Back to Journals » OncoTargets and Therapy » Volume 13
Synergistic Effect of 3-Bromopyruvate in Combination with Rapamycin Impacted Neuroblastoma Metabolism by Inhibiting Autophagy
Authors Gan L ![]() , Ren Y, Lu J, Ma J, Shen X, Zhuang Z
, Ren Y, Lu J, Ma J, Shen X, Zhuang Z
Received 11 August 2020
Accepted for publication 5 October 2020
Published 29 October 2020 Volume 2020:13 Pages 11125—11137
DOI https://doi.org/10.2147/OTT.S273108
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Lei Gan,* Yang Ren,* Jicheng Lu, Junzhe Ma, Xudong Shen, Zhixiang Zhuang
Department of Oncology, The Second Affiliated Hospital of Soochow University, Suzhou, Jiangsu Province 215004, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lei Gan; Zhixiang Zhuang
Department of Oncology, The Second Affiliated Hospital of Soochow University, Suzhou, Jiangsu Province 215004, People’s Republic of China
Email [email protected]; [email protected]
Background: Alterations in the cell metabolism, such as enhanced aerobic glycolysis, have been identified as a prominent hallmark of cancer cells. 3-Bromopyruvate (3-BrPA) is a proverbial hexokinase (HK)-II inhibitor, which can inhibit cancer cell energy metabolism. Rapamycin is a new type macrocyclic lactone, which can inhibit the serine/threonine protein kinase mTOR. In order to comprehend the influence of 3-BrPA on autophagy activity in vitro, we conducted a series of experiments using different human neuroblastoma (NB) cell lines.
Materials and Methods: The human NB cell lines were exposed to 3-BrPA and/or rapamycin, and the proliferation activity of the cells was detected by Cell Counting Kit-8 (CCK-8) assay. The mRNA expression of the cells treated with 3-BrPA and/or rapamycin was analyzed by quantitative real-time polymerase chain reaction (QPCR) assay. The protein expression of the cells was analyzed by Western Blotting (WB) assay. The effects of 3-BrPA and/or rapamycin treatment on cell cycle and cell apoptosis were analyzed by flow cytometry assay. Meanwhile, the cellular glucose absorption rate, lactate secretion rate and ATP content were also analyzed through the relevant metabolic analysis kits.
Results: Our results showed that 3-BrPA can induce growth inhibition in a dose-dependent pattern by cell apoptosis. 3-BrPA combined with rapamycin played a synergistic suppression role in NB cells, affected the cell apoptosis, cell cycle and the metabolic pathway. Up-regulated LC3-II accumulation was conscious in NB cells incubated with 3-BrPA and rapamycin. Rapamycin individually discourages the mTOR signaling pathway, while combined with 3-BrPA can enhance this phenomenon and influence cell metabolism of the NB cells.
Conclusion: The results suggested that 3-BrPA combined with rapamycin could induce cell apoptosis in NB cells by inhibiting mTOR activity. In conclusion, our research proposed that the dual inhibitory effect of the mTOR signaling pathway and the glycolytic activity may indicate a valid therapeutic tactic for NB chemoprevention.
Keywords: autophagy, metabolism, neuroblastoma, MYC, MCT1
Introduction
MYCN is a member of the MYC family, which is considered as an oncogenic transcription factor. The MYC family members also include MYC and MYCL. The principal function of MYCN is to improve cell growth and cell proliferation through transcriptional regulation, which is similar to other MYC family proteins.1,2 Neuroblastoma is a childhood malignant carcinoma, which is the most general pediatric solid carcinoma, derived from the sympathetic nervous system, accounting for roughly 7% of pediatric malignant tumors and 15% of pediatric malignant deaths.3 The most malignant neuroblastoma showed amplification of the oncogene MYCN, which was discovered in nearly 25% of neuroblastoma and nearly 40% of the high-risk individuals.4–6 Alternatively, in MYCN-nonamplified neuroblastoma, the expression of C-MYC protein is up-regulated, which is also correlated with poor prognosis.7 There are various strategies to treat neuroblastoma, including surgery, chemotherapy, radiotherapy, immunotherapy, autologous stem cell transplantation, ALK inhibitor and multiple combinations of such strategies.8 Nevertheless, the prognosis of advanced NB children is still disappointing, even with a battery of progressive adjuvant methods, especially for those children older than 1 year, whose long-term survival rate is usually below 40%.9 Therefore, the current treatment methods are limited, and more effective treatment options need to be explored.
The oncogene MYCN is ordinarily amplified in the high-risk neuroblastoma, and is related to the cellular metabolism of neuroblastoma, including oxidative glycolysis or Warburg metabolism.10,11 At present, it is difficult to target oncogene MYCN directly. Therefore, we chose the metabolic pathway activated by MYCN as a potential drug candidate target. The distinction in metabolism between neoplastic cells and normal cells has gradually become one of the targets of tumor therapies.11–14
The transport of monocarboxylates (such as lactate and pyruvate) is mediated by the proton-linked solute carrier 16 (SLC16) family membrane transport proteins, called monocarboxylate transporters (MCTs).15 At present, 14 MCT-related genes have been identified in mammals. Despite their sequence homology, only MCT1-MCT4 have been shown to be proton-dependent transporters of monocarboxylic acids, and the expression of MCT1-MCT4 is often elevated in human cancers. Birsoy et al have determined that MCT1, the SLC16A1 gene product, is the main determinant of 3-BrPA sensitivity. MCT1 is necessary and sufficient for cancer cells to take up 3-BrPA. In addition, SLC16A1 mRNA level is the best predictor of 3-BrPA sensitivity, with the highest level in glycolytic cancer cells.16
Our previous studies showed that 3-BrPA, a drug candidate that can inhibit cell glycolysis, preferentially induced massive cell death of the human neoplastic cells overexpressing the oncogene MYC, without obvious influences on those neoplastic cells with tiny or none MYC expression.17 In addition, we also found that the expression levels of MYC and MCT1 decreased in the neuroblastoma cells treated with 3-BrPA at the mRNA and protein levels.
Programmed cell death is a physiological and selective process, refers to the autonomous cell death controlled by a series of genes, which can maintain the stability of the internal environment. There are three familiar forms supposed to the programmed cell death, such as apoptosis, autophagy and necrosis. There are three dominating pathways of protein degradation in the eukaryotic cells, such as the ubiquitination pathway, the caspase pathway and the lysosome pathway. Autophagy is a lysosomal pathway, which can maintain the intracellular stability, response to stress, and mediate a dynamic process including degrade proteins, organelles, macromolecules and ribosomes.18 Under the stress, autophagy can generate nutrients and energy to maintain the cellular survival. There are many studies reported that autophagy is related to the pathological processes and pathogenical processes, containing infectious diseases, neurodegenerative diseases, autoimmune diseases, myopathy and malignant cancer diseases.19–21 The role of autophagy is constantly changing during the occurrence and development of different carcinomas.
