Back to Journals » International Journal of Nanomedicine » Volume 18
Synergistically Enhancing Immunotherapy Efficacy in Glioblastoma with Gold-Core Silica-Shell Nanoparticles and Radiation
Authors Chen SF ![]() , Kau M, Wang YC
, Kau M, Wang YC ![]() , Chen MH, Tung FI
, Chen MH, Tung FI ![]() , Chen MH, Liu TY
, Chen MH, Liu TY
Received 16 September 2023
Accepted for publication 12 December 2023
Published 14 December 2023 Volume 2023:18 Pages 7677—7693
DOI https://doi.org/10.2147/IJN.S440405
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. RDK Misra
Shuo-Fu Chen,1,* Min Kau,2,* Yu-Chi Wang,2 Ming-Hong Chen,3 Fu-I Tung,4,5 Mei-Hsiu Chen,6 Tse-Ying Liu2
1Department of Heavy Particles & Radiation Oncology, Taipei Veterans General Hospital, Taipei, Taiwan; 2Department of Biomedical Engineering, National Yang Ming Chiao Tung University, Taipei, Taiwan; 3Division of Neurosurgery, Department of Surgery, Far Eastern Memorial Hospital, New Taipei City, Taiwan; 4Department of Orthopedics, Yang-Ming Branch, Taipei City Hospital, Taipei, Taiwan; 5Department of Health and Welfare, College of City Management, University of Taipei, Taipei, Taiwan; 6Department of Internal Medicine, Far Eastern Memorial Hospital, New Taipei City, Taiwan
*These authors contributed equally to this work
Correspondence: Tse-Ying Liu, Department of Biomedical Engineering, National Yang Ming Chiao Tung University, No. 155, Sec. 2, Linong Street, Beitou District, Taipei, 112304, Taiwan, Tel +8862 28267923, Email [email protected]
Purpose: Glioblastoma is a highly aggressive brain tumor with universally poor outcomes. Recent progress in immune checkpoint inhibitors has led to increased interest in their application in glioblastoma. Nonetheless, the unique immune milieu in the brain has posed remarkable challenges to the efficacy of immunotherapy. We aimed to leverage the radiation-induced immunogenic cell death to overcome the immunosuppressive network in glioblastoma.
Methods: We developed a novel approach using the gold-core silica-shell nanoparticles (Au@SiO2 NPs) in combination with low-dose radiation to enhance the therapeutic efficacy of the immune checkpoint inhibitor (atezolizumab) in brain tumors. The biocompatibility, immune cell recruitment, and antitumor ability of the combinatorial strategy were determined using in vitro assays and in vivo models.
Results: Our approach successfully induced the migration of macrophages towards brain tumors and promoted cancer cell apoptosis. Subcutaneous tumor models demonstrated favorable safety profiles and significantly enhanced anticancer effects. In orthotopic brain tumor models, the multimodal therapy yielded substantial prognostic benefits over any individual modalities, achieving an impressive 40% survival rate.
Conclusion: In summary, the combination of Au@SiO2 NPs and low-dose radiation holds the potential to improve the clinical efficacy of immune checkpoint inhibitors. The synergetic strategy modulates tumor microenvironments and enhances systemic antitumor immunity, paving a novel way for glioblastoma treatment.
Keywords: brain cancer, silica, gold, atezolizumab, radiation therapy
Introduction
Glioblastoma, formerly known as glioblastoma multiforme (GBM), is the most aggressive and prevalent brain tumor primarily affecting adults. More than 50% of patients experience mortality within 3 months of diagnosis, and the median survival is only 14–16 months.1,2 The standard of treatment involves maximal surgical resection followed by radiation and temozolomide.3–5 However, their infiltrative nature presents challenges in achieving complete eradication.6 Additionally, the blood-brain barrier hinders the effectiveness of systemic delivery route.7–9 These difficulties have prompted extensive research to explore innovative treatment strategies.
Immune checkpoint inhibitors (ICIs), such as monoclonal antibodies targeting programmed cell death-1 (PD-1) and programmed cell death ligand-1 (PD-L1), have emerged as a promising avenue in cancer treatment.10,11 These agents block inhibitory pathways exploited by cancer cells to evade the immunosurveillance, restoring the immune system to mount a more potent attack against cancer cells.12,13 While ICIs have shown promise in various cancers,14–19 they remain limited for brain tumors due to the blood-brain barrier, the low immunogenic characteristics, and the immunosuppressive networks.20–23 To address these challenges, researchers are actively investigating the combination of different treatment modalities, such as photothermal therapy, sonodynamic therapy, and radiotherapy.24 These treatments have been suggested to induce cell damage, potentially triggering the release of damage-associated molecular patterns (DAMPs) and tumor-associated antigens.25,26 These molecules can either function as stress signals to alert the immune system or be recognized by antigen-presenting cells, which are responsible for capturing antigens and presenting them to cytotoxic T cells.27–29 Recent breakthroughs in nanomaterials have opened up new possibilities to amplify the synergistic effects between ICIs and these treatments.30–35 For example, a biomimetic cancer cell membrane-coated TiO2 nanoshell, developed by Ning et al, has shown effective tumor targeting and therapeutic efficiency through a combinatorial approach.36 Various emerging nanomaterials have been implemented in the photothermal therapy and sonodynamic therapy for glioblastoma.37–40 Additionally, the application of nanomedicine in radiotherapy adds another dimension to this innovative landscape.35 The integration of these nanotechnologies presents a promising avenue for advancing effective and targeted cancer therapies.41
Given the disappointing clinical trials combining immunotherapy with radiotherapy in glioblastoma to date,42,43 we propose a strategy of nanomedicine-assisted radiation to enhance the efficacy of immune checkpoint blockade on brain tumors. Radiation is known to induce intracellular damage by generating ROS through water ionization, and incorporation of gold nanoparticles (Au NPs) has been found to enhance radiation-induced ROS.44,45 When Au NPs are exposed to radiation, they tend to generate more secondary electrons due to their high electron density (high atomic number) and photoelectric absorption.46 The secondary electrons (Auger electrons) and photons, emitted from photoelectric effects, are expected to cause secondary water ionization, thereby producing additional ROS and leading to sensitizing effects.47 Au NPs can enhance water radiolysis upon irradiation in both intracellular and extracellular settings.44,46,48,49 To further amplify the immunogenic effects, we encapsulated the Au NPs with a silicon dioxide (SiO2) shell. Biodegradable materials like poly(lactic-co-glycolic acid) (PLGA) and SiO2 have been suggested to possess immune-stimulatory capabilities.50 The addition of a SiO2 shell serves a dual purpose—enhancing the immune stimulation while also acting as a structural barrier, preventing particle aggregation and ensuring sustained radio-sensitization effects. Such a novel treatment strategy has not yet been applied to brain cancer and warrants further investigation.
