Back to Journals » Drug Design, Development and Therapy » Volume 12
Synthesis and anticancer activity of dimeric podophyllotoxin derivatives
Authors Zi C, Yang L, Xu F, Dong F, Yang D, Li Y, Ding Z, Zhou J, Jiang Z ![]() , Hu J
, Hu J ![]()
Received 6 March 2018
Accepted for publication 18 July 2018
Published 9 October 2018 Volume 2018:12 Pages 3393—3406
DOI https://doi.org/10.2147/DDDT.S167382
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Cheng-Ting Zi,1–3,* Liu Yang,2,* Feng-Qing Xu,2 Fa-Wu Dong,2 Dan Yang,2 Yan Li,2 Zhong-Tao Ding,3 Jun Zhou,2 Zi-Hua Jiang,4 Jiang-Miao Hu2
1Key Laboratory of Pu-er Tea Science, Ministry of Education, College of Science, Yunnan Agricultural University, Kunming 650201, China; 2State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China; 3Key Laboratory of Medicinal Chemistry for Nature Resource, Ministry of Education, School of Chemical Science and Technology, Yunnan University, Kunming 650091, China; 4Department of Chemistry, Lakehead University, Thunder Bay, ON P7B 5E1, Canada
*These authors contributed equally to this work
Background: Podophyllotoxin is a potent cytotoxic agent and serves as a useful lead compound for the development of antitumor drugs. Several podophyllotoxin-derived antitumor agents, including etoposide, are currently in clinical use; however, their therapeutic efficacy is often limited due to side effects and the development of resistance by cancer cells. Previous studies have shown that 4β-1,2,3-triazole derivatives of podophyllotoxin exhibit more potent anticancer activity and better binding to topoisomerase-II than etoposide. The effect of dimerization of such derivatives on the anticancer activity has not been studied.
Methods: Two moieties of podophyllotoxin were linked at the C-4 position via 1,2,3-triazole rings to give a series of novel dimeric podophyllotoxin derivatives. 4β-Azido-substituted podophyllotoxin derivatives (23 and 24) were coupled with various dipropargyl functionalized linkers by utilizing the copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction to provide dimeric products in very good yield. The in vitro anticancer activity of the synthesized compounds was evaluated by MTT assay against a panel of five human cancer cell lines (HL-60, SMMC-7721, A-549, MCF-7, and SW480). The normal BEAS-2B (lung) cell line was also included for study in order to evaluate the cancer selectivity of the most active compound as compared with normal cells.
Results: A group of 16 dimeric podophyllotoxin derivatives with different linkers were synthesized and structurally characterized. Most compounds do not show significant cytotoxicity (IC50 > 40 mM) against all five cancer cell lines. However, one compound (29) which bears a perbutyrylated glucose residue on the glycerol linker is highly potent against all five cancer cell lines tested, with IC50 values ranging from 0.43 to 3.50 µM. This compound (29) also shows good selectivity towards cancer cell lines as compared with the normal BEAS-2B (lung) cell line, showing selectivity indexes from 4.4 to 35.7.
Conclusion: The anticancer activity of dimeric podophyllotoxin derivatives is generally speaking not improved as compared to their monomeric counterparts, and the potency of these dimeric derivatives can be largely affected by the nature of the linker between the two moieties. Among the synthesized derivatives, compound 29 is significantly more cytotoxic and selective towards cancer cells than etoposide and cisplatin, which are currently in clinical use. Compound 29 is a promising anticancer drug and needs further studies.
Keywords: podophyllotoxin, dimeric podophyllotoxin derivatives, CuAAC reaction, perbutytylated glucose, antitumor, synthesis
Introduction
Podophyllotoxin 1 (Figure 1) is the most abundant naturally occurring cyclolignan mainly isolated from podophyllum species and shows strong cytotoxic activity against various cancer cell lines by inhibiting microtubule assembly.1–3 Podophyllotoxin is not a clinically useful anticancer drug because of its high toxicity; however, several semisynthetic derivatives, such as etoposide (2) (Figure 1), are clinically used chemotherapeutic agents for a number of cancers, including small cell lung cancer, testicular carcinoma, lymphoma, and Kaposi’s sarcoma.4–6
 | Figure 1 Structures of podophyllotoxin (1), etoposide (2), 4β-1,2,3-triazolyl-podophyllotoxin derivatives (3), and dimeric 4β-1,2,3-triazolyl-podophyllotoxin derivatives (4). |
Earlier reports indicated that the β-configuration at C-4 of podophyllotoxin scaffold is not favorable for tubulin polymerization inhibition activity.7 However, the comparison of the crystal structures of tubulin-DMEP (4′-demethylepipodophyllotoxin) and tubulin-podophyllotoxin suggests that the C-4 β-configuration does not show any disadvantage for tubulin binding.8 For podophyllotoxin derivatives as topoisomerase-II inhibitors, structure–activity relationship (SAR) data show that 4β-substitution is essential for the anticancer activity.9,10
In the attempt to discover less toxic and more effective anticancer agents, many podophyllotoxin derivatives have been synthesized for biological studies.11,12 4β-1,2,3-Triazole derivatives of podophyllotoxin have been shown to exhibit more potent anticancer activity and better binding to topoisomerase-II than etoposide.13–15 Recently, we also reported a group of podophyllotoxin glycoconjugates linked via 4β-1,2,3-triazole functionality as potential antitumor agents.16–18 Our studies showed that podophyllotoxin derivatives with a perbutyrylated sugar residue displayed higher activity than their counterparts lacking butyryl groups.16,17 There have also been reports on the synthesis of dimeric podophyllotoxin derivatives19,20 which exhibited promising in vitro anticancer activity against different human tumour cell lines. In the present study, a group of dimeric podophyllotoxin derivatives 4 (Figure 1), with different linkers have been prepared using the Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction.21,22 Their synthesis and anticancer activity against five cancer cell lines are described.
Results and discussion
Chemical synthesis
The click reaction of copper(I)-catalyzed Huisgen 1,3-dipolar azide-alkyne cycloaddition (CuAAC) provides 1,4-disubstituted 1,2,3-triazoles, which is a powerful tool for the generation of novel pharmacophores.21,22 To access dimeric podophyllotoxin derivatives 4 (Figure 1), di-propargyl functionized linkers are required. Scheme 1 depicts the synthesis of symmetric 1,3-di-O-propargyl glycerol (10) and its glycosylated derivatives (12–14). Initially, we tried direct propargylation of glycerol with propargyl bromide in the presence of sodium hydride (NaH) to prepare 10. However, the reaction provided complicated products and the strategy was abandoned. Thus, Solketal 5 was treated with propargyl bromide and sodium hydride, and then with HCl in methanol solution to give diol 6 as previously described.23 The diol 6 was first converted to bis-silyl ether 7 by treatment with tert-butyldimethylsilyl chloride (TBSCl) and imidazole in anhydrous N,N-dimethylformamide (DMF). The more labile primary TBS ether in 7 was then selectively cleaved with pyridine-HF in pyridine.24 Etherification of the primary alcohol 8 with propargyl bromide gave nine in 56% yield. Treatment of 9 with tetrabutylammonium fluoride (TBAF) in tetrahydrofuran (THF) gave alcohol 10.25 Next, 10 was allowed to react with the α-glucose trichloroacetimidate derivative 1126 in the presence of BF3·Et2O at −78°C to provide, as expected, only the β-glycoside 12 in 63% yield. Then, removal of acetyl groups with CH3ONa in CH3OH produced 13, which was subjected to perbutyrylation with butyric anhydride in the presence of pyridine to give the perbutyrylated product 14 in good yield. Interestingly, the anomeric proton of the glucose residue was found significantly downfield and shifted in the 1H-NMR spectra of the peracetylated derivative 12 (δ 6.45 ppm, d, J=8.0 Hz) and perbubyrylated derivative 14 (δ 6.14 ppm, d, J=8.0 Hz), while it appears to be normal in non-acylated 13 (δ 4.35 ppm, d, J=8.0 Hz). The coupling constant of 8.0 Hz for the anomeric proton confirms a β-glycosidic linkage in 12–14.
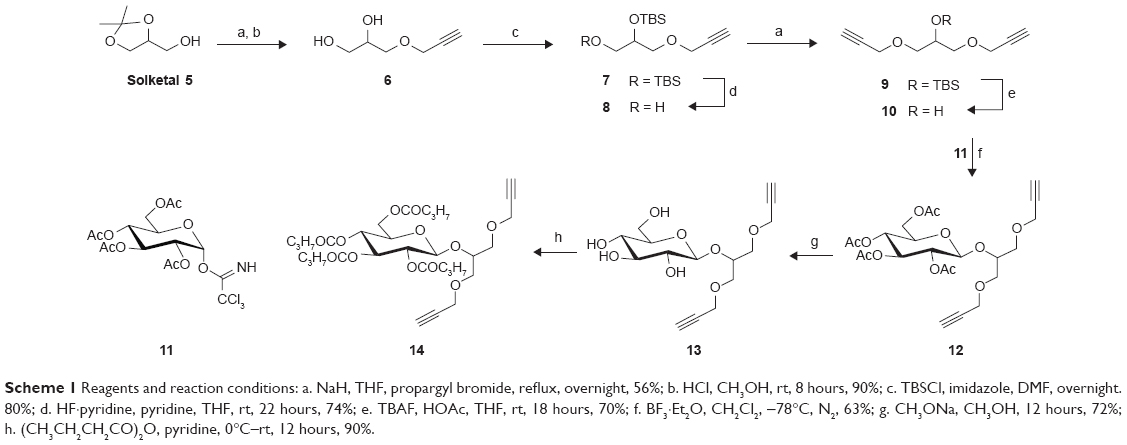 | Scheme 1 Reagents and reaction conditions: a. NaH, THF, propargyl bromide, reflux, overnight, 56%; b. HCl, CH3OH, rt, 8 hours, 90%; c. TBSCl, imidazole, DMF, overnight. 80%; d. HF·pyridine, pyridine, THF, rt, 22 hours, 74%; e. TBAF, HOAc, THF, rt, 18 hours, 70%; f. BF3·Et2O, CH2Cl2, −78°C, N2, 63%; g. CH3ONa, CH3OH, 12 hours, 72%; h. (CH3CH2CH2CO)2O, pyridine, 0°C–rt, 12 hours, 90%. |
The synthesis of dipropargyl functionalized linkers based on glucose scaffold is described in Scheme 2. The readily available 6-O-tritylated 1527 was treated with TBSCl and imidazole in anhydrous DMF to yield 16 in 80% yield. Acid catalyzed removal of the trityl group yielded 17 in 93% yield. Compound 17 was then treated with propargyl bromide and NaH to provide 18 (50%), which was treated with TBAF in the presence of acetic acid in THF to give triol 19 in 80% yield. Compound 19 was then subjected to benzylation, acetylation, or butyrylation by treatment with benzyl bromide/NaH, acetic anhydride/pyridine, or butyric anhydride/pyridine, respectively, to give 20, 21, or 22 in good to excellent yield.
