")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Synthesis and biological evaluation of a novel class of curcumin analogs as anti-inflammatory agents for prevention and treatment of sepsis in mouse model
Authors Zhao C , Zhang Y, Zou P, Wang J, He W, Shi D, Li H, Liang G, Yang S, Zhang X
Received 15 October 2014
Accepted for publication 20 November 2014
Published 18 March 2015 Volume 2015:9 Pages 1663—1678
DOI https://doi.org/10.2147/DDDT.S75862
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Chengguang Zhao,1,2,* Yali Zhang,1,2,* Peng Zou,1 Jian Wang,3 Wenfei He,2 Dengjian Shi,2 Huameng Li,2 Guang Liang,2 Shulin Yang1
1School of Environmental and Biological Engineering, Nanjing University of Science and Technology, Nanjing, 2Chemical Biology Research Center, School of Pharmaceutical Sciences, 3Department of Orthopedics, The 1st Affiliated Hospital, Wenzhou Medical University, Wenzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: A novel class of asymmetric mono-carbonyl analogs of curcumin (AMACs) were synthesized and screened for anti-inflammatory activity. These analogs are chemically stable as characterized by UV absorption spectra. In vitro, compounds 3f, 3m, 4b, and 4d markedly inhibited lipopolysaccharide (LPS)-induced expression of pro-inflammatory cytokines tumor necrosis factor-α and interleukin-6 in a dose-dependent manner, with IC50 values in low micromolar range. In vivo, compound 3f demonstrated potent preventive and therapeutic effects on LPS-induced sepsis in mouse model. Compound 3f downregulated the phosphorylation of extracellular signal-regulated kinase (ERK)1/2 MAPK and suppressed IκBα degradation, which suggests that the possible anti-inflammatory mechanism of compound 3f may be through downregulating nuclear factor kappa binding (NF-κB) and ERK pathways. Also, we solved the crystal structure of compound 3e to confirm the asymmetrical structure. The quantitative structure–activity relationship analysis reveals that the electron-withdrawing substituents on aromatic ring of lead structures could improve activity. These active AMACs represent a new class of anti-inflammatory agents with improved stability, bioavailability, and potency compared to curcumin. Our results suggest that 3f may be further developed as a potential agent for prevention and treatment of sepsis or other inflammation-related diseases.
Keywords: asymmetric mono-carbonyl analogs of curcumin (AMACs), stability, anti-inflammatory property, sepsis, QSAR
Introduction
Emerging evidence indicates that inflammation is involved not only in diseases caused by microbial pathogens but also in many chronic diseases such as heart disease, hypertension, cancer, and diabetes.1–3 Appropriate inflammatory response is beneficial for the host to protect against injuries caused by harmful stimuli, including pathogens and poisons, but uncontrolled inflammation leads to extensive tissue damage and manifestation of pathological states such as sepsis, which is initiated by Gram-negative bacteria and mediated by the pro-inflammatory cytokines and becomes the most common cause of morbidity and mortality in intensive care units.4,5 Pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-6 are critically involved in inflammation and related disorders.6 Overexpressions of TNF-α and IL-6 are responsible for the initiation and extension of pathological disorders including ulcerative colitis, diabetes, multiple sclerosis, atherosclerosis, and septic shock.7–9 Thus, anti-inflammatory agents that inhibit the overexpression of pro-inflammatory cytokines are of great interest for the clinical treatment of many inflammatory diseases including sepsis.
Curcumin (diferuloylmethane) (Figure 1), a major component of the popular spice turmeric (Curcuma longa) in Southeast Asia, has been found to have various bioactivities including anti-inflammatory, antioxidant, antiproliferative, immunomodulatory, and neuroprotective activities.10–12 In recent years, the anti-inflammatory properties of curcumin have been suggested on the basis of a number of in vitro and in vivo studies.13 Although curcumin exhibits good bioactivities and low toxicity, the clinical application of curcumin is limited due to its low stability and poor bioavailability.14,15 Thus, active and clinically promising curcumin analogs have been highly desired. During the last decade, synthetic curcumin analogs have been intensively investigated to enhance the bioactivities and bioavailability.13,16 Among the analogs, mono-carbonyl analogs of curcumin (MACs) were designed by deleting the reactive β-diketone moiety, which is considered to be the major contributor to the instability and rapid degradation of curcumin. This class of analogs has received much attention due to their improved chemical stability and pharmacokinetic profiles. Several representative MACs, such as FLLL12, PAC, UBS109, EF24, and HO-3867, have showed remarkable bioactivity both in vitro and in vivo (Figure 1).17–21
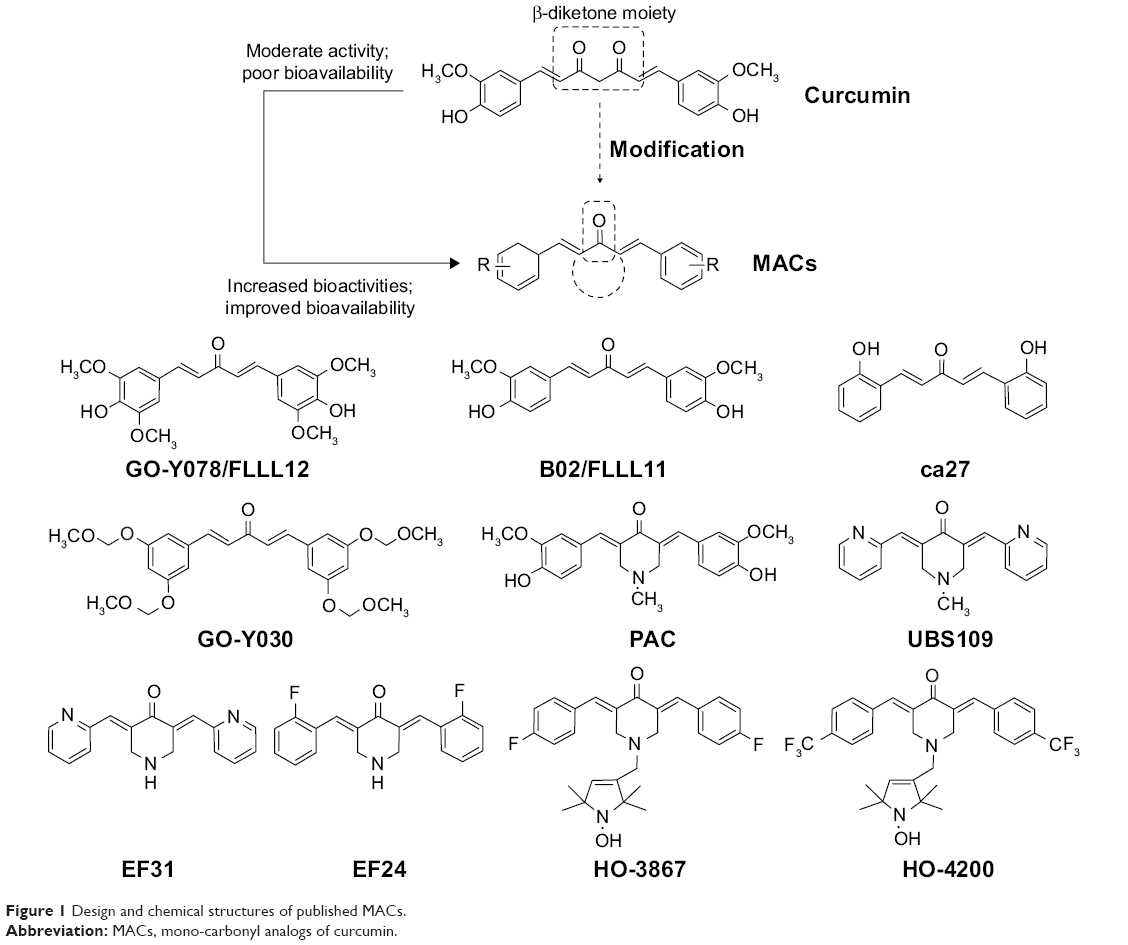 | Figure 1 Design and chemical structures of published MACs. |
This class of MACs is structurally symmetric. Studies also suggest that aromatic regions of these symmetric MACs might be critical for bioactivities.22–24 Although reports on biological evaluations of the symmetric MACs have been published in recent years, studies on the asymmetric MACs (AMACs) as anti-inflammatory agents remain scarce. It would be interesting to investigate the AMACs with two asymmetric aromatic moieties and different substituents for their anti-inflammatory effects and structure–activity relationship (SAR). Furthermore, the asymmetrical structure may improve the specificity of the active compound. Herein, a series of new AMACs with two different aromatic groups, separated by five carbon spacers, were designed, synthesized, and evaluated for their anti-inflammatory activities in vitro and in vivo.
Materials and methods
General procedure for the synthesis of compounds
All chemical reagents were obtained from Sigma-Aldrich, Fluka, or Aladdin, and were used without purification. Silica gel (GF254) for thin-layer chromatography and column chromatography (100–200 mesh and 200–300 mesh) were obtained from Aladdin. Melting points were determined on a Fisher-Johns melting apparatus and were uncorrected. 1H nuclear magnetic resonance (NMR) spectra were recorded on a Bruker 500-MHz instrument. The chemical shifts were presented in terms of parts per million with tetramethylsilane as the internal reference. Data of electron-spray ionization mass spectra (ESI-MS) in positive mode were recorded on a Bruker Esquire 3000+ spectrometer. All reagents, including newly synthesized compounds, have ≥95% purity as determined by high-performance liquid chromatography.
