Back to Journals » Drug Design, Development and Therapy » Volume 14
Synthesis and Biological Activity of Piperine Derivatives as Potential PPARγ Agonists
Authors Wang Y ![]() , Yao Y, Liu J, Wu L, Liu T
, Yao Y, Liu J, Wu L, Liu T ![]() , Cui J, Lee DYW
, Cui J, Lee DYW
Received 12 November 2019
Accepted for publication 11 May 2020
Published 26 May 2020 Volume 2020:14 Pages 2069—2078
DOI https://doi.org/10.2147/DDDT.S238245
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Yanli Wang,1– 3 Yuan Yao,4,5 Jing Liu,2,6 Lili Wu,7 Tonghua Liu,7 Jian Cui,1 David Yue-Wei Lee2
1School of Pharmacy, Minzu University of China, Beijing 100081, People’s Republic of China; 2Bio-Organic and Natural Products Laboratory, McLean Hospital, Harvard Medical School, Boston, MA 02478, USA; 3Key Laboratory of Plant Molecular Biology, Inner Mongolia Autonomous Region Institute of Biotechnology, Hohhot 010010, Inner Mongolia, People’s Republic of China; 4State Key Laboratory of Reproductive Regulation & Breeding of Grassland Livestock, School of Life Sciences, Inner Mongolia University, Hohhot 010070, People’s Republic of China; 5Department of Neurology, Inner Mongolia People’s Hospital, Hohhot 010017, Inner Mongolia, People’s Republic of China; 6Natural Pharmacia International Inc., Burlington, MA 01803, USA; 7School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing 100029, People’s Republic of China
Correspondence: David Yue-Wei Lee
Bio-Organic and Natural Products Laboratory, McLean Hospital, Harvard Medical School, Boston, MA 02478, USA
Tel +1617 855 2038
Email [email protected]
Jian Cui
School of Pharmacy, Minzu University of China, Beijing 100081, People’s Republic of China
Email [email protected]
Introduction: Peroxisome proliferator-activated receptor γ (PPARγ) plays a key role in glucose, which is a ligand-mediated transcription factor. The lipid homeostasis often serves as a pharmacological target for new drug discovery and development.
Materials and Methods: In the research, we synthesized a series of piperine derivatives and then used a fluorescence polarization-based PPARγ ligand screening assay to evaluate the agonistic activity of PPARγ. Then, we cultured human normal hepatocytes, which were treated with 100μM compounds 2a, 2t or 3d. Then, the levels of PPARγ gene were determined so as to show whether the compounds could activate or inhibit the expression of PPARγ.
Results: A total of 30 piperine derivatives were synthesized and evaluated. Compound 2a was identified as a potential PPARγ agonist with IC50 at 2.43 μM, which is 2 times more potent than the positive control rosiglitazone with IC50 at 5.61μM. The human hepatocytes cells were cultured and treated with compounds 2a, 2t or 3d as described in the “Materials and Methods” section. We found that compounds 2a, 2t and 3d could activate PPARγ by 11.8, 1.9 and 7.0 times compared with the “blank”, with compound 2a activation being the most significant. Molecular docking studies indicated that the piperine derivative 2a stably interacts with the amino acid residues of the PPARγ complex active site, which is consistent with the results of the in vitro PPARγ ligand screening assay.
