Back to Journals » International Journal of Nanomedicine » Volume 14
Synthesis and characterization of nanometer-sized liposomes for encapsulation and microRNA transfer to breast cancer cells
Authors Lujan H, Griffin WC, Taube JH ![]() , Sayes CM
, Sayes CM ![]()
Received 29 January 2019
Accepted for publication 4 May 2019
Published 11 July 2019 Volume 2019:14 Pages 5159—5173
DOI https://doi.org/10.2147/IJN.S203330
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Henry Lujan,1 Wezley C Griffin,2,3 Joseph H Taube,2,4 Christie M Sayes1,4
1Department of Environmental Science, Baylor University, Waco, TX, USA; 2Department of Biology, Baylor University, Waco, TX, USA; 3Department of Chemistry and Biochemistry, Baylor University, Waco, TX, USA; 4Institute for Biomedical Sciences, Baylor University, Waco, TX, USA
Introduction: The use of liposomes as a drug delivery carrier (DDC) for the treatment of various diseases, especially cancer, is rapidly increasing, requiring more stringent synthesis, formulation, and preservation techniques to bolster safety and efficacy. Liposomes otherwise referred to as phospholipid vesicles are self-assembled colloidal particles. When formed in either the micrometer or nanometer size range, they are ideal candidates as DDC because of their biological availability, performance, activity, and compatibility. Defining and addressing the critical quality attributes (CQAs) along the pharmaceutical production scale will enable a higher level of quality control for reproducibility. More specifically, understanding the CQAs of nanoliposomes that dictate its homogeneity and stability has the potential to widen applications in biomedical science.
Methods: To this end, we designed a study that aimed to define synthesis, characterization, formulation (encapsulation), preservation, and cargo delivery and trafficking as the major components within a target product profile for nanoliposomes. A series of synthetic schemes were employed to measure physicochemical properties relevant to nanomaterial drug product development, including concentration gradients, probe versus bath sonication, and storage temperature measured by microscopy (electron and light) and dynamic light scattering.
Results: Concentration was found to be a vital CQA as reducing concentrations resulted in nanometer-sized liposomes of <350 nm. Liposomes were loaded with microRNA and fluorescence spectroscopy was used to determine loading efficacy and stability over time. Lyophilization was used to create a dry powder formulation that was then assessed for stability for 6 months. Lastly, breast cancer cell lines were used to ensure efficacy of microRNA delivery and localization.
Conclusion: We conclude that microRNA can be loaded into nanometer-sized liposomes, preserved for months in a dried form, and maintain encapsulation after extended time periods in storage.
Keywords: liposome, microRNA, nanomaterial drug product, physicochemical characterization, drug delivery, breast cancer
Introduction
Liposomes are a vital component of the ever-evolving field of pharmaceutical drug development; they have demonstrated utility as a versatile drug delivery carrier (DDC) in many applications.1–3 More specifically, liposomes synthesized in the nanometer-size range (ie, <250 nm in diameter) are becoming a viable DDC for cancer therapeutics.4 Multiple liposome-based therapeutics are currently approved for use, including liposomal doxorubicin (Doxil®), approved for ovarian and multiple myeloma in the United States, and for metastatic breast cancer in Europe and Canada.5 The use of nanometer-sized liposomes is under extensive exploration for other biomedical applications, including theranostics (a molecular platform that simultaneously integrates diagnosis and therapy).6,7
Liposomes can be described as multi-functional non-biological complex drugs and have been shown to enhance drug solubility, offer protection from enzymatic degradation, overcome efflux pumps, and provide longer systemic circulation time than other clinical formulations.8–11 Because of the ability to engineer tunable size populations, liposomes are just as attractive as viruses for payload delivery to deep tissue sites, including primary tumors and sites of metastasis.12 However, when comparing liposomes to viral-based vectors, these synthetic delivery systems have multiple advantages including control of safe manufacturing processes, elimination of bioaerosol exposures, variable molecular composition, and increased tolerance for a variety of cargo sizes.13,14
The National Cancer Institute’s (NCI) Office of Cancer Nanotechnology Research has indicated RNA therapeutics, ie, small interfering RNA (siRNA), microRNA (miRNA), and messenger RNA (mRNA) as the next generation of nanotherapeutics to be reviewed by the FDA.15 Researchers have followed the lead of this organization and reported on the usefulness of liposome-encapsulated RNA as a potent therapeutic agent.1,16–18 Optimization of RNA-encapsulated liposomes can be accomplished in two ways. First, modifying liposome size and surface charge improves biodistribution and bioavailability. Second, RNA size and charge affect encapsulation efficacy and shelf-life. miRNA is composed of around 21 to 22 base pairs. This type of RNA species is unique (as compared to messenger, transfer, and ribosomal RNA) in its pharmaceutic application as well as its physicochemical properties (ie, size, charge, weight, and half-life). Producing RNA-loaded liposomes requires continuous monitoring of critical quality attributes (CQAs) specific to this type of payload.19 For example, cationic liposomes and liposomes prepared with cholesterol have been shown to have a better encapsulation efficiency (EE) for miRNA when compared to neutrally charged liposomes or liposomes prepared with other biomolecules.20,21 In addition, some cationic liposomes with encapsulated RNA have shown greater delivery efficiency to tumor sites.22 To enhance favorable pharmacokinetic properties, secondary coating such as poly(ethylene glycol) (PEG) can be added to improve stability and “shield” the liposomes while in the body.23 To increase the delivery of liposomes into cancer cells, targeting moieties (such as folate, transferrin, or antibodies) can be affixed to liposome surfaces to increase uptake into specific cell types.24,25
A ubiquitous challenge in RNA delivery is the low efficiency of delivery to tumor sites, in vivo.26,27 Liposomes can encapsulate either polar or non-polar molecules; size can be tailored either to induce cellular uptake or evade phagocytosis. Tumor-types with enhanced permeability retention (EPR) can be exploited to enable passive tissue uptake by engineering very small liposomes (<100 nm) to increase accumulation of therapeutic agent within a specific target site. Passive diffusion exploits the EPR effect while also lowering toxicity due to decreased circulation to unwanted areas in the body.28,29
microRNAs, cellular RNA species that prevent the production of a particular protein by binding to and targeting mRNA for destruction, are increasingly attractive targets for gene replacement therapy in cancer (and other progressive diseases).30–32 Due to their small size (ie, 21–22 base pairs), the mature, double-stranded form of microRNA mimics can be transferred into target cells without the need for viral-based expression systems.33 Synthetic miRNA mimics can be delivered without additional encapsulation but protection in nanocarriers, such as liposomes, offers the potential for greater stability over time.15,34,35 As liposomes have been under investigation for decades, they are well-positioned for rapid FDA approval as a miRNA delivery mechanism.