To the mixture of (E)-4-phenylbut-3-en-2-one (3 series) or (E)-4-(p-tolyl)but-3-en-2-one (4 series) (1.0 mmol) and aromatic aldehyde (1.0 mmol) in 6 mL EtOH and 3 mL H2O, was added 1.2 mL of 10% NaOH. The reaction was stirred at room temperature for 30 minutes and monitored by thin-layer chromatography to determine completion. Upon completion, ice water is added into the reaction mixture to precipitate the product. The solid products were purified by recrystallization. The oily compounds were purified by silica gel column chromatography (elutant: EtOAc/hexane). Their structures were determined by spectral data from ESI-MS and 1H NMR. The spectral data of the unreported compounds are shown below.
(1E,4E)-1-(2-Methoxyphenyl)-5-phenylpenta-1,4-dien-3-one (3a)
Yellow oil, 67.35% yield. 1H NMR (500 MHz, CDCl3) δ 8.10 (d, J =16.1 Hz, 1H, Ar (OCH3) −CH=), 7.76 (d, J =15.9 Hz, 1H, Ar–CH=), 7.66–7.64 (m, 3H, Ar–H2,6, Ar(OCH3)–H6), 7.44–7.39 (m, 4H, Ar–H3–5, Ar(OCH3) –H4), 7.16 (d, J =16.0 Hz, 2H, Ar–C=CH×2), 7.03 (d, J =7.5 Hz, 1H, Ar(OCH3)–H5), 6.97 (d, J =8.3 Hz, 1H, Ar(OCH3)–H3), 3.81 (s, 3H, OCH3). ESI-MS m/z: 264.9 (M+H)+, 286.9 (M+Na)+, calcd for C18H16O2: 264.12.
(1E,4E)-1-(2-Fluorophenyl)-5-phenylpenta-1,4-dien-3-one (3b)
Yellow oil, 56.42% yield. 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J =16.1 Hz, 1H, Ar(F)–CH=), 7.78 (d, J =16.0 Hz, 1H, Ar–CH=), 7.67−7.64 (m, 3H, Ar–H2,6, Ar(F)–H4), 7.45−7.38 (m, 4H, Ar–H3–5, Ar(F)–H6), 7.32 (m, 2H, Ar(F)–H3,5), 7.21 (d, J =16.1 Hz, 1H, Ar–C=CH), 7.12 (d, J =15.9 Hz, 1H, Ar(F)–C=CH). ESI-MS m/z: 252.9 (M+H)+, 274.9 (M+Na)+, calcd for C17H13FO: 252.10.
(1E,4E)-1-(2-Bromophenyl)-5-phenylpenta-1,4-dien-3-one (3c)
Yellow powder, 32.69% yield, mp 105.23°C–106.50°C. 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J =16.0 Hz, 1H, Ar(Br)–CH=), 7.78 (d, J =16.0 Hz, 1H, Ar–CH=), 7.75–7.69 (m, 3H, Ar–H2,6, Ar(Br) −H3), 7.68–7.59 (m, 6H, Ar–H3–5, Ar(Br)–H4–6), 7.15 (d, J =15.9 Hz, 1H, Ar–C=CH), 7.02 (d, J =16.0 Hz, 1H, Ar(Br)–C=CH). ESI-MS m/z: 334.9 (M+Na)+, calcd for C17H13BrO: 312.01.
(1E,4E)-1-(3,4-Dimethoxyphenyl)-5-phenylpenta-1,4-dien-3-one (3d)
Yellow oil, 56.24% yield. 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J =15.9 Hz, 1H, Ar–CH=), 7.72 (d, J =15.9 Hz, 1H, Ar–CH=), 7.64 (dd, J =6.5, 2.9 Hz, 2H, Ar–H2,6), 7.59–7.54 (m, 1H, Ar–H4), 7.49 (dd, J =8.2, 1.9 Hz, 1H, Ar(OCH3)–H2), 7.44 (d, J =1.8 Hz, 2H, Ar–H3,5), 7.23 (dd, J =8.3, 1.9 Hz, 1H, Ar(OCH3)–H6), 7.13 (d, J =15.9 Hz, 2H, CO–CH=×2), 7.02–6.98 (m, 1H, Ar(OCH3)–H5), 3.97 (s, 6H, −OCH3×2). ESI-MS m/z: 295.2 (M+H)+, 317.0 (M+Na)+ calcd for C19H18O3: 294.13.
(1E,4E)-1-(3,4-Dichlorophenyl)-5-phenylpenta-1,4-dien-3-one (3e)
Yellow powder, 86.72% yield, mp 131.2°C–132.57°C. 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J =16.0 Hz, 1H, Ar(Cl)–CH=), 7.73 (s, 1H, Ar–CH=), 7.66–7.63 (m, 3H, Ar–H2,6, Ar(Cl) −H6), 7.51 (d, J =8.3 Hz, 1H, Ar(Cl)–H5), 7.45 (m, 4H, Ar–H3–5, Ar(Cl)–H2), 7.10 (d, J =11.4 Hz, 1H, Ar–C=CH), 7.07 (d, J =11.5 Hz, 1H, Ar(Cl)–C=CH). ESI-MS m/z: 324.8 (M+Na)+, calcd for C17H12Cl2O: 302.03.
(1E,4E)-1-(2-Nitrophenyl)-5-phenylpenta-1,4-dien-3-one (3f)
Brick red powder, 46.65% yield, mp 90.4°C–91.57°C. 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J =16.0 Hz, 1H, Ar (NO2) −CH=), 7.75 (dd, J =7.8, 1.4 Hz, 1H, Ar (NO2)–H3), 7.71 (t, J =7.5 Hz, 1H, Ar(NO2)−H6), 7.67–7.64 (m, 2H, Ar (NO2)–H4, Ar–CH=), 7.62–7.56 (m, 1H, Ar(NO2)–H5), 7.46–7.44 (m, 5H, Ar–H), 7.15 (d, J =16.0 Hz, 1H, Ar(NO2)–C=CH), 6.94 (d, J =15.9 Hz, 1H, Ar–C=CH). ESI-MS m/z: 279.8 (M+H)+, 301.8 (M+Na)+. calcd for C17H13NO3: 279.09.
(1E,4E)-1-Phenyl-5-[4-(pyrrolidin-1-yl)phenyl]penta-1,4-dien-3-one (3i)
Brick red powder, 36.12% yield. mp 66.45°C−68.55°C. 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J =16.2 Hz, 2H, Ar–CH=×2), 7.73–7.67 (m, 4H, Ar-H2,6×2), 7.54–7.40 (m, 5H, Ar–H3–5, Ar(N) −H3,5), 7.19 (d, J =16.1 Hz, 2H, Ar–C=CH×2), 3.39 (t, J =5.6 Hz, 4H, N–CH2×2), 2.06 (m, 4H, CH2–CH2). ESI-MS m/z: 304.1 (M+H)+, calcd for C21H21NO: 303.16.
(1E,4E)-1-[4-(tert-Butyl)phenyl]-5-phenylpenta-1,4-dien-3-one (3j)
Yellow powder, 68.28% yield, mp 64.20°C−66.57°C. 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J =16.4 Hz, 2H, Ar–CH=×2), 7.63 (d, J =8.0 Hz, 2H, Ar(C(CH3)3−H2,6), 7.57 (d, J =8.0 Hz, 2H, Ar–H2,6), 7.51 (d, J =8.4 Hz, 2H, Ar–H3,5), 7.40 (m, 1H, Ar–H4), 7.32 (d, J =8.0 Hz, 2H, Ar(C(CH3)3−H3,5), 7.12 (d, J =15.6 Hz, 2H, Ar–C=CH×2), 1.36 (s, 9H, −C(CH3)3). ESI-MS m/z: 291.0 (M+H)+, calcd for C21H22O: 290.17.
(1E,4E)-1-phenyl-5-(thiophen-2-yl)penta-1,4-dien-3-one (3m)
Yellow powder, 76.2% yield, mp 87.0°C−89.95°C. [92°C−96°C, lit.25].
(1E,4E)-1-(2-Methoxyphenyl)-5-(p-tolyl)penta-1,4-dien-3-one (4a)
Yellow oil, 57.20% yield. 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J =16.2 Hz, 1H, Ar(OCH3)CH=), 7.74 (d, J =15.9 Hz, 1H, Ar(CH3)CH=), 7.64 (dd, J =7.7, 1.4 Hz, 1H, Ar(OCH3) −H6), 7.55 (d, J =8.0 Hz, 2H, Ar(CH3)–H2,6), 7.39 (d, J =6.9 Hz, 1H, Ar(OCH3)–H4), 7.24 (d, J =7.8 Hz, 2H, Ar(CH3)–H3,5), 7.17 (s, 1H, Ar(CH3)C=CH), 7.13 (d, J =6.2 Hz, 1H, Ar(OCH3)–H5), 7.02 (s, 1H, Ar(OCH3)–H3), 6.96 (d, J =16.3 Hz, 1H, Ar(OCH3)C=CH), 3.95 (s, 3H, −OCH3), 2.41 (s, 3H, −CH3). ESI-MS m/z: 279.0 (M+H)+, calcd for C19H18O2: 278.13.