Keywords: PPARγ, piperine derivatives, ligand screening assay, molecular docking
Introduction
The peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear receptors (NRs) which play a pivotal role in the regulation of genes associated with a wide range of physiological processes. In the chromatin, PPARs are a heterodimer with retinoid X receptor (RXR) and play a role of specific PPAR elements (PPRE) response to ligand binding. Ligand binding triggers the conformational change of LBD, which promotes the dissociation or recruitment of transcription co-regulators, mainly through ligand-dependent activation function 2 (AF2). Besides, post-translational modifications can modulate the affinity of the receptor for co-regulators to determine whether the target gene is induced or suppressed.1–3 There is no doubt that PPARs regulate genes involved in the metabolism of lipid and glucose. In addition, the PPARs are crucial to regulating some biological effects associated with vascular and inflammation feature.4–6
The PPARs also responding to the endogenous ligands including fatty acid metabolite and organic synthesis.7,8 PPARα, PPAR β/δ and PPARγ have been identified, which were significantly expressed in a tissue form. The most widely studied PPARγ isoforms are responsible for adipogenesis, glucose homeostasis, and regulation of lipid metabolism and gene transcription.9,10 PPARγ also has a critical role in insulin sensitization. Therefore, PPARγ is generally recognized as an ideal therapeutic measurement of diabetes and dyslipidemia. Thiazolidinediones (TZDs) such as rosiglitazone (RSG, Figure 1) and pioglitazone, are high-affinity ligands and full agonists of PPARγ, which have been successfully used to treat type II diabetes mellitus.11 TZDs have side effects in clinical application, we get the information of the PPARγ active mechanism and biological functions with the assistant of these synthetic ligands. TZDs as one type of PPARγ agonists have also been used as a potential drug candidate to develop effective and safe medicine.12–14
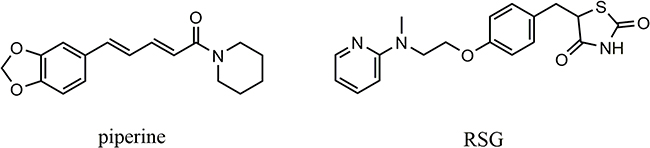 |
Figure 1 Chemical structures of piperine and RSG. |
Natural products have been served as rich sources for the discovery and development of modern medicine. Piperine (Figure 1) isolated from the traditional Mongolian medicine of piper longum L., which is well known for its extensive biological activities. It has been reported that piperine attenuates the differentiation of fat cell, which down-regulating the activity and expression of PPARγ, prohibiting the process of adipogenesis15,16 and increases the bioavailability of many other drugs.17,18 However, piperine has an acutely toxic effect,19 its biological applications are limited due to the poor solubility in aqueous environments. According to related reports that piperoyl-amino conjugates have better biological activity than piperine,20 In addition, the results of piperine pharmacokinetic studies showed that after the piperine metabolism by the human, it retains methylenedioxy ring and conjugated double bonds while the piperidine ring is modified to form a propionic acid group. Such piperine derivatives may have the advantages of structural diversity, improved solubility, and reduced toxicity. Therefore, we wish to utilize amino acids and methylenedioxy ring as the alternative surrogates to couple with the piperine.
This study describes the synthesis and biological evaluation of piperine derivatives with the FP-based PPARγ ligand screening assay. Compound 2a was identified as a potential PPARγ agonist with IC50 at 2.43 μM, which is 2 times more potent than the positive control Rosiglitazone with IC50 at 5.61μM. Molecular docking studies have shown that piperine derivative 2a fitted nicely with the amino acid residues at the active site of the PPARγ complex and is consistent with the result of in vitro ligand assay.
Materials and Methods
Chemical Synthesis
Synthesis of Piperic Acid
Piperine (64g, 219.9 mmol), KOH (113.5g, 17.9mmol) was refluxed with ethanol (1500mL) for 12h, the mixture was allowed to stand for cooling, suction filtration under reduced pressure, and the filter cake was washed with ethanol (95%) to pH=7, and filtered under reduced pressure to give crystals of brown piperic potassium salt. The crystal was dried to obtain the product. The piperic potassium salt was dissolved with water and then, gradually acidified with dilute hydrochloric acid, formed yellow precipitate, vacuum filtered, and washed with water (1400mL) and recrystallized from acetone to afford yellow crystalline compound.
General Procedure for the Synthesis of Piperic Conjugates (2a-2j)
Pre-dried dichloromethane (DCM) was added to Piperic acid (2.18g, 10mmol), and then added thionyl chloride (73.8mmol, 14.4mL), the contents were refluxed under nitrogen atmosphere for 2 hours. Removed the excess thionyl chloride under reduced pressure, and then got acid chloride of piperine. Amino acid ester hydrochloride (20mmol) in one portion to acid chloride of piperine in DCM; and Et3N in DCM under ice-cooling, stirred 2h at room temperature. Then added water (50 mL), washed the organic layer with water (2*25mL), dried over with anhydrous sodium sulfate, concentrated to give the crude product. The crude product was dissolved in ethyl acetate as few as possible, and the yellow solids were obtained by slowly recrystallization with petroleum ether to get the pure compounds (2a–2j).