There are multiple non-viral vectors, such as polyethylenimine (PEI) and polyamidoamine (PAMAM), which are currently under development for delivery of anti-cancer drugs and gene therapy.36,37 However, liposome-based methods offer several advantages to these approaches. The physicochemical and biophysical properties of liposomes can be easily manipulated to deliver a variety of therapeutic payloads (DNA, RNA, hydrophobic, hydrophilic, etc.) and can be fine-tuned to provide rapid or delayed release.38 Additionally, the inherent cytotoxicity of liposomes is reduced compared to PEI or PAMAM. The efficiency of payload delivery in PEI or PAMAM is directly dependent on the molecular weight of the polymers with higher molecular weights exhibiting greater efficiency.39 However, this increased efficiency comes at a price as the larger PEI or PAMAM polymers also have a higher cytotoxicity compared to liposomes which are considered more biologically inert.36,37,40 PEI and PAMAM are also less biodegradable, which is an additional contributing factor to in vivo toxicity. Lastly, there are multiple liposome-based therapeutic approaches approved or are currently under various stages of clinical trials which target a variety of cancer types providing further support for the approach.41
The objective of this study was twofold. First, nanoliposomes were produced and characterized against microliposomes for miRNA EE. Both liposome-types were subjected to a panel of storage conditions (including dehydration, decreased temperature, and time) and re-tested for miRNA content after reconstitution. The ethanol injection method along with bath sonication was successful in encapsulating, producing, and tuning the size of synthesized liposomes for systematic downstream testing and analysis. Second, breast cancer cells were exposed to the produced liposomes to assess cellular uptake and miRNA delivery. As triple-negative breast cancer cells show severely diminished expression of miR-203, we utilized a miR-203 mimic and two TNBC cell lines, MDA-MB-231 and Hs578t, as a convenient model of experimentation.42 We observed that both micro- and nanoliposomes effectively delivered miRNA into breast cancer cells and that nanoliposomes are likely internalized through the endocytic pathway.
Methods
Production of liposomal suspensions
Didodecyldimethylammonium bromide (DDAB; Millipore Sigma, St. Louis, MO, USA), ovine cholesterol (Avanti Polar Lipids, Alabaster, AL, USA), and tocopherol PEG 1000 succinate (TPGS; Millipore Sigma) were suspended in 100% ethanol in different vials at molar ratios of 12, 7, 1 M, respectively. In a new vial, 1 mL of each reagent was mixed yielding a 67 mM stock solution. This stock solution was then diluted by transferring this solution into 100% ethanol to make working solutions. Once the desired dilution was achieved, fluorescently tagged siRNA (Santa Cruz Biotechnology, Dallas, TX, USA) or synthetic microRNA (Millipore Sigma) was added and incubated at room temperature (22 ºC) for 30 mins. To a separate vial containing 4 mL of PBS, being magnetically stirred at 1,400 RPM, 1 mL of the working solution with RNA was pulled into a glass syringe and then rapidly injected. After 5 mins of vigorous stirring, the final concentration of the resultant RNA-loaded liposome suspension underwent dialysis in a Float-A-Lyzer (8–10 kDa size; Spectrum Laboratories, Houston, TX, USA) overnight.
Tuning particle size
After the liposomes were produced, a water bath sonicator (112xx Series Ultrasonic water bath; Fisherbrand, Houston, TX, USA) was then used to vary the liposome size. Samples were sonicated at 37 kHz for 10 mins to produce the nanoliposomes. Water temperature was maintained at 20ºC to avoid the generation of excessive heat while sonicating. Figure 1 depicts the procedure used to produce liposomes and control the size population.
 |
Figure 1 Synthesis scheme used to produce the nanoliposomes used in this study. Reagents were dissolved in ethanol, diluted with PBS, and incubated with RNA. After vigorous stirring, resultant liposomes undergo dialysis for 1 day. Samples are stored at 4°C (with or without sonication). (A and B) demonstrate the multi-model size population nature of synthesized liposomes. Scale bar represents 200 nm in both images. |
Preservation via lyophilization
The produced liposomes were placed in an −80°C freezer for 4 hrs. The frozen samples were then lyophilized overnight (−100 to −120°C at 185 mT) on a Virtis SP Scientific Benchtop Pro (SP Scientific, Warminster, PA, USA) until a powder was formed. Reconstitution at the original liposome production concentration was achieved with PBS.
Size population analyses – dynamic light scattering
To determine the hydrodynamic diameter (HDD) of the liposome populations, dynamic light scattering (Zeta Sizer Nano, Malvern Panalytical Ltd, Malvern, United Kingdom) was used. Briefly, 1 mL of the liposome suspension was placed in a zeta cell (Malvern Pananalytical, Westborough, MA, USA). The cell was placed in the Zeta Sizer Nano (Malvern Pananalytical) and set to calculate size and zeta potential at a refractive index of 1.450, absorption of 0, and suspension in PBS solution.
Size population analyses – nanoparticle tracking analysis
A ZetaView (Particle Metrix, Mebane, NC, USA) using nanoparticle tracking analysis used to view and measure liposome size in real time. Samples were diluted 1:10 in either water or PBS until single particles (liposomes) could be seen by the microscope. Between each dilution and when switching solutions (PBS to H2O), the system is flushed with excess solution corresponding to the solution that is going to be used during the analysis.
Size population analyses – liposomes in serum over time
Fresh liposomes synthesized as previously described were diluted into four 5 mL vials of PBS to a concentration of 500 μm to generate a stock that is quantifiable via DLS analysis. Two of the vials were then spiked with FBS to make a 10% FBS solution in PBS. The samples were labeled as follows: microliposomes in ultrapure water (UPH2O), microliposomes in PBS, microliposomes in 10% FBS, nanoliposomes in ultrapure water (UPH2O), nanoliposomes in PBS, and nanoliposomes in 10% FBS. The six samples were then incubated at 37°C for 1, 12, 16, 24, 48, and 168 hrs. At each time point, 1 mL of sample was collected and used for size analysis via DLS.
Morphological assessments – brightfield microscopy
The microliposomes were imaged via brightfield microscopy (CytoViva® Enhanced Microscope, Auburn, AL, USA). Briefly, 10 μL of each liposome suspension was placed onto a glass microscope slide and left to dry. Glass coverslips were used to increase image quality. The nanoliposome suspension was also imaged in the same manner, however, no particles were found. Electron microscopy was employed to assess morphology.
Morphological assessments – sample preparation
For electron microscopy imaging, liposome suspensions were centrifuged at 12,000 RPM for 10 mins and re-suspended in 2% osmium tetroxide (OsO4) in PBS fixation solution for 10 mins. OsO4 is a negative lipid-targeting stain that improves image quality. Next, liposomes were centrifuged and washed 3X with PBS to remove excess stain. Finally, liposomes were analyzed immediately after the last wash or lyophilized as described above before imaging.
Morphological assessments – scanning electron microscopy
Scanning electron microscopy (SEM) was used to analyze the liposome structures. The liposomes that did not undergo lyophilization were air-dried onto SEM stubs with carbon tape. These samples required sputter-coating with iridium at a thickness of 20 nm to eliminate electron charging. Lyophilized liposome powders were placed on an SEM stub with carbon tape. Excess powder was removed prior to imaging by gently tapping the stub. SEM images were taken on the Scanning Electron Microscope Versa 3D (FEI Company, Hillsboro, OR, USA).