In this study, we devised a novel approach for glioblastoma using gold-mesoporous silica nanoparticles (nanoparticles with Au core-SiO2 shell structure; Au@SiO2 NPs) together with X-ray irradiation (XR) to enhance the efficacy of anti-PD-L1 antibody (α-PD-L1) in brain tumors (Scheme 1). We investigated the effects of Au@SiO2 NPs in augmenting radiation-mediated oxidative stress, recruiting immune cells and eliciting immunogenic cell death using in vitro cell models. We further validated the ability of Au@SiO2 NPs in potentiating the synergy of low-dose radiation and α-PD-L1 using in vivo subcutaneous and orthotopic brain tumor models. In summary, the strategy involving Au@SiO2 NPs-assisted radiation therapy displays a powerful ability for antitumor immune activation, presenting a new direction in the exploration of effective treatment modalities for glioblastoma.
 |
Scheme 1 Schematic illustration of our synergistic strategy for glioblastoma treatment. |
Materials and Methods
Synthesis of Au@SiO2 NPs
Hexadecyl trimethyl ammonium bromide (CTAB) and tetraethoxysilane (TEOS) were obtained from Sigma-Aldrich. Hydrogen tetrachloroaurate(III) trihydrate (HAuCl4·3H2O) was purchased from Alfa Aesar. Au@SiO2 NPs were prepared using a one-step synthesis method. First, 250 mg CTAB was dissolved in deionized water (120 mL), followed by the addition of 3 mL NaOH (0.5 M) while stirring at 80 °C for 15 min. Subsequently, a mixture of 5 mL of 3.7 wt.% formaldehyde solution and 4 mL of HAuCl4·3H2O (0.05 M) was rapidly added, resulting in a color change from pale yellow to red. The pH of the solution was adjusted to 10, followed by the addition of 1 mL of TEOS and 3350 μL of ethanol (ETOH). The mixture was stirred for 1 h, and incubated at room temperature. The product was collected and washed several times with deionized water and ETOH.
Following centrifugation, the NPs were dissolved in a solvent composed of 7.5% (v/v) HCl-EtOH, subjected to ultrasonic oscillation, and then collected after ETOH washes. The final product was obtained by dissolving in a 5% (v/v) acetic acid-EtOH solvent to remove CTAB, followed by additional ultrasonic oscillation and ETOH washes.
Characterization of Au@SiO2 NPs
The morphologies and crystalline phase of Au@SiO2 NPs were analyzed using transmission electron microscopy (TEM, JEOL, JEM-2000 EX II, Japan), high-resolution TEM (HRTEM, JEOL, JEM-2010, Japan), and X-ray diffraction (XRD, Bruker D8-Advance X-ray powder diffractometer with Cu Kα radiation [λ=1.5406 Å]). The particle size distribution of the samples was determined using dynamic light scattering (DLS) at a scattering angle of 90° with a Zetasizer nano ZS90 (Malvern Instruments, Worcestershire, UK) at 25 °C.
Cell Lines
The Bioware Brite Cell Line GL261 Red-FLuc, a mouse glioma cell line, was obtained from PerkinElmer. The GL261 cell line was sourced from Deutsche Sammlung von Mikroorganismen und Zellkulturen. C8D1A (ATCC CRL-2541) murine type 1 astrocytes, C8S (ATCC CRL-2535) murine type 2 astrocytes, and b.end3 (ATCC CRL-2299) murine brain endothelial cells were purchased from the American Type Culture Collection (ATCC). RAW 264.7 (BCRC 60001) murine macrophages were obtained from the Bioresource Collection and Research Center (Taiwan). These cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, GE Healthcare, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone) and maintained at 37 °C in a 5% CO2 humidified incubator.
Biological Safety Evaluation of Au@SiO2 NPs: In vitro and in vivo Testing
The cell viability was evaluated using the MTT assay. The cells were co-cultured with various concentrations of Au@SiO2 NPs (50, 100, 200, and 400 μg/mL) for 24 h in a serum-based culture medium. Following PBS washing, 20× diluted PrestoBlue® reagent was added and incubated for 20 min at 37 °C to evaluate the cell viability. The supernatant was then transferred to a 96-well plate, and the absorbance was measured using a TECAN Sunrise ELISA Reader with excitation/emission set at 560/590 nm.
For in vivo acute toxicity assays, the mice were intra-peritoneally injected with Au@SiO2 NPs at different concentrations (2, 10, and 25 mg/kg) for eight consecutive days. The control group received PBS injections. The mice were sacrificed on the ninth day, and their liver, spleen, kidneys, lungs, and brains were harvested and fixed in 10% formalin solution for subsequent histopathological examination.
Intracellular ROS Measurement
The intracellular ROS production was determined using a fluorometric assay. Briefly, GL261 murine glioma cells were seeded in Ultra-Low Attachment Surface 6-well plates at a density of 1×106 cells per well. The cells were treated with Au@SiO2 NPs (100 μg/mL) for 24 h and then exposed to XR (ie, X-ray irradiation with a single fraction of 6 MeV photons at a dose of 2 Gray). Following the treatments, the cells were labeled with CellROX® Deep Red Reagent (5 μM, excitation/emission at 640/665 nm), H33342/DAPI (excitation/emission at 358/461 nm), and Alexa Fluor® 488 (excitation/emission at 493/519 nm). Stained cells were visualized using a confocal fluorescence microscope (Zeiss LSM 880).
Trans-Well Co-Culture with RAW 264.7 Macrophages
The trans-well system was utilized for both chemotaxis and caspases-3 assay. For the chemotaxis assay, RAW 264.7 (murine macrophages) were co-cultured with GL261 glioma cells using trans-well cell culture inserts with pored-filters. GL261 cells were seeded at 5×105 cells in 12-well plates and treated with Au@SiO2 NPs (100 μg/mL), followed by exposure to XR (ie, X-ray irradiation with a single fraction of 6 MeV photons at a dose of 2 Gray) on the next day. RAW 264.7 were labeled with Vybrant™ DiD Cell-Labeling Solution (Thermo Fisher) at 37 °C for 20 min. After centrifugation, RAW 264.7 cell pellets were resuspended in PBS, and 1×105 RAW 264.7 were placed on the upper layer of each 3 μm-pored trans-well membrane, co-cultured with GL261 cells for 24 h. After three PBS washes, the GL261 cells were fixed, permeabilized, and stained with Alexa Fluor® 488 and H33342/DAPI. The samples were then observed under a confocal microscope.
For the caspases-3 assay, the procedure was similar, but 0.4 μm-pored filters were used instead. GL261 cells were seeded at a density of 2×105 cells in six-well plates and co-cultured with RAW 264.7 cells for 2 days. The expression levels of caspase-3 were quantified using MetaMorph.
Animal Models
The six-to-eight-week-old female C57BL/6 black mice were purchased from BioLASCO Taiwan Co., Ltd. All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) of National Yang Ming Chiao Tung University (NYCU) and approved by IACUC of NYCU (NYMU-IACUC 1060814).
Subcutaneous Heterotopic Tumor Model
In the subcutaneous tumor model, C57BL/6 mice were injected with 5×106 GL261 glioma cells on their left flanks. Once 100 mm3 subcutaneous tumors were successfully induced, mice were randomly assigned to different treatments based on their tumor sizes, ensuring that each group had comparable average tumor sizes. The treatments included atezolizumab, Au@SiO2 NPs, and XR, either administered alone or in combination. Atezolizumab was purchased from selleckchem and administered through subcutaneous injection at a dosage of 10 mg/kg. Au@SiO2 NPs (30 μg/μL) were injected intratumorally one day before receiving XR (ie, X-ray irradiation with a single fraction of 6 MeV photons at a dose of 2 Gray). The changes in tumor sizes were monitored by measuring the surface diameters during the study, and the final tumor volumes were assessed after sacrificing the mice.