General Procedure for the Synthesis of Piperic Conjugates (2k–2t)
Mixed neutral alumina (15g) with KF (10 g) in 200 mL of water for the preparation of KF-Al2O3. Then removed water at 50–60°C in a rotary evaporator, further dried this reagent in a vacuum oven for 12h. Added Piperic amino acid ester conjugates (1.65mmol, 0.5g) to KF-Al2O3(3.13mol, 40% KF) and then stirred this dry mixture at room temperature in a round bottom flask to ensure uniform mixing of solid support KF-Al2O3 with the substrate. Transfer the uniformly dispersed powder to a 50mL eggplant bottle and heat and stir at 50°C for 2h. Then the reaction mixture was added water (5mL), stirred for 5min and then filtered. Neutralized the filtrate by adding aqueous HCl and obtained the precipitate filtered, dried it, obtained the pure piperic amino acid conjugates compounds.
General Procedure for the Synthesis of Derivatization of Bisphenol Piperic Amino Acid (Ester) Conjugates (3a–3e;3k–3o)
Dissolved piperic conjugates (2a-2e;2k-2o) with anhydrous DCM. Added BBr3 at −78 °C, stirred for 2h hours at 0°C, the solvent was removed under reduced pressure. The product was purified by column chromatography in DCM/MeOH (10:1) to get the pure compounds (3a-3e;3k-3o).
FP-Based PPARγ Ligand Screening Assay
We selected the PPARγ Ligand Screening Assay Kit, which provided a single-step assay to screen PPARγ ligands based on fluorescence polarization (FP).21,22 In this method, the ligand of PPARγ in conjunction with fluorescein and displacement probe. The fluorescent probes were replaced by PPARγ ligands, agonists and antagonists, resulting in the reduction of FP. Firstly, we prepared the assay cocktail and measured every ligand concentration in duplicate, covered the plate and used for 60–90 min at room temperature. Secondly, we read the data in the wavelengths of 470 nm and 530 nm, respectively. The results were calculated by FP. According to the kit protocol, we calculated IC50 with the mP-concentration displacement curve (Cayman).
Gene Expression Experiments to Activate PPARγ Assay
Cell Culture
Human normal hepatocytes (HL-7702[L-02]) purchased from the Biotechnology Research of Beijing BeNa Chuanglian in China. Quickly put the cryopreservation tube containing normal human hepatocytes into a 37 °C water bath and shake it constantly, so that the liquid in the tube will melt rapidly in 1–2 minutes. The cells suspension was transferred into the culture bottle adding CM 2–1 medium (90% RPMI-1640 + 10% FBS. RPMI-1640: 1640 medium, containing glutamine). And the cells were cultured in 5% CO2 incubator at 37 °C. The next day, the culture medium was changed for routine culture and passage. The cells grew well and were monolayer adherent. Depending on the number of cells, fresh culture medium should be replaced once every 2–3 days, and the passage should be 1:3 once every 4–5 days. When the cells grew to about 70% of the dish area, starvation culture was carried out. Then the cells were culture in RPMI-1640 medium without serum, which made the cells in a low nutritional state. It is for starvation culture. After 24 hours, the 100 μM compounds 2a, 2t or 3d was added in the cultures. The “blank” indicated without drugs.
Total RNA Extraction
Total RNAs of HL-7702[L-02] cells were isolated using the Trizol reagent kit (TRIzol™ Plus, Invitrogen, USA), following the manufacturer’s instructions and described before. RNA integrity, purity, and concentration were detected by electrophoresis and the NanoDrop 2000C spectrophotometer (NanoDrop Technologies, Thermo Scientific™, USA).23 The integrity and purity of RNA samples met the requirements of RT-qPCR analysis.24 http://dx.doi.org/10.17504/protocols.io.qafdsbn.
Relative Quantitative Real-Time PCR
The cDNA synthesis was performed with 1 μg of total RNA as template in a 20 μL reaction mixture including 4 μmol/L of each primer using the TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix Kit (TransGens, China) as described in the manufacture’s protocol. The primers for RT-qPCR were designed, meeting the requirements with Tm values of 60°C and products shorter than 200bp. The pairs of primer PPARγ-RT-F (TATCGACCAGCTGAATCCAGAG) and PPARγ-RT-R (GGGGGTGATGTGTTTGAACTTG); Human-actin-RT-F (CTCCATCCTGGCCTCGCTGT) and Human-actin-RT-R (GCTGTCACCTTCACCGTTCC) were used in RT-qPCR assay. The RT-qPCR assay was performed using the PerfectStart TM Green qPCR SuperMix kit (TransGens, China). And the Light Cycler 480 II Real-Time PCR System (Roche, Switzerland) was used as a quantitative analysis detector. The RT-qPCR procedure used default procedure (95 °C for 30 s, 45 cycles of 95°C for 10 s, 60 °C for 20 s) and the melting curves were performed immediately as described previously.23 The CT value of each reaction can be given automatically. The relative standard curve was made by template concentration (X-axis) and threshold cycles (Y-axis). Amplification efficiency (E) was calculated from equation 1, and gene dose was determined from equation 2, as described previously. Then, the target gene relative expressions were calculated by formula 2(-ΔΔCt).24 The β-actin gene was the reference gene. The expression level of target genes was normalized to this reference gene. The final data came from three test repetitions, each with three technical repetitions.
