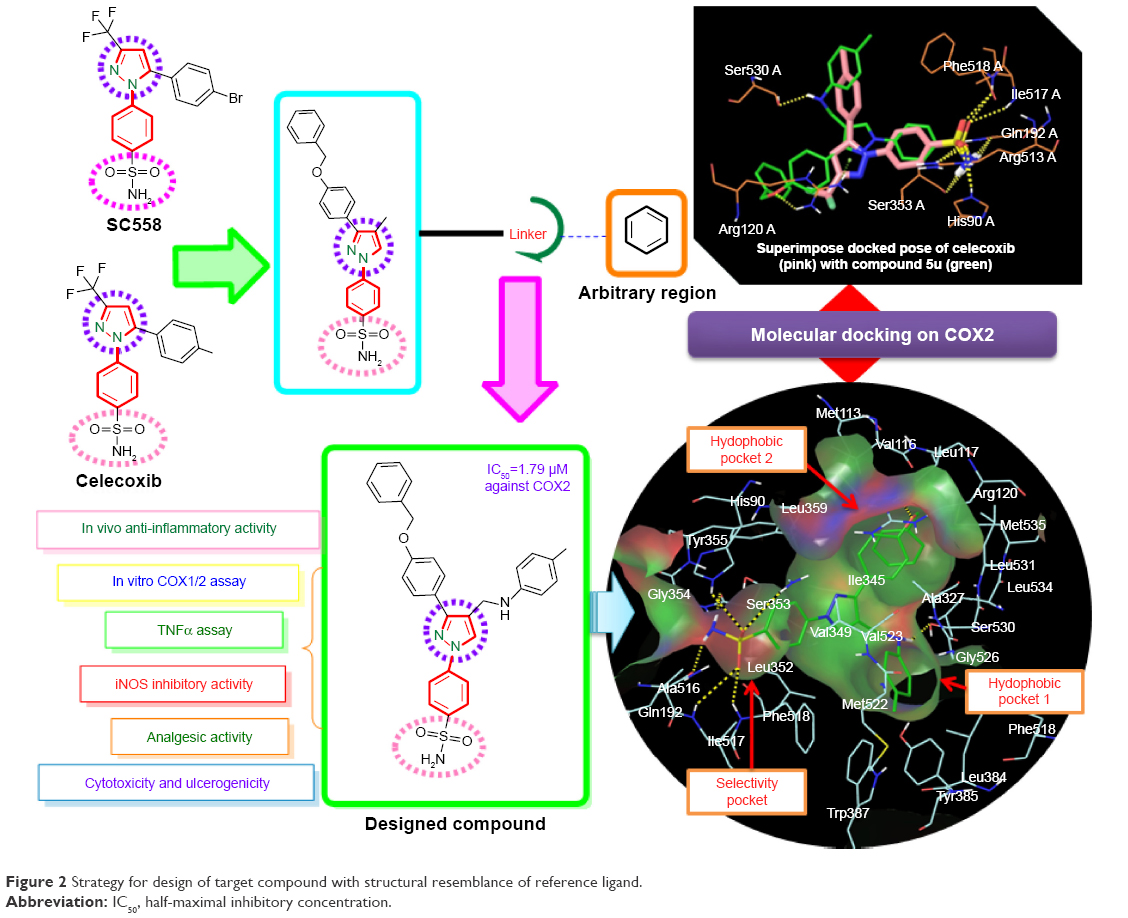 | Figure 2 Strategy for design of target compound with structural resemblance of reference ligand. |
Further the pyrazole moiety was linked to two aryl moieties, of which one was linked through the –CH2NH linker and the other through the –OCH2 linker. The significance of our study is in bringing together the pyrazole (excellent anti-inflammatory agent) and sulfonamide pharmacophore, known too for its wide range of biological activities, including anti-inflammatory activities,25,26 to obtain a derivative that is new and with a better pharmacological profile with fewer side effects. As part of our continuous research and efforts to design and develop a novel pyrazole-based anti-inflammatory agent with COX1/2-inhibition effect and continuation of our previous work, the present investigation describes the synthesis, anti-inflammatory, and analgesic activities of substituted pyrazole analogues with the aim of obtaining nonulcerogenic selective COX2 inhibitors as anti-inflammatory agents. Moreover, in order to increase affinity for COX2, the bulky hydrophobic benzyloxyphenyl group was incorporated in a manner similar to orientation of the trifluoromethyl group of celecoxib. Further, the importance of this bulky hydrophobic group was noticed by formation of one hydrogen bond with Arg120 in molecular docking studies. The high in vitro COX2-inhibition assay results of compounds 5u and 5s encouraged us to perform molecular docking studies to establish and understand the ligand–protein interactions. The in vitro COX assay and docking study results showed that the N1 substituent of the core pyrazole structure with benzene sulfonamide moiety is important for selective COX2-inhibitory activity. Docking studies also revealed that the SO2NH2 group of 5u and 5s, as well as celecoxib,27 formed hydrogen bonds with His90, Arg513, Phe518, Ser353, Gln192, and Ile517, and was completely inserted in the selective pocket of the COX2-active site.
Encouraged by all these facts, we designed a new series of hybrid pyrazole analogues (5a–5u) bearing the N-phenyly/p-sulfonamide phenyl group involving various electron-withdrawing and electron-releasing substituents and in vivo anti-inflammatory, analgesic, ulcerogenic, in vitro COX1/COX2 selective, TNFα, iNOS and cytotoxic activity.
Materials and methods
General chemistry
All the chemicals and reagents used were of laboratory-reagent grade and purchased from Sigma-Aldrich (St Louis, MO, USA), EMD Millipore (Billerica, MA, USA), SD Fine Chemicals Ltd (Mumbai, India), and Thermo Fisher Scientific (Waltham, MA, USA). Thin-layer chromatography (TLC) was performed to monitor the progress of the reaction using benzene, acetone, and toluene/ethyl acetate/formic acid (5:4:1) as eluent, and the spots were located under iodine vapors or ultraviolet light (254 nm). The melting points were determined by the open-capillary method with an electrical melting-point apparatus, and were uncorrected. Infrared (IR) spectra of the synthesized compounds were recorded in the 4,000–400 cm−1 range on a Shimadzu Fourier-transform (FT)-IR spectrometer by making KBr pellets, and 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance spectrometer at 300 and 75 MHz using dimethyl sulfoxide (DMSO)-d6 or CDCl3 as an NMR solvent. Ultraperformance liquid chromatography (UPLC) analysis of the compounds was performed on a Waters Acquity UPLC system (Waters Corporation, Milford, MA, USA). The mobile phase consisted of acetonitrile: 2 mmol ammonium acetate buffer (90:10, v/v). Synapt mass-spectrometry (MS) detection was carried out using UPLC MS (Waters Corporation) with an electrospray ionization (ESI) technique. Elemental analysis was performed on a CHNOS (carbon, hydrogen, nitrogen, oxygen, sulphur) elemental analyzer (Vario EL III) using sulfanilic acid as standard and tungsten(VI) oxide as combusting agent. Elemental analysis was ±0.4%, ie, within the theoretical values.
Synthesis of substituted (E)-1-(1-(4-(benzyloxy)phenyl)ethylidene)-2-phenylhydrazine (3a–3b)
A solution of 1-(4-(benzyloxy)phenyl)ethanone (1; 0.226 g, 10 mmol) and substituted phenyl hydrazine (2a–2b; 0.098 mL, 15 mmol) in 30 mL of absolute ethanol, and a glacial acetic acid of a catalytic amount (0.3 mL) was refluxed for 6–8 hours. The progress of the reaction was monitored by TLC using ethyl acetate:hexane (8:2) as a solvent system. On cooling, solid product was obtained, which was filtered, washed with ethanol, dried, and recrystallized from ethanol: white solid, yield 78%, melting point (MP) 123°C–125°C (3a); and off-white solid, yield 72%, MP 149°C–151°C (3b).
Synthesis of substituted 3-(4-(benzyloxy)phenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (4a–4b)
A solution of compound (3a–3b) in dry dimethylformamide at 0°C–5°C in a 100 mL round-bottom flask was treated with dropwise addition of POCl3 for 15–20 minutes. The reaction mixture was heated in a steam bath for 2 hours, cooled to room temperature, poured on crushed ice, and basified with sodium bicarbonate. The separated product was filtered, washed with water, dried, and recrystallized from ethanol to obtain yellow needle-like crystals of the synthesized compound.