Orthotopic Brain Tumor Model
In the orthotopic glioma model, the surgery was performed following the protocol developed by our group.51 GL261-Fluc-Red cells (5×106 cells/10 μL in Hank’s Balanced Salt Solution [HBSS]) were stereotactically injected into the mouse brain at 1 mm lateral from bregma and 2 mm deep from the cortical surface. The cell suspension (7 μL) was slowly injected into the mouse brain at a rate of 5 μL/min. After cell inoculation, successful tumor growth was confirmed using in vivo imaging system (IVIS, IVIS Lumina II, Perkin Elmer). Once the bioluminescence signals reached 106 photons, the mice with brain tumors were randomly divided into five treatment groups based on their tumor sizes. The treatment details remain as previously described, and the progression of the tumor was monitored using IVIS. Each treatment arm consisted of 6 mice in survival analysis, and all experiments were repeated at least three times.
Histologically Assessment of Treatment Responses
To investigate the post-treatment pathological changes and tumor-infiltrating immune cells, the surviving GL261-bearing mice were euthanized one week after the treatments. Following necropsy, major organs from each group were harvested and fixed in 10% formalin solution. Subsequently, the samples underwent trimming, paraffin embedding, sectioning, hematoxylin and eosin staining, and were microscopically examined by veterinary pathologists.
Statistical Analysis
Statistical analysis was conducted using Prism (Version 8, GraphPad Software, Inc., CA). All in vitro experiments were independently repeated at least three times (n = 3 biological replicates). The data are presented as mean ± SD, unless otherwise specified. Two groups of data were analyzed using two-way ANOVA. Statistical significance was defined as p < 0.05.
Results
Synthesis and Characterization of Au@SiO2 NPs
The synthesis of Au@SiO2 NPs is illustrated in Figure 1a. First, Au NPs were formed by adding a mixture of HAuCl4 and formaldehyde into a solution of CTAB and NaOH under constant stirring. Next, TEOS was introduced to create a silica layer around the Au NPs. Finally, the uniformly dispersed Au@SiO2 NPs were obtained by removing CTAB using an acidic solution. The average size of the Au@SiO2 NPs was 70 nm, as determined by Dynamic Light Scattering (DLS). The transmission electron microscopy (TEM) images provided clear evidence of the complete encapsulation of Au NPs by SiO2 shells, creating uniform core-shell nanostructures with strong contrast between the black cores and gray shells. Further analysis with high-resolution TEM (HR-TEM) showed the well-defined spherical morphology of the Au@SiO2 NPs. The lattice fringes of the NPs were observed with an average spacing of 0.24 nm, corresponding to the d-spacing of the Au (111) plane. The three distinct diffraction peaks in the X-ray Diffraction (XRD) pattern of Au@SiO2 NPs corresponded to the (111), (200), and (220) crystal planes of Au. In addition, a broad peak with low intensity was observed in the XRD pattern of Au@SiO2 NPs, which was assigned to the amorphous SiO2 layer. The observation in Figure 1 confirms the successful incorporation of Au NPs into the SiO2 shells.
 |
Figure 1 Morphologies and material characterization of Au@SiO2 NPs. Notes: (a) Schematic illustration of the preparation process and structure of Au@SiO2 NPs. (b) Transmission electron microscopy (TEM) images of Au@SiO2 NPs. Scale bar: 200 nm. (c) High-resolution (HR)-TEM images of Au NPs. Scale bar: 5 nm. (d) X-ray diffraction (XRD) patterns of Au@SiO2 NPs prepared in this study and Au crystals cited from the standard JCPDS file (JCPDS No. 04–0784). |
In vitro and in vivo Safety Profile of Au@SiO2 NPs
For cytotoxicity assessment, various types of cells found in brain were incubated with different concentrations of Au@SiO2 NPs (ranging from 0 to 400 μg/mL). The cell lines used in the study included GL261 (murine glioma cells), b.end3 (murine brain endothelial cells), C8D1A (murine type 1 astrocytes), and C8S (murine type 2 astrocytes). The MTT assay (Figure 2a) demonstrated the selective toxicity of Au@SiO2 NPs towards tumor cells. At a concentration of 50 μg/mL, no toxicity was observed in all normal cells, whereas 7.7% tumor cells were affected. The nanoparticles exhibited good biocompatibility for normal cells at a concentration of 100 μg/mL, in which only 78.1% of tumor cells survived. The cell viability significantly decreased when the concentration of Au@SiO2 NPs exceeded 200 μg/mL, with only 74.4% of C8D1A (normal cells) and 39.2% of GL261 cancer cells remaining viable. Based on these findings, the concentration of 100 μg/mL was selected for subsequent in vitro experiments.
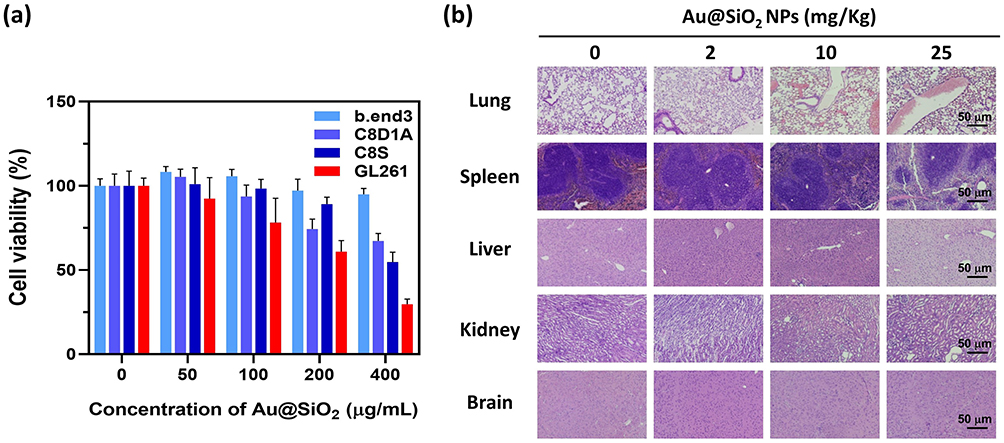 |
Figure 2 In vitro and in vivo safety profile of Au@SiO2 NPs. Notes: (a) In vitro cytotoxicity of Au@SiO2 NPs on normal cells (b.end3, C8D1A, C8S) and glioma cells (GL261) was measured using PrestoBlue Cell Viability Reagent. (b) In vivo tissue damage induced by Au@SiO2 NPs was observed through histological assessment. Scale bar: 50 μm. |
Furthermore, the acute toxicity profile of Au@SiO2 NPs was investigated using in vivo models. The mice were intra-peritoneally injected with Au@SiO2 NPs at different concentrations (0, 2, 10, and 25 mg/kg) for eight consecutive days. The mice were sacrificed on the 14th day, and their organs, including the lungs, spleen, liver, kidneys, and brain, were collected for histological examination. During the experiment, no adverse effects or deaths were observed in the mice. Figure 2b shows the results of the histological examination of tissue sections from different organs. It was found that at doses below 25 mg/kg, there were no significant pathological changes in the tissue sections of various organs. These preliminary findings suggest the safety of the Au@SiO2 NPs and their potential for clinical applications.