Morphological assessments – transmission electron microscopy
Transmission electron microscopy was also used to image liposomes (Figure 2). Briefly, 10 μL of each liposome suspension was placed on a strip of para-film. A formvar/carbon 200 mesh copper grid (Electron Microscopy Sciences, Hatfield, PA, USA) was placed on the surface of the liposome drop and left for 5 mins. The copper grid was then transferred to the surface of a uranyl acetate drop for an additional 5 mins. The grid was dabbed with an adsorbent pad to remove any excess stain. TEM images were then taken on a JEM-1010 (JEOL Inc., Peabody, MA, USA).
 |
Figure 2 Microscopy images of microliposomes and nanoliposomes. (A and B) Light micrographs of microliposomes show particles in a bimodal size distribution. (C) Scanning electron micrograph and (D) transmission electron micrograph of nanoliposomes in a monodisperse size population. |
RNA encapsulation efficiency
The efficiency of RNA encapsulation inside of micro- and nanoliposomes was measured using a Qubit microRNA assay (Molecular Probes, Thermo Fisher). First, liposomes were synthesized with non-specific siRNA (Santa Cruz Biotechnology) for encapsulation. This liposome suspension was then centrifuged at 12,000 RPM for 10 mins. The supernatant was then decanted into a new microcentrifuge tube while the pelleted liposomes were resuspended and left in lysis buffer consisting of 0.01 g sodium dodecyl sulfate (Thermo Scientific, Waltham, MA, USA), 200 μL 10% Triton X-100 solution (Thermo Scientific), and 1,800 μL Tris-EDTA Buffer (pH 8.0; G-Biosciences, St. Louis, MO, USA) for 30 mins. Second, GAPDH siRNA calibration standards in Qubit working solution were utilized to create a standard curve of siRNA in the Qubit software as detailed in the Qubit microRNA protocol. Lastly, samples were made in Qubit compatible tubes at a volume of 180 μL Qubit working solution and 20 μL of sample. Samples were run on the “microRNA assay” setting within the Qubit software.
To calculate the EE in terms of percentage (%), the following equation was used:
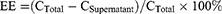
where CTotal is the total amount of RNA added to the liposome formulation during production and CSupernatant is the amount of RNA in the supernatant (ie, the portion not encapsulated in liposomes).
Cell culture
MDA-MB-231 and Hs578t cells (ATCC, Manassas, VA, USA) were cultured in DMEM (Corning, Corning, NY, USA) supplemented with 10% FBS (Gibco-ThermoFisher, Waltham, MA, USA), penicillin, and streptomycin (Lonza, Basel, Switzerland).
Fluorescent microscopy
Cells were seeded at 30,000 cells per well in an 8-chamber microscope slide. Control cells were transfected with 3 nM siRNA (FITC conjugate)-A (Santa Cruz Biotechnology) using FuGene® HD (Promega, Madison, WI, USA) following the manufacturer protocol. Unloaded and loaded liposomes in PBS were diluted to a final concentration of 75 µM in supplemented DMEM, added to the cells, and incubated for 4 hrs. Lysotracker Red DND-99 (100 µM) (ThermoFisher) was added to the cells 30 mins prior to fixation. Cells were washed 3 times with PBS followed by fixation with 4% paraformaldehyde solution. Fixation reaction was quenched with 1% glycine in PBS for 10 mins. Cells were then washed with PBS as described above and stained with DAPI. Cells were mounted with permount solution and stored at 4°C until visualization. Fluorescence confocal images were acquired with an Olympus FV-1000 confocal laser scanning microscope (Olympus Corp., Tokyo, Japan) and the FV10-ASW4.1 software package.
RNA extraction and quantitative RT-PCR
Cells exposed to 500 nM miRNA-containing liposomes were subjected to RNA extraction using Trizol following the manufacturer’s protocol (ThermoFisher); 200 ng of total RNA was subjected to miRNA-specific reverse transcription and amplification, conducted using miRNA-specific primers (Applied Biosystems, Foster City, CA, USA) and detected on a QuantStudio 5 (Applied Biosystems). Quantification of miRNA expression was performed using the delta-delta Ct method using small nucleolar U6 RNA as a normalizer.43
Results
The synthesis and characterization of liposome suspensions produced for biomedical applications are of interest to a variety of stakeholders, including drug developers, toxicologists, regulators, and clinicians. A streamlined approach for documenting synthesis schemes and physicochemical properties ought to be established to ensure drug product efficacy and safety. This is especially important for nanomaterial drug products since little information is known about their CQAs.
Here, we have systematically investigated the multiple controllable variables needed to produce high-quality RNA-loaded liposome suspensions. High quality, in this context, includes homogeneous diameter, encapsulation efficacy, stability over extended time periods, and low cytotoxicity. Table 1 lists the controllable parameters used to tune the quality of the resultant liposomes used in this study.
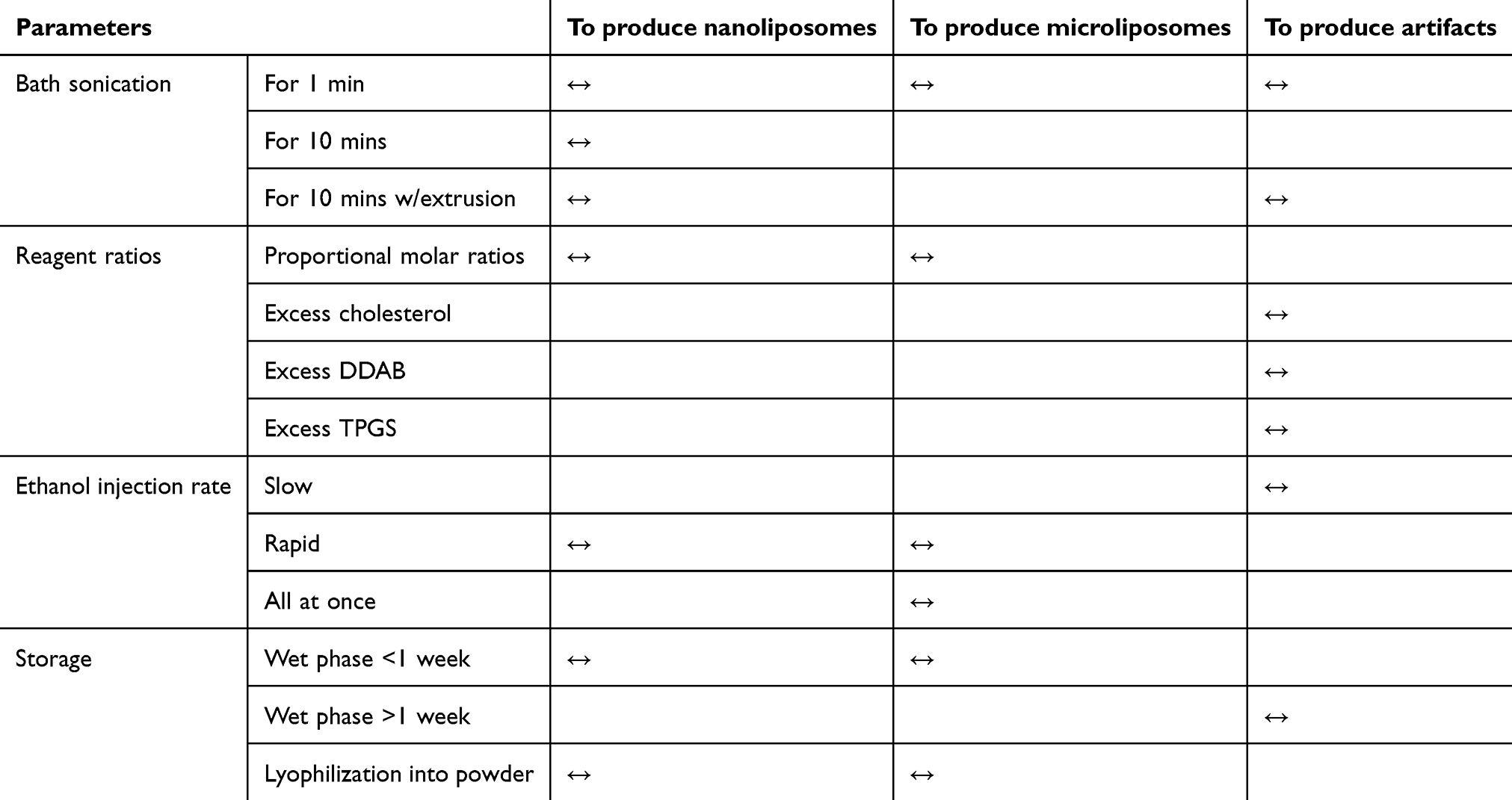 |
Table 1 List of controllable parameters used to tune the size of resultant liposomes |
The four main parameters controlled in this study included bath sonication, reagent ratio, ethanol injection rate, and storage conditions. The best combination employed to produce a high-quality nanoliposome suspension includes bath sonication for 10 mins with extrusion; the 12, 7, and 1 M ratios of DDAB, ovine cholesterol, and TPGS, respectively; rapid, but controlled, reagent injection into PBS, and storage in the wet phase (ie, in suspension) for <1 week or in the dry state (ie, lyophilized powder) up to 6 months.