3-(4-(Benzyloxy)phenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (4a)
White solid, yield 73%, MP 128°C–130°C, IR (KBr, cm−1) 2,935 (C–H aromatic), 1,710 (C=O), 1,670 (C=N), 1,350 (C=C), 1,255 (C–O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 5.12 (s, 2H, benzyloxy–CH2), 6.55 (d, 2H, Ar–H, J=8 Hz), 7.06 (d, 2H, Ar–H, J=8.4 Hz), 7.23 (d, 1H, Ar–H, J=8.2 Hz), 7.3–7.62 (m, 4H, Ar–H), 7.65 (d, 2H, Ar–H, J=8.8 Hz), 7.73–7.98 (m, 3H, Ar–H), 8.46 (s, 1H, pyrazole–CH), 9.86 (s, 1H, CHO). Analysis calculated for C23H18N2O2: C 77.95, H 5.12, N 7.9; found, C 77.83, H 5.17, N 7.83.
4-(3-(4-(Benzyloxy)phenyl)-4-formyl-1H-pyrazol-1-yl)benzenesulfonamide (4b)
White solid, yield 68%, MP 135°C–137°C, IR (KBr, cm−1) 3,420 (SO2NH stretching), 2,940 (C–H aromatic), 1,723 (C=O), 1,675 (C=N), 1,334 and 1,170 (S=O), 1,250 (C–O). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 5.34 (s, 2H, benzyloxy–CH2), 7.08 (d, 2H, Ar–H, J=8.4 Hz), 7.17 (bs, 2H, SO2NH2), 7.24–7.42 (m, 3H, Ar–H), 7.45 (d, 2H, Ar–H, J=8.6 Hz), 7.70–7.84 (m, 2H, Ar–H), 7.88 (d, 2H, Ar–H, J=7.8 Hz), 7.92 (d, 2H, Ar–H, J=8.6 Hz), 8.53 (s, 1H, pyrazole–CH), 9.82 (s, 1H, CHO). Analysis calculated for C23H19N3O4S: C 63.73, H 4.42, N 9.69; found, C 63.65, H 4.48, N 9.75.
Synthesis of N-((3-(4-(benzyloxy)phenyl)-1-phenyl-1H-pyrazol-4-yl)methyl)aniline derivatives (5a–5u)
A mixture of 3-(4-(benzyloxy)phenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (4a–4b); (10 mmol) and different substituted anilines (12 mmol) in 10 mL of methanol were added to a round-bottom flask and stirred at room temperature. I2 (0.4 mmol) was added slowly during stirring until all iodine was dissolved. Finally, NaBH4 (1.4 mmol) was added slowly to the resulting mixture and stirred further until completion of the reaction. The progress of the reaction was monitored by TLC using toluene:ethyl acetate:formic acid (5:4:1) as a solvent system. The solid precipitate obtained was filtered, washed with water, dried, and recrystallized from ethanol.28 Spectral characterization of compounds 5b–5u is covered in Supplementary materials.
N-((3-(4-(Benzyloxy)phenyl)-1-phenyl-1H-pyrazol-4-yl)methyl)aniline (5a)
White solid, yield 72%, MP 127°C–129°C, IR (KBr, cm−1) 3,396 (N–H str), 2,922 (C–H str aromatic), 1,670 (C=N), 1,517 (C=C), 1,240 (C–O), 1,008 (C–N). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 3.33 (bs, 1H, NH, D2O exchangeable), 4.3 (s, 2H, CH2–NH), 5.09 (s, 2H, CH2–benzyloxy), 6.55 (d, 2H, Ar–H, J=8 Hz), 7.07 (d, 2H, Ar–H, J=8.4 Hz), 7.28 (d, 1H, Ar–H, J=8.6 Hz), 7.42–7.62 (m, 5H, Ar–H), 7.68 (d, 2H, Ar–H, J=8.4 Hz), 7.73–7.84 (m, 3H, Ar–H), 7.93–8.24 (m, 4H, Ar–pyrazole), 8.61 (bs, 1H, pyrazole–H). 13C NMR (75 MHz, DMSO) δ (ppm): 41.05 (CH2–NH), 71.18 (benzyloxy–CH2), 113.12, 114.03, 114.37, 114.89, 119.42, 120.09, 120.49, 121.47, 125.71, 127.31, 127.9, 128.07, 128.67, 129.11, 129.46, 129.79, 130.1, 130.21, 131.33, 131.87, 131.99, 133.06, 137.17, 140.14 (C–phenylpyrazole), 152.31 (pyrazole), 153.17 (NH–phenyl), 159.62 (benzyloxy–C). ESI-MS (m/z): 432.30 (M++1). Analysis calculated for C29H25N3O, C 80.72, H 5.84, N 9.74; found, C 80.76, H 5.85, N 9.77.
Pharmacology
Anti-inflammatory activity
Carrageenan-induced rat-paw edema29 was used for the evaluation of in vivo anti-inflammatory activity of synthesized compounds. Wistar rats were procured from the Central Animal House facility of Jamia Hamdard, New Delhi, India (1141/CPCSEA), and adapted to room temperature in our laboratory in accordance with ARRIVE guidelines. This study was approved by CPCSEA (Committee for the Purpose of Control and Supervision on Experiments on Animals). The animals were fasted overnight (12 hours), weighed 150–200 g, and were divided into groups of six animals each. Group 1 served as control and received 0.5% w/v carboxymethyl cellulose, group 2 received the standard drug ibuprofen orally as a positive control at a dose of 20 mg/kg body weight, and the test groups were administered orally with an equimolar dose of the synthesized compounds as the standard drug. After 1 hour, all animals were injected with 0.1 mL of 1% carrageenan solution (prepared in 0.9% of 0.1 mL of saline solution) in the subplantar aponeurosis of the left hind paw, and the volume of the paw was measured by using a plethysmometer at intervals of 3 and 4 hours post-carrageenan treatment.
Analgesic activity
Acetic acid-induced writhing in mice was carried out using the method of Adeyemi et al.30 The writhing effect was induced by intraperitoneal injection of 0.6% acetic acid (v/v). Standard and test compounds were orally administered 30 minutes before chemical stimulus at an equimolar dose of 20 mg/kg body weight to groups of six animals each using ibuprofen as standard drug. The frequency of muscle contractions was counted for 20 minutes after acetic acid injection. Data represent the total number of writhes observed during the 20 minutes, and are expressed as writhing numbers.
Ulcerogenic activity
Test compounds with anti-inflammatory and analgesic activities comparable to celecoxib were further tested for acute ulcerogenic risk, as per Cioli et al’s method.31 The dose of the test and standard were three times the dose used for the estimation of the anti-inflammatory activity, ie, 60 mg/kg body weight. The control group received only 0.5% carboxymethyl cellulose. After drug treatment, the rats were fed a normal diet for 17 hours and then killed. Their stomachs were removed and opened along the greater curvature. The test and standard were compared after opening of the gastric mucosa, and the compounds had not caused any gastric ulceration or disruption of gastric epithelial cells at the aforementioned oral dose. Under microscopy, the effect of ulceration was examined. The mucosal damage in each stomach was assessed according to the following scoring system: 0.5, redness; 1, spot ulcer; 1.5, hemorrhagic streaks; 2, ulcers >3 but <5; 3, ulcers >5. The mean score of each treated group minus the mean score of control group was regarded as the severity index of gastric mucosal damage.
COX assay
Recombinant human COX2 has been expressed in an insect cell-expression system. The enzymes have been purified by using conventional chromatographic techniques. Enzymatic activities of COX2 were measured as per Copeland et al32 with slight modifications, using a chromogenic assay based on the oxidation of N, N, N, N,-tetramethyl-p-phenylenediamine (TMPD) during the reduction of PGG2 to PGH2. Briefly, the assay mixture contained Tris-HCl buffer (100 mM, Ph 8), hematin (15 mM), ethylenediaminetetraacetic acid (3 mM), enzyme (100 mg COX2), and the test compound. The mixture was preincubated at 25°C for 1 minute, and then the reaction was initiated by the addition of arachidonic acid and TMPD in a total volume of 1 mL. Enzyme activity was determined by estimating the velocity of TMPD oxidation for the first 25 seconds of the reaction by following the increase in absorbance at 603 nm. A low rate of nonenzymatic oxidation observed in the absence of COX2 was subtracted from the experimental value while calculating the percentage inhibition.