(1E,4E)-1-(2-Fluorophenyl)-5-(p-tolyl)penta-1,4-dien-3-one (4b)
Yellow powder, 72.93% yield, mp 68.80°C−69.76°C. 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J =16.1 Hz, 1H, Ar(F)CH=), 7.75 (d, J =15.9 Hz, 1H, Ar(CH3)CH=), 7.65 (td, J =7.6, 1.6 Hz, 1H, Ar(F)–H4), 7.55 (d, J =8.1 Hz, 2H, Ar(CH3)–H2,6), 7.40 (d, J =8.0 Hz, 1H, Ar(F)–H6), 7.25 (d, J =8.0 Hz, 2H, Ar(CH3)–H3,5), 7.22 (s, 1H, Ar(F)–H3), 7.19 (s, 1H, Ar(F)–H5),7.14 (d, J =16.4 Hz, 1H, Ar(CH3)C=CH), 7.08 (d, J =15.9 Hz, 1H, Ar(F)C=CH), 2.42 (s, 3H, –CH3). ESI-MS m/z: 266.9 (M+H)+, calcd for C18H15FO: 266.11.
(1E,4E)-1-(2-Bromophenyl)-5-(p-tolyl)penta-1,4-dien-3-one (4c)
Yellow oil, 69.43% yield. 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J =16.0 Hz, 1H, Ar(Br)CH=), 7.77 (d, 1H, Ar(CH3)CH=), 7.72 (dd, J =7.8, 1.6 Hz, 1H, Ar(Br)-H3), 7.66 (dd, J =8.0, 1.1 Hz, 1H, Ar(Br)–H5), 7.55 (d, J =8.1 Hz, 2H, Ar(CH3)–H2,6), 7.38 (t, J =7.3 Hz, 1H, Ar(Br)–H6), 7.27–7.26 (m, 1H, Ar(Br)–H4), 7.25 (d, J =8.0 Hz, 2H, Ar(CH3)–H3,5), 7.10 (d, J =15.9 Hz, 1H, Ar(CH3)C=CH), 7.01 (d, J =15.9 Hz, 1H, Ar(Br)C=CH), 2.42 (s, 3H, −CH3). ESI-MS m/z: 326.9 (M+H)+, calcd for C18H15BrO: 326.03.
(1E,4E)-1-(3,4-Dimethoxyphenyl)-5-(p-tolyl)penta-1,4-dien-3-one (4d)
Yellow powder, 39.29% yield, mp 128.50°C−129.40°C. 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J =15.7 Hz, 2H, ArCH=×2), 7.54 (d, J =8.1 Hz, 2H, Ar(CH3)–H2,6), 7.24 (d, J =8.1 Hz, 2H, Ar(CH3)–H3,5), 7.22 (d, J =1.8 Hz, 1H, Ar(OCH3)–H2), 7.17 (d, J =1.9 Hz, 1H, Ar(OCH3)–H6), 7.08 (d, J =15.9 Hz, 1H, Ar(CH3)C=CH), 6.97 (d, J =15.9 Hz, 1H, Ar(OCH3)C=CH), 6.92 (d, J =8.3 Hz, 1H, Ar(OCH3)–H5), 3.97 (s, 6H, −OCH3×2), 2.41 (s, 3H, −CH3). ESI-MS m/z: 309.0 (M+H)+, calcd for C20H20O3: 308.14.
(1E,4E)-1-(3,4-Dichlorophenyl)-5-(p-tolyl)penta-1,4-dien-3-one (4e)
Light yellow powder, 85.76% yield, mp 137.30°C−138.57°C. 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J =16.0 Hz, 2H, ArCH=×2), 7.63 (d, J =8.9 Hz, 1H, Ar(Cl)–H6), 7.54 (d, J =8.1 Hz, 2H, Ar(CH3)–H2,6), 7.51 (d, J =8.3 Hz, 1H, Ar(Cl)–H5), 7.45 (dd, J =8.3, 1.9 Hz, 1H, Ar(Cl)–H2), 7.25 (d, J =8.0 Hz, 2H, Ar(CH3)–H3,5), 7.09 (d, J =15.9 Hz, 1H, Ar(CH3)C=CH), 7.02 (d, J =15.9 Hz, 1H, Ar(Cl)C=CH), 2.42 (s, 3H, −CH3). ESI-MS m/z: 319.0 (M+H)+, 338.9 (M+Na)+, calcd for C18H14Cl2O: 316.04.
(1E,4E)-1-(2-Nitrophenyl)-5-(p-tolyl)penta-1,4-dien-3-one (4f)
Invisible green, 19.11.% yield, mp 106.63°C−107.97°C. 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J =15.9 Hz, 1H, Ar(NO2)CH=), 8.10 (d, J =8.1 Hz, 1H, Ar(NO2)–H3), 7.78 (d, J =15.9 Hz, 1H, Ar(CH3)CH=), 7.75 (d, J =6.7 Hz, 1H, Ar(NO2)–H6), 7.70 (t, J =7.4 Hz, 1H, Ar(NO2)–H4), 7.61–7.56 (m, 1H, Ar(NO2)–H5), 7.56 (d, J =7.9 Hz, 2H, Ar(CH3)–H2,6), 7.25 (d, J =7.9 Hz, 2H, Ar(CH3)–H3,5), 7.10 (d, J =15.9 Hz, 1H, Ar(CH3)C=CH), 6.94 (d, J =15.9 Hz, 1H, Ar(NO2)C=CH), 2.42 (s, 3H, −CH3). ESI-MS m/z: 294.0 (M+H)+, calcd for C18H15NO3: 293.11.
(1E,4E)-1-Phenyl-5-(p-tolyl)penta-1,4-dien-3-one (4g)
Yellow powder, 91.08% yield, mp 98.97°C−100.23°C. [110°C–111°C, lit.26].
(1E,4E)-1-(4-(diethylamino)phenyl)-5-(p-tolyl)penta-1,4-dien-3-one (4h)
Tangerine oil, 76.2% yield. 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J =16.2 Hz, 2H, ArCH=×2), 7.62 (d, J =8.3 Hz, 2H, Ar(N)–H2,6), 7.56 (d, J =8.4 Hz, 2H, Ar(CH3)–H2,6), 7.54–7.42 (m, 4H, Ar(N)–H3,5, Ar(CH3)–H3,5), 6.69 (d, J =15.6 Hz, 2H, ArC=CH×2), 3.44 (m, 4H, N–CH2×2), 2.38 (s, 3H, Ar–CH3), 1.22 (m, 6H, −CH3×2). ESI-MS m/z: 320.1 (M+H)+, calcd for C22H25NO: 319.19.
(1E,4E)-1-(4-(pyrrolidin-1-yl)phenyl)-5-(p-tolyl) penta-1,4-dien-3-one (4i)
Tangerine oil, 26.34% yield. 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J =16.4 Hz, 2H, ArCH=×2), 7.53–7.50 (m, 4H, Ar(N)–H2,6, Ar(CH3)–H2,6), 7.44 (d, J =8.0 Hz, 2H, Ar(CH3)–H3,5), 7.21 (d, J =7.8 Hz, 2H, Ar(N)–H3,5), 6.57 (d, J =15.6 Hz, 2H, ArC=CH×2), 3.39 (t, J =6.6 Hz, 4H, N–CH2×2), 2.38 (s, 3H, Ar–CH3), 1.96 (m, 4H, CH2–CH2). ESI-MS m/z: 318.1 (M+H)+, calcd for C22H23NO: 317.18.
(1E,4E)-1-[4-(tert-Butyl)phenyl]-5-(p-tolyl)penta-1,4-dien-3-one (4j)
Yellow powder, 78.95% yield, mp 140.97°C−142.27°C. 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J =15.8 Hz, 2H, ArCH=×2), 7.58 (d, J =8.3 Hz, 2H, Ar(C(CH3)3−H2,6), 7.54 (d, J =8.0 Hz, 2H, Ar(CH3)–H2,6), 7.46 (d, J =8.4 Hz, 2H, Ar(C(CH3)3−H3,5), 7.25 (d, J =8.0 Hz, 2H, Ar(CH3)−H3,5), 7.09 (d, J =15.6 Hz, 2H, ArC=CH×2), 2.42 (s, 3H, –CH3), 1.36 (s, 9H, −C(CH3). ESI-MS m/z: 305.1 (M+H)+, calcd for C22H24O: 304.18.
(1E,4E)-1-[4-(Benzyloxy)phenyl]-5-(p-tolyl)penta- 1,4-dien-3-one (4k)
Yellow powder, 24.28% yield, mp 143.13°C−149.17°C. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J =15.8 Hz, 2H, ArCH=×2), 7.60 (d, J =8.7 Hz, 2H, Ar(OCH2Ar)–H2,6), 7.54 (d, J =8.0 Hz, 2H, Ar(CH3)–H2,6), 7.46 (d, J =7.0 Hz, 2H, −OCH2Ar–H2,6), 7.43 (d, J =7.2 Hz, 2H, −OCH2Ar–H3,5), 7.38 (d, J =7.0 Hz, 1H, −OCH2Ar–H4), 7.24 (d, J =7.9 Hz, 2H, Ar(CH3)–H3,5), 7.06 (d, J =16.3 Hz, 2H, ArC=CH×2), 7.01 (d, J =7.9 Hz, 2H, Ar(OCH2Ar)–H3,5), 5.14 (s, 2H, ArOCH2Ar), 2.42 (s, 3H, −CH3). ESI-MS m/z: 355.1 (M+H)+, calcd for C25H22O2: 354.16.