Beclin, light chain 3 (LC3) A and B, SQSTM1/p62 and sirtuin 1 (SIRT1) are known as the autophagy-related proteins.22 Among them, LC3B and SQSTM1/p62 are autophagy markers widely used to monitor autophagy activity. LC3B is involved in the formation of the autophagosomes, while SQSTM1/p62 is used as a selective autophagy substrate, which has multiple domains that interact with autophagy machinery as an adaptor for the target cargo.23 The up-regulation or down-regulation of SQSTM1/p62 can play a tumorigenic or an anti-tumor effect in cancers.23
Therefore, our research was to explore whether HK-II inhibitor 3-BrPA can affect the metabolic level of the neuroblastoma cells by the autophagy pathway, which may provide a new therapeutic strategy for NB treatment.
Materials and Methods
Cell Culture and Materials
The neuroblastoma cell lines were purchased from Shanghai Genechem Co., LTD., and cultured in RPMI-1640 medium (Gibco) containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C of 5% carbon dioxide. The culture medium was removed once every two days. The neuroblastoma cell lines SK-N-BE2 and IMR-32 are MYCN-amplified, while SK-N-AS, SH-SY5Y and SK-N-SH are MYCN non-amplified cell lines. All the cells utilized in our experiments were confirmed to be free of mycoplasma. 3-BrPA powder was purchased from Sigma Aldrich. Rapamycin powder was purchased from MedChemExpress (MCE). All reagents were dissolved in dimethyl sulfoxide (DMSO, Sigma) and kept at −20°C. Antibodies were used as follows: N-Myc (13987, CST), C-Myc (5605S, CST), MCT1 (AB3538P, Millipore), p-mTOR (Ser2448) (5536S, CST), SQSTM1/p62 (88588S, CST), LC3B (83506S, CST) and β-actin (8457S, CST).
Cell Viability Assay
The cell proliferation of the NB cells treated with 3-BrPA and/or rapamycin was detected by Cell Counting Kit-8 (CCK-8, Bestbio) assay. The NB cell lines (IMR-32, SH-SY5Y and SK-N-SH) were sown in 96-well plates with 5×103 cells per well overnight. And the cells were treated with different concentrations (0µM, 25µM, 50µM, 75µM and 100µM) of 3-BrPA for 24h, whereas DMSO was used as the blank group. The supernatant were thoroughly removed, subsequently the CCK-8 solutions were joined in the wells and incubated for 4 hours at 37°C in dark place. Finally, we use a microplate reader to measure the absorbance of all the wells at 450 nm. Assays were performed on three independent experiments.
RNA Extraction and Quantitative Real-Time PCR
We use the TRIzol Reagent (15,596,018, Thermo Fischer Scientific) to extract total RNA of the cells, and then use a reverse transcriptase kit (R211-02, Vazyme) to synthesize cDNA according to the manufacturers’ protocols. Quantitative Real-time PCR (QPCR) was executed with the NovoStart® SYBR QPCR SuperMix Plus Kit (E096-01B, Novoprotein) and the Bio-Rad CFX 96 Real-time System. The relative mRNA expression was calculated by 2−ΔΔCt method and normalized to β-actin expression. Specific QPCR primer sequences are listed in Table 1.
 |
Table 1 Primers Used for Reverse Transcription-Quantitative PCR |
Western Blotting Analysis
The human neuroblastoma cells were treated with 3-BrPA and/or rapamycin, total proteins were isolated on ice with RIPA lysing buffer, and the protein concentration was measured using a BCA protein assay kit (Beyotime). A total of 40μg proteins were submitted to SDS-PAGE and transferred to the PVDF membrane. The PVDF membranes were blocked with 5% skimmed milk supplemented with 0.05% Tween 20 (Sigma Aldrich) in PBS for 1 hour, and then incubated with the primary antibody at 4°C overnight. The next day, the PVDF membranes were incubated with secondary antibody for 1 hour at the room temperature. Washing with TBST 3 times, the PVDF membranes were further developed with the ECL Plus Western Blotting Detection Reagents and Analysis System (BioTek). To control the equal amount of protein loading, the PVDF membranes were calibrated with β-actin antibody.
Annexin V-FITC/PI Staining
The Annexin V-FITC Apoptosis Kit (Biovision) was used to detect the cell apoptosis in neuroblastoma cells (SH-SY5Y and SK-N-SH) treated with 3-BrPA and/or rapamycin. We treated SH-SY5Y and SK-N-SH with 3-BrPA and/or rapamycin, collected the supernatant and the cells, centrifuged and resuspended, added 1×Binding Buffer and 5uL Annexin V-FITC, mixed gently and incubated on ice for 10 minutes in dark place, added 10uL PI staining solution, then gently mixed and incubated on ice for 5 minutes. The data were performed on the Accuri C6 (BD Biosciences), and the data were analyzed using Treestar FlowJo software.
Flow Cytometric Evaluation of Cell Cycle
The neuroblastoma cells (SH-SY5Y and SK-N-SH) were seeded into a 6-well plate at a density of 3×105 cells per well. The cells were treated with 3-BrPA and/or rapamycin, harvested with 0.25% trypsin (without EDTA), washed with pre-cooled PBS, fixed with 75% ethanol at 4°C for 2 hours or overnight, then washed twice with pre-cooled PBS, suspended in 100μL of 5 mg/mL RNase solution, incubated in darkness at the room temperature for 30 minutes, and then stained with 50 µg/mL Propidium Iodide (PI) solution for 30 minutes at 4°C in darkness. The analysis was performed with the FACS Calibur Flow Cytometer (BD Biosciences).
Metabolites Analysis
The neuroblastoma cells were cultured in RPMI-1640 medium with DMSO, 25μM rapamycin, 50μM 3-BrPA or 25μM rapamycin + 50μM 3-BrPA for 6 hours. The cellular glucose uptake, lactate production and ATP production were determined using respective assay kits obtained from BioVision (Milpitas). Data were an average of triplicate and presented as a percentage of the control group.
Statistical Analysis
Data were expressed as mean ± SD, and each result was detected from at least three independent experiments. The experimental results were analyzed by independent sample’s t-test and SPSS.21 (Armonk) was used for the analysis. All the residual significance analyses were performed using two-tailed Student’s t-test. P<0.05 was considered statistically significant.
Results
MCT1 Expression in Neuroblastoma
We detected the expression level of MCT1 in 5 neuroblastoma cell lines, such as MYCN-amplified neuroblastoma cell lines (SK-N-BE2 and IMR-32), MYC-amplified neuroblastoma cell lines (SK-N-AS and SH-SY5Y) and SK-N-SH (Figure 1A and C) at the mRNA level and the protein level by QPCR analysis and WB analysis. The expression level of genes related to glucose metabolism in these cells was also detected by QPCR analysis (Figure 1B). Our results exhibited that the expression of MCT1 was connected with the expression of N-Myc and C-Myc at the mRNA level, and also correlated with the expression of N-Myc and C-Myc at the protein level. In addition, we also found that the expression of SQSTM1/p62 in neuroblastoma was negatively correlated with N-Myc and C-Myc at the protein level. By QPCR analysis, we also found that the expression of genes related to glucose metabolism in five neuroblastoma cell lines themselves had little difference (Figure 1B, P>0.05). Our results showed that 3-BrPA treated in neuroblastoma was concentration-dependent, and the inhibitory effect was more pronounced in MCT1-expressed neuroblastoma cell lines (Figure 1D). When IMR-32 treated with 50µM 3-BrPA, the cell inhibition efficiency of IMR-32 was particularly obvious.