The size of liposomes in aqueous suspensions changes over time and temperature. Initial liposome production occurred at room temperature (21°C). At the time of synthesis, microliposomes are 4,959 nm in HDD and the nanoliposomes are 1,869±400 nm. After 48 and 168 hrs, the HDD of the microliposomes decrease to 1,295 and 1,374 nm, respectively, while the nanoliposomes increase in HDD to 1,932 and 2,410 nm, respectively. Overall, the microliposomes decrease in size by 72% and the nanoliposomes increase in size by 22%. Table 2 includes these data, as well as the size change when placed in 4°C, the surface charge changes at both 21°C and 4°C. Storage at 4°C results in a decrease in microliposome size (4,959–1,284 nm) but causes nanoliposomes to aggregate over time (1,869–4,227 nm). Neither time nor temperature causes significant changes to liposome surface charge (as measured by zeta potential).
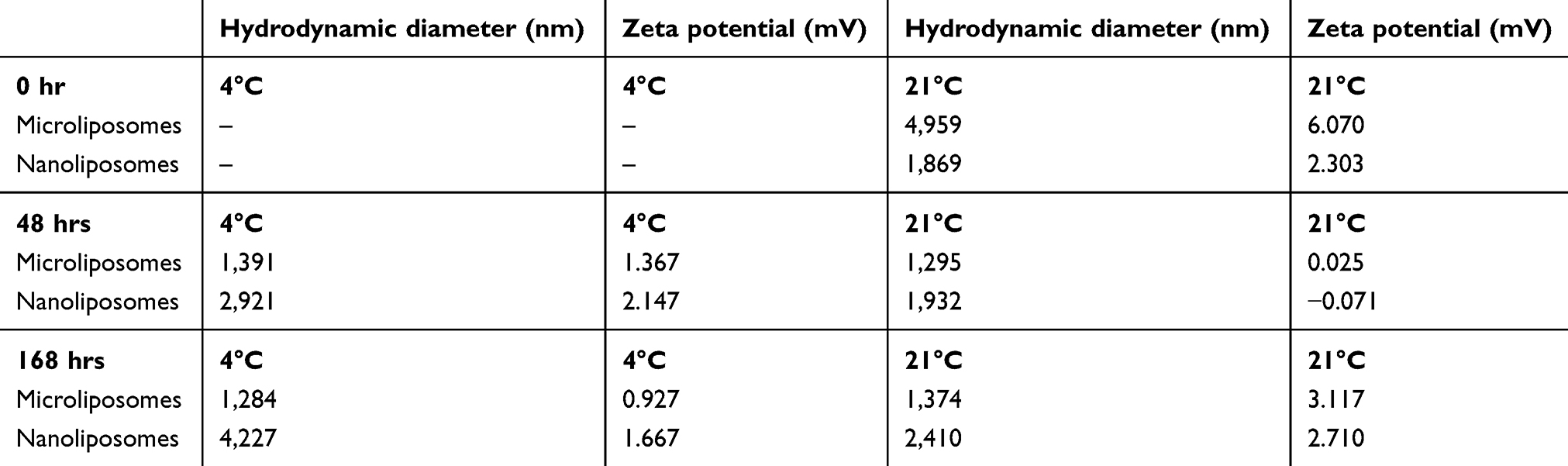 |
Table 2 Effects on miRNA-loaded liposome population size over time when changing temperature |
Interestingly, diluting the suspensions has a significant effect on the liposome size. Figure 3 shows that liposome size decreases when concentration decreases, for both micro- and nanoliposome samples. More specifically, when the microliposomes are diluted in ultrapure water, all particles are <1,500 nm in diameter, with a mean diameter of 787.6 nm. When microliposomes are diluted in PBS, all particles are also <1,500 nm in diameter but have a mean diameter of 550.4 nm. When the nanoliposomes are diluted in ultrapure water, all particles are <250 nm in diameter, with a mean diameter of 188.9 nm. When nanoliposomes are diluted in PBS, all particles are <350 nm in diameter, with a mean diameter of 168.2 nm.