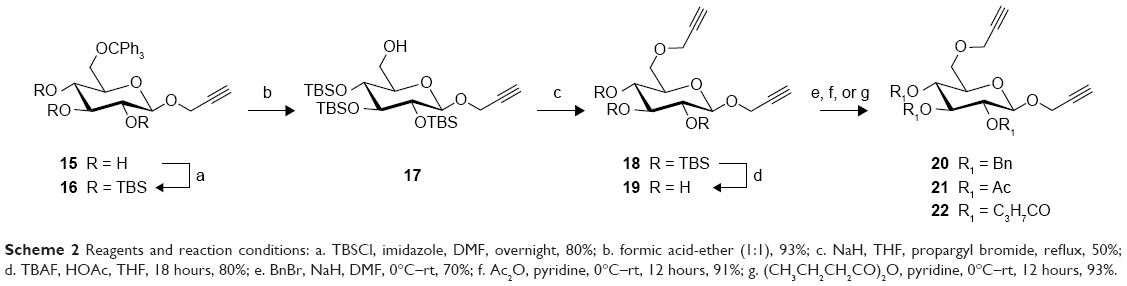 | Scheme 2 Reagents and reaction conditions: a. TBSCl, imidazole, DMF, overnight, 80%; b. formic acid-ether (1:1), 93%; c. NaH, THF, propargyl bromide, reflux, 50%; d. TBAF, HOAc, THF, 18 hours, 80%; e. BnBr, NaH, DMF, 0°C–rt, 70%; f. Ac2O, pyridine, 0°C–rt, 12 hours, 91%; g. (CH3CH2CH2CO)2O, pyridine, 0°C–rt, 12 hours, 93%. |
The azido-substituted podophyllotoxin derivatives needed for the click reaction, 4β-azido-4-deoxypodophyllotoxin 23 and 4β-azido-4-deoxy-4′-demethylpodophyllotoxin 24, can be readily prepared from podophyllotoxin according to a previous report.28 The azides 23 and 24 were allowed to react with dipropargyl functionalized linkers 10, 12–14, and 19–22 in the presence of CuSO4·5H2O, sodium ascorbate in t-butyl alcohol and water (1:1) at room temperature to obtain symmetric (25–32) and unsymmetric (33–40) dimeric podophyllotoxin derivatives in very good yield (Scheme 3).
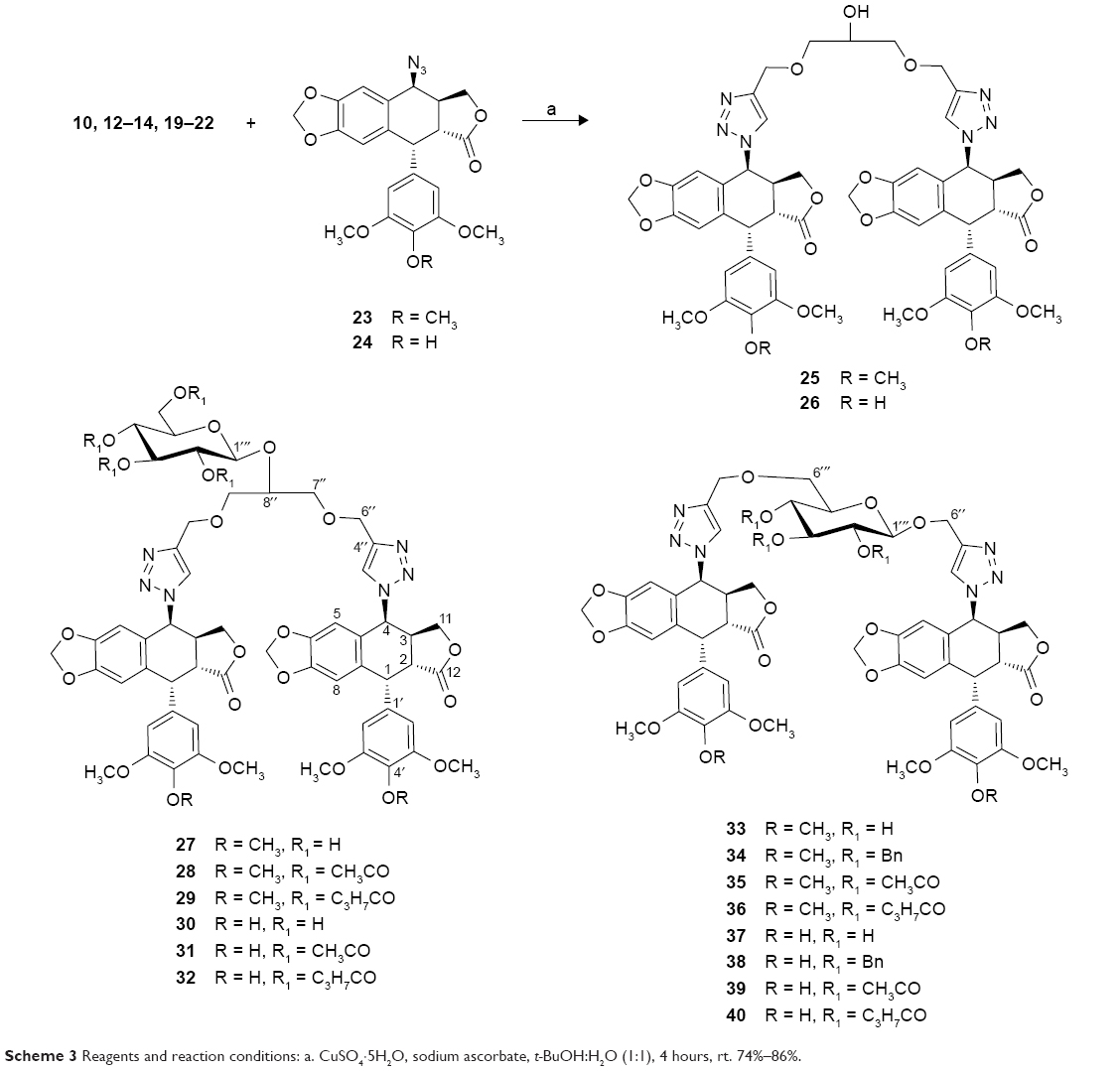 | Scheme 3 Reagents and reaction conditions: a. CuSO4·5H2O, sodium ascorbate, t-BuOH:H2O (1:1), 4 hours, rt. 74%–86%. |
All the products were characterized by 1H-NMR, 13C-NMR, ESI-MS, and HRESI-MS data. The presence of the triazole ring in these dimeric podophyllotoxin derivatives was confirmed by the proton signal at around δ 7.27–7.96 ppm (C5-H of the triazole ring) in the aromatic region of the 1H-NMR spectrum, as well as by a pair of carbon signals at around 145 ppm and 124 ppm in the 13C-NMR spectrum. The proton at C-4 of the podophyllotoxin scaffold of these derivatives appears to be doublet at 5.85–6.22 ppm, typically having J3,4 <5.0 Hz due to a cis relationship between H-3 and H-4. The two podophyllotoxin moieties in symmetric dimeric derivatives (25–32) are identical and give one set of NMR signals. On the other hand, the two podophyllotoxin moieties in unsymmetric dimeric derivatives (33–40) are not identical and produce two sets of NMR signals very close in chemical shifts. ESI-MS and HRESI-MS of all compounds showed the [M+Na]+ or [M+H]+ adduct as the molecular ion. Proton and carbon-13 NMR spectra for compounds 25–40 are available in the Supplementary Materials.
Anticancer activity
The in vitro anticancer activity of the synthesized dimeric podophyllotoxin derivatives 25–40 was evaluated using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay against five human cancer cell lines, including HL-60 (leukemia), SMMC-7721 (hepatoma), A-549 (lung cancer), MCF-7 (breast cancer), and SW480 (colon cancer). Etoposide (2) and cisplatin were taken as reference compounds and the IC50 (inhibition concentration with 50% cell growth relative to the control) of all compounds are presented in Table 1. Their IC50 values reveal that most of these derivatives are not active (IC50>40 μM). However, compound 29 is very active against all five cancer cell lines tested, with IC50 values ranging from 0.43 to 3.50 μM, which is significantly more potent than etoposide and cisplatin. Among the compounds based on a glucose linker (33–40), only 39 and 40 show moderate activity with IC50 values in the range of 14.00–30.45 μM.
 | Table 1 In vitro anticancer activity (IC50, μM) of dimeric podophyllotoxin derivatives 25–40 against human tumor cell lines |
The data in Table 1 indicate that the linking spacer between the two podophyllotoxin moieties can largely affect the activity of these compounds. Compound 29 which carries a perbutyrylated glucose residue displays much higher potency than those lacking a glucose residue (25), having a free glucose residue (27) or having a peracetylated glucose residue (28). This observation agrees with our earlier reports that several podophyllotoxin glycoconjugates containing perbutyrylated sugar residues show higher anticancer activity than those without butyryl groups.16,17 In comparison to 29, the 4′-demethylated analog 32 loses its activity, confirming the earlier observation that the substitution group on the 4′-position of podophyllotoxin scaffold can significantly affect the anticancer potency of podophyllotoxin derivatives.29 Previously, we reported several groups of monomeric podophyllotoxin derivatives bearing similar structure elements as those dimeric ones in the present study.16,17,29 Some of those monomeric derivatives show good anticancer activity with IC50 values in low μM range. Since most of the dimeric podophyllotoxin derivatives in this study display weak anticancer activity, dimerization might not be a good strategy for improving the potency of podophyllotoxin derivatives.
One major drawback of cancer chemotherapy is associated with the low-/non-selective nature of cytotoxic drugs, which attack cancer cells as well as normal cells, leading to serious side effects. To evaluate the degree of selectivity of 29, its growth inhibitory effect on a normal human bronchial epithelial cell line, BEAS-2B, was measured (Table 1). The selectivity index (SI) was expressed as the ratio of the IC50 value of the compound in normal BEAS-2B cell line over that in cancer cell line.30,31 A greater SI value indicates that the drug molecule displays higher selectivity towards cancer cells as compared with normal cells. As shown in Table 2, compound 29 has SI values ranging from 4.4 to 35.7 in all five cancer cell lines texted. Literature papers have considered that an SI value greater than 2.032 or 3.033 is an interesting selectivity index. Importantly, the selectivity indexes of 29 are much greater than those observed for both clinically used anticancer drugs, etoposide and cisplatin, except in the case of HL-60 cells where 29 and etoposide have similar SI values. These data suggest that 29 is significantly more cytotoxic to the cancer cell lines as compared with the normal cell line.