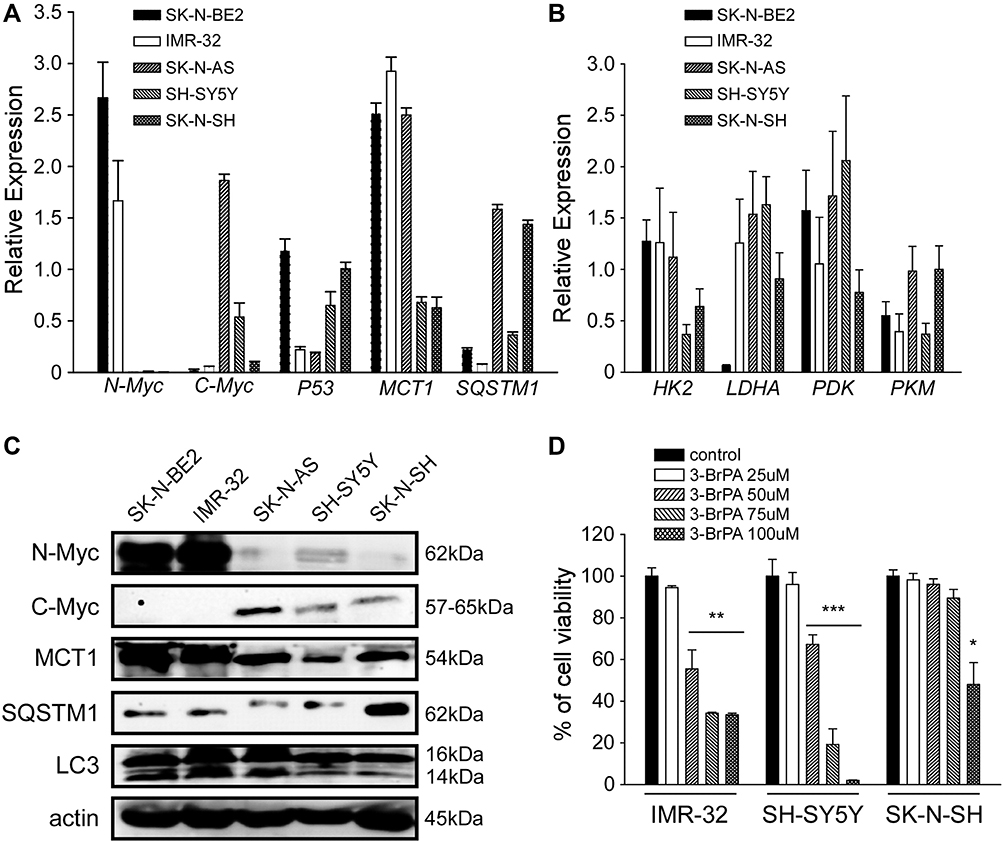 |
Figure 1 Gene expression level in un-treated NB cells and the cell viability of NB cells treated with 3-BrPA of different concentration. (A) The mRNA expression level of N-Myc, C-Myc, P53, MCT1 and SQSTM1 in NB cell lines (SK-N-BE2, IMR-32, SK-N-AS, SH-SY5Y and SK-N-SH) by QPCR analysis. (B) Metabolism related gene expression (HK2, LDHA, PDK and PKM) in NB cell lines by QPCR analysis. (C) The protein expression level of N-Myc, C-Myc, MCT1, SQSTM1 and LC3 (LC3-II/LC3-I) in NB cell lines by WB analysis. (D) The cell proliferation ability of 3-BrPA treated NB cells in different concentration by CCK-8 analysis (*P<0.05; **P<0.01; ***P<0.001). |
Expression of SQSTM1/P62 is Reduced in MYCN-Amplified Neuroblastoma Primary Tumors
To evaluate the relationship of MYCN and autophagy, we analyzed the expression of MYCN, MCT1 and autophagy relative genes in microarray data from 493 primary neuroblastoma tumors (GSE62564) respectively (Figure 2A and B). We found that the expression of MYCN and MCT1was significantly elevated in the MYCN-amplified neuroblastoma tumors compared with non-amplified neuroblastoma tumors (Figure 2A, Supplementary Figure 1), while sequestosome-1 (SQSTM1/p62) was lower expressed in the MYCN-amplified neuroblastoma tumors (Figure 2A). We found that the autophagy relative genes such as SQSTM1/p62, ATG5, BECLIN1, ATG7 and LC3B were negatively related with MYCN (P<0.001, Figure 2A). Meanwhile, the autophagy relative genes such as SQSTM1/p62, BECLIN1, ATG7 and LC3B were negatively related with MCT1 (P<0.001, Figure 2B). We also detected the expression of N-Myc, MCT1 and SQSTM1/p62 in primary neuroblastoma tumors by immunochemistry assay. We found that the expression of N-Myc, MCT1 was markedly elevated in MYCN-amplified tumors, whereas the expression of SQSTM1/p62 was reduced (Figure 2C). In MYCN-amplified neuroblastoma cell (IMR-32), we found that the protein expression levels of LC3-II/LC3-I and SQSTM1/p62 in the neuroblastoma cells treated with 3-BrPA of different concentration were increased (Figure 2D).