(1E,4E)-1-(furan-2-yl)-5-(p-tolyl)penta-1,4-dien-3-one (4l)
Lignt yellow powder, 83.16% yield, mp 81.10°C−82.20°C. 1H NMR (CDCl3) δ 7.66 (d, J =16.0 Hz, 1H, Ar–CH=C), 7.51 (d, J =15.7 Hz, 1H, Furan–CH=C), 7.49–7.54 (m, 3H, Furan–H5, Ar–H2,6), 7.38 (d, J =8.2 Hz, 2H, Ar–H3,5), 6.97 (d, J =15.7 Hz, 2H, Ar–C=CH×2), 6.71 (d, J =3.0 Hz, 1H, Furan–H3), 6.52 (s, 1H, Furan–H4), 2.42 (s, 3H,–CH3). ESI-MS m/z: 238.9 (M+H)+, calcd for C16H14O2: 238.10.
(1E,4E)-1-(thiophen-2-yl)-5-(p-tolyl)penta-1,4-dien-3-one (4m)
Yellow powder, 88.95% yield, mp 78.27°C−79.23°C. 1H NMR (CDCl3) δ 7.86 (d, J =15.5 Hz, 1H, Ar-CH=C), 7.70 (d, J =15.9 Hz, 1H, Thiophene–CH=C), 7.51–7.52 (d, J =8.0 Hz, 2H, Ar–H2,6), 7.41 (d, J =5.0 Hz, 1H, Thiophene–H5), 7.34 (d, J =3.5 Hz, 1H, Thiophene–H3), 7.22 (d, J =7.9 Hz, 2H, Ar–H3,5), 7.09 (dd, J =4.9, 3.7 Hz, 1H, Thiophene–H4), 6.98 (d, J =15.9 Hz, 1H, ArC=CH), 6.89 (d, J =15.5 Hz, 1H, ArC=CH), 2.39 (s, 3H, −CH3). ESI-MS m/z: 255.0 (M+H)+, calcd for C16H14OS: 254.08.
Animals
Male C57BL/6 mice weighing 18–22 g were obtained from the Animal Center of Wenzhou Medical University (Wenzhou, People’s Republic of China). Animals were housed at a constant room temperature with a 12-hour/12-hour light–dark cycle and fed with a standard rodent diet and water. The animals were acclimatized to the laboratory for at least 7 days before being used in the experiments. Protocols involving the use of animals were approved by the Wenzhou Medical University’s Animal Policy and Welfare Committee (approval documents: 2009/APWC/0031).
Reagents
Lipopolysaccharides (LPSs) were purchased from Sigma-Aldrich (St Louis, MO, USA). In addition, eBioscience (San Diego, CA, USA) was the source of the mouse IL-6 enzyme-linked immunosorbent assay (ELISA) kit and mouse TNF-α ELISA kit. Anti-glyceraldehyde 3-phosphate dehydrogenase, anti-IκBα (nuclear factor of kappa light-polypeptide gene enhancer in B-cells inhibitor alpha), and anti-extracellular signal-regulated kinase (ERK) antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Ultraviolet–visible (UV–vis) absorption spectra of curcumin and its analogs
Absorbance readings were taken from 250 nm to 600 nm on a spectraMax M5 (Molecular Devices, Sunnyvale, CA, USA). In the experiments where degradation of compounds was recorded, the absorption spectra were collected for over 25 minutes at 5-minute intervals. A stock solution of 1 mM curcumin or the new compounds was prepared and diluted with phosphate buffer solution (pH 7.4) to a final concentration of 20 μM. All spectral measurements were carried out in a 1 cm path-length quartz cuvette at 25°C.
Crystal structure determination of 3e by X-ray crystallography
Crystals of compound 3e suitable for X-ray diffraction were obtained by slowly evaporating a solution of the solid compound in mixed solvent at room temperature for 3 days. Single crystals were collected and mounted on the top of glass fibers in a random orientation. X-ray single crystal diffraction measurement was carried out at 293(2) K on a Bruker Smart CCD (charge-coupled-device) area diffractometer equipped with a graphite-monochromatic MoKα radiation (λ=0.71073 Å) for data collection. The unit cell dimensions were obtained with the least-squares refinements, and the structures were solved by direct methods with SHELXS-97.27 The final refinement was performed by full-matrix least-squares techniques with anisotropic thermal parameters for the nonhydrogen atoms on F2 (SHELXL-97).27 All the hydrogen atoms were placed in the calculated positions and constrained to ride on their parent atoms.
Mouse primary peritoneal macrophages preparation
C57BL/6 mice were stimulated by an intraperitoneal (ip) injection of 6% thioglycollate solution (0.3 g beef extract, 1 g tryptone, and 0.5 g NaCl dissolved in 100 mL ddH2O, and filtrated through 0.22 mm filter membrane, 3 mL per mouse) and kept in a pathogen-free condition for 3 days before mouse primary peritoneal macrophages (MPMs) isolation. Total MPMs were harvested by washing the peritoneal cavity with phosphate-buffered saline (PBS) containing 30 mM of EDTA (8 mL per mouse), centrifuged, and suspended in Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco/BRL life Technologies, Eggenstein, Germany) with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA), 100 U mL-1 penicillin and 100 mg/mL streptomycin. Non-adherent cells were removed by washing with medium 3 hours after seeding. Before treatment, MPMs were cultured in 35 mm plates (4×105 cells per plate with 1 mL RPMI-1640 medium) and incubated overnight at 37°C in a 5% CO2-humidified air.
MPM cell treatment and ELISA
LPS purchased from Sigma-Aldrich was dissolved in PBS. Curcumin and the obtained compounds were dissolved in dimethyl sulfoxide (DMSO) for in vitro experiments. 3f was prepared as a soluble preparation for in vivo experiments as described in our previous study.28 After cells were treated with each of the above compounds and LPS, the TNF-α and IL-6 levels in medium were determined with an ELISA kit (eBioscience) according to the manufacturer’s instructions. The total protein in cultural plates was collected, and the concentrations of protein were determined using Bio-Rad protein assay reagents. The total amount of the inflammatory factor in the media was normalized to the total amount of protein in the viable cell pellets.
Quantitative structure–activity relationship
The molecular structures of all the AMACs were built using Maestro (Version 9.1, Schrödinger, LLC). The full geometry of molecular structures was further optimized with MOPAC2009 version 9.01. All the calculations were based on the semiempirical parameterized model 6 (PM6) method.29 The molecular descriptor calculation was performed on MODEL (Molecular Descriptor Lab: http://jing.cz3.nus.edu.sg/cgi-bin/model/model.cgi), a web-based server for computing structural and physicochemical features of compounds, according to the methods described in the literature.30 The optimized molecular structures were uploaded to MODEL. About 4,000 molecular descriptors based on molecular 3D structures were obtained. After calculation, molecular descriptors remaining constant for all molecules were eliminated. Variable pairs with a correlation coefficient greater than 0.85 were classified as intercorrelated. Multiple linear regression analysis and leave-one-out cross-validation procedure of R program were used to build the quantitative SAR (QSAR) model. The details are provided in the Supplementary materials.
Western blot analysis
MPMs were pretreated with vehicle (DMSO) or the new compounds of interest at 10 μM for 2 hours followed by incubation with LPS (0.5 μg/mL) for 30 minutes. The protein levels of phosphorylated ERK (p-ERK), ERK, and IκB were detected by Western blot.
Compound 3f protection in mouse model of sepsis induced by LPS
Compound 3f was firstly dissolved with macrogol 15 hydroxystearate (a nonionic solubilizer for injection from Baden Aniline and Soda Factory [BASF]) with or without medium-chain triglycerides (MCTs, from BASF) in water bath at 37°C. The concentration of 3f was 2 mg/mL. The concentration of solubilizer ranged from 5% to 10%, and MCT from 0.5% to 2% in final solution. For the vehicle, the mixture of solubilizer and MCT was prepared at 10% and 2%, respectively. In preventive group (3f + LPS), male C57BL/6 mice weighing 18–22 g were pretreated with 3f in a water solution (10 mg/kg) by intravenous (iv) injection 15 minutes before the ip injection of LPS (20 mg/kg). In therapeutic treatment group (LPS + 3f), male mice received ip injection of LPS (20 mg/kg) 15 minutes prior to the treatment with 3f in a water solution (10 mg/kg) by iv injection. Control animals received a similar volume (100 μL) of vehicle. Mortality was recorded for 7 days.
Statistical analysis
The results are presented as means ± standard deviation. Student’s t-test was employed to analyze the differences between sets of data. Statistics were performed using GraphPad Prism software (GraphPad, San Diego, CA, USA). P-values less than 0.05 were considered indicative of significance. All in vitro experiments were repeated at least three times.