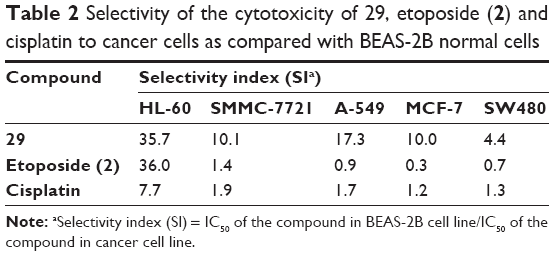 | Table 2 Selectivity of the cytotoxicity of 29, etoposide (2) and cisplatin to cancer cells as compared with BEAS-2B normal cells |
Conclusion
This paper describes the preparation of a group of dimeric podophyllotoxin derivatives linked via 1,2,3-triazole functional groups. 4β-Azido-podophyllotoxin/4′-demethylpodophyllotoxin reacts with various dipropargyl functionalized linkers by copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction to produce novel dimeric podophyllotoxin derivatives. MTT assay was used to evaluate the in vitro anticancer activity of these compounds against a panel of five human cancer cell lines including HL-60 (leukemia), SMMC-7721 (hepatoma), A-549 (lung cancer), MCF-7 (breast cancer), and SW480 (colon cancer). Most of the synthesized compounds do not show anticancer activity. Notably, compound 29, which bears a perbutyrylated glucose residue on the glycerol linker and is 4′-O-methylated on the E ring, is highly active against all five tested cancer cell lines with IC50 values ranging from 0.43 to 3.50 μM. As compared with the normal BEAS-2B (lung) cell line, compound 29 is significantly more selective towards all five tested cancer cell lines with selectivity indexes in the range of 4.4–35.7. Taken together, compound 29 is significantly more cytotoxic and selective towards cancer cells than the clinically used drug etoposide or cisplatin. Further studies are required to study the promising antitumor agent.
Experimental
General
Melting points were uncorrected. Mass spectroscopy (MS) data were obtained in the ESI mode using API Qstar Pulsar instrument. HRMS data were obtained in the ESI mode using LCMS-IT-TOF (Shimadzu, Kyoto, Japan). NMR spectra were acquired using Bruker AV-400 or DRX-500 or Bruker AVANCE III-600 (Bruker BioSpin GmbH, Rheinstetten, Germany) instruments, where tetramethylsilane (TMS) was used as an internal standard. Column chromatography (CC) was performed with flash silica gel (200–300 mesh; Qingdao Makall Group Co., Ltd; Qingdao; China). All reactions were monitored by thin-layer chromatography (TLC) and spots were visualized by spraying 10% H2SO4 in ethanol (EtOH) on warm silica gel plates. The human cancer cell lines HL-60, SMMC-7721, A-549, MCF-7, and SW480, and the normal BEAS-2B cell line were purchased from the American Type Culture Collection (ATCC).
1,2-Di-O-tert-butyldimethylsilyl-3-(prop-2-yn-1-yloxy)propan-1,2-diol (7)
Imidazole (3.8 g, 55.2 mmol) and TBSCl (7.2 g, 47.8 mmol) were added to a solution of diol 6 (2.4 g, 18.5 mmol) in dimethylformamide (DMF) (60 mL). The reaction mixture was stirred overnight, and then diluted with H2O (300 mL). The solution was extracted with ether (Et2O) (3×150 mL) and the combined organic layer was dried over sodium sulfate (Na2SO4). After removing the solvent in vacuo, the residue was purified by CC (Rf=0.20, petroleum ether: ethyl acetate=40:1) to give 7 (5.2 g, 80%) as a colorless oil. 1H-NMR (CDCl3, 400 MHz) δ 4.12 (d, 2 H, J=2.4 Hz, O-CH2), 3.84–3.78 (m, 1 H), 3.60–3.52 (m, 3 H), 3.45–3.41 (m, 1 H), 2.37 (t, 1 H, J=2.4 Hz, C≡CH), 0.87–0.86 (m, 18 H), 0.06–0.03 (m, 12 H); 13C-NMR (CDCl3, 100 MHz) δ 79.7 (C≡CH), 74.2 (C≡CH), 72.5 (O-CH), 71.6 (O-CH2), 64.8 (O-CH2), 58.4 (CH2-C≡CH), 25.9 (C-CH3), 25.8 (C-CH3), 25.6 (C-CH3), 18.2 (Si-C), 18.1 (Si-C), −4.7 (Si-CH3), −4.8 (Si-CH3), −5.4 (Si-CH3), −5.5 (Si-CH3); ESIMS was calculated for C18H38O3Si2Na [M+Na]+ 381 and was found to be 381.
2-O-tert-butyldimethylsilyl-3-(prop-2-yn-1-yloxy)propan-1,2-diol (8)
To a solution of 7 (6.8 g, 19.0 mmol) in dry THF (60 mL), the HF·pyridine complex (1.7 mL) and pyridine (10 mL) were added. The reaction mixture was stirred for 20 hours. After completion of the reaction (TLC monitoring), the solution was diluted with diethyl ether (100 mL), washed with 0.5 M HCl (2×50 mL) and saturated copper sulfate solution (50 mL), and dried over anhydrous Na2SO4. After removal of the solvents, the residue was purified by chromatography (Rf=0.50, petroleum ether: ethyl acetate=4:1) to give 8 (3.4 g, 74%) as a colorless liquid. 1H-NMR (CDCl3, 400 MHz) δ 4.16 (d, 2 H, J=2.4 Hz, O-CH2), 3.93–3.88 (m, 1 H), 3.67–3.63 (m, 1 H), 3.60–3.53 (m, 3 H), 2.44 (t, 1 H, J=2.4 Hz), 0.89 (s, 9 H) 0.10 (s, 6 H); 13C-NMR (CDCl3, 100 MHz) δ 79.7 (C≡CH), 74.2 (C≡CH), 72.5 (O-CH), 71.6 (O-CH2), 64.8 (HO-CH2), 58.4 (CH2-C≡CH), 25.9 (C-CH3), 18.2 (Si-C), 5.44 (Si-CH3); ESIMS was calculated for C12H24O3SiNa [M+Na]+ 267 and found to be 267.
2-O-tert-butyldimethylsilyl-1,3-di-(prop-2-yn-1-yloxy)propan-1,2-diol (9)
Suspension of NaH (253.2 mg, 6.3 mmol) in dry THF (5 mL) under N2 was added to a solution of 8 (1.0 g, 4.2 mmol) in dry tetrahydrofuran (THF) (15 mL) at 0°C. The mixture was stirred at room temperature for 0.5 hour, and then propargyl bromide (0.3 mL, 4.2 mmol) in THF (15 mL) was quickly added and the reaction was refluxed overnight. The reaction mixture was quenched with water, and then THF was removed in vacuo. The residue was extracted with CH2Cl2 (2×50 mL), and the organic layer was washed with brine (100 mL), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by CC (Rf=0.40, petroleum ether: ethyl acetate=10:1) to obtain 9 (1.2 g, 56%). 1H-NMR (CDCl3, 400 MHz) δ 4.33–4.32 (m, 2 H, O-CH2), 4.19–4.18 (m, 2 H, O-CH2), 3.79–3.75 (m, 1 H), 3.73–3.68 (m, 3 H), 3.62–3.58 (m, 1 H), 2.43–2.41 (m, 2 H, 2× C≡CH), 0.89 (s, 9 H), 0.06 (s, 9 H); 13C-NMR (CDCl3, 100 MHz) δ 77.7 (C≡CH), 74.9 (C≡CH), 69.5 (O-CH2), 62.4 (O-CH), 58.7 (O-CH2), 25.9 (C-CH3), 18.2 (Si-C), 5.44 (Si-CH3); ESIMS was calculated for C15H26O3SiNa [M+Na]+ 305 and was found to be 305.
1,3-Di-(prop-2-yn-1-yloxy)propan-2-ol (10)
Acetic acid (0.3 mL, 5.4 mmol) and tetra-butylammoniumfluoride trihydrate (1.5 mL, 5.4 mmol) were added to a solution of 9 (384.3 mg, 1.4 mmol) in dry THF (20 mL) at room temperature. The mixture was stirred for 18 hours, and then the solvent was evaporated under reduced pressure. The crude product was purified by passing through a short column (Rf=0.30, petroleum ether: ethyl acetate=2:1) to obtain 10 (160 mg, 70%) as a colorless liquid. 1H-NMR (CDCl3, 400 MHz) δ 4.40–4.28 (m, 2 H, O-CH2), 4.18–4.23 (m, 2 H, O-CH2), 3.83–3.76 (m, 1 H), 3.70–3.66 (m, 4 H), 2.48–2.45 (m, 2 H, 2× C≡CH); 13C-NMR (CDCl3, 100 MHz) δ 77.7 (C≡CH), 74.7 (C≡CH), 69.5 (O-CH2), 62.4 (O-CH), 58.7 (O-CH2); ESIMS was calculated for C9H12O3Na [M+Na]+ 191 and was found to be 191. The 1H-NMR and 13C-NMR data are in full agreement with those reported in the literature.25
1,3-Di-(prop-2-yn-1-yloxy)prop-2-yl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (12)
A solution of BF3·Et2O (87.2 μL, 0.7 mmol) in dichloromethane (1 mL) at −78°C was added dropwise to a solution of 1126 (225 mg, 0.5 mmol) and 10 (77 mg, 0.5 mmol) in dry CH2Cl2 (2 mL). The reaction mixture was brought to room temperature and stirred for 1 hour, and then Et3N (0.1 mL) was added to the mixture, followed by addition of AcOH (0.1 mL). The solvent was evaporated and the residue was purified by CC (Rf=0.60, petroleum ether: ethyl acetate=1:1) to obtain 12 (143.7 mg, 63%). 1H-NMR (CDCl3, 400 MHz) δ 6.45 (d, 1 H, J=8.0 Hz, C1-H), 5.20 (t, 1 H, J=10.0 Hz), 5.08 (t, 1 H, J=10.0 Hz), 5.03–4.97 (m, 1 H), 4.57–4.55 (m, 1 H), 4.29–4.28 (m, 2 H), 4.25–4.24 (m, 1 H), 4.18–4.15 (m, 2 H), 3.99–3.95 (m, 1 H), 3.89–3.86 (m, 1 H), 3.72–3.60 (m, 5 H), 2.45–2.43 (m, 2 H, 2× C≡CH), 2.09–2.00 (m, 12 H, 4× COCH3); 13C-NMR (CDCl3, 100 MHz) δ 170.7 (C=O), 170.3 (C=O), 170.2 (C=O), 169.4 (C=O), 101.0 (C-1), 76.2 (C≡CH), 74.8 (O-CH), 74.4 (C≡CH), 72.7, 71.8, 71.2, 69.2, 68.3 (O-CH2), 61.8 (C-6), 58.6 (O-CH2), 20.7 (COCH3), 20.7 (COCH3), 20.6 (COCH3), 20.5 (COCH3); ESIMS was calculated for C23H30O12Na [M+Na]+ 521 and was found to be 521.
1,3-Di-(prop-2-yn-1-yloxy)prop-2-yl β-D-glucopyranoside (13)
To a solution of 12 (115 mg, 0.2 mmol) in CH3OH (2 mL) sodium methoxide (4.2 mg, 0.07 mmol) was added. The resulting mixture was stirred for 24 hours (reaction monitored by TLC) and then the pH of the medium was adjusted to 7.0 by addition of HCl solution (1 M, H2O). The solvent was concentrated and the residue was purified by CC (Rf=0.20, CHCl3: CH3OH=9:1) to obtain the desired product 13 (54.9 mg, 72%). 1H-NMR (CD3OD, 400 MHz) δ 4.35 (d, 1 H, J=8.0 Hz, C1-H), 4.30–4.27 (m, 1 H), 4.19 (t, 4 H, J=2.4 Hz, 2× O-CH2), 4.00–3.86 (m, 3 H), 3.72–3.64 (m, 4 H), 3.35–3.27 (m, 2 H), 3.20–3.16 (m, 1 H), 2.87–2.85 (m, 2 H, 2× C≡CH); 13C-NMR (CD3OD, 100 MHz) δ 104.7 (C-1), 81.0 (C≡CH), 77.8 (C≡CH), 76.1, 75.9, 75.1, 75.0, 71.5 (O-CH2), 62.7 (C-6), 59.3 (O-CH2); ESIMS was calculated for C15H22O8Na [M+Na]+ 353 and was found to be 353.