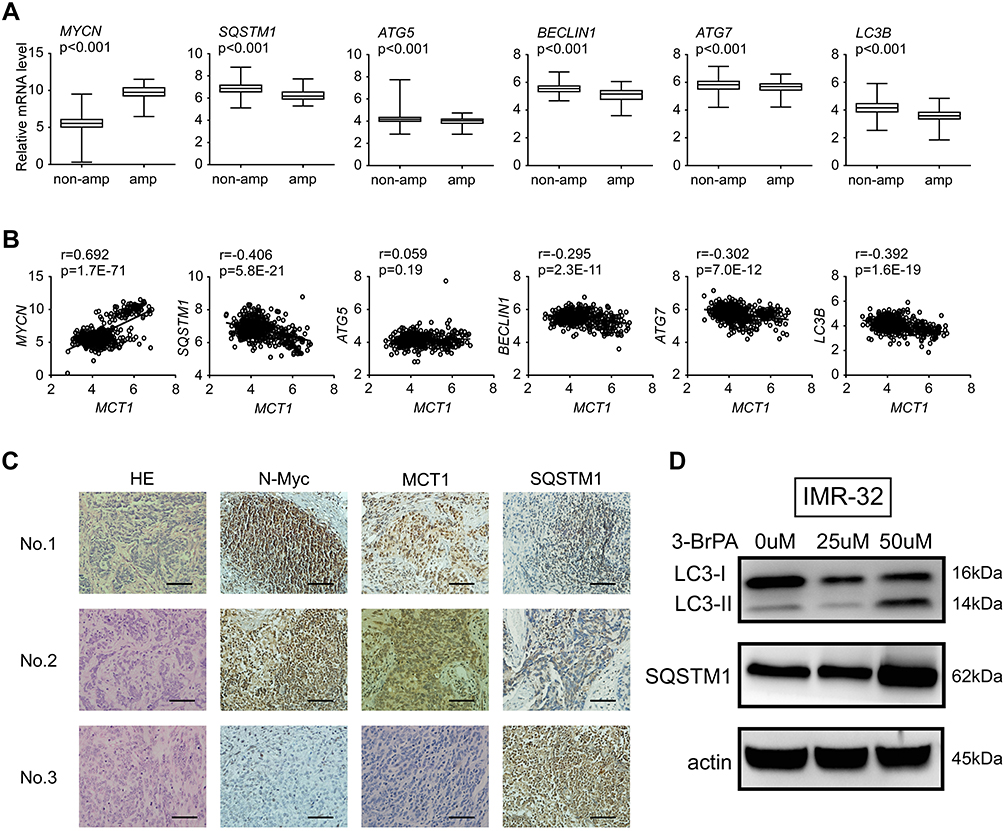 |
Figure 2 The expression of SQSTM1/p62 is reduced in MYCN-amplified neuroblastomas. (A) Relative expression of MYCN, SQSTM1, ATG5, BECLIN1, ATG7 and LC3B in 493 primary neuroblastoma tumors, non-amp: MYCN-nonamplified tumors (n=401); amp: MYCN-amplified tumors (n=92). (B) Correlation between mRNA levels of MCT1 and representative autophagy targets in MYCN-amplified neuroblastoma tumors. (C) Representative N-Myc, MCT1 and SQSTM1/p62 immunochemical staining in primary neuroblastoma tumors; sample No.1 and No.2: MYCN-amplified, high stage; sample No.3: MYCN-nonamplified, low stage. The scale bar represents 50μm. (D) The protein expression of LC3-II/LC3-I and SQSTM1/p62 in NB cells treated with 3-BrPA of different concentration by WB analysis. |
Influence of 3-BrPA and Rapamycin on NB Cells Proliferation
The effect of 3-BrPA, rapamycin, chloroquine (CQ) treated in NB cells on cell proliferation was evaluated. NB cell lines SH-SY5Y and SK-N-SH were treated with 50µM 3-BrPA, 25µM rapamycin and 25µM CQ. Cells were harvested, and the cell proliferation was measured using CCK-8 assay. As shown in Figure 3A, 3-BrPA combined with rapamycin significantly inhibited the proliferation of NB cells, especially in SH-SY5Y (P<0.001, 3-BrPA combined with rapamycin vs control). We also detected the cell proliferation of MYCN-amplified neuroblastoma cell lines SK-N-BE2 and IMR-32 treated with 50µM 3-BrPA, 25µM rapamycin and 25µM CQ (Supplementary Figure 2).
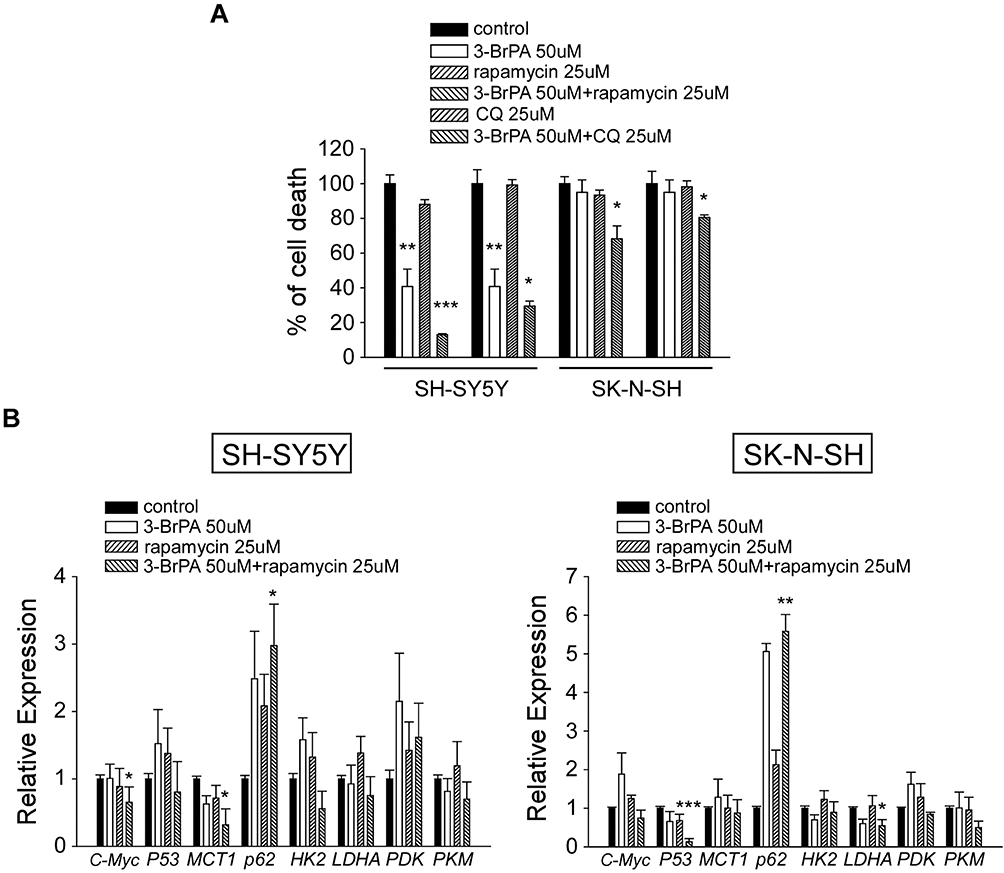 |
Figure 3 Effect of 3-BrPA and rapamycin in NB cells by CCK-8 analysis and QPCR analysis. (A) The cell proliferation effect of 3-BrPA, rapamycin, 3-BrPA + rapamycin, CQ, 3-BrPA + CQ treatment in SH-SY5Y and SK-N-SH by CCK-8 analysis. (B) Gene expression level in SH-SY5Y and SK-N-SH treated with control, 3-BrPA, rapamycin, 3-BrPA + rapamycin by QPCR analysis (*P<0.05; **P<0.01; ***P<0.001). |
3-BrPA and Rapamycin Down-Regulated the Expression of p-mTOR, Increased the Expression of LC3-II/LC3-I and SQSTM1/p62
For a more thorough analysis of the influence of 3-BrPA combined with rapamycin on autophagy and the related signaling pathways, we also analyzed the expression of related proteins by Western Blotting analysis. We found that p-mTOR levels were obviously reduced by 3-BrPA combined with rapamycin treatment compared to the control cells (P<0.05), LC3-II/LC3-I and SQSTM1/p62 mRNA and protein expression levels were dramatically increased in 3-BrPA combined with rapamycin treatment (Figures 3B and 4A–C). All experiments were conducted in triplicate. Together, these data strongly show that 3-BrPA combined with rapamycin inhibited autophagy ability via the mTOR pathway.