TNFα assay
Macrophage cells were developed in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum (FBS), 1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 2 μM glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin, which was obtained by passage through a stainless-steel mesh. Collected supernatants of cell culture from macrophage cells seeded with many concentrations of potent compounds were assayed for proinflammatory cytokine levels using Krishgen Biosystems enzyme-linked immunosorbent assay (ELISA) kits (KB2052; Mumbai, India). For this, macrophages were preincubated with cytochalasin D (2.5 μM/106 cells) for 30 minutes before the beginning of the experiment till the end. Cytokine levels were calculated according to the procedure recommended by the manufacturer. Cultured cells in 96-well plates at 2×106 cells/mL and cytokines were then measured from the supernatants by ELISA. The assay was performed according to the manufacturer’s instructions with multipoint analysis. Briefly, 100 μL of diluted capture antibody was added to each well in a 96-well plate and left to adhere overnight at 4°C. Plates were washed and then blocked with 1% phosphate-buffered saline (PBS) supplemented with 10% FBS for 1 hour at room temperature. The medium was induced with 100 μL/mL lipopolysaccharide (LPS), together with test samples or reference drug (celecoxib) at 20 μM concentrations, and incubated for 24 hours. The supernatant (50 μL) was then transferred into a 96-well ELISA plate, and TNFα level in macrophage-culture medium was quantified by ELISA kits. Plates were then sealed and incubated for 1 hour at room temperature. After being washed, 100 μL of a trimethylbenzidine substrate was added to each well. Stock solution (2N H2SO4) was finally added after incubation in the dark for 30 minutes at room temperature. Reading of sample absorbance was taken at 450 nm. The result was interpreted and values determined against the standard provided by the manufacturer.33
Nitric oxide (NO) assay
The NO assay was performed using mouse macrophages (RAW264.7, obtained from the American Type Culture Collection). Cultured cells were in phenol red free RPMI medium supplemented using 10% FBS, 100 U/mL penicillin G sodium, and 100 μg/mL streptomycin at 37°C in an atmosphere of 95% humidity and 5% CO2. Cells were seeded in 96-well plates at 104 cells/well and incubated for 24 hours. Test compounds diluted in serum-free medium were added to the cells. After 30 minutes of incubation, LPS (5 μg/mL) was added and the cells were additionally incubated for 24 hours. The concentration of NO was determined by measuring the level of nitrite released in the cell-culture supernatant by using Griess reagent (1% sulfanilamide and 0.1% N-[1-naphthyl]ethylenediamine dihydrochloride in 5% phosphoric acid). Percentage inhibition of nitrite production by the test compound as well as celecoxib as standard was calculated in comparison to the vehicle control. Optical density (OD) was measured with a microplate reader at 540 nm.34
MTT assay for cell viability
Cell culture was carried out in triplicate following the same protocol as that for the TNFα assay. RAW264.7 cells (2×105) were cultured in a 96-well plate containing Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS to get required confluence. These cells were stimulated with 20 μM test compounds in the presence of 100 μg/mL LPS for 24 hours. Afterward, the cells were washed twice with Dulbecco’s PBS and incubated with 100 μL of 0.5 mg/mL MTT (3-[4,5-dimethylthiazol-2-yl]2,5-diphenyltetrazolium bromide) for 2 hours at 37°C for testing cell viability. The medium was then discarded and 100 μL DMSO added. After 30 minutes’ incubation, OD was measured using an ELISA plate reader at 570 nm.35
Molecular docking
Molecular docking studies of all the compounds were carried out by taking X-ray crystal-structure data to understand the molecular interactions of the synthetic compounds within the binding site of COX1 (Protein Data Bank [PDB] ID 3KK6) and COX2 (PDB ID 1CVU). The molecular docking study was used to understand the possible best binding pose of the compounds (5a–5u) by which they could be sorted for identifying promising leads using Glide 7.0 XP Maestro 10.1 (Schrödinger, New York, NY, USA) running on a Linux 64 operating system.36 Molecular docking studies mainly involve selection and preparation of appropriate protein, grid generation, and ligand preparation, followed by analysis of docking output and their interaction. The ligands and the receptors were separately prepared by using LigPrep and Protein Preparation Wizard, respectively. The protein structure of both COX1 and COX2 was separately preprocessed using Protein Preparation Wizard, and missing hydrogen bonds were also added, then hydrogen bonds within the protein structure were optimized, followed by minimization of protein to a root-mean-square deviation of 0.3 Å for the default setup of Glide. The unwanted water molecules were also removed from the grid. The only chain A was selected in the refine tab. Docking scores are used to predict binding modes, binding affinity, and orientation of synthetic ligands at the active site of COX1 and COX2.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 Software. All data were analyzed by analysis of variance followed by Dunnett’s multiple comparison test for n=6: *P<0.05; **P<0.01. Relative-to-normal data were analyzed by paired Student’s t-test for n=6: ***P<0.001; ****P<0.005.
Results and discussion
Chemistry
The protocol for the synthesis of hybrid pyrazole analogues (5a–5u) is illustrated in Scheme 1. The intermediate compound 1 was synthesized by the reaction of 4-hydroxyacetophenone with benzyl chloride in the presence of Na2CO3/KI and acetone as a solvent with continuous stirring. Subsequently, compound 1 was further reacted with phenyl hydrazine/4-hydrazinylbenzenesulfonamide (2a–2b) to give compounds 3a–3b, which were further cycled in the presence of POCl3 and dry dimethylformamide to give 3-(4-(benzyloxy) phenyl)-1-phenyl/phenylsulfonamide-1H-pyrazole-4-carbaldehyde (4a–4b). Finally, different substituted anilines were treated through direct reductive amination using NaBH4/I2 as a reducing agent to give the desired compounds (5a–5u). All these compounds were reported for the first time and characterized by FTIR, 1H NMR, 13C NMR, and mass spectral data. In the 1H NMR spectrum of compound 4a, the aldehydic proton signal appeared singlet at δ=9.86 ppm, later disappeared, and a new signal of –CH2NH– peak appeared as a singlet at δ=4.3 ppm, integrating two protons, which suggested reductive amination to yield compound 5a. The D2O exchangeable NH peak appeared as a broad singlet at δ=3.33 ppm. Moreover, in the 13C NMR spectrum, the aldehydic peak was transformed into the –CH2NH– and appeared at δ=41.05 ppm. In 1H-NMR, the D2O exchangeable singlet peak was observed around δ=7.19 ppm, integrating two protons ascertained as free SO2NH2 in compound 5u. The FTIR spectra of compound 5u displayed the presence of the N–H characteristic absorption band at 3,410 and 3,360 cm−1, and SO2 characteristic absorption bands of SO2NH2 appeared at 1,342 and 1,172 cm−1. Mass spectrum of this prototype compounds showed a molecular ion peak, ie, (m/z) [M+] at 524.19, with reasonable intensity confirming the structures. The elucidation of physical and spectral data showed the successful synthesis of the desired compounds.