Molecular Docking
We used the crystal structure (PDB: 4EMA) of the PPARγ docking template combined with RSG. We used AutoDock 4.2.6 docking compounds 2a, 2t and 3d into the ligand-binding domain.25 Implemented AutodockTools 1.5.6 to build the autogrid box. Based on the known ligand, Set the grid center, and the grid contained 14×8×44 autogrid points, which is 0.375Å spacing. In the population, the number of individuals was 150, the maximum number of energy evaluations was 25 million and the generations were 27,000, respectively.
Results and Discussion
Chemistry
Following the synthetic scheme shown in Scheme 1, a total of 30 piperine derivatives were synthesized (see Supplementary materials). The basic skeleton of piperine consists of methylenedioxyphenyl (MDP) moiety, basic six-membered piperidine moiety attached to side-chain via an amide and linkage side-chain which consists of double bonds. This study aimed to establish SAR focusing on the side chain of piperine. A series of piperine derives shown in Table 1. Converted piperine into acid by hydrolysis used ethanolic KOH. Converted the acid into acid chloride used thionyl chloride, and then followed by condensation with the amine moiety of amino acid in dichloromethane. Basically, the part of piperine’s piperidine moiety was replaced by different amino acid esters and substituted aniline to obtain compounds 2a–2t. Boron tribromide can be used for the demethylation of aryl methyl ethers in the presence of many functional groups without affecting these, it does not affect the cleavage of methylenedioxy groups. Halogenation was done with boron tribromide to give piperine catechol 3a-3e, 3k-3o.
 |
Scheme 1 Reagents and conditions. Notes: (i) KOH, ethanol, reflux, 12h; HCl (ii) SOCl2, DCM, reflux, 12h (iii) DCM, (2a-2j), Et3N, rt, 2h (iv) KF- Al2O3, 50°C, 2h (v) BBr3, DCM, -78°C, 2h. |