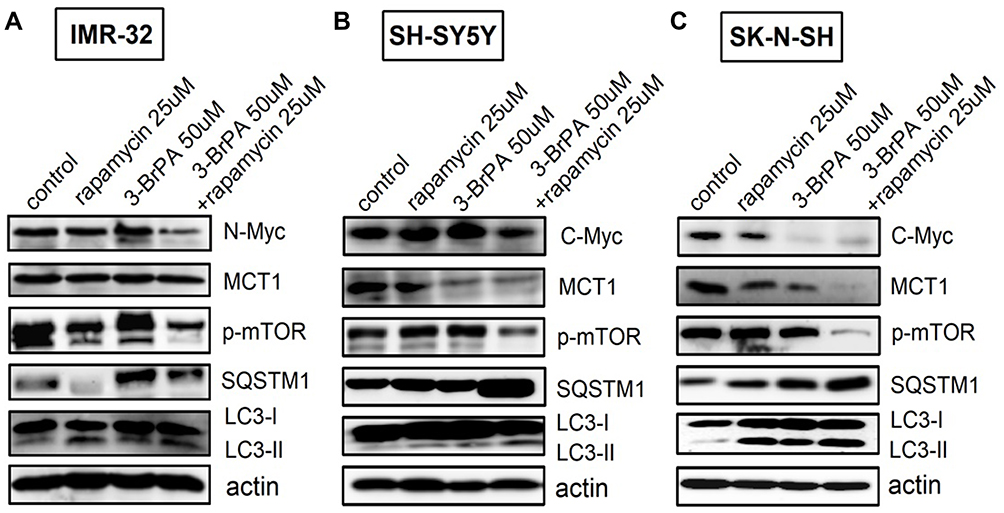 |
Figure 4 Effect of 3-BrPA and rapamycin in NB cells by Western Blotting analysis. Effect of 3-BrPA and rapamycin on the phosphorylation of mTOR and autophagy related proteins in (A) IMR-32 (B) SH-SY5Y and (C) SK-N-SH, cell lysates were suspended with rapamycin 25μM or 3-BrPA 50μM for 24 hours. And the cells were suspended with rapamycin and 3-BrPA of the same concentration concurrently for 24 hours. Actin was used as an internal control. |
Effect of 3-BrPA and Rapamycin Treatment on NB Cells Apoptosis
We treated SH-SY5Y and SK-N-SH of variant MCT1 expression with rapamycin alone, 3-BrPA alone, and 3-BrPA combined with rapamycin. Through the microscope, we found that the neuroblastoma cell morphology turned round and the number is lower than the group with rapamycin alone or 3-BrPA alone (Figure 5A, Supplementary Figure 3). We also utilized the cell apoptosis method, the results show that in the group with 3-BrPA and rapamycin, the cell apoptosis is significantly increased (Figure 5B and C).
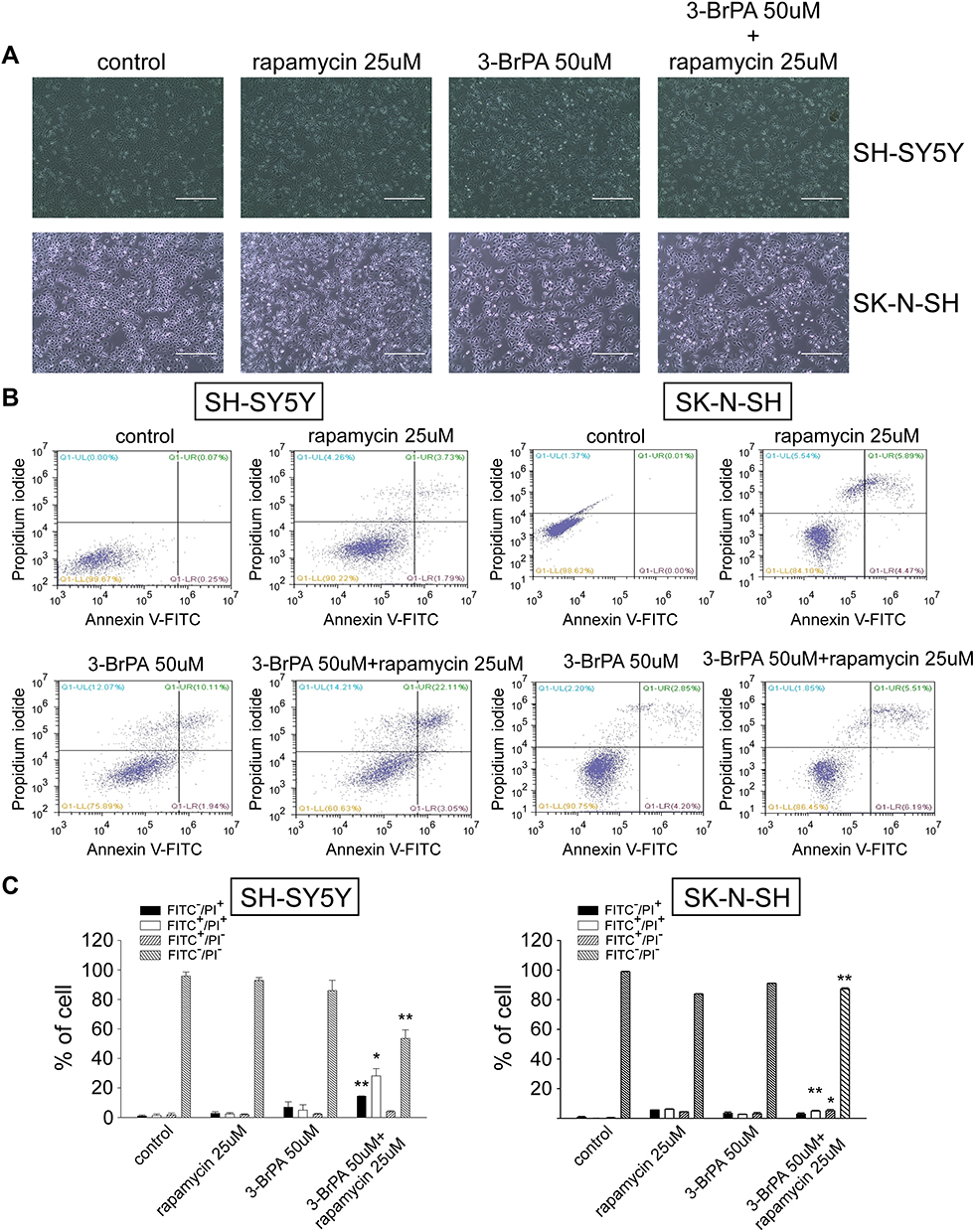 |
Figure 5 3-BrPA combined with rapamycin induced cell apoptosis in SH-SY5Y and SK-N-SH. (A) Morphological changes of NB cells were observed under a microscope. (B) The cell apoptosis ratio of SH-SY5Y and SK-N-SH treated with control, 25μM rapamycin, 50μM 3-BrPA, 50μM 3-BrPA + 25μM rapamycin was detected by flow cytometry analysis. (C) The bar chart shows cell apoptosis after drug treatment (control, 25μM rapamycin, 50μM 3-BrPA, 50μM 3-BrPA + 25μM rapamycin). *P<0.05; **P<0.01. |
Effect of 3-BrPA and Rapamycin Treatment on NB Cells Cycle
Cell proliferation analysis showed that 3-BrPA combined with rapamycin treatment remarkably inhibits the proliferation of NB cells. In order to evaluate whether the inhibitory effect on cell proliferation is related to cell cycle progression, we performed cell cycle analysis by FCM. As shown in Figure 6, FCM analysis showed that compared with the control group, 25μM rapamycin treatment resulted in an increase in the proportion of NB cells in the G0/G1 phase (P<0.05), and a decrease in the proportion of NB cells in the G2/M phase compared with the control group (P<0.05). Our results revealed that rapamycin can impede the cell cycle at the G0/G1 stage in NB cells. FCM analysis indicated that 50µM 3-BrPA treatment resulted in a reduce in the proportion of NB cells in the G0/G1 phase compared with the control group (P<0.05). Furthermore, the proportion of NB cells in the G2/M phase was extremely increased after 3-BrPA treatment compared with the control group (P<0.05). Our results revealed that 3-BrPA can impede the cell cycle at the G2/M stage in NB cells. In addition, 3-BrPA combined with rapamycin can induce significant cell apoptosis, especially in SH-SYSY (Figure 6A and B).