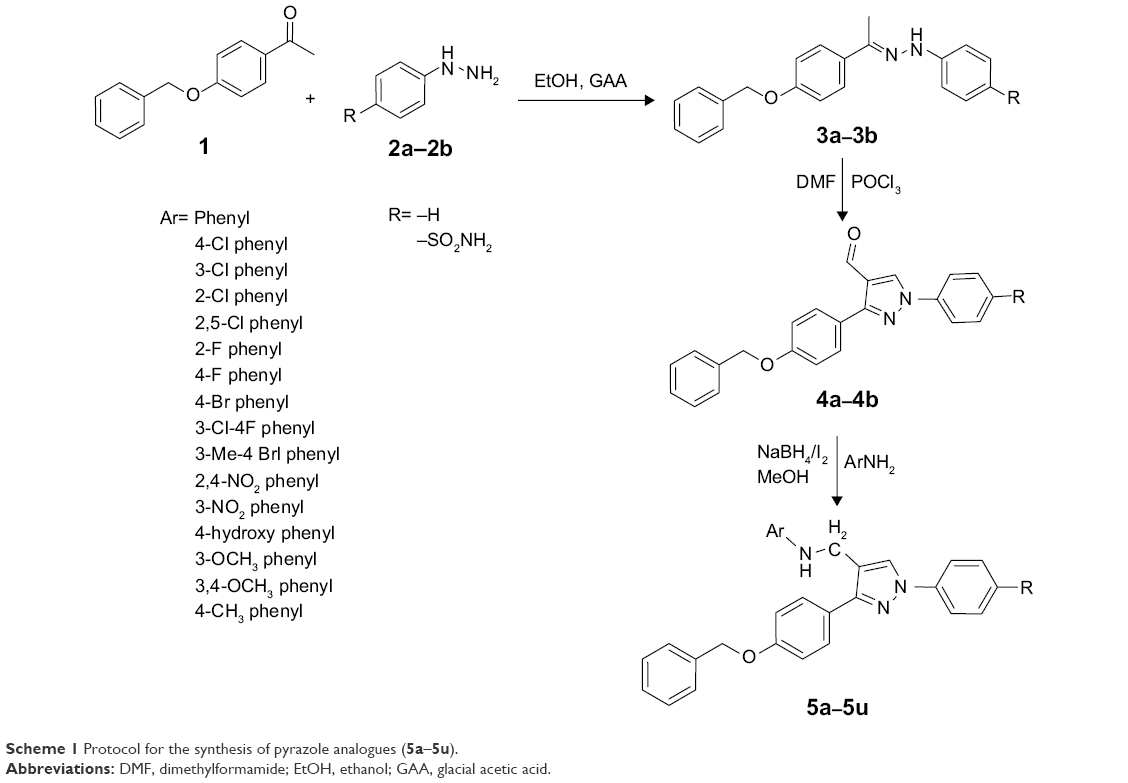 | Scheme 1 Protocol for the synthesis of pyrazole analogues (5a–5u). |
In vivo anti-inflammatory activity of synthesized derivatives (5a–5u)
Anti-inflammatory activity of all the synthesized compounds was evaluated by the carrageenan-induced hind-paw edema model.29 All the compounds showed decreased inhibition of inflammation at 3 and 4 hours (Table 1), and compounds 5s and 5u significantly decreased paw edema, showing inhibition of 80.87%–76.56% and 80.63%–78.09% at 3 and 4 hours compared to the standard drug ibuprofen, which showed 81.32%–79.23% inhibition at 3 and 4 hours, respectively. Compounds 5b, 5d, 5g, 5i, 5q, 5r, and 5t possessed moderate anti-inflammatory activity, whereas compounds 5a, 5c, 5e, 5j, 5k, and 5l exhibited low inhibition of inflammation compared with the standard drug ibuprofen. The anti-inflammatory activity of all the synthesized pyrazole derivatives (5a–5u) is given in Table 1.
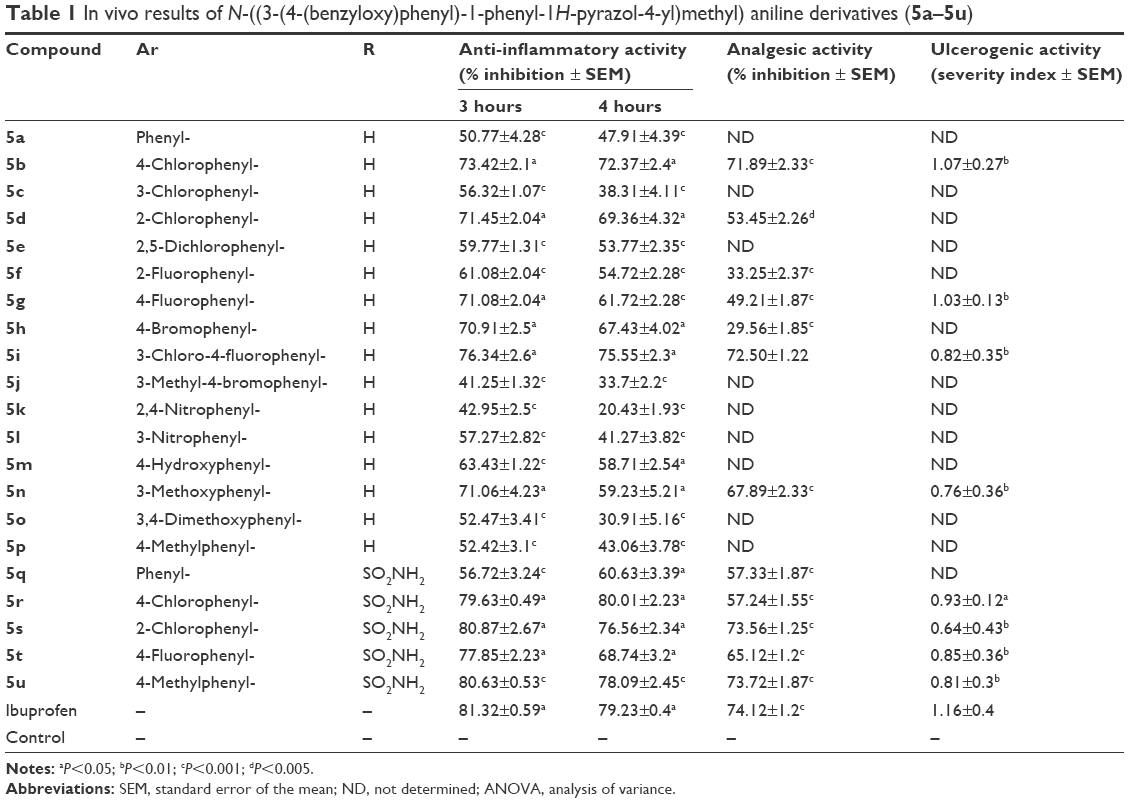 | Table 1 In vivo results of N-((3-(4-(benzyloxy)phenyl)-1-phenyl-1H-pyrazol-4-yl)methyl) aniline derivatives (5a–5u) |
Analgesic activity revealing 5s and 5u as potent derivatives
Pyrazole derivatives with favorable anti-inflammatory activity were selected for analgesic activity. Acetic acid was used intraperitoneally for abdominal constriction responses, which profoundly produces peripherally acting analgesics that may increase the level of PGE2 and PGF2α. The analgesic activity inhibition of 12 tested compounds was between 29.56% and 73.72%. The compounds 5i, 5b, 5n, 5q, 5r, 5d, and 5t showed significant analgesic activity. The analgesic activity of compounds 5s (73.56% inhibition) and 5u (73.72% inhibition) were found to be comparable with the standard drug ibuprofen (74.12% inhibition), as shown in Table 1.
Gastric ulceration studies of compounds 5s and 5u
Gastric ulceration is a major problem of NSAIDs.37 Therefore, compounds that showed significant anti-inflammatory and analgesic activities were selected for the gastric ulceration studies. It was found that the compounds 5s and 5u did not induce any gastric ulceration or rupture of the gastric mucosal layer at a drug concentration of 60 mg/kg, nor did ibuprofen result in any trace of damage or ulceration to the epithelial layer of stomachs of experimental rats. Histopathological results of rat stomachs treated with the standard drug (ibuprofen) and active compounds in comparison to healthy controls are shown in Figure 3.
 | Figure 3 Histopathological study of stomachs of rats treated with standard drug (ibuprofen) and active compounds in comparison to healthy controls. |
In vitro COX-inhibition assay
Compounds demonstrating substantial in vivo anti-inflammatory activity were evaluated further by in vitro COX-inhibition assay. The obtained data (Table 2) showed that all tested compounds were very weak inhibitors of the COX1 isoenzyme with half-maximal inhibitory concentration (IC50) =45.23–204.51 μM, but revealed significant selective COX2 isoenzyme-inhibitory activity with IC50=1.79–9.63 μM. Compounds 5u, 5s, 5r, and 5t displayed significant selective COX2 inhibition, with selective index (SI) values of 74.92, 72.95, 64.40, and 22.21, respectively, compared to celecoxib, with an SI of 78.06. The SI may be defined as the ratio of drug concentration (IC50) that inhibits the COX1 and COX2 isoforms. Compounds 5b, 5i, 5g, 5n, 5d, and 5q showed moderate SI values of 47.45, 32.34, 33.79, 21.97, 19.60, and 14.87, respectively. SI values of compounds 5u, 5s, 5r, and 5t showed their selective nature toward the COX2 isoform (Table 2).