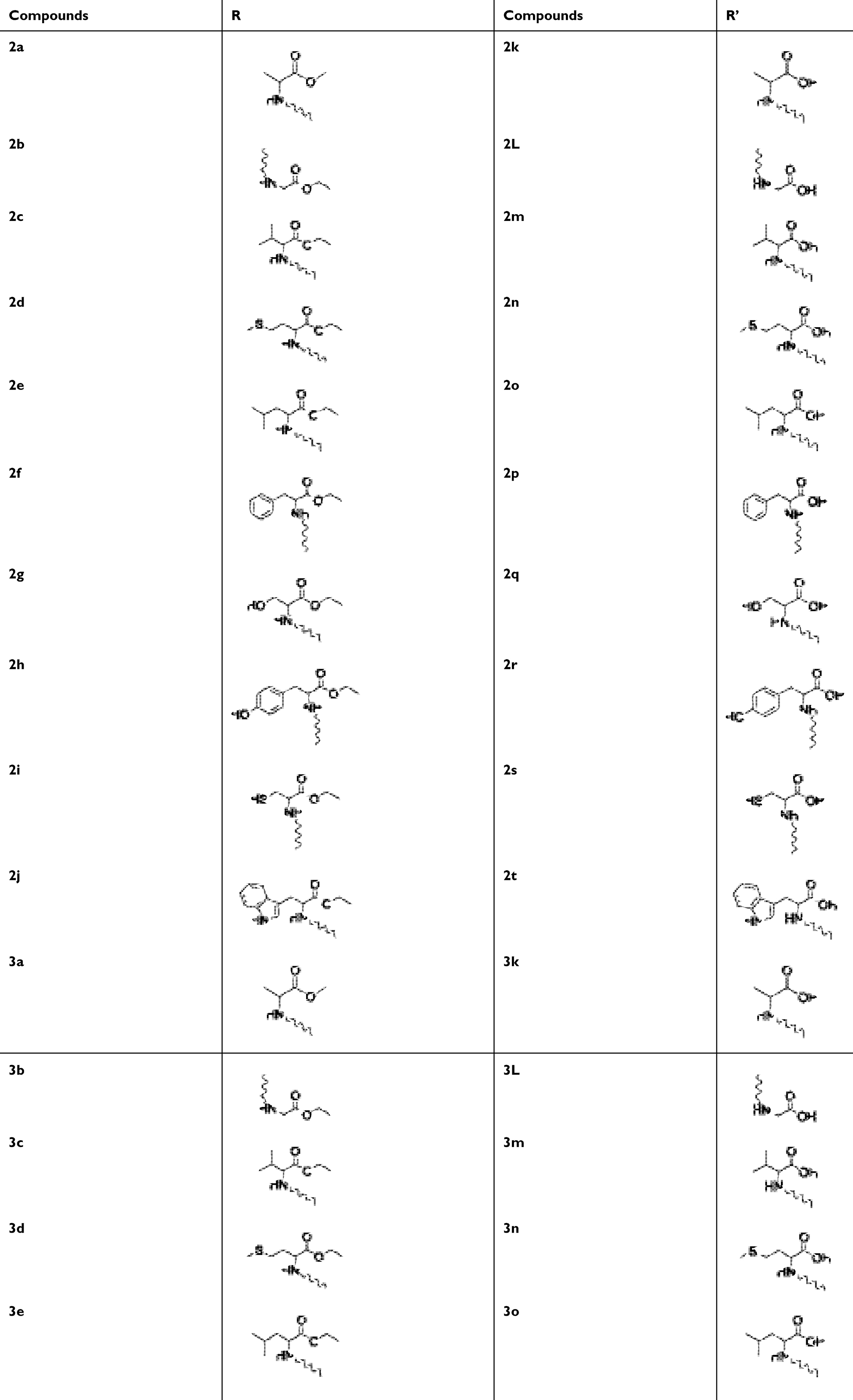 |
Table 1 Synthesis of Piperine Derivatives |
FP-Based PPARγ Ligand Screening Assay
We used a convenient FP-based PPARγ ligand screening assay to assess the agonistic potency of 30 piperine derivatives.26,27 We chose RSG as a PPARγ agonist as a positive control. The addition of large amounts of a PPARγ ligand would result in a larger reduction in the MP value of the well. So, plotting mP vs ligand concentration allowed the construction of an IC50 curve with a broad dynamic range. As shown in Figure 2, thirteen compounds (2a, 2e, 2i, 2m, 2n, 2p, 2q, 2r, 2s, 2t, 3a, 3b, 3c, 3d) were found to exhibit potent agonistic activity. As shown in Figure 3, the MP values of three compounds (2a, 2t, 3d) decreased significantly with the increasing of the logarithm of the concentration, and this trend was consistent with the positive control. However, the MP values of negative control (DMSO) were consistent with the change of concentration. As shown in Table 2, the compound 2t (IC50=1.03 μM) showed the most potent effects of FP, the compound 2a (IC50=2.43 μM) showed the remarkable agonistic activity on PPARγ, they were both more potent than RSG (IC50=5.61 μM) and piperine(IC50=18.35 μM). Besides, the compound 3d (IC50=79.32μM) showed moderate agonistic activity on PPARγ. Among the 30 small-molecule piperine derivatives (2a-2t, 3a-3e and 3k-3o), piperine derivatives exhibited biological activity better than piperine. Especially, we founded the group of the amino acid-modified piperine contribute the most to the agonist potency (2t > 2a> 3d). Compounds 2t and 2a are being investigated in vivo and results will be reported separately.