Enhanced Radiation-Induced ROS Production by Au@SiO2 NPs
Reactive oxygen species (ROS)-mediated DNA damage is the primary mechanism of radiation-induced cell death.52,53 To assess the capacity of Au@SiO2 NPs in enhancing ROS production upon irradiation, cancer cells were co-cultured with Au@SiO2 NPs and exposed to XR (ie, X-ray irradiation with a single fraction of 6 MeV photons at a dose of 2 Gray). We established a 3D spheroid model using Ultra-Low Attachment Surface wells to simulate the three-dimensional nature of in vivo tumors. As shown in Figure 3, GL261 (murine glioma cells) cells exhibited a minimal amount of ROS when treated with Au@SiO2 NPs alone (Figure 3b and f) or exposed to XR alone (Figure 3c and g). On the contrary, the combination of Au@SiO2 NPs and XR resulted in significantly higher levels of ROS production (Figure 3d and h). The observation suggested that Au@SiO2 NPs exhibited a powerful ability to amplify ROS production and radiation-induced oxidative stress.
 |
Figure 3 The effect of Au@SiO2 NPs and X-ray irradiation (XR) on intracellular production of reactive oxygen species (ROS). Notes: (a–d) Merged fluorescence images of 3D cell pallets of GL261 cells receiving different treatments. ROS production, nucleus, and cytoskeleton of live cells were labeled with CellROX Deep Red Reagent (pink), H33342 (blue), and Phalloidin (green), respectively. (e–h) Fluorescence images showing ROS (pink) produced from the 3D cell pallets of GL261 cells receiving different treatments. The experimental groups were divided into four: (1) Control, (2) Au@SiO2 NPs alone (100 μg/mL), (3) XR alone (X-ray irradiation with a single fraction of 6 MeV photons at a dose of 2 Gray), and (4) Au@SiO2 NPs + XR. |
Recruitment of Macrophages by Au@SiO2 NPs in Combination with Radiotherapy
To investigate whether our treatment strategy could successfully recruit macrophages towards tumor cells for antigen presentation, which is a key mechanism in immunotherapy, an in vitro chemotaxis assay was set up as illustrated in Figure 4a. In this assay, cancer cells were placed on the lower layer, whereas murine macrophages (RAW264.7) were positioned above the trans-well membranes with multiple 3 μm-sized pores. These pores were designed to enable the migration of macrophages through the membrane. The migration of macrophages was visualized using confocal microscopy.
 |
Figure 4 The effect of GL261 cells receiving Au@SiO2 NPs and X-ray irradiation (XR) on the chemotactic migration of RAW264.7 macrophages. Notes: (a) Experimental scheme using a 3 μm trans-well, on which RAW264.7 macrophages were observed. (b) Confocal images showing RAW264.7 macrophages (labeled in red) chemotactically migrating to GL261 cells receiving Au@SiO2 NPs and/or XR. RAW264.7 macrophages, nucleus, and cytoskeleton were labeled with Cas Vybrant™ DiD Cell-Labeling Solution (red), DAPI (blue), and Phalloidin (green), respectively. Scale bars in all images were 100 µm. The experimental groups were divided into four: (1) Control, (2) Au@SiO2 NPs alone (100 μg/mL), (3) XR alone (X-ray irradiation with a single fraction of 6 MeV photons at a dose of 2 Gray), and (4) Au@SiO2 NPs + XR. |
As shown in Figure 4b, a significant chemotactic migration of macrophages (labeled in red) was triggered by the combination of Au@SiO2 NPs and XR (ie, X-ray irradiation with a single fraction of 6 MeV photons at a dose of 2 Gray), which was not shown in other groups (ie, control, Au@SiO2 alone, and XR alone). This finding demonstrates that the treatment strategy could effectively mobilize the macrophages towards the cancer cells, which plays a critical role in promoting macrophage-mediated cancer cell apoptosis and recruitment of more T cells to mount a robust immune response against cancer cells.
Enhanced Caspase-3 Expression by Au@SiO2 NPs in Combination with Radiotherapy
In addition to chemotaxis, we further explored the potential of Au@SiO2 NPs in boosting systemic antitumor immunity through interactions with macrophages. This investigation was significant in understanding how macrophages contribute to cancer cell damage even when they do not make direct contact with the cancer cells, which is the most common scenario for brain tumors. To achieve this, we set up a trans-well system where cancer cells were positioned on the lower layer and macrophages (RAW264.7) were placed on the upper layer. Unlike the previous setup, the trans-well membrane used in this experiment has a pore size of 0.4 μm, which effectively prevents the migration of macrophages to the lower layer.
Caspase-3, an important marker of apoptosis, is commonly utilized to assess the efficacy of cancer therapy. In our study, the caspase-3 expression served as an indicator of cell damage based on our earlier identification of apoptosis involvement in the treatment.51 In the absence of macrophage, the caspase-3 expression did not show a significant increase when treated with either Au@SiO2 NPs or XR alone (Figure 5). The caspase-3 expression in cancer cells treated with both Au@SiO2 NPs and XR was only comparable to the sum of effects induced by individual treatments. These findings indicate that even with elevated ROS levels, the combination of Au@SiO2 NPs and XR might not effectively boost caspase-3 in the absence of macrophages.
 |
Figure 5 The effect of RAW264.7 macrophages on the production of caspase-3 in GL261 cells receiving Au@SiO2 NPs and/or X-ray irradiation (XR). Notes: The production of caspase-3 protein (labeled in red) in GL261 cells was observed through immunofluorescence staining and confocal microscopic imaging. (a) Experimental scheme using a 0.4 μm trans-well. (b) Confocal microscopic images of GL261 cells co-cultured without or with RAW264.7 macrophages. (c) Quantitative measurement of caspase-3 protein levels in each confocal microscopic image. The experimental groups were divided into four: (1) Control, (2) Au@SiO2 NPs alone (100 μg/mL), (3) XR alone (X-ray irradiation with a single fraction of 6 MeV photons at a dose of 2 Gray), and (4) Au@SiO2 NPs + XR. ****p < 0.0001. |
On the other hand, in the presence of macrophages, a substantial rise in caspase-3 expression was found when cancer cells were treated with both Au@SiO2 NPs and XR. The increase was even three times higher compared to the groups without any of the three components, indicating a strong synergistic effect among these elements. Collectively our results highlighted that the addition of nanoparticles could potentiate radiation-induced antitumor immunity through the promotion of macrophage-mediated cancer cell damage.
Enhanced Therapeutic Efficacy in GL261-Bearing Mice
The combination of Au@SiO2 NPs and XR has been demonstrated to enhance radiation-induced oxidative stress, chemotaxis and systemic immune response in in vitro studies. Based on these findings, we hypothesized that Au@SiO2 NPs may augment the clinical efficacy of the atezolizumab in animal models. To ensure a robust evaluation, we carefully selected the lowest dose for each treatment component: atezolizumab (anti-PD-L1 antibody; α-PD-L1) at a dose of 10 mg/kg, Au@SiO2 NPs at a dose of 1 mg/kg, and XR with a single fraction of 6 MeV photons at a dose of 2 Gray. During our preliminary tests, these controlled dosages were determined to have no significant acute toxicity or radiation-enhancing effects. This allowed us to assess and understand the genuine synergistic effects of the combination therapy.