Results and discussion
Chemistry
These AMACs (3a–m, 4a–m) were prepared via aldol condensation of substituted aromatic aldehydes and the intermediates (E)-4-phenylbut-3-en-2-one (2a) or (E)-4-(p-tolyl)but-3-en-2-one (2b), which were obtained by coupling the commercially available benzaldehyde (1a) or p-tolualdehyde (1b) with acetone in an alkaline medium, respectively. Different substituents with opposing electronic properties in the benzene rings were designed and used. The structures of compounds 3a–m and 4a–m are shown in Figure 2. The whole reaction gave the respective products in a yield ranging from 19.11% to 91.08%. The synthetic yields, melting points, 1H NMR, and ESI-MS analysis of unpublished compounds are described in the “Materials and methods” section. Analytical and spectral data of all synthesized compounds are in full agreement with the proposed structures. The purities of all newly synthesized compounds were analyzed by high-performance liquid chromatography, with the purity of all being higher than 95%. In addition, the single crystal of compound (3e) suitable for X-ray diffraction study was obtained from CH2Cl2–CH3CH2OH mixture (v/v =3:1) by a slow-evaporation method at room temperature. The 3D crystal structure of 3e was determined using X-ray crystallography. The selected bond lengths and bond angles are described in Figure 3. Importantly, 3e crystal structure confirmed the expected trans–trans confirmation in the structure of AMAC.
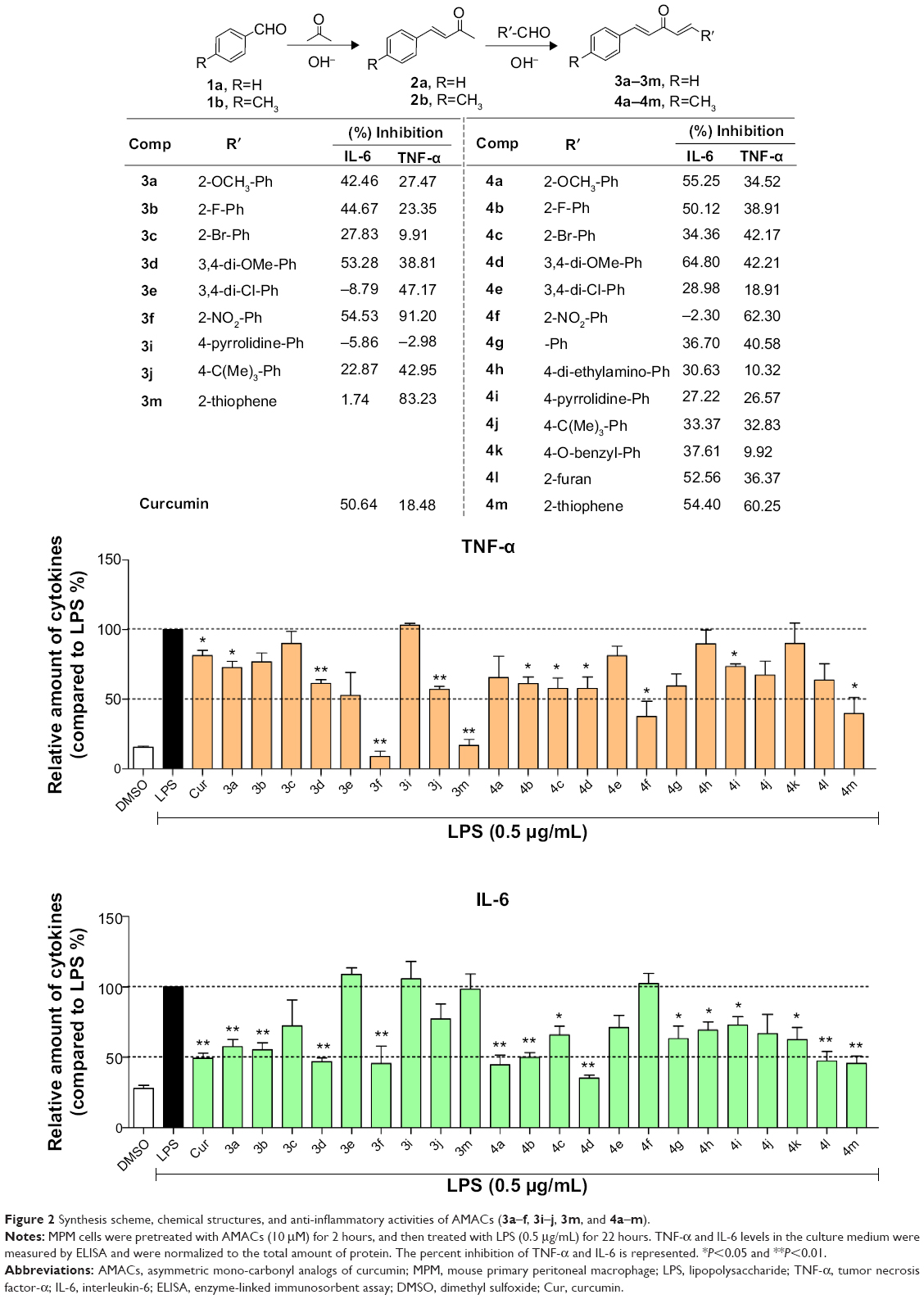 | Figure 2 Synthesis scheme, chemical structures, and anti-inflammatory activities of AMACs (3a–f, 3i–j, 3m, and 4a–m). |
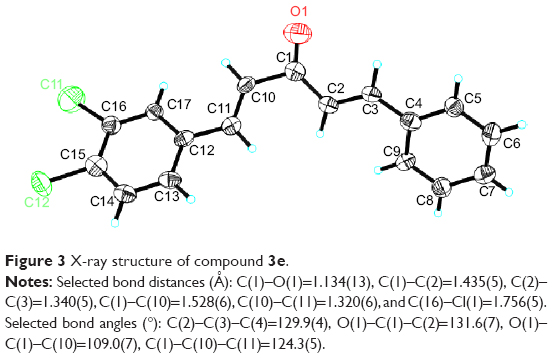 | Figure 3 X-ray structure of compound 3e. |
UV–vis absorption spectra of curcumin and its analogs in phosphate buffer
It is reported that curcumin undergoes rapid decomposition in the physiological buffer solution (pH 7.4), which limits its bioavailability in clinical application.31 To investigate the stability of these AMACs, we measured the UV–vis absorption spectra of representative AMACs in phosphate buffer, pH 7.4. Figure 4 shows the UV–vis absorption spectra of curcumin and active compounds 3f, 3m, 4b, and 4d. The intensity of UV spectra of these AMACs demonstrated very little change over time in phosphate buffer solution (PBS, pH 7.4). For comparison, the UV absorption intensity of curcumin decreased significantly over time in the buffer solution (pH 7.4). Within 25 minutes of incubation, curcumin lost more than 60% of its original intensity (Figure 4). These results indicate that AMACs are stable in the buffer at physiological pH 7.4 (Figure 4). Our previously reported MACs also showed chemical stability and demonstrated significantly improved pharmacokinetic profiles.13,32 As such, these new AMACs could significantly improve their bioavailability as potential anti-inflammation therapeutic agents.