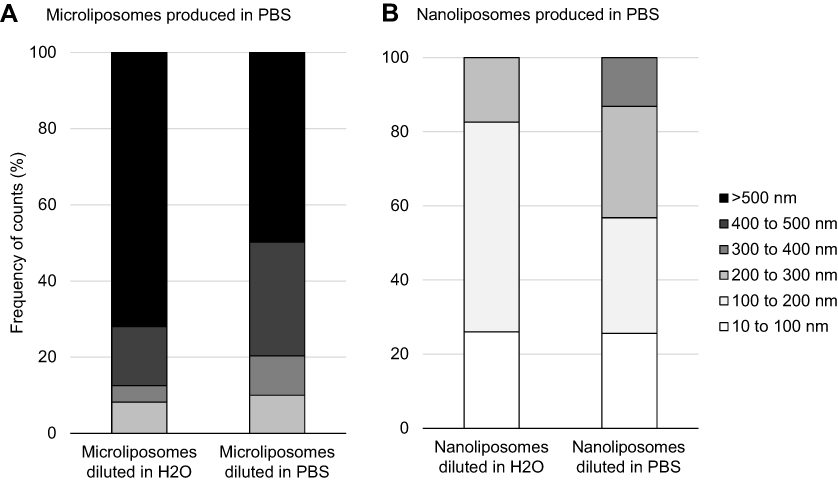 |
Figure 3 Effects on liposome population size when changing concentration and ionic strength of suspension media. (A) Particle size distribution of microliposomes. (B) Particle size distribution of nanoliposomes. Samples were diluted 100X, resulting in 1 mg/mL concentrations. For both micro- and nanoliposome samples, size decreases when concentration decreases. |
Liposomes used for biomedical applications are primarily administered while in aqueous suspension. The suspension matrix is often a buffer, such as PBS. However, the stability of produced liposomes while suspended in an aqueous suspension is short-lived; liposome suspensions degrade, through dissolution, after approximately 1 week. Refrigeration extends the shelf-life of liposomes, when kept at 4°C, suspensions remain stable up to an additional week. Because of these short stable storage time periods, the utility of liposomes in biomedical applications is limited. Notably, drug formulators have employed spray-dried or freeze-dried techniques to pharmaceuticals that are most effective as suspensions. In order to ascertain the effect of lyophilization on liposomes, we imaged dried liposomes using SEM. Figure 4 shows the SEM images of micro- and nanoliposomes lyophilized into dry powders. Liposomes maintain their homogeneous average sizes and spheroidal shape without degrading the particles or introducing other artifacts.
 |
Figure 4 Scanning electron micrographs of (A and B) microliposomes and (C and D) nanoliposomes after lyophilization and stored for 168 hrs. |
Table 3 provides further evidence of the ability to extend the shelf-life of liposomes. In this table, RNA EE is reported. During the initial production of liposomes, 98% of RNA used as a starting reagent is encapsulated in the microliposome sample. For nanoliposomes, 96% is encapsulated. After the microliposomes are lyophilized and re-suspended, 94.6% of the RNA remains encapsulated in the microliposomes and 83.4% remains encapsulated in the nanoliposomes.
 |
Table 3 RNA encapsulation efficacy in liposomes at time of production versus after resuspending stored liposomes |
To determine the stability of liposomes once introduced into a serum rich environment, liposomes were incubated in a PBS solution spiked with 10% fetal bovine serum (FBS) solution at 37°C. Measuring the physicochemical properties in this environment gives information regarding the possible changes to the liposomes after in vivo administration. The effect of FBS on the size and charge of the micro- and nanoliposomes over time (1–168 hrs) was compared to liposomes in ultra-pure water (UPH2O) as well as liposomes in PBS.
Figure 5A and B shows that the microliposomes in 10% FBS consistently maintained the smallest particle size (as measured by HDD) and most negative surface charge (as measured by zeta potential) over time. When microliposomes are suspended in UPH2O, the particle size and surface charge decreased at a faster rate as compared to the microliposomes suspended in PBS or 10% FBS. The decrease in size and charge when suspended in UPH2O is probably due to liposome collapse. The contents within the liposomes tend to diffuse to the surrounding matrix in an effort to maintain equilibrium. However, liposome size and charge increase slightly in the PBS and FBS suspensions. The increase is likely due to the formation of a corona, ie, the adsorption of proteins (in the case of FBS) or association with ions (in the case of PBS). Since the microliposomes have a positively charged surface, the PBS and FBS help to maintain charge stability and membrane integrity.
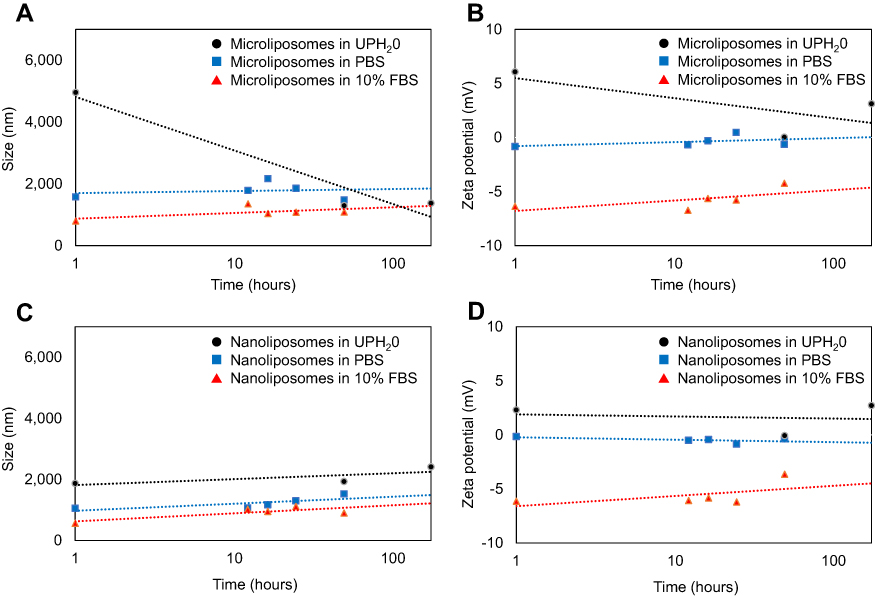 |
Figure 5 Effect on liposome population size after incubation in ultrapure water (UPH2O), PBS solution, and 10% FBS over time. The size (A) and the zeta potential (B) of the microliposomes both decrease significantly over time when in UPH2O, while the size and zeta potential of the microliposomes remain relatively constant in PBS or 10% FBS over time. However, the size (C) and zeta potential (D) of the nanoliposomes suspended in UPH2O, PBS, and FBS remain constant over time. |
Figure 5C and D plots the changes in nanoliposome particle size and surface charge. Similarly, these smaller liposomes suspended in 10% FBS consistently maintained the smallest particle size (as measured by HDD) and most negative surface charge (as measured by zeta potential) over time. However, nanoliposomes maintained relatively consistent size and charge when suspended in UPH2O and PBS. When suspended in UPH2O, nanoliposomes are less vulnerable to diffusion equilibrium because these liposomes have a smaller payload concentration. Furthermore, because nanoliposomes are smaller than microliposomes, there is less surface area on a per particle basis for proteins to adsorb or ions to associate.
We next applied synthesized and miRNA-encapsulated micro- and nano-liposomes to triple-negative breast cancer cell lines in order to ascertain their capacity to deliver miRNA and their intracellular trafficking. MDA-MB-231 and Hs578t cells, cultured in vitro and exposed to miR-203-loaded micro- and nanoliposomes, experienced a 20- to 40-fold increase in the relative amount of miR-203 compared to cells exposed to non-loaded micro- and nanoliposomes (Figure 5). As the concentration of liposomes and the concentration of loaded miRNA were held constant between micro- and nanoliposomes, we show here that the size of the liposome did not affect the amount of microRNA transferred into cells.
In order to track the internalization of liposome-loaded cargo into cells, we next visualized a fluorescently labeled siRNA, which was incorporated into nanoliposomes and then added to cultured MDA-MB-231 cells. Transferred siRNA were detected adjacent to cell nuclei and partially overlapping with endocytic vesicles, labeled with Lysotracker. Thus, nanoliposomes are sufficient to deliver detectable miRNA mimic into cultured human cells.