1,3-Di-(prop-2-yn-1-yloxy)prop-2-yl 2,3,4,6-tetra-O-butyryl-β-D-glucopyranoside (14)
Butyric anhydride (0.3 mL, 2 mmol) was added to a solution of 13 (66.0 mg, 0.2 mmol) in pyridine (2 mL) at 0°C. The reaction mixture was stirred for 12 hours and then was diluted with water (5 mL) and extracted with ethyl acetate (3×5 mL). The organic layer was washed with 10% aqueous hydrochloric acid (10 mL) and brine (10 mL). The organic layer was dried over magnesium sulfate and evaporated to give a residue, which was purified by CC (Rf=0.20, petroleum ether: ethyl acetate=9:1) to obtain 14 (109.8 mg, 90%). 1H-NMR (CDCl3, 400 MHz) δ 6.14 (d, 1 H, J=8.0 Hz, C1-H), 5.23 (t, 1 H, J=9.2 Hz), 5.10 (t, 1 H, J=9.2 Hz), 5.05–4.99 (m, 1 H), 4.58–4.55 (m, 1 H), 4.28 (d, 2 H, J=2.4 Hz, O-CH2), 4.21–4.19 (m, 1 H), 4.16 (d, 2 H, J=2.4 Hz, O-CH2), 3.98–3.85 (m, 2 H), 3.72–3.70 (m, 4 H), 3.63–3.60 (m, 2 H, 2× C≡CH), 2.44–2.41 (m, 2 H, COCH2), 2.33–2.20 (m, 6 H, 3× COCH2), 1.66–1.54 (m, 8 H, 4× CH2CH3), 0.95–0.88 (m, 12 H, 4× CH2CH3); 13C-NMR (CDCl3, 100 MHz) δ 173.3 (C=O), 172.7 (C=O), 172.0 (C=O), 171.9 (C=O), 101.0 (C-1), 76.0 (C≡CH), 74.8 (O-CH), 74.4 (C≡CH), 71.9, 70.9, 70.1, 69.3, 68.0 (O-CH2), 68.0 (O-CH2), 61.7 (C-6), 58.6 (O-CH2), 35.9 (COCH2), 35.8 (COCH2), 35.8 (COCH2), 35.8 (COCH2), 18.2 (CH2CH3), 18.2 (CH2CH3), 18.2 (CH2CH3), 18.2 (CH2CH3), 13.6 (CH2CH3), 13.5 (CH2CH3), 13.5 (CH2CH3), 13.5 (CH2CH3); ESIMS was calculated for C31H46O12Na [M+Na]+ 633 and was found to be 633.
Prop-2-yn-1-yl 2,3,4-tri-O-tert-butyldimethylsilyl-6-O-trityl-β-D-glucopyranoside (16)
Imidazole (2.5 g, 36.0 mmol) and TBSCl (4.9 g, 32.5 mmol) were added to a solution of 1527 (4.2 g, 9.0 mmol) in DMF (50 mL). The reaction mixture was stirred overnight, and then diluted with H2O (200 mL). The solution was extracted with CH2Cl2, and the organic layer dried over Na2SO4. After removing the solvent in vacuo, the residue was purified by CC (Rf=0.30, petroleum ether: ethyl acetate=30:1) to give 16 (5.9 g, 80%) as a colorless oil. 1H-NMR (CDCl3, 400 MHz) δ 7.52–7.24 (m, 15 H, Ar-H), 4.57 (d, 1 H, J=6.8 Hz, C1-H), 4.54 (d, 2 H, J=2.4 Hz, O-CH2), 3.56–3.52 (m, 1 H), 3.45–3.38 (m, 4 H), 3.25–3.21 (m, 1 H), 2.49 (t, 1 H, J=2.4 Hz, C≡CH), 0.95–0.65 (s, 27 H), 0.20–0.05 (s, 18 H); 13C-NMR (CDCl3, 100 MHz) δ 144.1, 128.8, 127.8, 126.9, 100.3 (C-1), 86.3, 78.7 (C≡CH), 78.7 (C≡CH), 76.2, 75.3, 74.9, 71.5, 63.8 (C-6), 55.3 (O-CH2), 26.0 (Si-C), 25.9 (Si-C), 25.8 (Si-C), 18.3 (C-CH3), 18.2 (C-CH3), 17.9 (C-CH3), −3.8 (Si-CH3), −3.9 (Si-CH3), −4.0 (Si-CH3), −4.7 (Si-CH3), −4.7 (Si-CH3), −5.2 (Si-CH3); ESIMS was calculated for C46H70O6Si3Na [M+Na]+ 825 and was found to be 825.
Prop-2-yn-1-yl 2,3,4-tri-O-tert-butyldimethylsilyl-β-D-glucopyranoside (17)
A solution of formic acid in ether (30 mL: 30 mL) was added dropwise to a solution of 16 (1.2 g, 1.5 mmol) in diethyl ether (30 mL) at room temperature. The mixture was stirred for 3 hours, then diluted with water (30 mL), and quenched via careful addition of solid potassium carbonate (K2CO3). The layers were separated, and the aqueous layer was extracted with Et2O (3×30 mL), and the resulting organic layers were combined and concentrated in vacuo. The residue was dissolved in CH3OH (40 mL), treated with K2CO3 (1.1 g) and stirred for 5 minutes. The resulting mixture was concentrated, and the residue was purified by CC (Rf=0.40, petroleum ether: ethyl acetate=9:1) to obtain 17 (779.3 mg, 93%). 1H-NMR (CDCl3, 400 MHz) δ 4.57 (d, 1 H, J=7.6 Hz, C1-H), 4.35 (d, 2 H, J=2.4 Hz, O-CH2), 3.89–3.85 (m, 1 H), 3.71–3.66 (m, 1 H), 3.54–3.43 (m, 2 H), 3.36–3.30 (m, 2 H), 2.45 (t, 1 H, J=2.4 Hz, C≡CH), 0.90–0.88 (m, 27 H), 0.15–0.11 (m, 18 H); 13C-NMR (CDCl3, 100 MHz) δ 101.2 (C-1), 78.8 (C≡CH), 78.0 (C≡CH), 76.3, 75.1, 75.0, 70.7, 62.5 (C-6), 56.2 (O-CH2), 25.9 (C-CH3), 25.9 (C-CH3), 25.8 (C-CH3), 18.2 (Si-C), 18.2 (Si-C), 18.2 (Si-C), −3.9 (Si-CH3), −3.9 (Si-CH3), −4.0 (Si-CH3), −4.7 (Si-CH3), −4.7 (Si-CH3), −5.0 (Si-CH3); ESIMS was calculated for C27H56O6Si3Na [M+Na]+ 583 and was found to be 583.
Prop-2-yn-1-yl 6-O-(prop-2-yn-1-yl)-2,3,4-tri-O-tert-butyldimethylsilyl-β-D-glucopyranoside (18)
A solution of 17 (536.3 mg, 1 mmol) in dry THF (10 mL) was added at 0°C to a suspension of NaH (60 mg, 2.5 mmol) in dry THF (3 mL) under N2. The mixture was stirred at room temperature for 0.5 hour, then propargyl bromide (1 mL, 0.2 mmol) in THF (5 mL) was quickly added and the reaction mixture was refluxed overnight. The reaction mixture was quenched with water (20 mL), and then THF was removed in vacuo. The residue was extracted with CH2Cl2 (2×20 mL), and the organic layer was washed with brine (20 mL) and dried over Na2SO4, concentrated under reduced pressure. The residue was purified by CC (Rf=0.50, petroleum ether: ethyl acetate=30:1) to obtain 18 (299.2 mg, 50%). 1H-NMR (CDCl3, 400 MHz) 4.42 (d, 1 H, J=7.6 Hz, C1-H), 4.37–4.35 (m, 1 H), 4.24–4.19 (m, 4 H, 2× O-CH2), 3.87–3.84 (m, 1 H), 3.66–3.62 (m, 1 H), 3.55–3.50 (m, 1 H), 3.46–3.40 (m, 1 H), 3.36–3.32 (m, 1 H), 2.42–2.40 (m, 2 H, 2× C≡CH), 0.90–0.89 (m, 27 H), 0.15–0.11 (m, 18 H); 13C-NMR (CDCl3, 100 MHz) δ 100.3 (C-1), 79.7 (C≡CH), 78.8 (C≡CH), 78.2 (C≡CH), 76.0, 75.0, 74.8, 74.6 (C≡CH), 70.7, 68.8 (C-6), 58.6 (CH2-C≡CH), 55.6 (CH2-C≡CH), 26.0 (C-CH3), 25.9 (C-CH3), 25.9 (C-CH3), 18.2 (Si-C), 18.2 (Si-C), 18.2 (Si-C) −3.8 (Si-CH3), −3.9 (Si-CH3), −4.0 (Si-CH3), −4.8 (Si-CH3), −4.7 (Si-CH3), −5.0 (Si-CH3); ESIMS was calculated for C30H58O6Si3Na [M+Na]+ 621 and was found to be 621.
Prop-2-yn-1-yl 6-O-(prop-2-yn-1-yl)-β-D-glucopyranoside (19)
To a solution of 18 (119.7 mg, 0.2 mmol) in dry THF (5 mL), TBAF (0.2 mL, 0.7 mmol) was added at room temperature. The mixture was stirred for 18 hours, and then the solvent was evaporated under reduced pressure and the crude product was purified by CC (Rf=0.50, CHCl3: CH3OH=20:1) to obtain the desired product 19 (41.0 mg, 80%) as a colorless syrup. 1H-NMR ((CD3)2SO, 600 MHz) 5.97 (d, 1 H, J=7.6 Hz, C1-H), 5.93–5.89 (m, 1 H), 5.13–4.91 (m, 2 H), 4.56 (dd, 1 H, J=1.8 Hz, 11.4 Hz), 4.31–4.29 (m, 1 H), 4.00–3.97 (m, 4 H, 2× O-CH2), 3.85–3.76 (m, 1 H), 2.38–2.35 (m, 2 H, 2× C≡CH); 13C-NMR ((CD3)2SO, 150 MHz) δ 100.8 (C-1), 80.5 (C≡CH), 79.8 (C≡CH), 77.4 (C≡CH), 77.1 (C≡CH), 76.5, 75.4, 73.1, 70.0, 69.1 (C-6), 57.6 (O-CH2), 57.5 (O-CH2); ESIMS was calculated for C12H16O6Na [M+Na]+ 279 and was found to be 279.