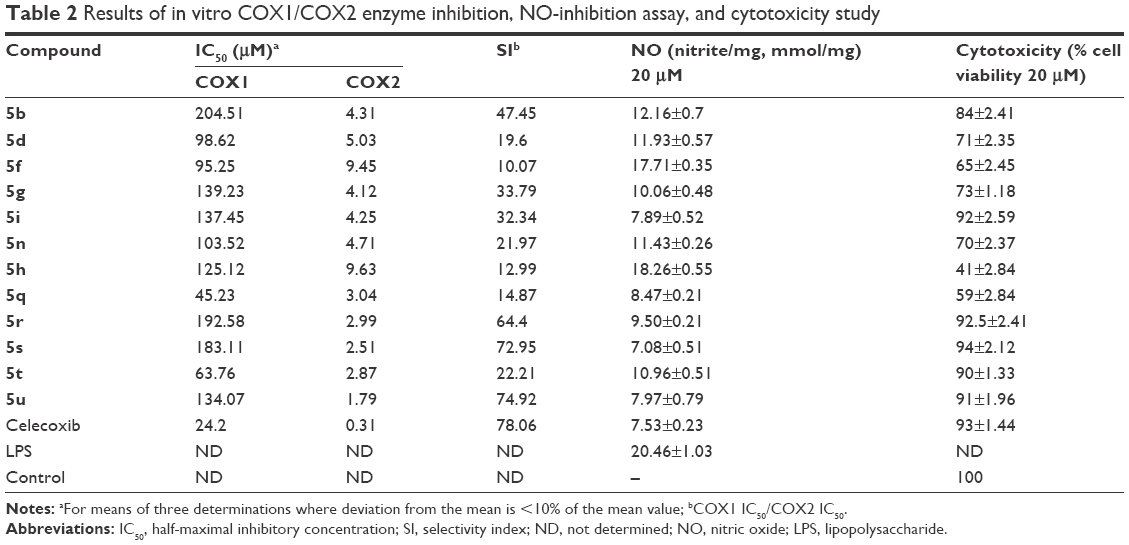 | Table 2 Results of in vitro COX1/COX2 enzyme inhibition, NO-inhibition assay, and cytotoxicity study |
In vitro TNFα assay
Compounds displaying good in vivo anti-inflammatory activities were further evaluated for their in vitro TNFα activity. LPS-induced cells elevated the level of TNFα markedly, which was evaluated on RAW264.7 cells. All selected 12 compounds showed TNFα inhibition at 20 μM drug concentrations (Figure 4). It was found that the level of TNFα post-LPS induction was significantly controlled by compounds 5i, 5u, 5j, and 5r. These compounds exhibited potent results, with good inhibitions of 75.49%, 71.52%, 70.54%, and 69.91%, respectively, while the standard drug celecoxib showed inhibition of 71.97% at 20 μM drug concentrations.
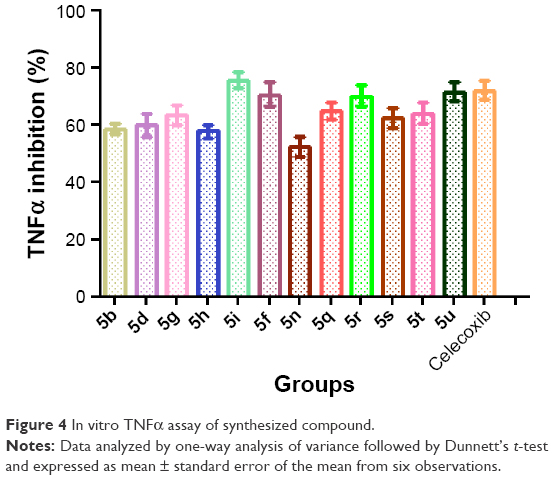 | Figure 4 In vitro TNFα assay of synthesized compound. |
iNOS assay
An important signaling molecule of NO is formed during the inflammatory response from macrophages and activated cells.38 Higher NO concentration has been observed previously in the synovial fluid of those suffering from rheumatoid arthritis.39 Our aim was to check the formation and elevation of NO levels in carrageenan-induced inflammation in the rat. A significant rise in NO levels was observed in carrageenan-treated rats compared with controls. Few selected compounds decreased nitrite levels significantly, which was responsible for the formation of NO (Table 2). The compounds 5s, 5i, 5u, and 5q consequently suppressed the increase in NO level to 7.08, 7.89, 7.97, and 8.47 mmol/mg, respectively, while the standard drug celecoxib suppressed the increase in NO level to 7.53 mmol/mg.
Cytotoxicity test through MTT assay
Cytotoxicity study of active anti-inflammatory compounds was evaluated by MTT assay. The results for the compounds are shown in Table 2. The effects clearly demonstrated that five compounds – 5s, 5r, 5i, 5u, and 5t possessed more than 90% cell viability at 20 μM drug concentration; therefore, these compounds did not cause any normal cell death and would be assumed safe. The compounds 5b, 5g, 5d, and 5n showed significantly low cytotoxic effect, with 70%–85% cell viability, but the remaining tested compounds were found to be cytotoxic.
Molecular docking study
The promising in vitro COX2-inhibition results of compounds 5u and 5s encouraged us to perform molecular docking studies to establish and understand the ligand–protein interactions. For this study, the crystal structures of COX1 and COX2 enzyme complexes with a cocrystal (http://www.pdb.org) were selected as the protein target used for the docking study. Before analyzing the docking studies, we must understand the COX1 and COX2 pocket structure, since we know that the COX2 active site is approximately 25% larger than COX1, which forms a new side pocket called a selectivity pocket and can accommodate bigger structure. That is why arylsulfonamide derivatives could not be docked for COX1, because these compounds (arylsulfonamide) are not able to reach the active site of COX1 or accommodate/interact in the smaller space of the COX1 site. The volume of the selectivity pocket is reduced, due to the presence of Ile523 in COX1, while the presence of Val523 in COX2 provides enough space for possible interaction and stable binding of COX2 inhibitors.40 This fact and prediction was validated by crystallographic structure (PDB ID 3KK6). Among all the synthesized molecules, the most promising COX2 inhibitors 5u and 5s exhibited high docking scores of −12.907 and −12.18, respectively, whereas the docking score of celecoxib was found to be −9.924. The results of docking studies showed that all the compounds exhibited higher docking scores than celecoxib (Table 3). The orientation and conformation of docked compounds 5u and 5s were similar to the celecoxib. The two-dimensional LigPlot diagram of compounds 5u and 5s with COX2 is represented in Figure 5. The methylene amino (–CH2–NH–) group between the pyrazole scaffold and aryl ring increases flexibility, and due to this, the aryl ring can be accommodated more favorably toward the binding pocket of COX2. The –NH– of p-methylaniline (5u) and 2-chloroaniline (5s) functionality participated in hydrogen bonding with the side chain of Ser530 residue of the binding pocket of COX2 at distances of 2.35 Å (NH·OH) and 2.44 Å (NH·OH), respectively (Figure 6). The binding interaction with Ser530 is the key important feature for COX2-inhibitory activity, and also contributed to stabilize the ligand–enzyme complexes. In order to enhance affinity for COX2, the bulky benzyloxyphenyl moiety in the pyrazole ring was incorporated. The interaction of docked compounds 5u and 5s with COX2 depicted in Figure 6A and C shows that the oxygen atom of the benzyloxy group at the para-position of the phenyl ring attached to the pyrazole core had one important H-bonding interaction with Arg120 at distances of 3.01 Å (O·HN– guanidine) and 2.88 Å (O·HN– guanidine), respectively. The 4-methylbenzene ring of 5u was occupied in a hydrophobic cavity, like the 4-methylbenzene ring of celecoxib, and surrounded by amino acid residues Phe518, Trp387, Phe381, Tyr385, and Leu381, displayed in gray CPK representation (Figure 7B). The benzyloxyphenyl group attached to the C3 position of the pyrazole ring resides in another hydrophobic pocket, surrounded by Tyr355, Val349, Ile345, Leu359, Leu531, Leu117, and Val116 displayed in gray CPK representation (Figure 7B). Furthermore, the docked pose of 5u and 5s revealed that the hydrogen atoms of the sulfonamide pharmacophore (SO2NH2) formed two hydrogen bonds with the backbone of Ser353 (1.87 Å, H·O=C; 1.82 Å, H·O=C) and side chain of Gln192 (1.81 Å, H·O=C; 1.84 Å, H·O=C) in the selectivity pocket of COX2. Finally, the oxygen atoms of the sulfonamide group inside the selectivity pocket assumed a favorable conformation in which one of the oxygen atoms formed two hydrogen bonds with the side chain of His90 (2.24 Å, S=O·HN; 2.49 Å, S=O·HN) and side chain of Arg513 (3.06 Å, S=O·HN; 2.58 Å, S=O·HN), and the other oxygen atom formed a hydrogen bond with the backbone of Phe518 (2.44 Å, S=O·H; 2.49 Å, S=O·H) in compounds 5u and 5s, respectively. Moreover, one oxygen atom of the sulfonamide group of 5u showed an additional hydrogen-bond interaction with Ile517 (O·HN, 3.05 Å) in the selective binding pocket, which was not observed in celecoxib. The orientation and interaction of the sulfonamide pharmacophore of 5u and 5s in the docked pose were similar to the sulfonamide pharmacophore of celecoxib, which was further validated by superimposed docked pose (Figure 7A). Further zooming the docked pose of 5u revealed six hydrogen bonds with amino acid residues of His90, Arg513, Phe518, Ser353, Gln192, and Ile517 in the selectivity pocket of COX2 (Figure 6B). These types of interactions are essential pharmacophoric requisites, and play a crucial role for designing selective COX2-inhibitory activity. The receptor-surface model of compound 5u bound to COX2, showing hydrophobic pocket 1, hydrophobic pocket 2, and the selectivity pocket in which p-methylaniline, benzyloxyphenyl, and sulfonamide were accommodated, respectively (Figure 8).