Discussion
Over the last 30 years, liposomes have been proposed and researched as efficient carriers for drugs, diagnostics, and vaccines. This field of study (ie, liposomology) is truly interdisciplinary; it is populated with scientists, engineers, and clinicians from diverse backgrounds who specialize in various components of the liposome drug development pipeline. The pipeline spans initial liposome synthesis to stability during storage with drug encapsulation, mass-production, and therapeutic assessment in-between (Figure 7).44 Many techniques and processes have been established (and subsequently revised) over time to balance the costs versus the benefits of liposome manufacture.13,45 This paper demonstrates the effects of various environmental conditions (such as changes in sonication and injection rates, reagent ratios, and storage parameters) on the production, stability, and efficacy of liposomes as microRNA carriers, in an effort to highlight the need for extensive physicochemical characterization of liposome drug products over the entire product life cycle.
The size of a liposome defines its utility in a biomedical application. When using micrometer or nanometer-sized liposomes in biomedical applications, the physical, chemical, and biological characterization of critical characteristics must always be considered. The design of the liposome drug product is guided by the known desired attributes. Specific applications are attainable due to the tunable characteristics of liposomes including, but not limited to, the ability to incorporate water- or lipid-soluble agents; to target specific sites in the body via number of lamellae, surface charge and functionalization; and to control particle size through subtle changes in synthesis procedures. For example, breast cancer cells and tumors exhibit the EPR effect which dictates that a DDC is in the nanometer range (around 100 nm) with a prolonged circulation.46 The improved pharmacokinetics have shown accumulation of the liposomes in the breast cancer tumors after achieving 100 nm size and utilizing PEG to increase circulation time.47 In contrast, liposomes larger than 1,000 nm deliver a higher payload concentration, but stay in systemic circulation for extended time periods (weeks).47 Research has shown that liposomes between 20 and 200 nm are optimized for enhanced drug solubility, protection from enzymatic degradation, overcoming efflux pumps, and effective circulation time.28 The best performing liposome for systemic circulation time is the nanoliposome (ie, a liposome with particle dimensions <200 nm in diameter and <0.2 in (poly)dispersity index (PdI)). Nanoliposomes, as with a plethora of other nanoparticles, easily cross the cytoplasmic membrane.48,49 With respect to liposome size, lower concentrations tend to produce small HDD measurements, due to the decreased agglomeration from fewer particles in the system.
The success of micro- and nanoliposomes in biomedical applications lies in the EE, targeted drug delivery, and prolonged stability of active pharmaceutical ingredients (API). As new drug candidates, nanoliposomes should not only demonstrate the clear advantage of the liposome carrier for the intended purpose, but also prove practical and financial feasibility of the formulation over conventional pharmaceuticals. For delivery applications, nanoliposomal formulations should possess the following CQAs:
- Narrow size distributions,
- High API entrapment efficiencies,
- Long-term stability in circulation as well as in storage,
- Precise (and measurable) release properties that are based on the intended administration, and
- Low toxicity induced by the carrier itself or other inactive ingredients.
To achieve practical and financial feasibility, nanoliposomal formulations should also possess the following qualities:
- Flexible preparation method to account for a wide range of payload ingredients,
- Ability to be subjected to sterilization procedures without significant loss in production yield, and
- Predictable cost estimates for liposome production on large scales.
The central strategy needed when producing reproducible, commercial amounts of liposomes are not necessarily assembling the membranes, but instead, focusing on the process that forms vesicles of the intended size and structure that also entrap APIs with high efficiency and without leakage.
The most common and conventional liposome production method includes suspending lipids in an organic solvent, injecting the lipids into an aqueous media, purifying the resultant liposomes, and then characterizing the final product. However, this technique results in two risky artifacts. First, solvent residues increase toxicity and, second, heterogenous size and shape decrease efficacy. Solvents (such as chloroform, ether, ethanol, or methanol) have the potential to chemically react with the entrapped API and remain present in the final liposome formulation. This contamination has been shown to increase toxicity, decrease API efficacy, and decrease the stability of the vesicles.50 Therefore, final liposome products must be subjected to decontamination and sterilization procedures. Filtration, dialysis, chemical modifications, and heat are the standard manufacturing parameters that are routinely used in pharmaceutical production processes. However, these processes contribute to the immediate degradation of the liposome drug product. Research addressing this challenge is ongoing in both academic and industrial settings.13 Figure 6 presents the liposomal drug development pipeline used today.
 |
Figure 6 Liposomes are internalized and deliver detectable levels of microRNA in the breast cancer cell lines. (A) Quantitative RT-PCR was performed on the indicated cell lines 4 hrs after addition of miR-203 loaded liposomes at a concentration of 500 nM. MCF7 cells, expressing abundant miR-203, are used as a reference cell line. Small nucleolar RNA U6 was used as a normalization control. (B) Fluorescence confocal microscopy was performed on MDA-MB-231 cells 4 hrs after exposure to the indicated liposomes or transfection reagent, mixed with FITC-labeled siRNA. Counterstaining with DAPI and LysoTracker Red was then performed. Scale bar represents 15 µm for major panels and 5 µm for insets. Arrows indicate regions with distributed extra-endosomal FITC-siRNA. Arrowhead indicates region expanded for inset. |
 |
Figure 7 The liposomal drug product development pipeline highlights the main steps of synthesis, formulation, sterilization, storage, resuspension, and administration. Additional considerations are also included to provide more details along the flowchart. |
The formation of the vesicle is only the first step in liposome drug development; subsequent steps involve the incorporation of API in the mixture. Incorporation of pharmaceutical ingredients into liposomes can be achieved by one of the three routes: adding the drug to (1) the initial reaction scheme along with the liposomal ingredients; (2) the reaction scheme after the initial temperature has dropped to (but not below) the phase transition temperature of the lipids; or (3) the liposomes after they are prepared (ie, room temperature incubation of DNA with empty liposomes). The transition temperatures of the lipids that comprise the final liposome product are a critical physicochemical property important to the overall synthetic scheme.51 Lipids have a characteristic phase transition temperature (Tm); they exist in different physical states above and below the Tm. In general, lipids tend to exist as a rigid, well-ordered arrangements when below the Tm. Alternatively, when above the Tm, lipids are in a liquid-crystalline. Liposome fluidity is tuned by the use of phospholipids with different phase transition temperatures. Cholesterol affects the fluidity of the vesicles, allowing the synthesis scheme used in this study to remain at room temperature (21°). Furthermore, at high concentrations (ie, >30 M%), cholesterol can decrease fluidity at a temperature greater than Tm.52–55 Semple et al reported that this phenomenon creates a more stable formulation while circulating in physiological fluids.54
The characteristics of the liposomes are not the only suite of factors that change the drug EE. The characteristics of the API itself also contribute to liposome drug stability.29,56,57 While one of the advantages of using nanoliposomes as carriers for drug delivery application is the ability to load the vesicle with either a water- or lipid-soluble API, water-soluble drugs are generally more amicable to entrapment in a lipid bilayer structure. Sharma et al showed that entrapment of a lipophilic drug is more efficacious when using a multi-laminar vesicle as the carrier.58 Of course, one of the most efficient ways to encapsulate a significant amount of API is to create a large unilamellar vesicle loaded with lipophilic molecules, but since the stability of these particle-types is low and the size of the particle is too high for optimized systemic performance.