Prop-2-yn-1-yl 6-O-(prop-2-yn-1-yl)-2,3,4-tri-O-benzyl-β-D-glucopyranoside (20)
Suspension (60%) of NaH in paraffin (28.0 mg, 0.7 mmol) was added to a solution of 19 (51.2 mg, 0.2 mmol) in anhydrous DMF (3 mL) at 0°C under argon atmosphere. The resulting solution was stirred for 30 minutes at room temperature. Benzyl bromide (0.2 mL, 1.1 mmol) was added drop-wise at 0°C followed by a catalytic amount of tetra-n-butyl ammonium iodide (20 mg). The resulting reaction mixture was stirred at room temperature under argon for 10 hours. After completion of the reaction (as judged by TLC), excess NaH was quenched with methanol (0.5 mL) followed by ice water (10 mL) and extracted with ether (3×10 mL). The combined organic layer was washed with water, brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude residue was purified by CC (Rf=0.20, petroleum ether: ethyl acetate=30:1) to obtain 20 (73.6 mg, 70%). 1H-NMR (CDCl3, 400 MHz) δ 7.38–7.26 (m, 15 H, Ar-H), 4.99–4.92 (m, 1 H), 4.88–4.79 (m, 1 H), 4.70–4.64 (m, 6 H, 3× O-CH2), 4.45 (d, 1 H, J=6.8 Hz, C1-H), 4.43–4.40 (m, 1 H), 4.27–4.15 (m, 1 H), 3.86–3.82 (m, 1 H), 3.78–3.75 (m, 1 H), 3.70–3.61 (m, 2 H, O-CH2), 3.50–3.46 (m, 2 H, O-CH2), 2.47 (s, 1 H, C≡CH), 2.39 (s, 1 H, C≡CH); 13C-NMR (CDCl3, 100 MHz) δ 138.6, 138.3, 138.1, 128.5, 128.4, 128.4, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.8, 127.7, 127.6, 101.4 (C-1), 84.5, 81.9, 79.6 (C≡CH), 79.0 (C≡CH), 75.7, 75.0, 74.9, 74.8 (C≡CH), 74.8 (C≡CH), 68.1 (C-6), 58.6 (O-CH2), 56.0 (O-CH2); ESIMS was calculated for C33H34O6Na [M+Na]+ 549 and was found to be 549.
Prop-2-yn-1-yl 6-O-(prop-2-yn-1-yl)-2,3,4-tri-O-acetyl-β-D-glucopyranoside (21)
To a solution of 20 (51.2 mg, 0.2 mmol) in pyridine (2 mL) at 0°C, acetic anhydride (0.2 mL, 2 mmol) was added. The reaction mixture was stirred at room temperature for 12 hours (TLC monitoring). The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (3×10 mL). The organic layer was washed with 10% aqueous hydrochloric acid (10 mL) and brine (10 mL). The organic layer was dried over Na2SO4 and evaporated to give a residue, which was purified by CC (Rf=0.20, petroleum ether: ethyl acetate=4:1) to obtain 21 (69.5 mg, 91%). 1H-NMR (CDCl3, 400 MHz) δ 4.78 (t, 1 H, J=9.4 Hz), 4.66 (d, 1 H, J=8.0 Hz, C1-H), 4.37–4.36 (m, 2 H), 4.24–4.20 (m, 2 H), 3.87 (dd, 1 H, J=1.7 Hz, 10.8 Hz), 3.68–3.43 (m, 4 H, 2× O-CH2), 2.43–2.42 (m, 2 H, 2× C≡CH), 2.15–2.10 (m, 9 H, 3× COCH3); 13C-NMR (CDCl3, 100 MHz) δ 171.0 (C=O), 169.9 (C=O), 169.8 (C=O), 100.4 (C-1), 79.5 (C≡CH), 78.7 (C≡CH), 76.2 (C≡CH), 76.0 (C≡CH), 74.9, 74.7, 73.9, 71.9, 68.5 (C-6), 58.7 (O-CH2), 55.6 (O-CH2), 25.9 (COCH3), 25.9 (COCH3), 25.8 (COCH3); ESIMS was calculated for C18H22O9Na [M+Na]+ 405 and found to be 405.
Prop-2-yn-1-yl 6-O-(prop-2-yn-1-yl)-2,3,4-tri-O-butyryl-β-D-glucopyranoside (22)
Butyryl anhydride (0.3 mL, 2 mmol) was added to a solution of 20 (51.2 mg, 0.2 mmol) in pyridine (2 mL) at 0°C. The reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (3×10 mL). The organic layer was washed with 10% aqueous hydrochloric acid (10 mL) and brine (10 mL). The organic layer was dried over Na2SO4 and evaporated to give a residue, which was purified by CC (Rf=0.20, petroleum ether: ethyl acetate=10:1) to obtain 22 (86.7 mg, 93%). 1H-NMR (CDCl3, 400 MHz) δ 4.92 (t, 1 H, J=9.3 Hz), 4.47 (d, 1 H, J=8.0 Hz, C1-H), 4.38–4.36 (m, 2 H), 4.19–4.17 (m, 3 H), 3.65–3.44 (m, 4 H, 2× O-CH2), 2.43–2.41 (m, 2 H, 2× C≡CH), 2.36–2.32 (m, 6 H, 3× COCH2), 1.70–1.63 (m, 6 H, 3× CH2CH3), 0.98–0.89 (m, 9 H, 3× CH2CH3); 13C-NMR (CDCl3, 100 MHz) δ 173.2 (C=O), 173.2 (C=O), 173.1 (C=O), 100.4 (C-1), 79.3 (C≡CH), 78.6 (C≡CH), 76.7 (C≡CH), 76.2 (C≡CH), 75.2, 74.9, 73.3, 71.9, 68.7 (C-6), 58.7 (O-CH2), 55.8 (O-CH2), 36.2 (COCH2), 36.2 (COCH2), 36.1 (COCH2), 18.4 (CH2CH3), 18.2 (CH2CH3), 18.2 (CH2CH3), 13.7 (CH2CH3), 13.6 (CH2CH3), 13.6 (CH2CH3); ESIMS was calculated for C24H34O9Na [M+Na]+ 489 and was found to be 489.
Click reaction – general procedure for the preparation of dimeric podophyllotoxin derivatives 25–40
Copper (II) sulfate pentahydrate (0.01 mmol) and sodium ascorbate (1.0 M in H2O, 3 d) were added to a solution of a terminal-alkyne 10, 12–14, or 19–22 (0.1 mmol) and a 4β-azido-podophyllotoxin analog 23 or 2428 (0.1 mmol) in t−BuOH-H2O (1:1, 1 mL) at room temperature. The reaction mixture was stirred at room temperature for 4 hours until the starting material disappeared as indicated by TLC. Then, the mixture was diluted with water (10 mL) and extracted with ethyl acetate (3×10 mL), and the combined organic layer was dried over Na2SO4. The solvent was evaporated and the residue was purified by CC to obtain the cycloaddition product.
1,3-Di-[1-(4-deoxypodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methoxy]-propan-2-ol (25)
Rf=0.60 (CHCl3: CH3OH=30:1). White amorphous powder, yield 86%; mp. 164°C–166°C (CH2Cl2); [α]D23.4: −117.9 (c 0.28, Pyridine); 1H-NMR (CDCl3, 400 MHz) δ 7.35 (s, 1 H), 7.32 (s, 1 H), 6.62–6.61 (m, 4 H), 6.31 (s, 4 H), 6.08–5.99 (m, 6 H), 4.75–4.71 (m, 2 H), 4.68–4.60 (m, 6 H), 4.38–4.35 (m, 2 H), 3.81 (s, 6 H), 3.76 (s, 12 H), 3.70–3.69 (s, 1 H), 3.64–3.60 (m, 4 H), 3.24–3.22 (m, 2 H), 3.14–3.11 (m, 2 H); 13C-NMR (CDCl3, 100 MHz) δ 173.2, 152.8, 149.4, 148.0, 145.4, 145.0, 137.5, 134.3, 133.2, 124.6, 123.0, 110.5, 108.8, 108.1, 102.0, 79.1, 70.3, 70.1, 67.4, 60.7, 58.7, 56.3, 43.6, 41.5, 37.1; ESIMS: m/z 1069 [M+Na]+, HRESIMS was calculated for C53H54N6NaO17 [M+Na]+ 1069.3443 and was found to be 1069.3437.
1,3-Di-[1-(4-deoxy-4′-demethylpodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methoxy]-propan-2-ol (26)
Rf=0.40 (CHCl3: CH3OH=30:1). White amorphous powder, yield 82%; mp. 190°C–192°C (CH3OH); [α]D23.5: −200.6 (c 0.11, Pyridine); 1H-NMR (C2D6SO, 400 MHz) δ 7.96 (s, 1 H), 7.95 (s, 1 H), 6.73 (s, 2 H), 6.64 (s, 2 H), 6.25 (s, 4 H), 6.22 (d, 2 H, J=5.2 Hz), 6.02–5.96 (m, 4 H), 4.67–4.64 (m, 2 H), 4.62–4.58 (m, 4 H), 4.48–4.47 (m, 2 H), 4.36–4.33 (m, 2 H), 3.63 (s, 12 H), 3.53–3.26 (m, 5 H), 3.24–3.18 (m, 2 H), 2.95–2.91 (m, 2 H); 13C-NMR (C2D6SO, 100 MHz) δ 173.7, 148.1, 147.3, 146.9, 144.5, 143.9, 134.9, 133.3, 129.7, 126.1, 124.7, 124.6, 109.9, 108.7, 108.4, 101.6, 78.7, 69.6, 67.2, 60.7, 57.5, 56.0, 42.8, 40.9, 36.5; ESIMS: m/z 1041 [M+Na]+, HRESIMS was calculated for C51H50N6NaO17 [M+Na]+ 1041.3130 and was found to be 1041.3127.
1,3-Di-[1-(4-deoxypodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methoxy]-prop-2-yl β-D-glucopyranoside (27)
Rf=0.50 (CHCl3: CH3OH=9:1). White amorphous powder, yield 76%; mp. 140°C–142°C (CH2Cl2); [α]D23.6: −53.7 (c 0.19, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.49 (s, 1 hour), 7.41 (s, 1 H), 6.59–6.58 (m, 2 H), 6.55 (m, 2 H), 6.30 (s, 4 H), 6.04–5.93 (m, 6 H), 4.71–4.67 (m, 5 H), 4.53–4.50 (m, 2 H), 4.33–4.29 (m, 4 H), 3.95–3.92 (m, 1 H), 3.77 (s, 6 H), 3.74 (s, 12 H), 3.65–3.52 (m, 9 H), 3.37–3.18 (m, 5 H); 13C-NMR (CDCl3, 100 MHz) δ 173.6, 173.5, 152.7, 149.2, 147.9, 144.8, 144.5, 137.3, 134.4, 133.2, 124.8, 110.4, 108.8, 108.1, 103.2, 102.0, 77.2, 76.4, 75.9, 73.4, 69.7, 67.5, 64.3, 64.2, 62.6, 61.5, 60.7, 58.6, 56.3, 43.6, 41.4, 37.0; ESIMS: m/z 1209 [M+H]+, HRESIMS was calculated for C59H64N6NaO22 [M+Na]+ 1231.3971 and was found to be 1231.3962.