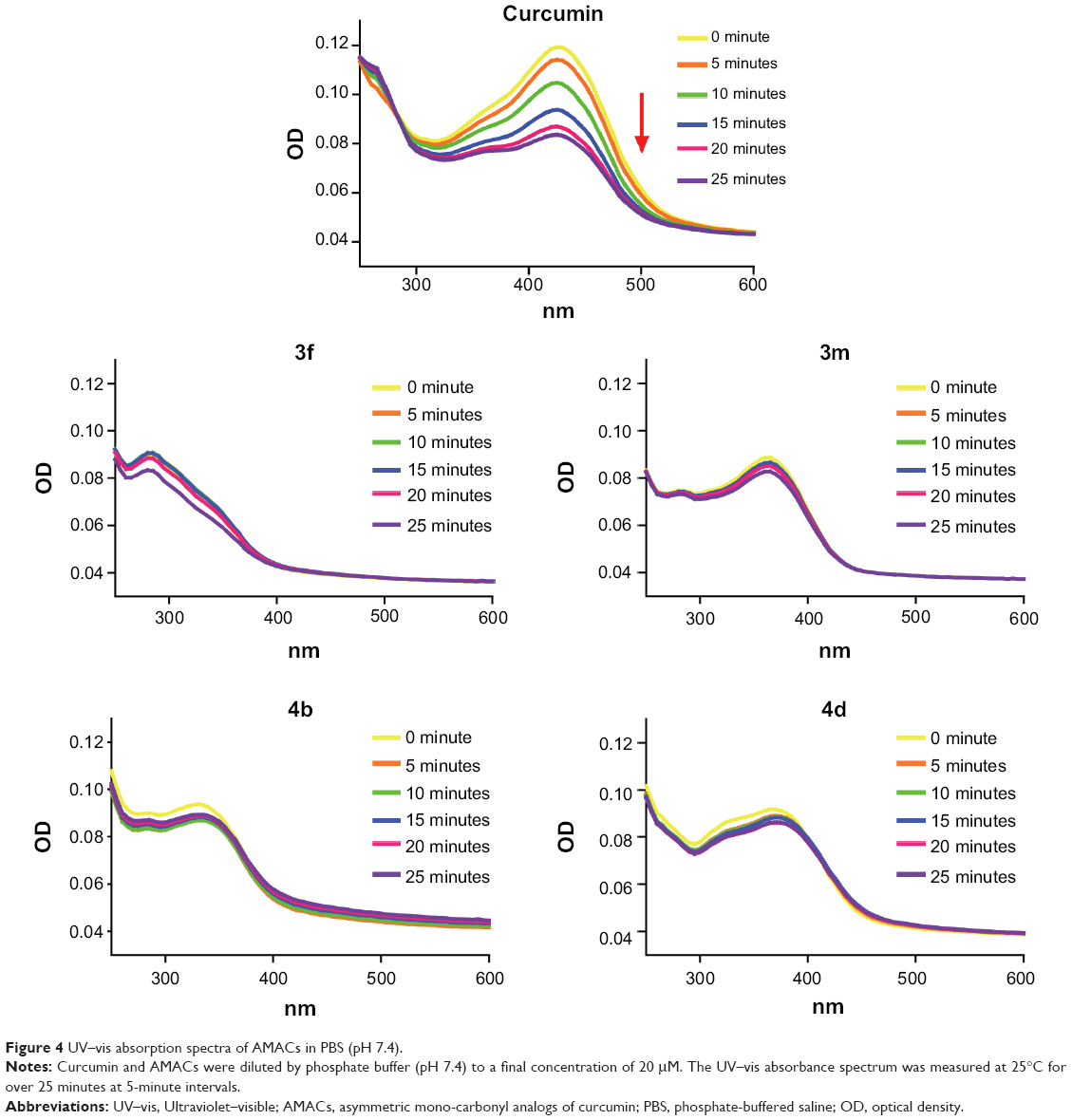 | Figure 4 UV–vis absorption spectra of AMACs in PBS (pH 7.4). |
Inhibitory screening against LPS-induced TNF-α and IL-6 release
We screened these obtained novel AMACs for their inhibitory activity on the TNF-α and IL-6 expression stimulated by LPS. MPM cells extracted from C57BL/6 mice were used for in vitro anti-inflammatory studies. Briefly, MPM cells were pretreated with curcumin, analogs (10 μM), or vehicle control (DMSO, 0.1%) for 2 hours, followed by the treatment with LPS (0.5 μg/mL) for 22 hours. The levels of TNF-α and IL-6 in culture medium were determined through ELISA. As shown in Figure 2, among these 22 compounds, the majority demonstrated various degrees of inhibition of TNF-α and IL-6 expression induced by LPS. Compounds 3f, 3m, 4f, and 4m exhibited a high degree of inhibitory effects on TNF-α expression, and compounds 3d, 3f, 4a, 4b, 4d, 4l, and 4m showed remarkable downregulation (>50%) of IL-6 expression stimulated by LPS. The most potent compound 3f, a 2-nitro-substituted compound, showed an inhibition rate of 54.53% and 91.20% on LPS-induced IL-6 and TNF-α, respectively. For a comparison, compounds 3f, 3m, 4f, and 4m showed significantly more potent (greater than threefold) inhibition on TNF-α expression than curcumin. The analogs are more potent than curcumin (Figure 2).
Quantitative structure–activity relationship
To analyze the SAR of different chemical groups of these compounds and their biological activity, a QSAR model was calculated (Figure 5). In QSAR model study, the structure–properties of compounds are typically represented by their molecular descriptors.33 Moran, solvent-accessible surface (SAS), weighted holistic invariant molecular (WHIM), and Geary molecular descriptors were used in our QSAR model. A more detailed description of these molecular descriptors is provided in the Supplementary materials. The statistically significant QSAR models, Equations 1 and 2, were obtained, with regression coefficients (R2) of 0.85 and 0.83, respectively. These two models fit relatively well the observed inhibitory activities of these compounds against TNF-α and IL-6 expressions. The SAR analysis revealed that compounds with an electron-withdrawing substituent (3e, 3f, and 4f) on aromatic ring are likely to exert higher TNF-α inhibition than those with an electron-donating group (3i, 4h, and 4k). Compounds with highly electron-withdrawing nitro group showed the highest inhibitory activity (3f). Substitution of benzene ring with thiophene (3m, 4m) in compounds also increased the inhibitory activity against TNF-α. In addition, the QSAR results indicate that the molecular surface may play a significant role in the anti-inflammatory activity of the AMACs. As for the correlation between chemical structures of these compounds and their inhibitory effects on IL-6 expression, however, the SAR model is not statistically significant from the present data.
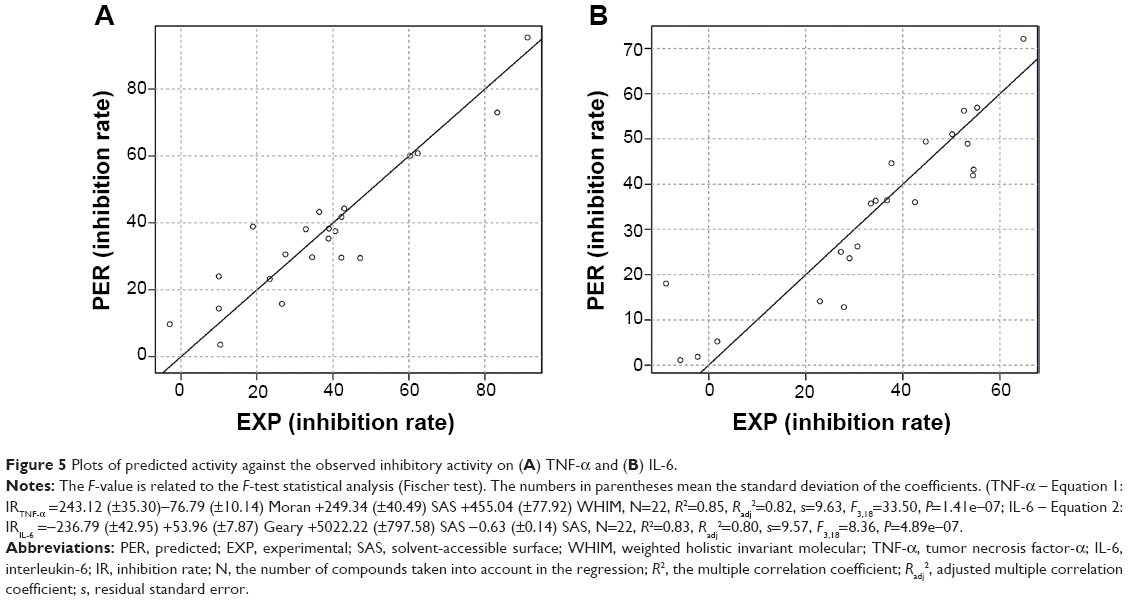 | Figure 5 Plots of predicted activity against the observed inhibitory activity on (A) TNF-α and (B) IL-6. |
Active compounds inhibit TNF-α and IL-6 release in a dose-dependent manner
Four active AMAC compounds, 3f, 3m, 4b, and 4d, which showed low cytotoxicity in human umbilical vein endothelial cells (data not shown), were chosen for further biological evaluation in a dose-dependent manner. MPM cells were pretreated with 3f, 3m, 4b, or 4d in escalating doses (2.5 μM, 5.0 μM, 10 μM, or 20 μM) for 2 hours. After treatment, the cells were then incubated with LPS (0.5 μg/mL) for 22 hours. As shown in Figure 6, compounds 3f, 3m, 4b, and 4d dose-dependently inhibited LPS-induced TNF-α and IL-6 release in macrophage with IC50 values in low micromolar range. Compound 3f demonstrated the most potent activity for inhibiting TNF-α and IL-6 expression. Consistent with our previous report,34 these analogs are more potent than curcumin. The results suggest that these compounds effectively downregulated the expression of both TNF-α and IL-6 and could be potential anti-inflammatory agents.
 | Figure 6 AMAC compounds 3f, 3m, 4b, and 4d inhibited LPS-induced TNF-α and IL-6 release in MPMs in a dose-dependent manner. |
Effects of active compounds on the MAPK/ERK signaling pathway
Studies have shown that elevated expressions of IL-6 and TNF-α induced by LPS are associated with the activation of multiple signal transduction pathways, including ERK1/2 and transcriptional factor nuclear factor kappa binding (NF-κB).35,36 In NF-κB signaling, IκB degradation frees NF-κB p65 subunit and allows it to translocate into the nucleus, followed by activating the transcription of inflammatory genes. Curcumin has been shown to inhibit the activation of ERK1/2 and NF-κB and exert anti-inflammatory or anticancer effects in various cells.37,38 To study the molecular mechanism underlying the downregulation of these signaling pathways, we investigated the inhibitory effects of these AMAC compounds (3f, 3m, 4b, and 4d) on ERK1/2 and NF-κB stimulated by LPS. MPM cells were pretreated with compounds 3f, 3m, 4b, and 4d (10 μM) or DMSO (vehicle control) for 2 hours, respectively, followed by the treatment with LPS (0.5 μg/mL) for 30 minutes. Western blot assay was used to detect the levels of ERK1/2 phosphorylation and IκB degradation. As shown in Figure 7, 3f significantly inhibited LPS-induced ERK1/2 phosphorylation and IκB degradation, while 3m, 4b, and 4d showed no effects on the activation of ERK1/2 and NF-κB. The anti-inflammatory activity of compound 3f is likely via downregulating NF-κB and ERK pathways, and blocking IκB degradation. However, the bioactivities of 3m, 4b, and 4d may be NF-κB/ERK independent. Curcumin has been shown to serve as a multi-target agent to modulate multiple signaling pathways implicated in inflammation and cell proliferation. Our results demonstrated that these active AMACs, though sharing similar structural skeleton, exert anti-inflammatory activities by different mechanisms, which are worth further investigation.