Amphiphilic drugs (which contain a hydrophobic head and a lipophilic tail) are often unstable after systemic delivery into physiological fluids due to their high propensity to diffuse out of the liposomes when experiencing subtle changes in pH.59 However, recent research by Clerc et al has shown that when the active loading technique is used, ie, intentional controlling the pH of liposome interior to be either significantly higher or lower than the pH of circulation environment; then, high EE and prolonged systemic stability can be achieved.59 The notable advantage of increasing EE is decreasing the number of particles needed in the drug product to observe desired therapeutic effect. When the number of nanoliposomes is lower, then induced toxicological response is also lowered.
The last challenge in liposome drug development is the stability of the drug system when in storage. Liposomes dispersed in an aqueous suspension become unstable after long-term storage.60 Hydrolysis and oxidation of the lipids, liposome aggregation, and API leakage are the most cited causes of liposome instabilities. There are many examples in the literature demonstrating the increased stability of liposome drug products after lyophilization or freeze-drying.29,60–62 Lyophilization preserves liposomes in a dried form which can then be reconstituted with water or buffer for injection prior to administration.61 In some cases, cryoprotectants, such as lactose, sucrose, may need to be added to maintain particle size after rehydration. Lammers et al have reported that the gene-silencing efficiency of siRNA-loaded liposomes in solution showed 80% reduction following 1 month of storage at room temperature.63 Interestingly, rehydrated lyophilized liposomes showed 100% gene-silencing efficiency following the same time and temperature. Although therapeutic efficiency of liposome-based drugs may vary depending on the choice of lipids, the preparation technique, and physicochemical characteristics of the liposomes and API, lyophilization is useful for the long-term storage of liposome-based drugs.
Conclusion
The shelf-life stability of liposome drug products is largely determined by the CQAs identified by the pharmaceutical team. Characteristics such as the uniformity of size distribution, EE, and minimal drug leakage from the nano-carrier should be measured and reported. By optimizing the size distribution, pH, ionic strength, and storage conditions, liposome formulations have the potential to be distributed worldwide and remain stable for years.
Acknowledgments
CMS thanks the Department of Environmental Science. JHT thanks the Department of Biology. This study was supported in part by funds from the Young Investigator Development Program to JHT and by the Vice Provost for Research Postdoctoral support to WG through Baylor University.
Disclosure
The authors have no conflicts of interest in this work.
References
1. Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. doi:10.1126/science.1095833
2. Song XL, Ju RJ, Xiao Y, et al. Application of multifunctional targeting epirubicin liposomes in the treatment of non-small-cell lung cancer. Int J Nanomedicine. 2017;12:7433–7451. doi:10.2147/IJN.S141787
3. Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30(11):592–599. doi:10.1016/j.tips.2009.08.004
4. Miele E, Spinelli GP, Di Fabrizio E, Ferretti E, Tomao S, Gulino A. Nanoparticle-based delivery of small interfering RNA: challenges for cancer therapy. Int J Nanomedicine. 2012;7:3637–3657. doi:10.2147/IJN.S23696
5. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108
6. Wang LS, Chuang MC, Ho JA. Nanotheranostics – a review of recent publications. Int J Nanomedicine. 2012;7:4679–4695. doi:10.2147/IJN.S33065
7. Janib S, Moses A, MacKay J. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Deliv Rev. 2010;62(11):1052–1063. doi:10.1016/j.addr.2010.08.004
8. Sayes CM, Aquino GV, Hickey AJ. Nanomaterial drug products: manufacturing and analytical perspectives. Aaps J. 2017;19(1):18–25. doi:10.1208/s12248-016-0008-x
9. Sayes CM, Staats H, Hickey AJ. Scale of health: indices of safety and efficacy in the evolving environment of large biological datasets. Pharm Res. 2014;31(9):2256–2265. doi:10.1007/s11095-014-1415-2
10. Patil ML, Zhang M, Minko T. Multifunctional triblock nanocarrier (PAMAM-PEG-PLL) for the efficient intracellular siRNA delivery and gene silencing. ACS Nano. 2011;5(3):1877–1887. doi:10.1021/nn102711d
11. Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release. 2014;194:238–256. doi:10.1016/j.jconrel.2014.09.001
12. Goldman E, Zinger A, da Silva D, et al. Nanoparticles target early-stage breast cancer metastasis in vivo. Nanotechnology. 2017;28(43):43LT01. doi:10.1088/1361-6528/aa8a3d
13. Mozafari MR. Liposomes: an overview of manufacturing techniques. Cell Mol Biol Lett. 2005;10(4):711–719.
14. Vemuri S, Rhodes CT. Preparation and characterization of liposomes as therapeutic delivery systems: a review. Pharm Acta Helv. 1995;70(2):95–111.
15. Naseri Z, Oskuee R, Jaafari M, Moghadam M. Exosome-mediated delivery of functionally active miRNA-142-3p inhibitor reduces tumorigenicity of breast cancer in vitro and in vivo. Int J Nanomedicine. 2018;13:7727–7747. doi:10.2147/IJN.S182384
16. Kawasaki ES, Player A. Nanotechnology, nanomedicine, and the development of new, effective therapies for cancer. Nanomedicine. 2005;1(2):101–109. doi:10.1016/j.nano.2005.03.002
17. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. doi:10.1038/nrd2742
18. Seviour EG, Sehgal V, Mishra D, et al. Targeting KRas-dependent tumour growth, circulating tumour cells and metastasis in vivo by clinically significant miR-193a-3p. Oncogene. 2017;36(10):1339–1350. doi:10.1038/onc.2016.308
19. Peng Q, Zhang S, Yang Q, et al. Preformed albumin corona, a protective coating for nanoparticles based drug delivery system. Biomaterials. 2013;34(33):8521–8530. doi:10.1016/j.biomaterials.2013.07.102
20. Pardridge WM. shRNA and siRNA delivery to the brain. Adv Drug Deliv Rev. 2007;59(2–3):141–152. doi:10.1016/j.addr.2007.03.008
21. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. doi:10.1038/35078107
22. Thurston G, McLean JW, Rizen M, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest. 1998;101(7):1401–1413. doi:10.1172/JCI965
23. Li W, Szoka FC. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007;24(3):438–449. doi:10.1007/s11095-006-9180-5
24. Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41(2):147–162.
25. Steichen SD, Caldorera-Moore M, Peppas NA. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci. 2013;48(3):416–427. doi:10.1016/j.ejps.2012.12.006
26. Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11(1):59–67. doi:10.1038/nrc2966
27. Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2(12):711–719. doi:10.1038/nchembio839
28. Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318.
29. Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2012;7:49–60. doi:10.2147/IJN.S26766
30. Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137(6):1005–1017. doi:10.1016/j.cell.2009.04.021
31. Bader AG, Brown D, Stoudemire J, Lammers P. Developing therapeutic microRNAs for cancer. Gene Ther. 2011;18(12):1121–1126. doi:10.1038/gt.2011.79
32. Zhang B, Farwell MA. microRNAs: a new emerging class of players for disease diagnostics and gene therapy. J Cell Mol Med. 2008;12(1):3–21. doi:10.1111/j.1582-4934.2007.00196.x
33. Kaboli PJ, Rahmat A, Ismail P, Ling KH. MicroRNA-based therapy and breast cancer: a comprehensive review of novel therapeutic strategies from diagnosis to treatment. Pharmacol Res. 2015;97:104–121. doi:10.1016/j.phrs.2015.04.015
34. Gutiérrez-Puente Y, Tari AM, Stephens C, Rosenblum M, Guerra RT, Lopez-Berestein G. Safety, pharmacokinetics, and tissue distribution of liposomal P-ethoxy antisense oligonucleotides targeted to Bcl-2. J Pharmacol Exp Ther. 1999;291(2):865–869.
35. Ozpolat B, Sood AK, Lopez-Berestein G. Liposomal siRNA nanocarriers for cancer therapy. Adv Drug Deliv Rev. 2014;66:110–116. doi:10.1016/j.addr.2013.12.008
36. Li J, Liang H, Liu J, Wang Z. Poly (amidoamine) (PAMAM) dendrimer mediated delivery of drug and pDNA/siRNA for cancer therapy. Int J Pharm. 2018;546(1–2):215–225. doi:10.1016/j.ijpharm.2018.05.045
37. Cheng W, Chen L, Ho H, Lin H, Sheu M. Stearyl polyethylenimine complexed with plasmids as the core of human serum albumin nanoparticles noncovalently bound to CRISPR/Cas9 plasmids or siRNA for disrupting or silencing PD-L1 expression for immunotherapy. Int J Nanomedicine. 2018;13:7079–7094. doi:10.2147/IJN.S181440
38. Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front Pharmacol. 2015;6:286. doi:10.3389/fphar.2015.00286
39. Kircheis R, Kichler A, Wallner G, et al. Coupling of cell-binding ligands to polyethylenimine for targeted gene delivery. Gene Ther. 1997;4(5):409–418. doi:10.1038/sj.gt.3300418
40. Goyal P, Goyal K, Vijaya Kumar SG, Singh A, Katare OP, Mishra DN. Liposomal drug delivery systems–clinical applications. Acta Pharm. 2005;55(1):1–25.
41. Allen T, Cullis P. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi:10.1016/j.addr.2012.09.037
42. Taube JH, Malouf GG, Lu E, et al. Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Sci Rep. 2013;3:2687. doi:10.1038/srep02687
43. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108.
44. Sayes CM, Aquino GV, Hickey AJ. Nanomaterial drug products: manufacturing and analytical perspectives. Aaps J. 2017;19(1):1–8.
45. Maherani B, Arab-Tehrany E, Mozafari M, Gaiani C, Linder M. Liposomes: a review of manufacturing techniques and targeting strategies. Curr Nanosci. 2011;7(3):436–452. doi:10.2174/157341311795542453
46. Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63(3):170–183. doi:10.1016/j.addr.2010.10.008
47. Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994;54(4):987–992.
48. Li W, Chen C, Ye C, et al. The translocation of fullerenic nanoparticles into lysosome via the pathway of clathrin-mediated endocytosis. Nanotechnology. 2008;19(14):145102. doi:10.1088/0957-4484/19/14/145102
49. Berg J, Ho S, Hwang W, et al. Internalization of carbon black and maghemite iron oxide nanoparticle mixtures leads to oxidant production. Chem Res Toxicol. 2010;23(12):1874–1882. doi:10.1021/tx100307h
50. Grodowska K, Parczewski A. Organic solvents in the pharmaceutical industry. Acta Pol Pharm. 2010;67(1):3–12.
51. Blok MC, van der Neut-Kok EC, van Deenen LL, de Gier J. The effect of chain length and lipid phase transitions on the selective permeability properties of liposomes. Biochim Biophys Acta. 1975;406(2):187–196. doi:10.1016/0005-2736(75)90003-6
52. Coderch L, Fonollosa J, De Pera M, Estelrich J, De La Maza A, Parra JL. Influence of cholesterol on liposome fluidity by EPR. Relationship with percutaneous absorption. J Control Release. 2000;68(1):85–95.
53. López-Pinto JM, González-Rodríguez ML, Rabasco AM. Effect of cholesterol and ethanol on dermal delivery from DPPC liposomes. Int J Pharm. 2005;298(1):1–12. doi:10.1016/j.ijpharm.2005.02.021
54. Semple SC, Chonn A, Cullis PR. Influence of cholesterol on the association of plasma proteins with liposomes. Biochemistry. 1996;35(8):2521–2525. doi:10.1021/bi950414i
55. Kunastitchai S, Sarisuta N, Panyarachun B, Müller BW. Physical and chemical stability of miconazole liposomes prepared by supercritical aerosol solvent extraction system (ASES) process. Pharm Dev Technol. 2007;12(4):361–370. doi:10.1080/10837450701369352
56. Patel P, Pailla SR, Rangaraj N, Cheruvu HS, Dodoala S, Sampathi S. Quality by design approach for developing lipid-based nanoformulations of gliclazide to improve oral bioavailability and anti-diabetic activity. AAPS PharmSciTech. 2019;20(2):45. doi:10.1208/s12249-018-1214-x
57. George M, Ghosh I. Identifying the correlation between drug/stabilizer properties and critical quality attributes (CQAs) of nanosuspension formulation prepared by wet media milling technology. Eur J Pharm Sci. 2013;48(1–2):142–152. doi:10.1016/j.ejps.2012.10.004
58. Sharma A, Straubinger RM. Novel taxol formulations: preparation and characterization of taxol-containing liposomes. Pharm Res. 1994;11(6):889–896.
59. Clerc S, Barenholz Y. Loading of amphipathic weak acids into liposomes in response to transmembrane calcium acetate gradients. Biochim Biophys Acta. 1995;1240(2):257–265.
60. Chen C, Han D, Cai C, Tang X. An overview of liposome lyophilization and its future potential. J Control Release. 2010;142(3):299–311. doi:10.1016/j.jconrel.2009.10.024
61. El-Nesr OH, Yahiya SA, El-Gazayerly ON. Effect of formulation design and freeze-drying on properties of fluconazole multilamellar liposomes. Saudi Pharm J. 2010;18(4):217–224. doi:10.1016/j.jsps.2010.07.003
62. Glavas-Dodov M, Fredro-Kumbaradzi E, Goracinova K, et al. The effects of lyophilization on the stability of liposomes containing 5-FU. Int J Pharm. 2005;291(1–2):79–86. doi:10.1016/j.ijpharm.2004.07.045
63. Lammers T, Hennink WE, Storm G. Tumour-targeted nanomedicines: principles and practice. Br J Cancer. 2008;99(3):392–397. doi:10.1038/sj.bjc.6604483
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.