 |
Table 2 IC50s of Piperine Derivatives in FP-Based PPARγ Ligand Screening Assay |
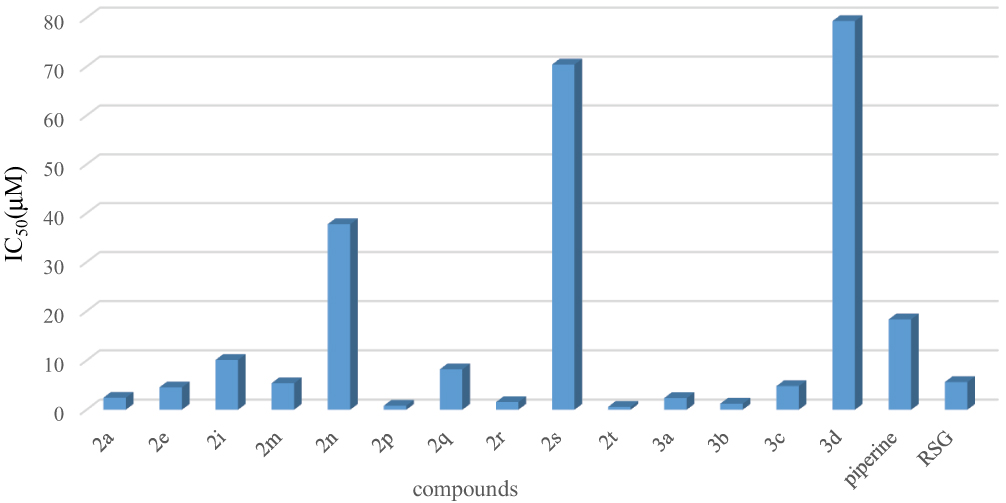 |
Figure 2 FP-based PPARγ ligand screening assay of piperine derivatives. |
 |
Figure 3 Compounds 2a, 2t, 3d showed potent effects of FP comparable to RSG. |
The Compounds 2a, 2t, and 3d Can Activate the Gene Expression of PPARγ
We cultured human normal hepatocytes, which were treated with 100 μM compounds 2a, 2t or 3d. And then the levels of PPARγ gene expression were determined. So as to show whether the compounds could activate or inhibit the expression of PPARγ. The human hepatocytes HL-7702[L-02] cells were cultured and treated with compounds 2a, 2t or 3d as described in the method section. We found that the relative expression of the PPARγ activity increased by 11.8, 1.9 and 7.0 times in compounds 2a, 2t and 3d, respectively, compared with the “blank” (Figure 4). These results indicated that compounds 2a, 2t, and 3d could activate PPARγ, in which three compounds, compound 2a activation was the most significant.
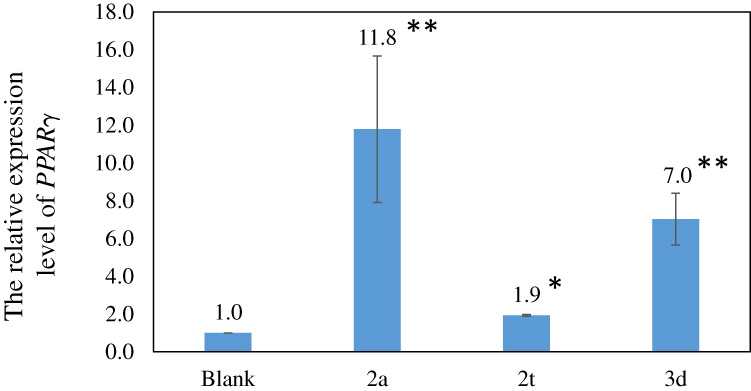 |
Figure 4 The relative expression level of PPARγ in compound of 2a, 2t and 3d. Notes: The values are the average of three experiments. The standard error bars are as indicated. The significance of discrepancy between data by single factor analysis of variance. The “*” showed significant differences between blank and compounds, “*”p-value<0.05; “**”p-value<0.001. |
Molecular Docking
Firstly, according to the PPARγ complex’s X-ray structure, the binding mode of RSG has been analyzed. The TZD of RSG and the three amino acid residues (His323, Ser289, and Tyr473) forms an H-bonding interaction. The RSG’s hydrophobic tail interacts with the residues of Ile341, Val339, Phe363, Leu353, Phe368, Met364 and Leu330. So, we have chosen low binding energy and a similar binding mode for the study to dock poses. In Table 3, the in vitro agonistic activity showed good correlations with the molecular properties. The potent compounds proved useful for examining the piperine derivatives because they displayed high values of tPSA and LogP, we did the experiment to identify the complex for the docking study. The binding energy of RSG was at −5.3 kcal/mol, while that of 2a was at −1.2 kcal/mol. This discrepancy showed that the chemical structure of piperine derivative and RSG play a significant role in the binding pocket and it is conceivable that the receptor was differentially sensitized by the binding moieties of RSG and piperine. There was a good correlation between piperine derivatives with similar chemical structures. But, among the three agonists (2a, 2t, and 3d), compound, 2a exhibited the highest PPARγ activity and the highest binding energy. The 2a interacted with Ser289’s residues to form the PPARγ active sites’ hydrogen bonds and its hydrophobic tail was encompassed from Val339, Leu340, Ile341, Try327, Leu330 by a surface formed. The 2t interacted with residues of Tyr473, His449, Ser342 to form the PPARγ active sites’ hydrogen bonds. The 3d interacted with residues of Ser289 to form hydrogen bonds to the PPARγ active sites His449. (Figure 5A–E).