First, we assessed the anticancer effects using a subcutaneous tumor model. After subcutaneous implantation of GL261 cancer cells (day 0), successful tumor engraftment was confirmed on day 7. Following the tumor size screening, mice with comparable tumor sizes were selected and assigned to one of the five treatment groups: (1) control; (2) Au@SiO2 NPs plus α-PD-L1; (3) XR plus α-PD-L1; (4) Au@SiO2 NPs plus XR and α-PD-L1; and (5) α-PD-L1 alone. The experimental design is depicted in Figure 6a. Following the administration of α-PD-L1 on day 14, the tumor growth was monitored by measuring the in vivo surface diameters of the tumors. The tumors continued to enlarge in the control group, and were successfully inhibited by most of the treatments. Notably, the group treated with Au@SiO2 NPs, XR, and α-PD-L1 demonstrated the slowest rate of tumor growth (Figure 6b).
 |
Figure 6 In vivo therapeutic efficacy of different treatments was evaluated using mice bearing subcutaneous GL261 tumors. Notes: (a) Scheme of experimental design. (b) Exterior observation of tumor growth (Day 14 to 26 after implantation) and the final resected tumor volume (Day 28) from mice receiving different treatments. Four representative mice were selected from each treatment arm. Red circles mark the inoculated tumors. (c) Relative tumor volume measurements in mice receiving different treatments, with day 14 volume (V0) as the reference point. α-PD-L1 at a dose of 10 mg/kg, Au@SiO2 NPs at a dose of 1 mg/kg, and X-ray irradiation (XR) with a single fraction of 6 MeV photons at a dose of 2 Gray. *p < 0.05. |
After sacrificing the mice on day 28 after inoculation, the final tumor volumes were reassessed through surgical resection. Among them, the combined approach yielded the most significant decrease in tumor sizes (Figure 6c). While other treatment groups combining only two of the components also showed tumor reduction, the extent of reduction was not as substantial as when all three components were combined. In conclusion, this novel combination is effective in inhibiting tumor proliferation in animal models, providing insights into future strategies for brain tumors.
Improved Prognosis in GL261-Brain Tumor Model
To verify the in vivo therapeutic efficacy of the combined strategy against brain tumors, we established an intracranial orthotopic mouse glioma model. The tumor growth was confirmed through in vivo imaging system (IVIS) on day 7 and day 14 after stereotactic intracranial implantation. Thirty mice with successfully induced brain tumors of similar sizes were included and then allocated to five different treatment groups: (1) control; (2) Au@SiO2 NPs plus α-PD-L1; (3) XR plus α-PD-L1; (4) Au@SiO2 NPs plus XR and α-PD-L1; and (5) α-PD-L1 alone. The experimental workflow is illustrated in Figure 7a. In the Au@SiO2 NPs plus XR and α-PD-L1 group, mice were administered α-PD-L1 (10 mg/Kg) via intraperitoneal injection on day 14, followed by intratumoral Au@SiO2 NPs injection on the next day, and XR at 24 hours after the nanoparticle injection.
 |
Figure 7 In vivo therapeutic efficacy of different treatments in mice bearing intracranial GL261-Fluc-red tumors. Notes: (a) Scheme of experimental design. (b) IVIS images showing the growth of GL261-Fluc-red tumors on Day 14 (treatment initiation) and Day 21 after tumor inoculation. (c) Kaplan-Meier survival curves of mice receiving different treatments. α-PD-L1 at a dose of 10 mg/kg, Au@SiO2 NPs at a dose of 1 mg/kg, and X-ray irradiation (XR) with a single fraction of 6 MeV photons at a dose of 2 Gray. |
Figure 7b presents the IVIS images of three representative mice per treatment arm on day 14 and day 21 after tumor implantation. The mice treated with α-PD-L1 alone exhibited minimal change in the bioluminescent signal, indicating limited tumor response. The addition of either nanoparticles or radiation alone to the α-PD-L1 did not significantly improve the treatment responses. However, it is noteworthy that the combination of XR with Au@SiO2 NPs significantly enhanced the therapeutic effects of α-PD-L1. The bioluminescent signal showed a marked decrease, and one tumor even disappeared by day 21.
All mice were monitored for 50 days for Kaplan-Meier survival analysis. Our study showed that the combination of Au@SiO2 NPs, α-PD-L1, and XR resulted in significantly improved survival compared to each treatment modality alone (Figure 7c). Specifically, long-term survival (beyond 50 days after implantation) was observed exclusively in the combination group, with a 40% survival rate on day 50. These results provided compelling evidence for the in vivo synergistic effects and prognostic benefits of this combined strategy.
Activation of Systemic Antitumor Immunity
To elucidate the mechanisms underlying the treatment effects, the brains and spleens of the mice were surgically removed for histopathological examination. The control group (Figure 8a) exhibited extensive tumor invasion and tissue destruction, whereas the mice receiving Au@SiO2 NPs plus α-PD-L1 (Figure 8b) showed a diffuse decrease in neoplasm cell density, with few residual tumor cells at the inferior frontal lobe. Notably, the regenerative process was evident at the initial tumor site, along with increased CD8+ cells infiltration. On the other hand, the XR plus α-PD-L1 group (Figure 8c) showed persistent hypercellular infiltrative lesions spreading throughout the frontal lobe.
 |
Figure 8 Hematoxylin and eosin (H&E) staining of whole brain sagittal sections obtained from mice receiving different treatments. Notes: (a) Control; (b) Au@SiO2 NPs + α-PD-L1; (c) XR+ α-PD-L1; (d) Au@SiO2 NPs + XR + α-PD-L1; (e) α-PD-L1. α-PD-L1 at a dose of 10 mg/kg, Au@SiO2 NPs at a dose of 1 mg/kg, and X-ray irradiation (XR) with a single fraction of 6 MeV photons at a dose of 2 Gray. Black boxes magnify the inoculation sites in forebrains, and red arrows denote the residual tumor cells. Scale bar: 250 μm. |
The most significant improvement was observed in the group receiving all three components (Au@SiO2 NPs plus XR and α-PD-L1; Figure 8d), where most tumor cells were eliminated. The synergistic benefits led to higher levels of both CD8+ and CD4+ cell infiltration compared to the groups using only two components (Table S1 and Figure S1). Moreover, the combined strategy also triggered a substantial increase in CD8+ T cells infiltration within the spleens of mice (Figure S2). This observation suggests that the systemic influx of CD8+ effector T cells plays a central role in the treatment effects. In contrast, the group treated with α-PD-L1 alone (Figure 8e) showed persistent tumor invasion without significant tissue healing and immune cell infiltration.
In summary, the introduction of Au@SiO2 NPs (Figure 8b and d) markedly reduced tumor density and promoted regenerative processes and immune activity. Our combinatorial approach has shown promise as a novel treatment strategy to stimulate systemic immune response against brain tumors and improve clinical outcomes.