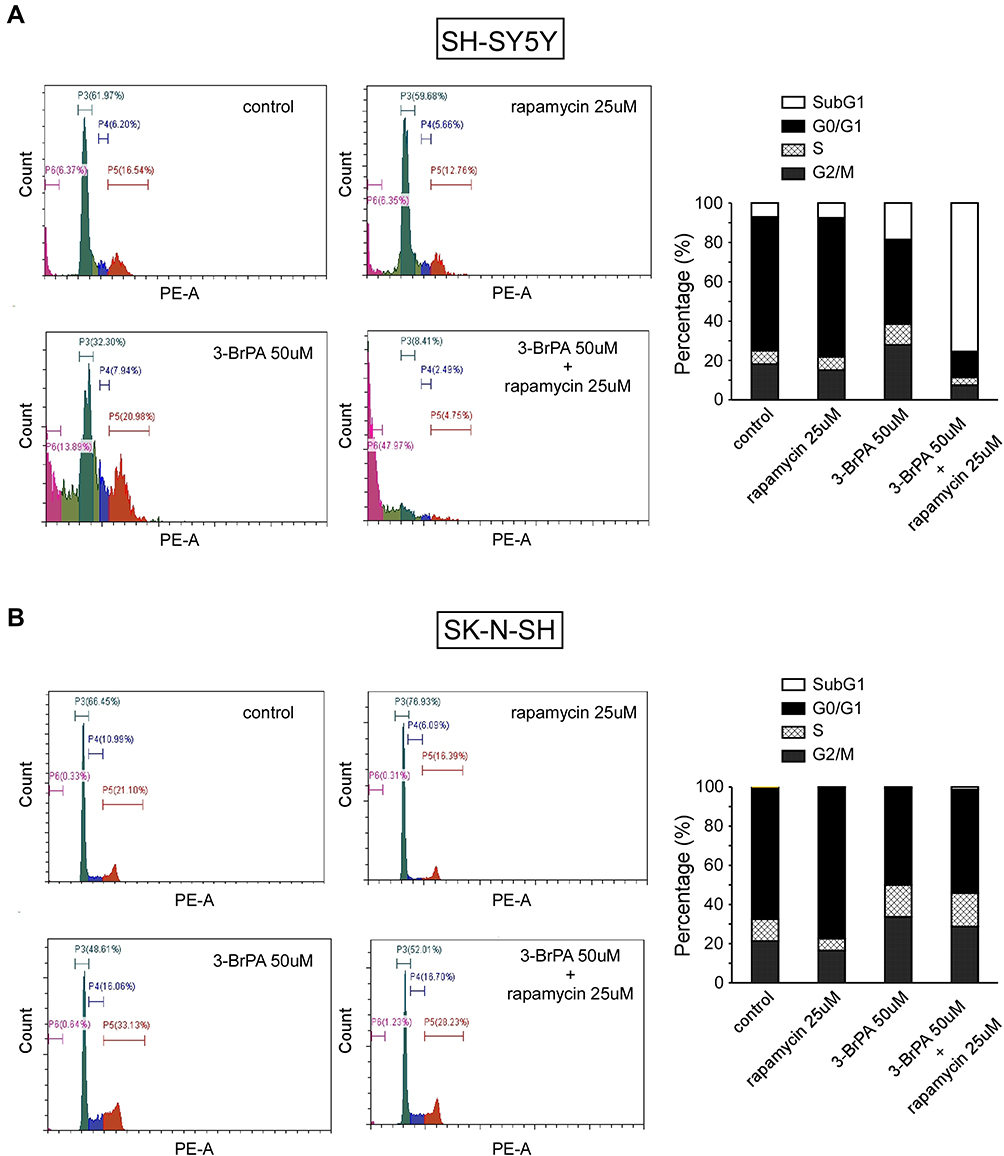 |
Figure 6 Cell cycle analysis of 3-BrPA and rapamycin in NB cells compared with the control cells analyzed by flow cytometry. (A) Cell cycle analysis in SH-SY5Y. (B) Cell cycle analysis in SK-N-SH. |
3-BrPA inhibits aerobic glycolysis, which can cause a serious metabolic dysfunction when combined with rapamycin. In order to comprehend how 3-BrPA and rapamycin execute MYC-overexpressing cells, we observed their effects on the cell metabolism of SH-SY5Y and SK-N-SH. Figure 7A briefly shows the process of glucose producing lactic acid and producing acetyl-CoA to participate in the tricarboxylic acid (TCA) cycle. Administration of 3-BrPA combined with rapamycin caused a substantial decrease in the cellular glucose uptake and lactate production related with an apparent decline in the intracellular adenosine triphosphate (ATP) production (Figure 7B–D).