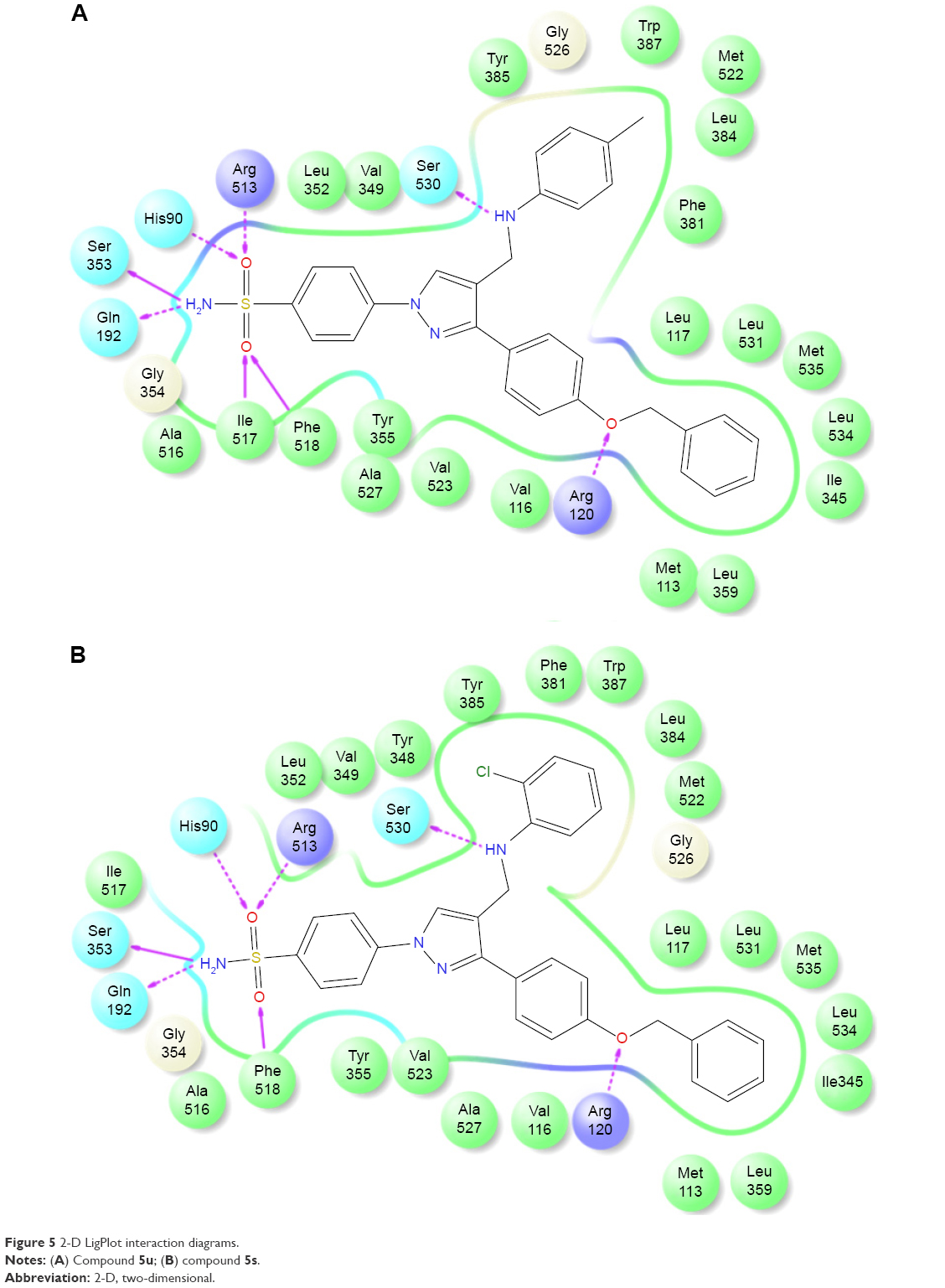 | Figure 5 2-D LigPlot interaction diagrams. |
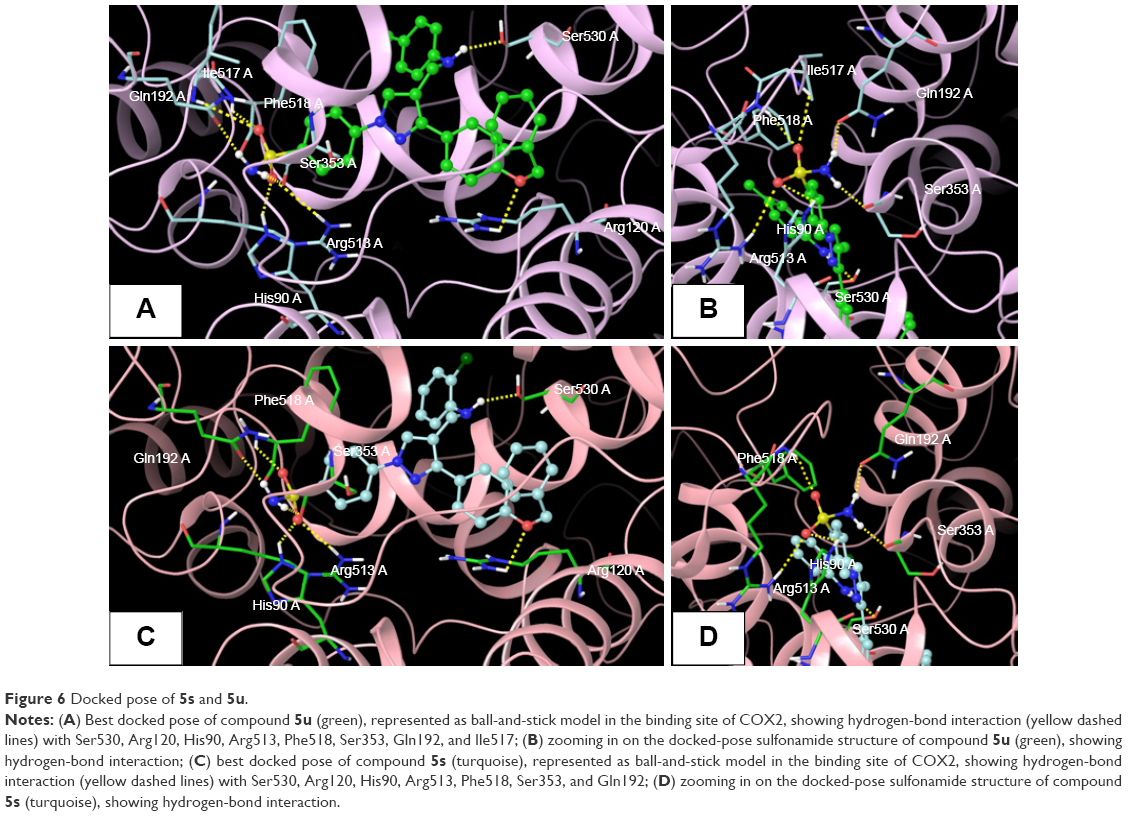 | Figure 6 Docked pose of 5s and 5u. |
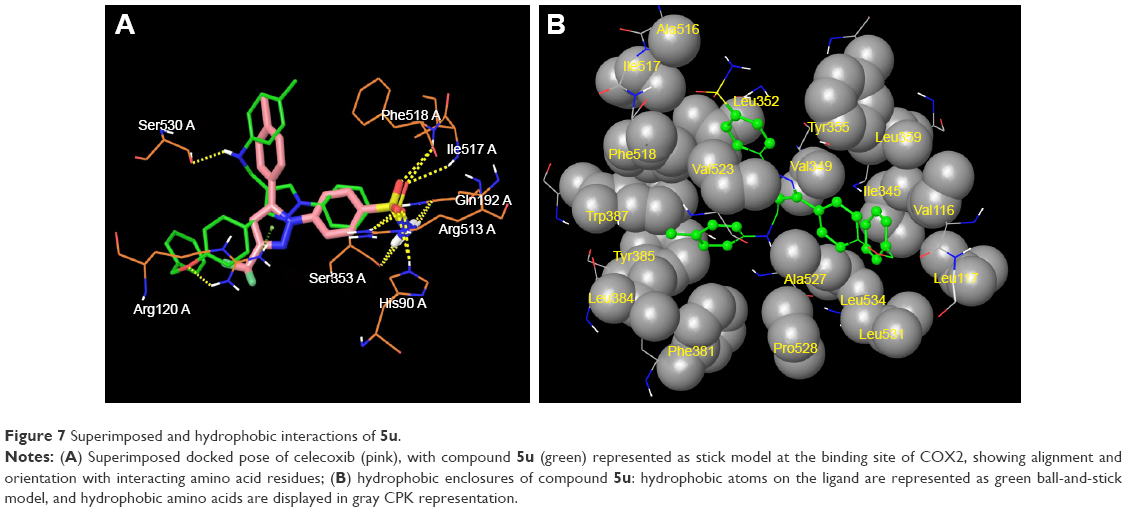 | Figure 7 Superimposed and hydrophobic interactions of 5u. |
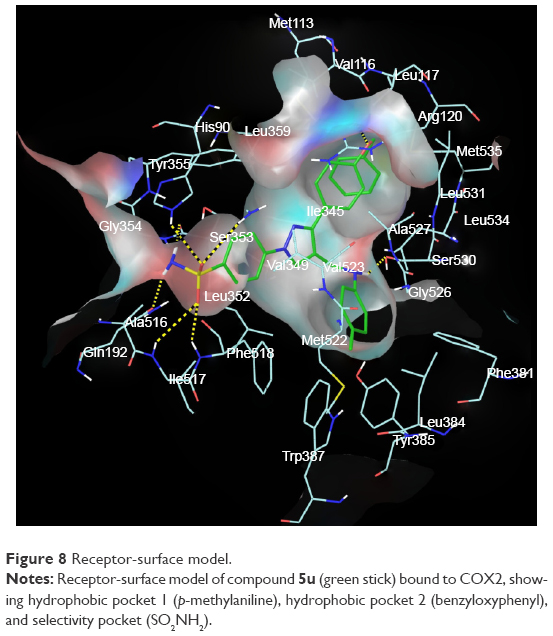 | Figure 8 Receptor-surface model. |
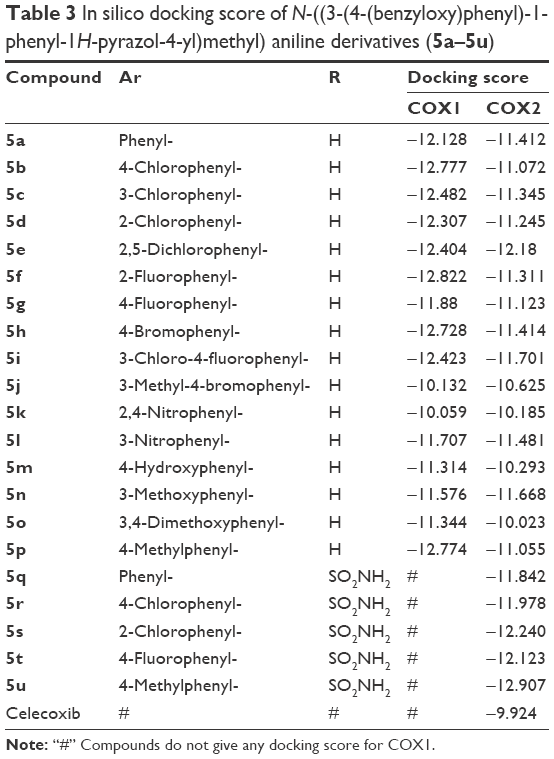 | Table 3 In silico docking score of N-((3-(4-(benzyloxy)phenyl)-1-phenyl-1H-pyrazol-4-yl)methyl) aniline derivatives (5a–5u) |
Structure–activity relationship
The structure–activity relationship of pyrazole analogues was as follows:
- both electron-withdrawing and electron-releasing groups showed good docking scores and imparted good biological effects
- among compounds 5a–5u, compounds 5q–5u, with a sulfonamide substitution on the para-position of the N-phenyl ring, displayed higher COX2-inhibitory activity than those without para-substitution derivatives in compounds 5a–5p
- pyrazole derivatives bearing a methyl group at the para-position at the aniline ring and the sulfonamide group at the N-phenyl ring showed higher COX2-inhibitory activity than those with Cl or F substituents at the same position
- COX2-inhibitory activity was slightly decreased in compounds with one chloro-group at ortho-position on the aniline ring in sulfonamide containing derivative 5s compared to compound 5u, while the same substituent at the same position in compounds without having sulfonamide moiety showed a remarkable decrease in COX2-inhibitory activity