Discussion
Glioblastoma is a highly devastating cancer with unfavorable prognosis. While immunotherapy has revolutionized the treatment landscape of various types of cancer, their effectiveness in glioblastoma is hindered by the unique immune microenvironment. Radiotherapy holds promise in inducing immunogenic cell death and altering the blood-brain barrier,54,55 making it a potential partner for immunotherapy in glioblastoma treatment.26,56 Moreover, recent evidence challenging the traditional concept of central nervous system (CNS) immune privilege has opened up avenues for enhancing the immunological activities within the brain through radiation to effectively combat glioblastoma.57,58
In this study, we developed an innovative strategy aiming to synergistically enhance the immunogenic effects of radiation by combining the Au NP core with a SiO2 shell. Our approach has effectively strengthened radiation-induced damage in glioblastoma cells through robust ROS production. Our preliminary in vitro experiment has shown that the combination of Au@SiO2 NPs and XR can enhance DNA damage in glioblastoma cells,51 aligning with Joh et al’s finding.59 The pronounced cell damage coupled with the release of tumor antigens can serve as stress signals to reprogram the tumor microenvironment and elicit a robust systemic immune response against tumors. It has been suggested that ROS play a role in activating microglia and evoking inflammatory reactions, facilitating the trafficking of immune cells across the blood-brain barrier.60,61 In line with these findings, our study revealed that tumor cells treated with Au@SiO2 NPs and XR exhibit an enhanced ability to attract macrophages more effectively compared to any single modality arm. These infiltrating macrophages can be polarized into either M1 (pro-inflammatory) or M2 (anti-inflammatory, tumor-associated macrophages) phenotypes.62,63 M1-macrophages are known for their roles in orchestrating the cancer cell apoptosis and anti-tumor properties. Our study revealed that the combination of Au@SiO2 NPs and XR significantly boosts caspase-3 expression in the presence of macrophages. This suggested that the combined approach synergistically drives the polarization of macrophages towards the M1 phenotype, thereby promoting macrophage-mediated clearance of cancer cells.
In vivo, our nanoparticles have demonstrated efficacy in both subcutaneous and in situ brain tumor models. In this study, we used α-PD-L1 (atezolizumab) in view of the prominent expression of PD-L1 in glioblastoma.64–66 This combined approach effectively reduced tumor size, necrotic area, and prevented tumor infiltration into normal brain tissue in mice. The improvement was remarkably pronounced than other treatment groups that combined only two of the three components. Pathologically, the antitumor response is mainly mediated by the systemic expansion of the cytotoxic CD8+ T cell population. These findings unveiled the potential of our strategy to amplify systemic antitumor immunity against CNS neoplasms, overcoming the immunosuppressive environment of the tumor. Upon reviewing the H&E stains (Figure S1), some tumor cells did exhibit characteristics of necrosis. This suggests that the involvement of systemic immunity, which are absent in in vitro studies, significantly impacts cell death pathways, involving not only apoptosis but also other cell death pathways.67
In preclinical murine glioma models, various radiation doses and fractionations have been integrated with multimodal immunotherapy.68 Zeng et al observed that focal stereotactic radiotherapy (10-Gray single-fraction) combined with PD-1 blockade resulted in long-term survival and increased cytotoxic T cell infiltration.69 Similarly, Belcaid et al demonstrated that combining stereotactic radiotherapy (10-Gray single-fraction) with anti-CTLA-4 antibody and 4–1BB agonist significantly improved the prognosis.70 Combining stereotactic radiotherapy (10-Gray single-fraction) with PD-1 blockade and TIM-3 blockade also achieved significantly superior overall survival.71 On the other hand, the combination of radio-enhancers, radiation (12 Gray per fraction, 3 fractions in total) and PD-1 blockade has enhanced immunogenic cell death in several solid cancer models,72,73 but its efficacy in brain tumors remains unexplored. It is worth noting that these investigations primarily relied on high-dose radiation (10–12 Gray per fraction), which may be cytotoxic to infiltrating immune cells.20,74 On the contrary, low-dose radiation offers several advantages as it avoids the production of immunosuppressive factors that can harm immune cells and polarize M2 macrophages.74,75 While low dose radiation as a standalone treatment may not be fully sufficient for tumor eradication, it can incite danger signaling by releasing tumor-associated antigens.76 This process can reactivate the immune system and sensitize cold tumors, rendering them more responsive to immune checkpoint inhibitors.77 Our study also found that low-dose radiation could induce a shift towards M1 macrophages, aligned with the findings by Klug et al.78
However, the combination of low-dose radiation with immunotherapy remains largely unexplored to date, possibly due to concerns of risk of local recurrence. To intensify local control while harnessing the benefits of low-dose radiation, we incorporated Au@SiO2 NPs as radio-enhancers into our combinatorial approach. We demonstrated that this proposed strategy is technically feasible with a favorable safety profile and encouraging antitumor ability. The selective toxicity of Au@SiO2 NPs to tumor cells is consistent with our earlier observations of their preferential accumulation within glioblastoma cells.51 In clinical practice, the intratumoral administration can serve as part of adjuvant therapy in combination with radiotherapy and immunotherapy following surgical resection.5 By delivering these nanoparticles to the surgical bed (the areas with the highest recurrence risk), we can ensure the effective accumulation within the target and reduce systemic adverse events. Moreover, this approach can also achieve distant cell injury through activation of a systemic anti-tumor immunity, without the necessity of direct nanoparticle delivery to individual tumor cells. Instead, these nanoparticles can act as sensitizing signals spreading throughout the body, effectively priming the immune cells to target and eliminate cancer cells. This innovative strategy shows great promise in glioblastoma and warrants further investigation in both preclinical and clinical studies.
Several limitations should be acknowledged. First, as the isolation and primary microglia culture is viable to variable processing issues, we utilized macrophages in our study to minimize potential confounding factors. Considering the microenvironment of glioblastoma is influenced by both resident microglia and recruited macrophages, studying these macrophages would provide additional insights into the impact of our strategy on circulating immune cells. Secondly, since the preliminary experiments has revealed suboptimal synergy between Au@SiO2 NPs and XR in the absence of immunotherapy, further animal studies with this combination were not pursued in view of animal welfare. Thirdly, in this study, we selected caspase-3 expression as an indicator of cell damage based on prior discovery of apoptosis involvement in our treatment.51 While various forms of cell injuries may be activated upon irradiation in vivo,79 our focus was not on emphasizing specific cell death pathways. Instead, we aimed to demonstrate that our combined approach can enhance cell damage and elicit anti-tumor immune responses against brain tumors. Future research is needed to clarify the dominant cell death pathways involved.
Conclusion
In summary, our study demonstrated that combining low-dose XR, Au@SiO2 NPs and PD-L1 blockade presents a feasible and well-tolerated approach in glioblastoma treatments with prognostic benefits. The synergistic effects significantly enhanced radiation-induced oxidative stress, macrophage chemotaxis, reactivated the immune microenvironment and promoted effective immune responses against cancer cells, which was not achieved in any single modality. The combinatorial strategy offers valuable insights for developing novel and effective treatments against this devastating cancer.