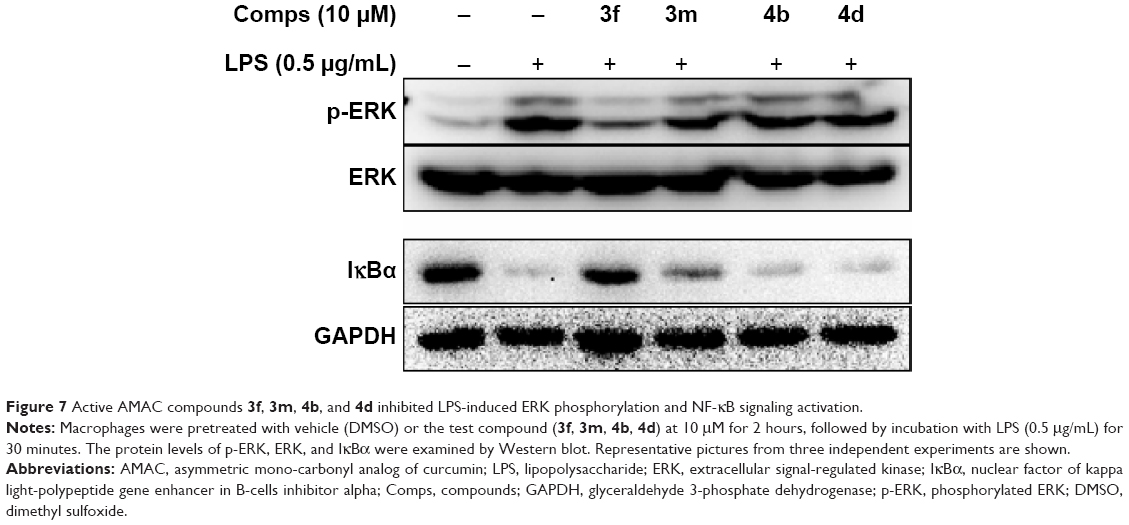 | Figure 7 Active AMAC compounds 3f, 3m, 4b, and 4d inhibited LPS-induced ERK phosphorylation and NF-κB signaling activation. |
Preventive and therapeutic effects of 3f on mice with the LPS-induced sepsis
In vivo, we evaluated the anti-inflammatory effects of lead compound 3f in mouse model of sepsis. LPS, a commonly used endotoxin, was used to induce sepsis. C57BL/6 male mice were randomly divided into three groups: vehicle (iv), LPS (20 mg/kg, ip), and LPS + 3f (10 mg/kg 3f, iv). To test therapeutic effect, mice received ip injection of LPS (20 mg/kg), followed 15 minutes later by the injection of 3f. The survival was recorded for 7 days after the LPS injection at the interval of 1 day. As shown in Figure 8A, all animals treated with LPS alone died within 3 days as a result of the septic shock, while 50% of the 3f-treated animals (LPS + 3f group) survived (therapeutic effect). To further evaluate the preventive effect of 3f on acute sepsis, mice were treated with 3f (10 mg/kg) 15 minutes prior to the LPS injection. Figure 8B shows that only 10% of the 3f-untreated mice (LPS group) survived after 7 days of LPS injection, while around 60% of the 3f-treated animals (3f + LPS group, note the order is reversed here) survived. The survival rates were significantly increased as compared to that of the control group (≥50% survivals in both the prevention group and the therapeutic treatment group, P<0.01 in both groups vs LPS group). In both therapeutic treatment and preventive treatment experiments, 3f markedly improved the survival in mice with LPS-induced sepsis. In the previous in vitro experiments, 3f inhibited the LPS-induced TNF-α and IL-6 expression via NF-κB and ERK pathways. The in vivo results suggest that 3f treatment may effectively reduce TNF-α and IL-6 expression induced by LPS, which results in a prolonged survival in mice model. Thus, 3f treatment effectively prolongs survival in mice model with the acute inflammatory shock. The results provide further in vivo evidence for the anti-inflammatory effects of the novel AMACs.
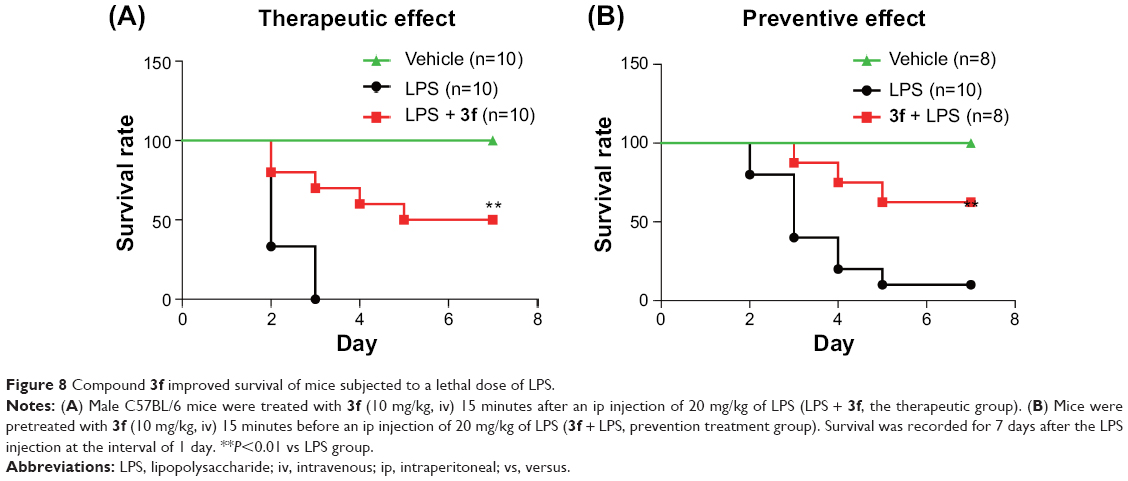 | Figure 8 Compound 3f improved survival of mice subjected to a lethal dose of LPS. |
Conclusion
In this study, we designed and synthesized a series of novel AMACs, and evaluated their anti-inflammatory activity both in vitro and in vivo. Compounds 3f, 3m, 4b, and 4d significantly suppressed the production of TNF-α and IL-6 in LPS-stimulated macrophages. Further investigation into the possible mechanism reveals that the anti-inflammatory activity of 3f might be associated with its inhibition against LPS-induced NF-κB and ERK pathway activation. In vivo, treatment with 3f effectively prolonged the survival in mice model of LPS-induced sepsis. These AMACs show improved stability, bioavailability, and potency compared to curcumin. Our results suggest that lead compound 3f, a novel AMAC, may serve as a potential agent targeting NF-κB and ERK pathways for prevention and treatment of sepsis or other inflammation-related diseases.
Acknowledgments
This work was supported by the National Natural Science Funding of China (21272179, 81302642, and 81202462), High-Level Innovative Talent Funding of Zhejiang Department of Health (GL), Zhejiang Natural Science Funding (LY13H060006 and LQ12H30002), Project of Zhejiang Provincial Key Constructive Subject (Traditional Chinese Medicine, 2012-XK-A28), and Zhejiang Key Group Project in Scientific Innovation (2010R50042) and China Postdoctoral Science Foundation (grants 20090461121 and 201003591).
Disclosure
The authors report no conflicts of interest in this work.