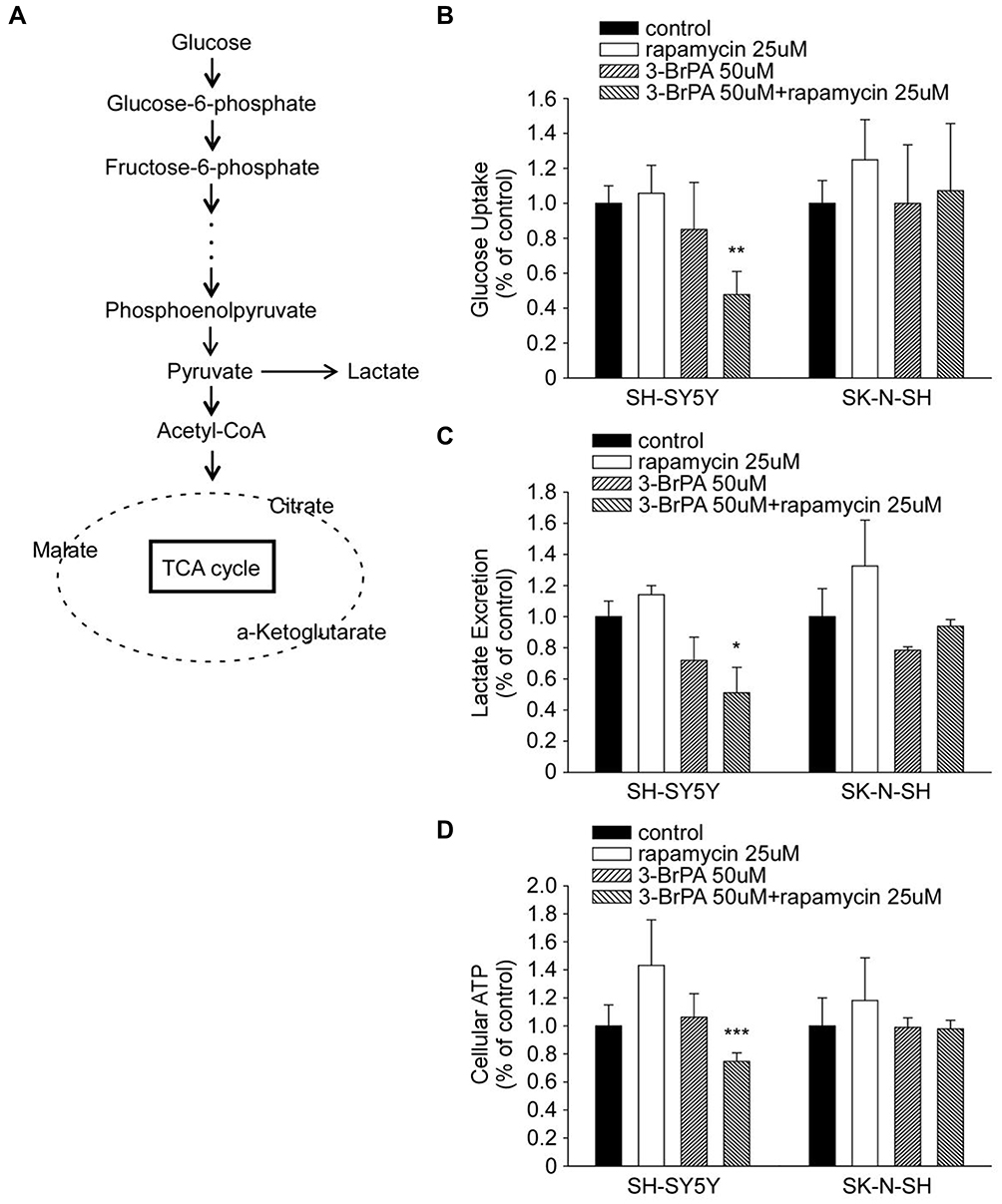 |
Figure 7 3-BrPA and rapamycin administration induced metabolic changes in NB cells. (A) Diagram depicting glucose metabolism via aerobic glycolysis and tricarboxylic acid (TCA) cycle. (B–D) Relative changes in the cellular glucose uptake, lactate production and intracellular ATP content in SH-SY5Y and SK-N-SH cells upon 3-BrPA, rapamycin or 3-BrPA + rapamycin treatment. Data shown are averages of triplicate. *P<0.05; **P<0.01; ***P<0.001. |
Discussion
Neuroblastoma is the most well-known and fatal solid tumor in children, the majority of the NB children have metastatic disease typically characterized by prompt overgrowth and diffuse infiltration. Although the use of enhanced diagnostic methods and assorted multimodal therapies has made progress to improve the cure rate of other pediatric tumors, the survival rate for NB patients is still daunting.24,25 Therefore, it remains essential for researchers to explore neoteric treatment strategies for NB children.
Metabolic reprogramming is one hallmark of cancer cells.26 Even in the exposure of oxygen, malignant cancer cells priorly rely on glycolysis, instead of oxidative phosphorylation, for adenosine triphosphate (ATP) production. The metabolic abnormity is generally referred to as the “Warburg effect” (aerobic glycolysis) and confers a proliferative advantage to malignant cancer cells.27,28
For a long time, selective targeting of tumor glucose metabolism has been considered a potential treatment method. In our previous study, we have revealed that 3-BrPA preferentially induced cell death in human malignant cancer cells strictly dependent on MYC overexpression.17 Overall, our results identified a novel mechanism whereby MYC sensitizes malignant cancer cells to metabolic inhibitors and confirm 3-BrPA as a potential MYC-selective cancer therapy.
Qianwen Zhang found that HK-II inhibitor 3-BrPA can induce autophagy by stimulating ROS formation in human breast cancer cells.29 Ganapathy-Kanniappan S found that 3-BrPA can induce endoplasmic reticulum stress, overcome autophagy and cause cell apoptosis in human HCC cell lines.30 In addition, we found that autophagy activity was different in neuroblastomas with different levels of MYCN amplification, and the glucose metabolism inhibitor 3-BrPA treated in neuroblastoma cells could induce autophagy activity.
Autophagy is a response of cells to metabolic stress and plays an important role in tumorigenesis,31 which is an ancient catabolic process used by cells to eliminate excess or dysfunctional organelles or large subcellular structures and thus performs an important housekeeping role for the cells. Autophagy can degrade and eliminate misfolded/aggregated proteins and damaged organelles through the lysosomal pathway, thereby maintaining the homeostasis of the cells.32,33 Autophagy is vital for the majority of normal tissues, malignant cancer cells appear to be especially dependent on autophagy for survival under ischemic stress or therapeutic stress, and in response to loss of matrix attachment. In cancers, autophagy can modulate tumor suppression as well as tumor promotion.34,35 Autophagy is up-regulated markedly in cancers as they progress to malignancy.
In the context of human cancer, as tumors develop into invasive and malignant tumors, autophagy increases significantly.36 Studies showed that increased LC3B staining was associated with increased metastasis and poor prognosis in breast cancer and melanoma.37–39
Rapamycin, which can be used as immunosuppressant, is a new type macrocyclic lactone, isolated from Streptomyces hygroscopicus.40 Preceding investigations showed that rapamycin down-regulated MYCN protein expression, which can prohibit neuroblastoma cells proliferation, and this process was intimately correlated with the PI3K/Akt/mTOR pathway.41–43 Rapamycin has been generally utilized as an anti-proliferative drug and immunosuppressant agent in the clinics. Recently, generous researches have focused on the functions of rapamycin in malignant tumors. Xiaokun Lin showed that rapamycin restrains the cell proliferation of neuroblastoma cells and induces autophagy.44 Our research primarily explored the influence of rapamycin in human neuroblastoma cell lines. The cell proliferation experiments such as CCK-8 assay showed that rapamycin hampered the proliferation of human neuroblastoma cells. We notarized that cell growth restriction is the foundation of rapamycin’s anti-tumor capacity, and also proved that rapamycin can play a synergistic character in tumor inhibitory. Cell cycle regulation dysregulation is another distinguishing feature of malignant tumor, causing cancer cells proliferate indefinitely. We detected the G0/G1 stage, S stage and G2/M stage in human neuroblastoma cells treated with 3-BrPA and rapamycin. Cell cycle experiments suggested that rapamycin prevented the cell cycle in the G0/G1 stage of SH-S5Y and SK-N-SH, thereby inhibiting DNA replication and cancer cell proliferation.