Funding
We are grateful to the Ministry of Science and Technology (MOST 111-2221-E-A49-051-MY2, MOST 111-2811-E-A49A-007-MY2, MOST 111-2314-B-038-094, MOST 110-2622-E-A49A-504, MOST 109-2221-E-010-020-MY2, and MOST 108-2314-B-010-035-MY3) and the Department of Health, Taipei City Government (11201-62-004, and 11101-62-009) for financial support.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest. 2017;127(2):415–426. doi:10.1172/JCI89587
2. Fritz L, Dirven L, Reijneveld JC, et al. Advance care planning in glioblastoma patients. Cancers. 2016;8(11):102. doi:10.3390/cancers8110102
3. Mirimanoff R-O, Gorlia T, Mason W, et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 Phase III randomized trial. J Clin Oncol. 2006;24(16):2563–2569. doi:10.1200/JCO.2005.04.5963
4. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi:10.1016/S1470-2045(09)70025-7
5. Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi:10.1056/NEJMoa043330
6. Brandes AA, Tosoni A, Franceschi E, et al. Recurrence pattern after temozolomide concomitant with and adjuvant to radiotherapy in newly diagnosed patients with glioblastoma: correlation with MGMT promoter methylation status. J Clin Oncol. 2009;27(8):1275–1279. doi:10.1200/JCO.2008.19.4969
7. Muldoon LL, Alvarez JI, Begley DJ, et al. Immunologic privilege in the central nervous system and the blood–brain barrier. J Cerebl Blood Flow Metabol. 2013;33(1):13–21. doi:10.1038/jcbfm.2012.153
8. Engelhardt B. Molecular mechanisms involved in T cell migration across the blood–brain barrier. J Neural Transm. 2006;113(4):477–485. doi:10.1007/s00702-005-0409-y
9. Dubois L, Campanati L, Righy C, et al. Gliomas and the vascular fragility of the blood brain barrier. Front Cell Neurosci. 2014;8:418. doi:10.3389/fncel.2014.00418
10. Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–327. doi:10.1038/nri2744
11. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239
12. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi:10.1016/j.ccell.2015.03.001
13. Pitt JM, Vétizou M, Daillère R, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and-extrinsic factors. Immunity. 2016;44(6):1255–1269. doi:10.1016/j.immuni.2016.06.001
14. Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N Engl J Med. 2017;377(20):1919–1929. doi:10.1056/NEJMoa1709937
15. Choueiri TK, Tomczak P, Park SH, et al. Adjuvant pembrolizumab after nephrectomy in renal-cell carcinoma. N Engl J Med. 2021;385(8):683–694. doi:10.1056/NEJMoa2106391
16. Kelly RJ, Ajani JA, Kuzdzal J, et al. Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer. N Engl J Med. 2021;384(13):1191–1203. doi:10.1056/NEJMoa2032125
17. Janjigian YY, Shitara K, Moehler M, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, Phase 3 trial. Lancet. 2021;398(10294):27–40. doi:10.1016/S0140-6736(21)00797-2
18. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745
19. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017;390(10105):1853–1862. doi:10.1016/S0140-6736(17)31601-X
20. Wang X, Guo G, Guan H, Yu Y, Lu J, Yu J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J Exper Clin Cancer Res. 2019;38:1–13.
21. Huang J, Liu F, Liu Z, et al. Immune checkpoint in glioblastoma: promising and challenging. Front Pharmacol. 2017;8:242. doi:10.3389/fphar.2017.00242
22. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol. 2018;9:1004. doi:10.3389/fimmu.2018.01004
23. Hu F, a Dzaye OD, Hahn A, et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol. 2015;17(2):200–210. doi:10.1093/neuonc/nou324
24. Bausart M, Préat V, Malfanti A. Immunotherapy for glioblastoma: the promise of combination strategies. J Exper Clin Cancer Res. 2022;41(1):35. doi:10.1186/s13046-022-02251-2
25. Guo S, Yao Y, Tang Y, et al. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Signal Transduct Target Ther. 2023;8(1):205. doi:10.1038/s41392-023-01462-z
26. Rajani KR, Carlstrom LP, Parney IF, Johnson AJ, Warrington AE, Burns TC. Harnessing radiation biology to augment immunotherapy for glioblastoma. Front Oncol. 2019;8:656. doi:10.3389/fonc.2018.00656
27. Zhu M, Yang M, Zhang J, et al. Immunogenic cell death induction by ionizing radiation. Front Immunol. 2021;12:705361. doi:10.3389/fimmu.2021.705361
28. Golden EB, Apetoh L. Radiotherapy and Immunogenic Cell Death. Elsevier; 2015:11–17.
29. Liu T, Pei P, Shen W, Hu L, Yang K. Radiation‐induced immunogenic cell death for cancer radioimmunotherapy. Small Methods. 2023;7(5):2201401. doi:10.1002/smtd.202201401
30. Yang Y, Huang J, Liu M, et al. Emerging sonodynamic therapy‐based nanomedicines for cancer immunotherapy. Adv Sci. 2023;10(2):2204365. doi:10.1002/advs.202204365
31. Sang W, Zhang Z, Dai Y, Chen X. Recent advances in nanomaterial-based synergistic combination cancer immunotherapy. Chem Soc Rev. 2019;48(14):3771–3810. doi:10.1039/c8cs00896e
32. Wang J, Li Z, Wang Z, et al. Nanomaterials for combinational radio–immuno oncotherapy. Adv Funct Mater. 2020;30(30):1910676. doi:10.1002/adfm.201910676
33. Xu P, Liang F. Nanomaterial-based tumor photothermal immunotherapy. Int J Nanomedicine. 2020;Volume 15:9159–9180. doi:10.2147/IJN.S249252
34. Liang S, Yao J, Liu D, Rao L, Chen X, Wang Z. Harnessing nanomaterials for cancer sonodynamic immunotherapy. Adv Mater. 2023;35(33):2211130. doi:10.1002/adma.202211130
35. Goel S, Ni D, Cai W. Harnessing the power of nanotechnology for enhanced radiation therapy. ACS nano. 2017;11(6):5233–5237. doi:10.1021/acsnano.7b03675
36. Ning S, Dai X, Tang W, et al. Cancer cell membrane-coated C-TiO2 hollow nanoshells for combined sonodynamic and hypoxia-activated chemotherapy. Acta Biomater. 2022;152:562–574. doi:10.1016/j.actbio.2022.08.067
37. Liu D, Dai X, Zhang W, et al. Liquid exfoliation of ultrasmall zirconium carbide nanodots as a noninflammatory photothermal agent in the treatment of glioma. Biomaterials. 2023;292:121917. doi:10.1016/j.biomaterials.2022.121917
38. Guo Q, Yin M, Fan J, et al. Peroxidase-mimicking TA-VOx nanobranches for enhanced photothermal/chemodynamic therapy of glioma by inhibiting the expression of HSP60. Mater Des. 2022;224:111366. doi:10.1016/j.matdes.2022.111366
39. Yang Y, Wang X, Qian H, Cheng L. Titanium-based sonosensitizers for sonodynamic cancer therapy. Appl Mater Today. 2021;25:101215. doi:10.1016/j.apmt.2021.101215
40. Sun L, Wang X, Gong F, et al. Silicon nanowires decorated with platinum nanoparticles were applied for photothermal-enhanced sonodynamic therapy. Theranostics. 2021;11(19):9234. doi:10.7150/thno.58755
41. Guo Q-L, Dai X-L, Yin M-Y, et al. Nanosensitizers for sonodynamic therapy for glioblastoma multiforme: current progress and future perspectives. Milit Med Res. 2022;9(1):1–18. doi:10.1186/s40779-022-00386-z
42. Omuro A, Brandes AA, Carpentier AF, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase III trial. Neuro Oncol. 2023;25(1):123–134. doi:10.1093/neuonc/noac099
43. Lim M, Weller M, Idbaih A, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022;24(11):1935–1949. doi:10.1093/neuonc/noac116
44. Shrestha S, Cooper LN, Andreev OA, Reshetnyak YK, Antosh MP. Gold nanoparticles for radiation enhancement in vivo. Jacobs J Rad Oncol. 2016;3(1):26.