- compounds with halide groups were active against TNFα and reduced inflammation during in vivo anti-inflammatory activity
- significant in vivo anti-inflammatory and in vitro selective COX2 activity was observed in the sulfonamide derivatives 5u and 5s.
Conclusion
In this work, we designed, synthesized, and evaluated a new series of pyrazole-bearing methylamine derivatives by reductive amination. Compounds showing significant anti-inflammatory activity were subjected to COX2- and TNFα-inhibition studies. Compounds 5s and 5u exhibited potent anti-inflammatory activity of 80.87% and 80.63% inhibition compared with the standard drug ibuprofen, which showed 81.32%. The optimal COX2-inhibitory potency of 5s and 5u (IC50=2.51 and 1.79 μM) and SI =72.95 and 74.92 was comparable with celecoxib, with SI =78.06. This provides information regarding promising TNFα-inhibitory potency, with 75.49% and 71.52% inhibition for 5i and 5u. The structure–activity relationships showed that sulfonamide substituted with methylamine pyrazole derivatives has the basic geometry to provide potent and selective inhibition of the COX2 isoenzyme and exhibited excellent anti-inflammatory activities. Outcomes of the in vitro COX assay, docking analysis, and binding mode of pyrazole derivatives for good COX2-inhibition activity revealed that the compounds must have the –SO2NH2 group to bind perfectly in the selective pocket of the COX2 enzyme. Finally, molecular modeling and biological studies gave us valuable ideas for the design of novel and more potent COX2 inhibitors as anti-inflammatory agents.