Our study also found that autophagy protein SQSTM1/p62 was down-regulated in MYCN-amplified neuroblastoma, suggesting that autophagy activity was high in high-risk neuroblastoma and might play a carcinogenic role. However, with the addition of the glucose metabolism inhibitor 3-BrPA, the autophagy pathway of neuroblastoma was damaged. The combination of rapamycin and 3-BrPA can exert a synergistic inhibitory effect on tumor, which not only affects the relevant genes in the autophagy pathway, but also affects the intracellular glucose metabolism level. This study provides a new therapeutic strategy for the combination of 3-BrPA and rapamycin in the treatment of high-risk neuroblastoma. Therefore, there might be a coupling relationship between metabolic reprogramming and autophagy ability. The notion that cancer cells reprogram their metabolism to meet the biosynthetic challenges of cell growth and cell proliferation may influence autophagy ability, which may provide opportunities for manipulating cell metabolism in the direction of cancer therapy.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (grant number: 81803553) and the National Natural Science Foundation Pre-research Program of China (SDFEYGJ1608).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. doi:10.1016/j.cell.2012.03.003
2. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. 2013;3(10):a014415. doi:10.1101/cshperspect.a014415
3. George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975–978. doi:10.1038/nature07397
4. Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362(23):2011–2202. doi:10.1056/NEJMra0804577
5. Brodeur GM, Seeger RC, Schwab M, et al. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224(4653):112–114. doi:10.1126/science.6719137
6. Cohn SL, Pearson AD, London WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009;27(2):289–297. doi:10.1200/JCO.2008.16.6785
7. Liu X, Mazanek P, Dam V, et al. Deregulated Wnt/β-catenin program in high-risk neuroblastomas without MYCN amplification. Oncogene. 2008;27(10):1478–1488. doi:10.1038/sj.onc.1210769
8. Mei H, Wang Y, Lin Z, Tong Q. The mTOR signaling pathway in pediatric neuroblastoma. Pediatr Hematol Oncol. 2013;30(7):605–615. doi:10.3109/08880018.2013.798058
9. Berthold F, Boos J, Burdach S, et al. Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemo-therapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. Lancet Oncol. 2005;6(9):649–658. doi:10.1016/S1470-2045(05)70291-6
10. Aminzadeh S, Vidali S, Sperl W, et al. Energy metabolism in neuroblastoma and Wilms tumor. Transl Pediatr. 2015;4(1):20–32.
11. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123(9):3685–3692. doi:10.1172/JCI69741
12. Chourasia AH, Boland ML, Macleod KF. Mitophagy and cancer. Cancer Metabol. 2015;3:1–11. doi:10.1186/s40170-015-0130-8
13. Dejure FR, Eilers M. MYC and tumor metabolism: chicken and egg. EMBO J. 2017;36(23):3409–3420. doi:10.15252/embj.201796438
14. Chandel N. Four key questions about metformin and cancer. BMC Biol. 2014;12:85.
15. Halestrap AP, Wilson MC. The monocarboxylate transporter family-role and regulation. IUBMB Life. 2012;64(2):109–119. doi:10.1002/iub.572
16. Birsoy K, Wang T, Possemato R, et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat Genet. 2013;45(1):104–108. doi:10.1038/ng.2471
17. Gan L, Xiu R, Ren P, et al. Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporter. Oncogene. 2016;35(23):3037–3048. doi:10.1038/onc.2015.360
18. Berardi DE, Campodónico PB, Díaz Bessone MI, et al. Autophagy: friend or foe in breast cancer development, progression, and treatment. Int J Breast Cancer. 2011;2011:595092. doi:10.4061/2011/595092
19. Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–730.
20. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi:10.1038/nature09782
21. Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016;14:15–23. doi:10.1016/j.ebiom.2016.10.034
22. Qian HR, Yang Y. Functional role of autophagy in gastric cancer. Oncotarget. 2016;7(14):17641–17651. doi:10.18632/oncotarget.7508
23. Islam MA, Sooro MA, Zhang P. Autophagic regulation of p62 is critical for cancer therapy. Int J Mol Sci. 2018;19(5):1405. doi:10.3390/ijms19051405
24. Brodeur GM, Iyer R, Croucher JL, et al. Therapeutic targets for neuroblastomas. Expert Opin Ther Targets. 2014;18(3):277–292. doi:10.1517/14728222.2014.867946
25. Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and therapy. Annu Rev Med. 2015;66:49–63. doi:10.1146/annurev-med-011514-023121
26. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674.
27. Tekade RK, Sun X. The Warburg effect and glucose-derived cancer theranostics. Drug Discov Today. 2017;22(11):1637–1653. doi:10.1016/j.drudis.2017.08.003
28. Chen Z, Zuo X, Zhang Y, et al. MiR-3662 suppresses hepatocellular carcinoma growth through inhibition of HIF-1α-mediated Warburg effect. Cell Death Dis. 2018;9(5):549. doi:10.1038/s41419-018-0616-8
29. Zhang Q, Zhang Y, Zhang P, et al. Hexokinase II inhibitor, 3-BrPA induced autophagy by stimulating ROS formation in human breast cancer cells. Genes Cancer. 2014;5(3–4):100–112. doi:10.18632/genesandcancer.9
30. Ganapathy-Kanniappan S, Geschwind JF, Kunjithapatham R, et al. 3-Bromopyruvate induces endoplasmic reticulum stress, overcomes autophagy and causes apoptosis in human HCC cell lines. Anticancer Res. 2010;30(3):923–935.
31. Zhi X, Zhong Q. Autophagy in cancer. F1000 Prime Rep. 2015;7:18. doi:10.12703/P7-18
32. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
33. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159(6):1263–1276. doi:10.1016/j.cell.2014.11.006
34. Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155(6):1216–1219. doi:10.1016/j.cell.2013.11.019
35. Gump JM, Staskiewicz L, Morgan MJ, et al. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol. 2014;16(1):47–54. doi:10.1038/ncb2886
36. Mowers EE, Sharifi MN, Macleod KF. Autophagy in cancer metastasis. Oncogene. 2017;36(12):1619–1630.
37. Lazova R, Klump V, Pawelek J. Autophagy in cutaneous malignant melanoma. J Cutan Pathol. 2010;37(2):256–268. doi:10.1111/j.1600-0560.2009.01359.x
38. Lazova R, Camp RL, Klump V, et al. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18(2):370–379. doi:10.1158/1078-0432.CCR-11-1282
39. Zhao H, Yang M, Zhao J, et al. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol. 2013;30(1):475. doi:10.1007/s12032-013-0475-1
40. Li KL, Wang YF, Qin JR, et al. Rapamycin enhances the anti-angiogenesis and anti-proliferation ability of YM155 in oral squamous cell carcinoma. Tumour Biol. 2017;39(6):1010428317706213.
41. Zhang L, Smith KM, Chong AL, et al. In vivo antitumor and antimetastatic activity of sunitinib in preclinical neuroblastoma mouse model. Neoplasia. 2009;11(5):426–435. doi:10.1593/neo.09166
42. Moreno-Smith M, Lakoma A, Chen Z, et al. p53 non-genotoxic activation and mTORC1 inhibition lead to effective combination for neuroblastoma therapy. Clin Cancer Res. 2017;23(21):6629–6639. doi:10.1158/1078-0432.CCR-17-0668
43. Johnsen JI, Segerström L, Orrego A, et al. Inhibitors of mammalian target of Rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene. 2008;27(20):2910–2922. doi:10.1038/sj.onc.1210938
44. Lin X, Han L, Weng J, et al. Rapamycin inhibits proliferation and induces autophagy in human neuroblastoma cells. Biosci Rep. 2018;38(6):BSR20181822. doi:10.1042/BSR20181822
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.