45. Choi J, Kim G, Cho SB, Im H-J. Radiosensitizing high-Z metal nanoparticles for enhanced radiotherapy of glioblastoma multiforme. J Nanobiotechnol. 2020;18(1):1–23. doi:10.1186/s12951-020-00684-5
46. Misawa M, Takahashi J. Generation of reactive oxygen species induced by gold nanoparticles under x-ray and UV Irradiations. Nanomedicine. 2011;7(5):604–614. doi:10.1016/j.nano.2011.01.014
47. Kuncic Z, Lacombe S. Nanoparticle radio-enhancement: principles, progress and application to cancer treatment. Phys Med Biol. 2018;63(2):02TR01. doi:10.1088/1361-6560/aa99ce
48. Her S, Jaffray DA, Allen C. Gold nanoparticles for applications in cancer radiotherapy: mechanisms and recent advancements. Adv Drug Delivery Rev. 2017;109:84–101. doi:10.1016/j.addr.2015.12.012
49. Gilles M, Brun E, Sicard-Roselli C. Quantification of hydroxyl radicals and solvated electrons produced by irradiated gold nanoparticles suggests a crucial role of interfacial water. J Colloid Interface Sci. 2018;525:31–38. doi:10.1016/j.jcis.2018.04.017
50. Gu L, Ruff LE, Qin Z, Corr M, Hedrick SM, Sailor MJ. Multivalent porous silicon nanoparticles enhance the immune activation potency of agonistic CD40 antibody. Adv Mater. 2012;24(29):3981–3987. doi:10.1002/adma.201200776
51. Chiang C-S, Shih I-J, Shueng P-W, et al. Tumor cell-targeting radiotherapy in the treatment of glioblastoma multiforme using linear accelerators. Acta Biomater. 2021;125:300–311. doi:10.1016/j.actbio.2021.02.019
52. Tominaga H, Kodama S, Matsuda N, Suzuki K, Watanabe M. Involvement of reactive oxygen species (ROS) in the induction of genetic instability by radiation. J Radiat Res. 2004;45(2):181–188. doi:10.1269/jrr.45.181
53. Riley P. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65(1):27–33. doi:10.1080/09553009414550041
54. Odé Z, Derieppe MP, Groenink L, et al. Blood-brain barrier permeability following conventional photon radiotherapy–A systematic review and meta-analysis of clinical and preclinical studies. Clin Transl Radiat Oncol. 2022;35:44–55. doi:10.1016/j.ctro.2022.04.013
55. van Vulpen M, Kal HB, Taphoorn MJ, El Sharouni SY. Changes in blood-brain barrier permeability induced by radiotherapy: implications for timing of chemotherapy? Oncol Rep. 2002;9(4):683–688.
56. Nesseler JP, Schaue D, McBride WH, et al. Irradiation to improve the response to immunotherapeutic agents in glioblastomas. Adv Radiat Oncol. 2019;4(2):268–282. doi:10.1016/j.adro.2018.11.005
57. Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–341. doi:10.1038/nature14432
58. Fecci PE, Heimberger AB, Sampson JH. Immunotherapy for primary brain tumors: no longer a matter of privilege. Clin Cancer Res. 2014;20(22):5620–5629. doi:10.1158/1078-0432.CCR-14-0832
59. Joh DY, Sun L, Stangl M, et al. Selective targeting of brain tumors with gold nanoparticle-induced radiosensitization. PLoS One. 2013;8(4):e62425. doi:10.1371/journal.pone.0062425
60. Seo S-J, Han S-M, Cho J-H, et al. Enhanced production of reactive oxygen species by gadolinium oxide nanoparticles under core–inner-shell excitation by proton or monochromatic X-ray irradiation: implication of the contribution from the interatomic de-excitation-mediated nanoradiator effect to dose enhancement. Radiat Environ Biophys. 2015;54(4):423–431. doi:10.1007/s00411-015-0612-7
61. Patel JP, Frey BN. Disruption in the blood-brain barrier: the missing link between brain and body inflammation in bipolar disorder? Neural Plast. 2015;2015:1–12. doi:10.1155/2015/708306
62. Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther. 2021;6(1):127. doi:10.1038/s41392-021-00506-6
63. Murray PJ. Macrophage polarization. Ann Rev Physiol. 2017;79(1):541–566. doi:10.1146/annurev-physiol-022516-034339
64. Berghoff AS, Kiesel B, Widhalm G, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17(8):1064–1075. doi:10.1093/neuonc/nou307
65. Nduom EK, Wei J, Yaghi NK, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2015;18(2):195–205. doi:10.1093/neuonc/nov172
66. Xue S, Hu M, Iyer V, Yu J. Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. J Hematol Oncol. 2017;10(1):1–10. doi:10.1186/s13045-017-0455-6
67. Montico B, Nigro A, Casolaro V, Dal Col J. Immunogenic apoptosis as a novel tool for anticancer vaccine development. Int J Mol Sci. 2018;19(2):594. doi:10.3390/ijms19020594
68. Awada H, Paris F, Pecqueur C. Exploiting radiation immunostimulatory effects to improve glioblastoma outcome. Neuro Oncol. 2023;25(3):433–446. doi:10.1093/neuonc/noac239
69. Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–349. doi:10.1016/j.ijrobp.2012.12.025
70. Belcaid Z, Phallen JA, Zeng J, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS One. 2014;9(7):e101764. doi:10.1371/journal.pone.0101764
71. Kim JE, Patel MA, Mangraviti A, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124–136. doi:10.1158/1078-0432.CCR-15-1535
72. Hu Y, Paris S, Barsoumian H, et al. Radiation therapy enhanced by NBTXR3 nanoparticles overcomes anti-PD1 resistance and evokes abscopal effects. Int J Radiat Oncol Biol Phys. 2021;111(3):647–657. doi:10.1016/j.ijrobp.2021.06.041
73. Hu Y, Paris S, Bertolet G, et al. NBTXR3 improves the efficacy of immunoradiotherapy combining nonfucosylated anti-CTLA4 in an anti-PD1 resistant lung cancer model. Front Immunol. 2022;13:1022011. doi:10.3389/fimmu.2022.1022011
74. Yovino S, Kleinberg L, Grossman SA, Narayanan M, Ford E. The etiology of treatment-related lymphopenia in patients with malignant gliomas: modeling radiation dose to circulating lymphocytes explains clinical observations and suggests methods of modifying the impact of radiation on immune cells. Cancer Invest. 2013;31(2):140–144. doi:10.3109/07357907.2012.762780
75. Sheng Y, Zhang B, Xing B, et al. Cancer-associated fibroblasts exposed to high-dose ionizing radiation promote M2 polarization of macrophages, which induce radiosensitivity in cervical cancer. Cancers. 2023;15(5):1620. doi:10.3390/cancers15051620
76. Lumniczky K, Impens N, Armengol G, et al. Low dose ionizing radiation effects on the immune system. Environ Int. 2021;149:106212. doi:10.1016/j.envint.2020.106212
77. Herrera FG, Ronet C, Ochoa de Olza M, et al. Low-dose radiotherapy reverses tumor immune desertification and resistance to immunotherapy. Cancer Discov. 2022;12(1):108–133. doi:10.1158/2159-8290.CD-21-0003
78. Klug F, Prakash H, Huber PE, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602. doi:10.1016/j.ccr.2013.09.014
79. Wouters BG. Cell death after irradiation: how, when and why cells die. In: Basic Clinical Radiobiology.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.