1,3-Di-[1-(4-deoxypodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methoxy]-prop-2-yl 2,3,4,6-tera-O-acetyl-β-D-glucopyranoside (28)
Rf=0.50 (CHCl3: CH3OH=30:1). White amorphous powder, yield 80%; mp. 140°C–142°C (CH2Cl2); [α]D23.8: −42.9 (c 0.14, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.44 (s, 1 H), 7.35 (s, 1 H), 6.63 (s, 2 H), 6.62–6.61 (m, 2 H), 6.32 (s, 4 H), 6.08 (d, 2 H, J=2.0 Hz), 6.01–6.00 (m, 4 H), 5.90 (d, 1 H, J=7.2 Hz), 5.22–5.17 (m, 1 H), 5.11–5.09 (m, 1 H), 5.00–4.96 (m, 1 H), 4.76–4.73 (m, 4 H), 4.71–4.66 (m, 2 H), 4.58–4.52 (m, 3 H), 4.39–4.25 (m, 3 H), 4.15–4.11 (m, 1 H), 3.94–3.91 (m, 1 H), 3.81 (s, 6 H), 3.77 (s, 12 H), 3.65–3.58 (m, 4 H), 3.31–3.21 (m, 4 H), 2.07–2.01 (m, 12 H, 4× COCH3); 13C-NMR (CDCl3, 100 MHz) δ 173.3, 173.2, 170.6, 170.2, 169.5, 169.5, 152.8, 149.3, 148.0, 145.2, 144.9, 137.5, 134.3, 133.2, 124.7, 123.2, 110.5, 108.8, 108.2, 101.9, 100.9, 77.2, 72.6, 71.8, 71.2, 70.2, 68.3, 67.4, 64.7, 61.8, 60.8, 58.6, 56.2, 43.4, 41.5, 37.1, 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3), 20.6 (COCH3); ESIMS: m/z 1377 [M+H]+, HRESIMS was calculated for C67H72N6NaO26 [M+Na]+ 1399.4394 and was found to be 1399.4391.
1,3-Di-[1-(4-deoxypodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methoxy]-prop-2-yl 2,3,4,6-tera-O-butyryl-β-D-glucopyranoside (29)
Rf=0.30 (petroleum ether: ethyl acetate=1:3). White amorphous powder, yield 78%; mp 110°C–112°C (CH2Cl2); [α]D23.8: −40.1 (c 0.22, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.44 (s, 1 H), 7.37 (s, 1 H), 6.63 (s, 2 H), 6.61 (s, 2 H), 6.32 (s, 4 H), 6.08–5.99 (m, 6 H), 5.88 (d, 1 H, J=3.2 Hz), 5.25–5.21 (m, 1 H), 5.14–5.08 (m, 1 H), 5.02–4.95 (m, 1 H), 4.79–4.76 (m, 4 H), 4.72–4.66 (m, 2 H), 4.57–4.51 (m, 3 H), 4.38–4.31 (m, 2 H), 4.21–4.17 (m, 2 H), 3.91–3.88 (m, 1 H), 3.81 (s, 6 H), 3.76 (s, 12 H), 3.63–3.57 (m, 4 H), 3.54–3.48 (m, 2 H), 3.31–3.21 (m, 2 H), 2.30–2.18 (m, 8 H, 4× COCH2), 1.63–1.53 (m, 8 H, 4× CH2CH3), 0.92–0.87 (m, 12 H, 4× CH2CH3); 13C-NMR (CDCl3, 100 MHz) δ 173.2 (2C), 172.6, 172.4, 172.0, 172.0, 152.7 (2C), 149.3, 148.0, 145.3, 145.2, 137.5, 134.3, 133.0, 124.7, 123.2, 110.5, 108.8, 108.2, 101.9, 100.7, 77.2, 72.2, 72.0, 71.0, 68.0, 67.3, 64.7, 61.7, 60.7, 56.3, 56.0, 43.6, 41.5, 37.1, 35.9 (COCH2), 35.8 (COCH2), 35.7 (COCH2), 18.3 (CH2CH3), 18.2 (CH2CH3), 18.1 (CH2CH3), 13.6 (CH2CH3), 13.5 (CH2CH3), 13.5 (CH2CH3); ESIMS: m/z 1489 [M+H]+, HRESIMS was calculated for C75H88N6NaO26 [M+Na]+ 1511.5646 and was found to be 1511.5639.
1,3-Di-[1-(4-deoxy-4′-demethylpodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methoxy]-prop-2-yl β-D-glucopyranoside (30)
Rf=0.40 (CHCl3: CH3OH=9:1). White amorphous powder, yield 79%; mp 173°C–174°C (CHCl3); [α]D23.3: −60.0 (c 0.14, CH3OH+CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.44 (s, 1 H), 7.36 (s, 1 H), 6.56–6.55 (m, 2 H), 6.54 (s, 2 H), 6.26 (s, 4 H), 6.03–5.92 (m, 6 H), 4.69–4.66 (m, 5 H), 4.52 (d, 2 H, J=4.0 Hz), 4.31–4.27 (m, 2 H), 4.24–4.19 (m, 2 H), 3.93–3.90 (m, 1 H), 3.80–3.77 (m, 2 H), 3.72 (s, 12 H), 3.64–3.54 (m, 7 H), 3.36–3.18 (m, 5 H); 13C-NMR (CDCl3, 100 MHz) δ 173.9, 173.8, 149.3, 147.9, 146.7, 144.8, 144.6, 134.4, 133.4, 129.6, 124.6, 123.6, 110.4, 108.76, 107.8, 101.3, 101.9, 77.2, 76.2, 75.9, 73.4, 73.3, 69.9, 67.4, 64.4, 63.2, 58.7, 56.4, 43.3, 41.5, 36.9; ESIMS: m/z 1181 [M+H]+, HRESIMS was calculated for C57H60N6NaO22 [M+Na]+ 1203.3658 and was found to be 1203.3637.
1,3-Di-[1-(4-deoxy-4′-demethylpodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methoxy]-prop-2-yl 2,3,4,6-tera-O-acetyl-β-D-glucopyranoside (31)
Rf=0.50 (CHCl3: CH3OH=15:1). White amorphous powder, yield 81%; mp 158°C–160°C (CHCl3); [α]D23.2: −49.7 (c 0.15, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.52 (s, 1 H), 7.41 (s, 1 H), 6.65–6.64 (m, 2 H), 6.37–6.35 (m, 6 H), 6.10 (d, 2 H, J=4.2 Hz), 6.04–6.03 (m, 4 H), 6.00 (d, 1 H J=7.2 Hz), 5.24–5.20 (m, 1 H), 5.15–5.00 (m, 1 H), 5.02–4.96 (m, 1 H), 4.79–4.70 (m, 6 H), 4.60–4.55 (m, 3 H), 4.40–4.37 (m, 1 H), 4.32–4.27 (m, 2 H), 4.17–4.15 (m, 1 H), 3.96–3.92 (m, 1 H), 3.78–3.70 (m, 2 H), 3.66–3.60 (m, 2 H), 3.27–3.20 (m, 4 H), 2.09–2.03 (m, 12 H, 4× COCH3); 13C-NMR (CD3OD, 100 MHz) δ 173.4, 173.3, 170.7, 170.3, 169.5, 169.5, 149.3, 147.9, 146.6, 145.2, 144.9, 134.4, 133.4, 130.0, 124.7, 123.3, 110.5, 108.8, 107.8, 101.9, 100.9, 77.0, 72.6, 71.9, 71.2, 70.2, 70.1, 68.3, 67.4, 64.8, 61.8, 58.7, 56.6, 43.5, 41.7, 37.1, 20.8 (COCH3), 20.7 (COCH3), 20.6 (COCH3), 20.6 (COCH3); ESIMS: m/z 1349 [M+H]+, HRESIMS was calculated for C65H68N6NaO26 [M+Na]+ 1371.4081 and was found to be 1371.4072.
1,3-Di-[1-(4-deoxy-4′-demethylpodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methoxy]-prop-2-yl 2,3,4,6-tera-O-butyryl-β-D-glucopyranoside (32)
Rf=0.20 (petroleum ether: ethyl acetate=1:3). White amorphous powder, yield 78%; mp 138°C–139°C (CH2Cl2); [α]D23.8: −50.8 (c 0.14, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.52 (s, 1 H), 7.40 (s, 1 H), 6.62 (s, 2 H), 6.61 (s, 2 H), 6.33 (s, 4 H), 6.07–5.97 (m, 7 H), 5.26–5.21 (m, 1 H), 5.16–5.12 (m, 1 H), 5.02–4.96 (m, 1 H), 4.78–4.73 (m, 4 H), 4.70–4.68 (m, 2 H), 4.57–4.51 (m, 3 H), 4.36–4.28 (m, 2 H), 4.21–4.17 (m, 2 H), 3.92–3.88 (m, 1 H), 3.80 (s, 12 H), 3.74–3.69 (m, 2 H), 3.61–3.58 (m, 2 H), 3.54–3.47 (m, 2 H), 3.32–3.27 (m, 2 H), 2.31–2.20 (m, 8 H, 4× COCH2), 1.65–1.53 (m, 8 H, 4× CH2CH3), 0.92–0.87 (m, 12 H, 4× CH2CH3); 13C-NMR (CDCl3, 100 MHz) δ 173.2, 172.3, 172.1, 172.0, 172.0, 148.2, 148.0, 146.6, 145.1, 145.0, 134.3, 133.4, 129.8, 124.7, 123.2, 110.5, 108.8, 107.8, 101.9, 100.4, 77.2, 72.2, 72.1, 71.0, 68.0, 67.3, 65.2, 64.7, 61.7, 58.6, 56.5, 43.5, 41.6, 37.0, 36.5, 36.0 (COCH2), 35.9 (COCH2), 35.9 (COCH2), 18.3 (CH2CH3), 18.2 (CH2CH3), 18.1 (CH2CH3), 13.6 (CH2CH3), 13.6 (CH2CH3), 13.5 (CH2CH3); ESIMS: m/z 1461 [M+H]+, HRESIMS was calculated for C73H84N6NaO26 [M+Na]+ 1483.5333 was found to be 1483.5317.
1,6-Di-O-[1-(4-deoxypodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methyl]-β-D-glucopyranose (33)
Rf=0.60 (CHCl3: CH3OH=10:1). White amorphous powder, yield 72%; mp 200°C–202°C (CHCl3-CH3OH); [α]D22.9: −52.7 (c 0.18, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 7.50 (s, 1 H), 7.40 (s, 1 H), 6.60 (s, 2 H), 6.59–6.57 (m, 2 H), 6.33 (s, 4 H), 6.06–5.96 (m, 6 H), 4.89 (d, 1 H, J=8.0 Hz), 4.74–4.70 (m, 4 H), 4.64–4.56 (m, 2 H), 4.44–4.42 (m, 2 H), 4.16–4.12 (m, 2 H), 3.81 (s, 6 H), 3.77 (s, 12 H), 3.73–3.71 (m, 2 H), 3.49–3.36 (m, 4 H), 3.20–3.18 (m, 4 H); 13C-NMR (CDCl3, 150 MHz) δ 173.5, 173.5, 152.8, 149.2, 147.9, 144.7, 144.3, 137.5, 134.4, 133.2, 124.8, 123.5, 110.4, 108.8, 108.2, 102.4, 101.9, 76.4, 75.2, 73.3, 69.8, 69.6, 67.4, 64.4, 62.5, 60.7, 58.6, 56.3, 43.6, 41.5, 37.0; ESIMS: m/z 1135 [M+H]+, HRESIMS was calculated for C56H58N6NaO20 [M+Na]+ 1157.3604 was found to be 1157.3592.