Acknowledgments
The authors are thankful to Jamia Hamdard, New Delhi, India, for providing facilities for research work and spectral and elemental studies. Md Jahangir Alam expresses thanks to the University Grant Commission (UGC), New Delhi, for the award of Moulana Azad National Fellowship as financial assistance (201213-MANF-2012-13-MUS-BIH-17183). Ozair Alam is also thankful to DST-SERB (SB/FT/LS-203-2012), New Delhi, for providing funds to purchase Schrödinger software (molecular modeling).
Disclosure
The authors report no conflicts of interest in this work.
References
Botting JH. Nonsteroidal antiinflammatory agents. Drugs Today (Barc). 1999;35:225–235. | ||
Grosser T, Fries S, Fitzgerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. | ||
Girgis AS, Ellithey M. Facile synthesis of non-steroidal anti-inflammatory active bisbenzamide-containing compounds. Bioorg Med Chem. 2006;14:8527–8532. | ||
Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1993;268:6610–6614. | ||
Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N Engl J Med. 2000;343:1520–1528. | ||
Bayly CI, Black WC, Léger S, Ouimet N, Ouellet M, Percival MD. Structure-based design of COX-2 selectivity into flurbiprofen. Bioorg Med Chem Lett. 1999;9:307–312. | ||
Allaj V, Guo C, Nie D. Non-steroid anti-inflammatory drugs, prostaglandins, and cancer. Cell Biosci. 2013;3:8. | ||
Motawi TK, Elgawad HM, Shahin NN. Modulation of indomethacin-induced gastric injury by spermine and taurine in rats. J Biochem Mol Toxicol. 2007;21:280–288. | ||
Wolfe MM, Lichtenstein RD, Singh GN. Gastrointestinal toxicity of non-steroidal anti-inflammatory drugs. N Engl J Med. 1999;340:1888–1899. | ||
Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. | ||
Sun Y, Li YH, Wu XX, et al. Ethanol extract from Artemisia vestita, a traditional Tibetan medicine, exerts anti-sepsis action through down-regulating the MAPK and NF-κB pathways. Int J Mol Med. 2006;17:957–962. | ||
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–979. | ||
Karima R, Matsumoto S, Higashi H, Matsushima K. The molecular pathogenesis of endotoxic shock and organ failure. Mol Med Today. 1999;3:123–132. | ||
Birkedal-Hansen H. Role of cytokines and inflammatory mediators in tissue destruction. J Periodontal Res. 1993;28:500–510. | ||
Park HA, Lee K, Park SJ, et al. Identification of antitumor activity of pyrazole oxime ethers. Bioorg Med Chem Lett. 2005;15:3307–3312. | ||
Balbi A, Anzaldi M, Macciò C, et al. Synthesis and biological evaluation of novel pyrazole derivatives with anticancer activity. Eur J Med Chem. 2011;46:5293–5309. | ||
Saleem S, Shaharyar MA, Khusroo MJ, et al. Anticancer potential of rhamnocitrin 4′-β-D-galactopyranoside against N-diethylnitrosamine-induced hepatocellular carcinoma in rats. Mol Cell Biochem. 2013;384:147–153. | ||
Storer R, Ashton CJ, Baxter AD, et al. The synthesis and antiviral activity of 4-fluoro-1-β-D-ribofuranosyl-1H-pyrazole-3-carboxamide. Nucleosides Nucleotides. 1999;18:203–216. | ||
Kaushik D, Khan SA, Chawla G, Kumar S. N′-[(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)methylene] 2/4-substituted hydrazides: synthesis and anticonvulsant activity. Eur J Med Chem. 2010;45:3943–3949. | ||
Tanitame A, Oyamada Y, Ofuji K, et al. Synthesis and antibacterial activity of a novel series of potent DNA gyrase inhibitors: pyrazole derivatives. J Med Chem. 2004;47:3693–3696. | ||
Chen HS, Li ZM, Han YF. Synthesis and fungicidal activity against Rhizoctonia solani of 2-alkyl (alkylthio)-5-pyrazolyl-1,3,4-oxadiazoles (thiadiazoles). J Agric Food Chem. 2000;48:5312–5315. | ||
Palomer A, Pascual J, Cabré M, et al. Structure-based design of cyclooxygenase-2 selectivity into ketoprofen. Bioorg Med Chem Lett. 2002;12:533–537. | ||
Perrone MG, Scilimati A, Simone L, Vitale P. Selective COX-1 inhibition: a therapeutic target to be reconsidered. Curr Med Chem. 2010;17:3769–3805. | ||
Abdel-Aziz AA, El Tahir KE, Asiri YA. Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: Molecular docking study. Eur J Med Chem. 2011;46:1648–1655. | ||
Sondhi SM, Rani R, Roy P, Agrawal SK, Saxena AK. Microwave-assisted synthesis of N-substituted cyclic imides and their evaluation for anticancer and anti-inflammatory activities. Bioorg Med Chem Lett. 2009;19:1534–1538. | ||
Lima LM, Castro P, Machado AL, et al. Synthesis and anti-inflammatory activity of phthalimide derivatives, designed as new thalidomide analogues. Bioorg Med Chem. 2002;10:3067–3073. | ||
Penning TD, Talley JJ, Bertenshaw SR, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib). J Med Chem. 1997;40:1347–1365. | ||
Ali MR, Kumar S, Afzal O, Shalmali N, Sharma M, Bawa S. Development of 2-(substituted benzylamino)-4-methyl-1,3-thiazole-5-carboxylic acid derivatives as xanthine oxidase inhibitors and free radical scavengers. Chem Biol Drug Des. 2015;87:508–516. | ||
Winter CA, Risley EA, Nuss GW. Carrageenin-induced edema in hind paw of the rat as an assay for anti-inflammatory drugs. Proc Soc Exp Biol Med. 1962;3:544–547. | ||
Adeyemi O, Okpo OS, Okpak O. The analgesic effect of the methanolic extract of Acanthus montanus. J Ethnopharmacol. 2004;90:45–48. | ||
Cioli V, Pothole S, Rossi V, Barcellona PS, Corradino C. The role of direct tissue contact in the production of gastrointestinal ulcers by anti-inflammatory drugs in rats. Toxicol Appl Pharmacol. 1979;50:283–289. | ||
Copeland RA, Williams JM, Giannaras J, et al. Mechanism of selective inhibition of the inducible isoform of prostaglandin G/H synthase. Proc Natl Acad Sci U S A. 1999;91:11202–11206. | ||
Malik F, Singh J, Khajuria A, et al. A standardized root extract of Withania somnifera and its major constituent withanolide-A elicit humoral and cell-mediated immune responses by up regulation of Th1-dominant polarization in BALB/c mice. Life Sci. 2007;80:1525–1538. | ||
Quang DN, Harinantenaina L, Nishizawa T, et al. Inhibition of nitric oxide production in RAW 264.7 cells by azaphilones from xylariaceous fungi. Biol Pharm Bull. 2006;29:34–37. | ||
Tewtrakul S, Subhadhirasakul S. Effects of compounds from Kaempferia parviflora on nitric oxide, prostaglandin E2 and tumor necrosis factor-alpha productions in RAW264.7 macrophage cells. J Ethnopharmacol. 2008;120:81–84. | ||
Schrödinger. Maestro version 10.1 [software]. New York: Schrödinger; 2016. | ||
Haider S, Nazreen S, Alam MM, Hamid H, Alam MS. Anti-inflammatory and anti-nociceptive activities of Platanus orientalis Linn. and its ulcerogenic risk evaluation. J Ethnopharmacol. 2012;143:236–240. | ||
Shen HM, Kennedy JL, Ou DW. Inhibition of cytokine release by cocaine. Int J Immunopharmacol. 1994;16:295–300. | ||
Hassan MQ, Hadi RA, Al-Rawi ZS, Padron VA, Stohs SJ. The glutathione defense system in the pathogenesis of rheumatoid arthritis. J Appl Toxicol. 2001;21:69–73. | ||
Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.