References
Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood). 2008;233(6):674–688. | ||
Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature. 2008;454(7203):463–469. | ||
Griffioen AW, Molema G. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol Rev. 2000;52(2):237–268. | ||
Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. | ||
Hofer N, Kothari R, Morris N, Muller W, Resch B. The fetal inflammatory response syndrome is a risk factor for morbidity in preterm neonates. Am J Obstet Gynecol. 2013;209(6):542.e1–542.e11. | ||
Bolton CH, Downs LG, Victory JG, et al. Endothelial dysfunction in chronic renal failure: roles of lipoprotein oxidation and pro-inflammatory cytokines. Nephrol Dial Transplant. 2001;16(6):1189–1197. | ||
Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis. 2000;148(2):209–214. | ||
Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96(9):939–949. | ||
Neuman MG. Immune dysfunction in inflammatory bowel disease. Transl Res. 2007;149(4):173–186. | ||
Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci. 2009;30(2):85–94. | ||
Fan X, Zhang C, Liu DB, Yan J, Liang HP. The clinical applications of curcumin: current state and the future. Curr Pharm Des. 2013;19(11):2011–2031. | ||
Liu JY, Chen SY, Lv L, Song L, Guo SR, Huang ST. Recent progress in studying curcumin and its nano-preparations for cancer therapy. Curr Pharm Des. 2013;19(11):1974–1993. | ||
Zhao CG, Liu ZG, Liang G. Promising curcumin-based drug design: mono-carbonyl analogues of curcumin (MACs). Curr Pharm Des. 2013;19(11):2114–2135. | ||
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4(6):807–818. | ||
Prasad S, Tyagi AK, Aggarwal BB. Recent developments in delivery, bioavailability, absorption and metabolism of curcumin: the golden pigment from golden spice. Cancer Res Treat. 2014;46(1):2–18. | ||
Anand P, Thomas SG, Kunnumakkara AB, et al. Biological activities of curcumin and its analogues (congeners) made by man and mother nature. Biochem Pharmacol. 2008;76(11):1590–1611. | ||
Lin L, Hutzen B, Ball S, et al. New curcumin analogues exhibit enhanced growth-suppressive activity and inhibit AKT and signal transducer and activator of transcription 3 phosphorylation in breast and prostate cancer cells. Cancer Sci. 2009;100(9):1719–1727. | ||
Al-Hujaily EM, Mohamed AG, Al-Sharif I, et al. PAC, a novel curcumin analogue, has anti-breast cancer properties with higher efficiency on ER-negative cells. Breast Cancer Res Treat. 2011;128(1):97–107. | ||
Nagaraju GP, Zhu S, Wen J, et al. Novel synthetic curcumin analogues EF31 and UBS109 are potent DNA hypomethylating agents in pancreatic cancer. Cancer Lett. 2013;341(2):195–203. | ||
Thomas SL, Zhong D, Zhou W, et al. EF24, a novel curcumin analog, disrupts the microtubule cytoskeleton and inhibits HIF-1. Cell Cycle. 2008;7(15):2409–2417. | ||
Tierney BJ, McCann GA, Cohn DE, et al. HO-3867, a STAT3 inhibitor induces apoptosis by inactivation of STAT3 activity in BRCA1-mutated ovarian cancer cells. Cancer Biol Ther. 2012;13(9):766–775. | ||
Zhou JM, Geng GY, Shi QW, Sauriol F, Wu JH. Design and synthesis of androgen receptor antagonists with bulky side chains for overcoming antiandrogen resistance. J Med Chem. 2009;52(17):5546–5550. | ||
Chakraborti S, Das L, Kapoor N, et al. Curcumin recognizes a unique binding site of tubulin. J Med Chem. 2011;54(18):6183–6196. | ||
Wu J, Li J, Cai Y, et al. Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J Med Chem. 2011;4(23):8110–8123. | ||
Rule NG, Detty MR, Kaeding JE, Sinicropi JA. Syntheses of 4h-thiopyran-4-one 1,1-dioxides as precursors to sulfone-containing analogs of tetracyanoquinodimethane. J Org Chem. 1995;60(6):1665–1673. | ||
Chen GLJ, Duan H, Li T. Improved ultrasound-induced synthesis 1, 5-diaryl-1, 4-pentadien-3-ones. Chem J Int. 2004;6:7–10. | ||
Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. | ||
Zhao C, Cai Y, He X, et al. Synthesis and anti-inflammatory evaluation of novel mono-carbonyl analogues of curcumin in LPS-stimulated RAW 264.7 macrophages. Eur J Med Chem. 2010;45(12):5773–5780. | ||
Stewart JJP. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model. 2007;13(12):1173–1213. | ||
Li ZR, Han LY, Xue Y, et al. MODEL – molecular descriptor lab: a web-based server for computing structural and physicochemical features of compounds. Biotechnol Bioeng. 2007;97(2):389–396. | ||
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4(6):807–818. | ||
Liang G, Shao L, Wang Y, et al. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg Med Chem. 2009;17(6):2623–2631. | ||
Xue Y, Li H, Ung CY, Yap CW, Chen YZ. Classification of a diverse set of tetrahymena pyriformis toxicity chemical compounds from molecular descriptors by statistical learning methods. Chem Res Toxicol. 2006;19(8):1030–1039. | ||
Liang G, Zhou H, Wang Y, et al. Inhibition of LPS-induced production of inflammatory factors in the macrophages by mono-carbonyl analogues of curcumin. J Cell Mol Med. 2009;13(9B):3370–3379. | ||
Barnes PJ, Larin M. Mechanisms of disease – nuclear factor-kappa b – a pivotal transcription factor in chronic inflammatory diseases. New Engl J Med. 1997;336(15):1066–1071. | ||
Mosallanejad K, Sekine Y, Ishikura-Kinoshita S, et al. The DEAH-box RNA helicase DHX15 activates NF-kappaB and MAPK signaling downstream of MAVS during antiviral responses. Sci Signal. 2014;7(323):ra40. | ||
Dong HJ, Shang CZ, Peng DW, et al. Curcumin attenuates ischemia-like injury induced IL-1beta elevation in brain microvascular endothelial cells via inhibiting MAPK pathways and nuclear factor-kappaB activation. Neurol Sci. 2014;35(9):1387–1392. | ||
Pan Y, Huang Y, Wang Z, et al. Inhibition of MAPK-mediated ACE expression by compound C66 prevents STZ-induced diabetic nephropathy. J Cell Mol Med. 2014;18(2):231–241. |
Supplementary materials
Quantitative structure–activity relationship
Multiple linear regression analysis
Multiple linear regression (MLR) analysis is a statistical technique that uses several explanatory variables to predict the outcome of a response variable.1–3 The goal of MLR is to model the relationship between the explanatory and response variables. In our present study, MLR was performed using R program, a powerful tool for statistical computing and graphics, to derive quantitative structure-activity relationship models. The biological data used in this study were their tumor necrosis factor (TNF)-α or interleukin (IL)-6 inhibitory rates when compared to lipopolysaccharide-alone group. Compounds with negative values were abandoned because of their pro-inflammatory activities. The inhibition rates against TNF-α and IL-6 release, named as IRTNF-α and IRIL-6, respectively, were used as dependent variables in the linearization procedure. Subsequently, stepwise MLR was used to select the significant descriptors. The most relevant descriptors were used as independent variables.
Validation of the models
Validation of the lineal models is required for testing the predictive ability and generalizing the methods by cross-validation. The leave-one-out (LOO) procedure was employed. When a data point was removed from the analyzed set, the regression was recalculated, and then the predicted value for that point was compared to its actual value. This process was repeated until each datum had been omitted once, and then the sum of squares of these deletion residuals could be used to calculate q2, an equivalent statistic to R2.
Results
One-variable and multivariable regressions between the different activities and the abundant descriptors were studied. The statistically significant models were obtained with three variables for anti-TNF-α and anti-IL-6 activities (Equations 1 and 2 in Figure 5). The involved molecular descriptors and their corresponding definition are listed in Table S1.
Equation 1 was the best quantitative structure–activity relationship model obtained with the Moran, SAS, and WHIM descriptors, which had a high adjusted squared regression coefficient (R2adj=0.82). The variables in Model 1 included the topological descriptors and geometrical molecular descriptors. Two of those, SAS and WHIM descriptor, belong to geometrical molecular descriptors. Another variable is Moran descriptor that is an autocorrelation index descriptor. Analogously, Equation 2 was obtained for anti-IL-6 activities. Other three molecular descriptors were employed to construct Model 2. Geary topological autocorrelation descriptors are classified as topological descriptors. Molecular volume and molecular surface-derived descriptors, labeled as SAS in Table S1, belong to geometrical molecular descriptors.
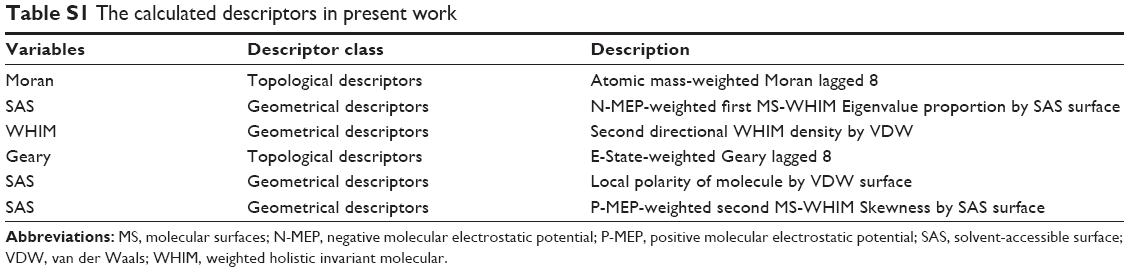 | Table S1 The calculated descriptors in present work |
Based on the statistical analysis, two satisfactory models for anti-TNF-α and anti-IL-6 activities were obtained. The correlation between compound activities and structure information could be illustrated from different perspectives by using various descriptors. R2 was a common parameter to test the validity of models, while it could be increased artificially by adding more variables (descriptors). To better test the validity of the models, the q2 values were adopted as a more reliable statistic parameter, which means the square of the correlation coefficient of the cross-validation and are calculated from LOO test. The q2 values could be used as a measure of the predictive ability of a regression equation. The q2 values of Equations 1 and 2 are 0.71 and 0.70, respectively, indicating the stability of these models. The scatter plot of predicted vs experimental values is illustrated in Figure X. As can be seen, Equations 1 and 2 have the modest quality, and the variables used in these equations can explain the variance in the anti-TNF-α and anti-IL-6 activities of the asymmetric mono-carbonyl analogs of curcumin.
References
Stewart JJ. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model. 2007;13:1173–1213. | ||
Li Z, Han L, Xue Y, et al. NMODEL – molecular descriptor lab: a web-based server for computing structural and physicochemical features of compounds. Biotechnol Bioeng. 2007;97: 389–396. | ||
Frick WE, Ge Z, Zepp RG. Nowcasting and forecasting concentrations of biological contaminants at beaches: a feasibility and case study. Environ Sci Technol. 2008;42:4818–4824. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.