1,6-Di-O-[1-(4-deoxypodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methyl]-2,3,4-tri-O-benzyl-β-D-glucopyranose (34)
Rf=0.50 (CHCl3: CH3OH=30:1). White amorphous powder, yield 86%; mp 150°C–153°C (CHCl3); [α]D23.4: −48.2 (c 0.14, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.32–7.24 (m, 17 H), 6.60 (s, 2 H), 6.57–6.55 (m, 2 H), 6.31 (s, 4 H), 6.07–5.88 (m, 6 H), 4.85–4.79 (m, 7 H), 4.73–4.62 (m, 6 H), 4.57–4.48 (m, 2 H), 4.38–4.34 (m, 1 H), 4.29–4.26 (m, 1 H), 3.81 (s, 6 H), 3.76 (s, 12 H), 3.67–3.61 (m, 2 H), 3.51–3.43 (m, 3 H), 3.23–3.12 (m, 4 H); 13C-NMR (CDCl3, 100 MHz) δ 173.2, 153.6, 152.8, 149.3, 149.2, 148.0, 148.0, 138.4, 138.3, 137.9, 137.5, 134.2, 133.1, 128.5, 128.4, 128.4, 127.9, 127.9, 127.9, 127.7, 124.7, 124.7, 110.5, 108.8, 108.2, 103.0, 101.9, 84.5, 82.0, 75.7, 75.7, 74.9, 69.6, 67.3, 64.9, 60.8, 58.6, 56.3, 43.5, 41.5, 37.1; ESIMS: m/z 1405 [M+H]+, HRESIMS was calculated for C77H76N6NaO20 [M+Na]+ 1427.5012 and was found to be 1427.4997.
1,6-Di-O-[1-(4-deoxypodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methyl]-2,3,4-tri-O-acetyl-β-D-glucopyranose (35)
Rf=0.30 (CHCl3: CH3OH=15:1). White amorphous powder, yield 80%; mp 140°C–141°C (CH2Cl2); [α]D23.1: −7.2 (c 0.12, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 7.51 (s, 1 H), 7.41 (s, 1 H), 6.72–6.71 (m, 2 H), 6.39–6.38 (m, 4 H), 6.25–6.24 (m, 2 H), 6.01–5.98 (m, 4 H), 5.87 (d, 1 H, J=4.8 Hz), 5.85 (d, 1 H, J=4.8 Hz), 5.20 (t, 1 H, J=9.6 Hz), 5.11 (t, 1 H, J=9.6 Hz), 4.99–4.96 (m, 1 H), 4.91 (d, 1 H, J=8.0 Hz), 4.80–4.78 (m, 1 H), 4.71–4.69 (m, 1 H), 4.65–4.61 (m, 5 H), 4.56–4.49 (m, 2 H), 4.41–4.38 (m, 2 H), 3.85 (s, 6 H), 3.82 (s, 12 H), 3.67–3.59 (m, 2 H), 3.55–3.50 (m, 2 H), 2.01–1.96 (m, 9 H, 3× COCH3); 13C-NMR (CDCl3, 150 MHz) δ 177.6, 177.4, 170.2, 169.5, 169.4, 153.7, 148.5, 147.8, 145.0, 144.3, 138.1, 137.2, 130.3, 125.9, 123.9, 110.2, 106.6, 104.7, 101.6, 99.5, 73.1, 72.9, 71.2, 69.1, 68.0, 65.0, 62.4, 60.9, 59.2, 56.3, 45.3, 44.7, 38.3, 20.7 (COCH3), 20.6 (COCH3), 20.6 (COCH3); ESIMS: m/z 1261 [M+H]+, HRESIMS was calculated for C62H64N6NaO23 [M+Na]+ 1283.3921 and was found to be 1283.3918.
1,6-Di-O-[1-(4-deoxypodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methyl]-2,3,4-tri-O-butyryl-β-D-glucopyranose (36)
Rf=0.40 (petroleum ether: ethyl acetate=10:1). White amorphous powder, yield 75%; mp 151°C–153°C (CHCl3); [α]D22.8: −55.3 (c 0.17, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 7.38 (s, 1 H), 7.30 (s, 1 H), 6.65–6.62 (m, 4 H), 6.35–6.34 (m, 4 H), 6.00 (d, 2 H, J=4.2 Hz), 6.03–6.01 (m, 4 H), 5.24 (t, 1 H, J=9.6 Hz), 5.13–5.08 (m, 1 H), 5.04–5.00 (m, 1 H), 4.97–4.87 (m, 2 H), 4.81–4.76 (m, 2 H), 4.72–4.65 (m, 4 H), 4.59–4.57 (m, 2 H), 4.39–4.36 (m, 2 H), 3.84 (s, 6 H), 3.79 (s, 12 H), 3.74–3.69 (m, 1 H), 3.61–3.58 (m, 1 H), 3.25–3.21 (m, 1 H), 3.14–3.11 (m, 1 H), 2.23–2.18 (m, 6 H, 3× COCH2), 1.59–1.54 (m, 6 H, 3× CH2CH3), 0.92–0.88 (m, 9 H, 3× CH2CH3); 13C-NMR (CDCl3, 150 MHz) δ 173.2, 173.1, 172.6, 172.1, 172.0, 152.8, 149.4, 148.1, 137.8, 134.3, 133.2, 124.7, 123.2, 110.5, 108.8, 108.2, 102.0, 100.3, 73.4, 72.3, 71.0, 69.3, 68.3, 67.3, 65.3, 63.0, 60.8, 58.6, 56.3, 43.6, 41.5, 37.1, 35.9 (COCH2), 35.9 (COCH2), 35.8 (COCH2), 18.3 (CH2CH3), 18.2 (CH2CH3), 18.2 (CH2CH3), 13.6 (CH2CH3), 13.5 (CH2CH3), 13.4 (CH2CH3); ESIMS: m/z 1345 [M+H]+, HRESIMS was calculated for C68H76N6NaO23 [M+Na]+ 1367.4860 and was found to be 1367.4844.
1,6-Di-O-[1-(4-deoxy-4′-demethylpodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methyl]-β-D-glucopyranose (37)
Rf=0.20 (CHCl3: CH3OH=9:1). White amorphous powder, yield 76%; mp 195°C–196°C (CHCl3-CH3OH); [α]D23.5: −124.6 (c 0.14, Pyridine); 1H-NMR (C5D5N, 600 MHz) δ 8.20 (s, 1 H), 8.17 (s, 1 H), 6.85 (m, 4 H), 6.80–6.79 (m, 4 H), 6.55 (d, 1 H, J=4.8 Hz), 6.50 (d, 1 H, J=4.8 Hz), 5.95 (s, 4 H), 5.93 (d, 1 H, J=7.8 Hz), 5.33–5.31 (m, 1 H), 5.14–5.12 (m, 1 H), 4.99–4.94 (m, 5 H), 4.41–4.37 (m, 1 H), 4.15–4.10 (m, 1 H), 4.00–3.95 (m, 1 H), 3.83–3.76 (m, 3 H), 3.73 (s, 12 H), 3.67–3.61 (m, 3 H), 3.44–3.36 (m, 4 H); 13C-NMR (C5D5N, 150 MHz) δ 174.0, 173.9, 149.0, 148.6, 148.0, 145.6, 145.4, 137.3, 134.3, 129.9, 126.3, 124.5, 110.6, 109.6, 109.1, 104.3, 102.2, 78.3, 77.0, 74.7, 71.4, 71.2, 67.8, 65.3, 63.2, 58.6, 56.4, 44.0, 41.9, 37.8; ESIMS: m/z 1107 [M+H]+, HRESIMS was calculated for C54H54N6NaO20 [M+Na]+ 1129.3291 and was found to be 1129.3281.
1,6-Di-O-[1-(4-deoxy-4′-demethylpodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methyl]-2,3,4-tri-O-benzyl-β-D-glucopyranose (38)
Rf=0.20 (CHCl3: CH3OH=30:1). White amorphous powder, yield 85%; mp 174°C–176°C (CHCl3); [α]D23.5: −58.8 (c 0.20, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.32–7.24 (m, 17 H), 6.59 (s, 2 H), 6.56–6.54 (m, 2 H), 6.32 (s, 4 H), 6.06 (d, 1 H, J=4.0 Hz), 6.01 (d, 1 H, J=4.0 Hz), 5.97–5.87 (m, 4 H), 4.91–4.79 (m, 7 H), 4.72–4.67 (m, 4 H), 4.63 (d, 2 H, J=4.0 Hz), 4.57–4.55 (m, 1 H), 4.50–4.48 (m, 1 H), 4.34–4.31 (m, 1 H), 4.26–4.24 (m, 1 H), 4.14–4.09 (m, 1 H), 3.78 (s, 12 H), 3.69–3.61 (m, 2 H), 3.51–3.40 (m, 3 H), 3.22–3.11 (m, 4 H); 13C-NMR (CDCl3, 100 MHz) δ 173.3, 149.3, 148.0, 147.9, 146.6, 144.8, 138.5, 138.3, 137.9, 134.4, 129.8, 128.5, 128.4, 128.4, 127.9, 127.9, 127.9, 127.7, 124.8, 123.0, 110.5, 110.4, 107.8, 103.0, 101.8, 84.5, 81.9, 75.7, 74.9, 74.7, 69.6, 67.4, 65.0, 63.2, 58.5, 56.5, 43.4, 41.6, 37.0; ESIMS: m/z 1377 [M+H]+, HRESIMS was calculated for C75H72N6NaO20 [M+Na]+ 1399.4699 was found to be 1399.4683.
1,6-Di-O-[1-(4-deoxy-4′-demethylpodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methyl]-2,3,4-tri-O-acetyl-β-D-glucopyranose (39)
Rf=0.50 (CHCl3: CH3OH=15:1). White amorphous powder, yield 84%; mp 183°C–184°C (CHCl3); [α]D23.3: −55.9 (c 0.18, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.33 (s, 1 H), 7.27 (s, 1 H), 6.62–6.59 (m, 4 H), 6.32–6.31 (m, 4 H), 6.09–6.07 (m, 2 H), 6.01–5.98 (m, 4 H), 5.16 (t, 1 H, J=9.6 Hz), 5.04 (t, 1 H, J=9.6 Hz), 4.96–4.92 (m, 1 H), 4.80 (d, 1 H, J=8.0 Hz), 4.76–4.72 (m, 2 H), 4.64–4.54 (m, 7 H), 4.36–4.32 (m, 2 H), 3.78 (s, 12 H), 3.70–3.67 (m, 1 H), 3.60–3.56 (m, 1 H), 3.26–3.19 (m, 2 H), 1.98–1.93 (m, 9 H, 3× COCH3); 13C-NMR (CDCl3, 100 MHz) δ 173.3, 173.1, 170.2, 169.4, 169.4, 149.3, 148.0, 146.6, 144.8, 144.4, 134.3, 133.4, 129.8, 124.6, 123.3, 110.5, 108.8, 107.8, 101.9, 100.0, 73.1, 72.7, 71.2, 69.1, 68.6, 67.3, 65.0, 62.8, 58.6, 56.5, 43.4, 42.1, 41.6, 37.0, 20.6 (COCH3), 20.6 (COCH3), 20.5 (COCH3); ESIMS: m/z 1317 [M+H]+, HRESIMS was calculated for C66H72N6NaO23 [M+Na]+ 1339.4547 and was found to be 1339.4526.