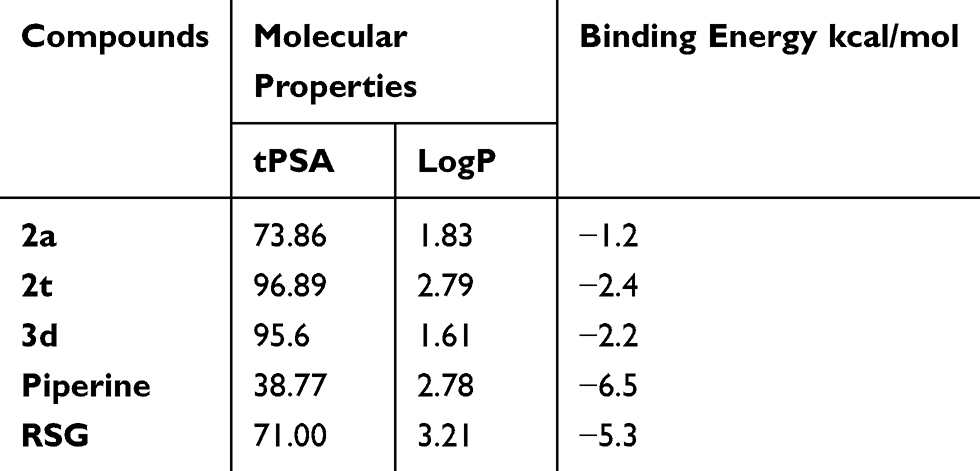 |
Table 3 The Molecular Properties and Binding Energy of Selected Piperine Derivatives |
 |
Figure 5 The binding modes of agonists with PPARγ. Notes: H-binding interactions of RSG and other agonists were shown in yellow dotted line, respectively. (A) 2a, (B) 2t, (C) 3d, (D) piperine, (E) RSG. |
Conclusion
PPARγ is considered as a therapeutic target for the treatment of diabetes and dyslipidemia, which modulates the transcription of genes responsible for glucose homeostasis, adipose differentiation, and lipid metabolism. In this study, we synthesized 30 piperine derivatives and evaluated with quick FP-based PPARγ ligand screening assay to agonistic activity with an easy. A preliminary structure-activity relationship was established. Among the derivatives tested, ligand 2a and 2t exhibited potent PPARγ agonistic activity than RSG and piperine. By test the gene expression of PPARγ in human normal hepatocytes, we found that compound 2a, 2t, and 3d could activate PPARγ, compound 2a activation was the most significant. Molecular docking studies indicated that the piperine derivative 2a stably interacts with the amino acid residues of the PPARγ complex active site, which is consistent with the results of the in vitro PPARγ ligand screening assay. The compound of 2a’s further chemical modifications and in vivo evaluation is in progress.
Acknowledgments
This research was supported by the Bio-Organic and Natural Products Laboratory, McLean Hospital, Harvard Medical School and the China Scholarship Council.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi:10.1146/annurev.med.53.082901.104018
2. Nettles KW, Greene GL. Ligand control of coregulator recruitment to nuclear receptors. Annu Rev Physiol. 2005;67:309–333. doi:10.1146/annurev.physiol.66.032802.154710
3. Perissi V, Rosenfeld MG. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol. 2005;6:542.
4. Vanden Berghe W, Vermeulen L, Delerive P, De Bosscher K, Staels B, Haegeman G. A paradigm for gene regulation: inflammation, NF-kappaB and PPAR. Adv Exp Med Biol. 2003;544:181–196.
5. Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. BBA. 1812;2011:1007–1022.
6. Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23:351–363. doi:10.1016/j.tem.2012.05.001
7. Krey G, Braissant O, L Horset F, et al. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol. 1997;11:779–791. doi:10.1210/mend.11.6.0007
8. Marion-Letellier R, Savoye G, Ghosh S. Fatty acids, eicosanoids and PPAR gamma. Eur J Pharmacol. 2016;785:44–49.
9. Ahmadian M, Suh JM, Hah N, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;99:557–566. doi:10.1038/nm.3159
10. Tripathi AC, Gupta SJ, Fatima GN, Sonar PK, Verma A, Saraf SK. 4-thiazolidinones: the advances continue. Eur J Med Chem. 2014;72:52–77. doi:10.1016/j.ejmech.2013.11.017
11. da Silva FMC, Dos Santos JC, Campos JLO, et al. Structure-based identification of novel PPAR gamma ligands. Bioorg Med Chem Lett. 2013;23:5795–5802. doi:10.1016/j.bmcl.2013.09.010
12. Ohashi M, Gamo K, Oyama T, Miyachi H. Peroxisome proliferator-activated receptor gamma (PPARγ) has multiple binding points that accommodate ligands in various conformations: structurally similar PPARγ partial agonists bind to PPARγ LBD in different conformations. Bioorg Med Chem Lett. 2015;25:2758–2762. doi:10.1016/j.bmcl.2015.05.025
13. Ohashi M, Oyama T, Miyachi H. Different structures of the two peroxisome proliferator-activated receptor gamma (PPARγ) ligand-binding domains in homodimeric complex with partial agonist, but not full agonist. Bioorg Med Chem Lett. 2015;25:2639–2644. doi:10.1016/j.bmcl.2015.04.076
14. Kim KJ, Lee MS, Jo K, Hwang JK. Piperidine alkaloids from piper retrofractum Vahl. protect against high-fat diet-induced obesity by regulating lipid metabolism and activating AMP-activated protein kinase. Biochem Biophys Res Commun. 2011;411:219–225. doi:10.1016/j.bbrc.2011.06.153
15. Park U-H, Jeong H-S, Jo E-Y, et al. Piperine, a component of black pepper, inhibits adipogenesis by antagonizing PPARc activity in 3T3-L1 cells. J Agric Food Chem. 2012;60:3853–3860. doi:10.1021/jf204514a
16. Atal CK, Dubey RK, Singh J. Biochemical basis of enhanced drug bioavailability by piperine: evidence that piperine is a potent inhibitor of drug metabolism. J Pharmacol Exp Ther. 1985;232:258–262.
17. Patil UK, Singh A, Chakraborty AK. Role of piperine as a bioavailability enhancer. Int J Rec Adv Pharma Res. 2011;4:416–423.
18. Pawinee P, Glinsukon T, Toskulkao C. Acute and subacute toxicity of piperine in mice, rats and hamsters. Toxicol Lett. 1983;16:351–359.
19. Singh IP, Jain SK, Kaur A, et al. Synthesis and antileishmanial activity of piperoyl-amino acid conjugates. Eur J Med Chem. 2010;45:3439–3445. doi:10.1016/j.ejmech.2010.04.033
20. Ferre P. The biology of peroxisome proliferator– activated receptors relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53:S43–S50. doi:10.2337/diabetes.53.2007.S43
21. Vidal-Puig A, Jimenez-Liñan M, Lowell BB, et al. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest. 1996;97:2553. doi:10.1172/JCI118703
22. Yao Y, Fan L, Shi Y, Odsbu I. Morigen. A spatial control for correct timing of gene expression during the Escherichia coli cell cycle. Genes. 2016;8:1. doi:10.3390/genes8010001
23. Fleige S, Pfaffl MW. RNA integrity and the effect on the real-time qRT-PCR performance. Mol Aspects Med. 2006;27:126–139. doi:10.1016/j.mam.2005.12.003
24. Mihaela Š, Veronika O, Špela J, et al. Improved determination of plasmid copy number using quantitative real-time PCR for monitoring fermentation processes. Microb Cell Fact. 2008;7:1–12.
25. Usui S, Suzuki T, Hattori Y, et al. Design, synthesis, and biological activity of novel PPARγ ligands based on rosiglitazone and 15d-PGJ2. Bioorg Med Chem Lett. 2005;15:1547–1551. doi:10.1016/j.bmcl.2005.01.074
26. Fang M, Webster TF, Ferguson PL, Stapleton HM. Characterizing the peroxisome proliferator-activated receptor (PPARγ) ligand binding potential of several major flame retardants, their metabolites, and chemical mixtures in house dust. Environ Health Perspect. 2015;123:166–172. doi:10.1289/ehp.1408522
27. Rossi AM, Taylor CW. Analysis of protein-ligand interactions by fluorescence polarization. Nat Protoc. 2011;6(3):365–387. doi:10.1038/nprot.2011.305
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.