1,6-Di-O-[1-(4-deoxy-4′-demethylpodophyllotoxin-4β-yl)-1,2,3-triazol-4-yl-methyl]-2,3,4-tri-O-butyryl-β-D-glucopyranose (40)
Rf=0.20 (petroleum ether: ethyl acetate=10:1). White amorphous powder, yield 82%; mp 184°C–186°C (CHCl3); [α]D23.4: −56.4 (c 0.28, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 7.35 (s, 1 hour), 7.28 (s, 1 H), 6.60 (s, 2 H), 6.59–6.58 (m, 2 H), 6.32–6.31 (m, 4 H), 6.06–5.97 (m, 6 H), 5.21 (t, 1 H, J=9.6 Hz), 5.05 (t, 1 H, J=9.6 Hz), 5.00–4.95 (m, 1 H), 4.82–4.78 (m, 2 H), 4.74–4.71 (m, 2 H), 4.65–4.62 (m, 4 H), 4.56–4.52 (m, 2 H), 4.33–4.30 (m, 2 H), 3.77 (s, 12 H), 3.71–3.65 (m, 1 H), 3.59–3.53 (m, 1 H), 3.20–3.16 (m, 1 H), 3.09–3.04 (m, 1 H), 2.20–2.13 (m, 6 H, 3× COCH2), 1.56–1.50 (m, 6 H, 3× CH2CH3), 0.89–0.81 (m, 9 H, 3× CH2CH3); 13C-NMR (CDCl3, 100 MHz) δ 173.3, 173.2, 172.6, 172.0, 171.9, 149.3, 149.3, 147.9, 144.9, 144.5, 134.3, 133.3, 129.7, 124.6, 123.3, 100.4, 108.7, 107.8, 101.9, 100.2, 73.3, 72.3, 70.9, 68.3, 67.3, 65.2, 62.9, 58.6, 56.5, 43.4, 41.5, 37.0, 35.9 (COCH2), 35.8 (COCH2), 35.8 (COCH2), 18.3 (CH2CH3), 18.2 (CH2CH3), 18.2 (CH2CH3), 13.6 (CH2CH3), 13.5 (CH2CH3), 13.4 (CH2CH3); ESIMS: m/z 1233 [M+H]+, HRESIMS was calculated for C60H650N6NaO23 [M+Na]+ 1255.3608 and was found to be 1255.3595.
Cell culture and cytotoxicity assay
The following human tumor cell lines were used: HL-60, SMMC-7721, A-549, MCF-7, and SW480. All the cells were cultured in RMPI-1640 or DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum (Hyclone) at 37°C in a humidified atmosphere with 5% CO2. Cell viability was assessed by conducting colorimetric measurements of the amount of insoluble formazan formed in living cells based on the reduction of 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich Co., St Louis, MO, USA). Briefly, adherent cells (100 μL) were seeded into each well of a 96-well cell culture plate and allowed to adhere for 12 hours before drug addition, while suspended cells were seeded just before drug addition, both with an initial density of 1×105 cells/mL in 100 μL of medium. Each tumor cell line was exposed to the test compound at various concentrations in triplicate for 48 hours. After the incubation, MTT (100 μg) was added to each well, and the incubation continued for 4 hours at 37°C. The cells lysed with SDS (200 μL) after removal of 100 μL of medium. The OD of lysate was measured at 595 nm in a 96-well microtiter plate reader (Bio-Rad 680).
Acknowledgments
This work was financially supported by Yunnan province (grant nos 2015HB093 and 2015FB168) and the National Key Research and Development Program of China (grant no 2017YFD0201402). The authors thank the staff of analytical group of the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, for measurements of all spectra.
Disclosure
The authors report no conflicts of interest in this work.
References
Castro MA, del Corral JM, García PA, et al. Synthesis and biological evaluation of new podophyllic aldehyde derivatives with cytotoxic and apoptosis-inducing activities. J Med Chem. 2010;53(3):983–993. | ||
Sk UH, Dixit D, Sen E. Comparative study of microtubule inhibitors – estramustine and natural podophyllotoxin conjugated PAMAM dendrimer on glioma cell proliferation. Eur J Med Chem. 2013;68:47–57. | ||
Ma Y, Fang S, Li H, et al. Biological evaluation and molecular modelling study of podophyllotoxin derivatives as potent inhibitors of tubulin polymerization. Chem Biol Drug Des. 2013;82(1):12–21. | ||
Jordan A, Hadfield JA, Lawrence NJ, McGown AT. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med Res Rev. 1998;18(4):259–296. | ||
Wang ZQ, Kuo YH, Schnur D, et al. Antitumor agents. 113. New 4 beta-arylamino derivatives of 4′-O-demethylepipodophyllotoxin and related compounds as potent inhibitors of human DNA topoisomerase II. J Med Chem. 1990;33(9):2660–2666. | ||
Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34(10):1514–1521. | ||
Brewer CF, Loike JD, Horwitz SB, Sternlicht H, Gensler WJ. Conformational analysis of podophyllotoxin and its congeners. Structure – activity relationship in microtubule assembly. J Med Chem. 1979;22(3):215–221. | ||
Niu L, Wang Y, Wang C, et al. Structure of 4′-demethylepipodophyllotoxin in complex with tubulin provides a rationale for drug design. Biochem Biophys Res Commun. 2017;493(1):718–722. | ||
Sackett DL. Podophyllotoxin, steganacin and combretastatin: natural products that bind at the colchicine site of tubulin. Pharmacol Ther. 1993;59(2):163–228. | ||
Moraes RM, Dayan FE, Canel C. The lignans of Podophyllum. Stud Nat Prod Chem. 2002;26:149–182. | ||
Yu X, Che Z, Xu H. Recent Advances in the Chemistry and Biology of Podophyllotoxins. Chem Eur J. 2017;23(19):4467–4526. | ||
Kamal A, Hussaini SM, Malik MS. Recent developments towards podophyllotoxin congeners as potential apoptosis inducers. Anticancer Agents Med Chem. 2015;15(5):565–574. | ||
Bhat BA, Reddy PB, Agrawal SK, Saxena AK, Kumar HM, Qazi GN. Studies on novel 4beta-[(4-substituted)-1,2,3-triazol-1-yl] podophyllotoxins as potential anticancer agents. Eur J Med Chem. 2008;43(10):2067–2072. | ||
Reddy DM, Srinivas J, Chashoo G, Saxena AK, Sampath Kumar HM. 4β-[(4-Alkyl)-1,2,3-triazol-1-yl] podophyllotoxins as anticancer compounds: design, synthesis and biological evaluation. Eur J Med Chem. 2011;46(6):1983–1991. | ||
Chen H, Zuo S, Wang X, et al. Synthesis of 4β-triazole-podophyllotoxin derivatives by azide-alkyne cycloaddition and biological evaluation as potential antitumor agents. Eur J Med Chem. 2011;46(9):4709–4714. | ||
Zi CT, Xu FQ, Li GT, et al. Synthesis and anticancer activity of glucosylated podophyllotoxin derivatives linked via 4β-triazole rings. Molecules. 2013;18(11):13992–14012. | ||
Zi CT, Liu ZH, Li GT, et al. Design, synthesis, and cytotoxicity of perbutyrylated glycosides of 4β-triazolopodophyllotoxin derivatives. Molecules. 2015;20(2):3255–3280. | ||
Zi CT, Yang L, Gao W, et al. Click glycosylation for the synthesis of 1, 2, 3-triazole-linked picropodophyllotoxin glycoconjugates and their anticancer activity. Chemistry Select. 2017;2(18):5038–5044. | ||
Kamal A, Laxman E, Khanna GB, et al. Design, synthesis, biological evaluation and QSAR studies of novel bisepipodophyllotoxins as cytotoxic agents. Bioorg Med Chem. 2004;12(15):4197–4209. | ||
Passarella D, Peretto B, Blasco y Yepes R, et al. Synthesis and biological evaluation of novel thiocolchicine-podophyllotoxin conjugates. Eur J Med Chem. 2010;45(1):219–226. | ||
Bock VD, Hiemstra H, van Maarseveen JH. CuI-Catalyzed Alkyne-Azide “Click” Cycloadditions from a Mechanistic and Synthetic Perspective. Eur J Org Chem. 2006;2006(1):51–68. | ||
Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40(11):2004–2021. | ||
Nielsen MM, Dimitrov I, Takamuku S, Jannasch P, Jankova K, Hvilsted S. Dendronized polymer architectures for fuel cell membranes. Fuel Cells. 2013;13(3):342–354. | ||
Nazaré M, Waldmann H. Synthesis of the (9S, 18R) diastereomer of cyclamenol A. Angew Chem Int Ed Engl. 2000;39(6):1125–1128. | ||
Smith AB, Ott GR. Total Synthesis of (−)-Macrolactin A. J Am Chem Soc. 1996;118(51):13095–13096. | ||
Matsuo I, Isomura M, Miyazaki T, Sakakibara T, Ajisaka K. Chemoenzymatic synthesis of the branched oligosaccharides which correspond to the core structures of N-linked sugar chains. Carbohydr Res. 1997;305(3–4):401–413. | ||
Sureshkumar G, Hotha S. Gold mediated glycosylations: selective activation of propargyl 1,2-orthoesters in the presence of aglycones containing a propargyl moiety. Chem Commun (Camb). 2008;(36):4282–4284. | ||
Hansen HF, Jensen RB, Willumsen AM, et al. New compounds related to podophyllotoxin and congeners: synthesis, structure elucidation and biological testing. Acta Chem Scand. 1993;47(12):1190–1200. | ||
Zi CT, Yang D, Dong FW, et al. Synthesis and antitumor activity of novel per-butyrylated glycosides of podophyllotoxin and its derivatives. Bioorg Med Chem. 2015;23(7):1437–1446. | ||
de Oliveira PF, Alves JM, Damasceno JL, Oliveira RAM, Dias HJ, Crotti AEM. Cytotoxicity screening of essential oils in cancer cell lines. Rev Bras Farmacogn. 2012;22:88–93. | ||
Shi JF, Wu P, Jiang ZH, Wei XY. Synthesis and tumor cell growth inhibitory activity of biotinylated annonaceous acetogenins. Eur J Med Chem. 2014;71:219–228. | ||
Suffness M, Pezzuto JM. Assays related to cancer drug discovery. In: Hostettmann K, editor. Methods in Plant Biochemistry: Assays for Bioactivity. London: Academic Press; 1990:71–133. | ||
Bézivin C, Tomasi S, Lohézic-Le Dévéhat F, Boustie J. Cytotoxic activity of some lichen extracts on murine and human cancer cell lines. Phytomedicine. 2003;10(6–7):499–503. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.