")
Back to Journals » Cancer Management and Research » Volume 11
Synergistic cytotoxicity of homoharringtonine and etoposide in acute myeloid leukemia cells involves disrupted antioxidant defense
Authors Zhang J, Geng H, Liu L, Zhang H
Received 14 September 2018
Accepted for publication 14 November 2018
Published 22 January 2019 Volume 2019:11 Pages 1023—1032
DOI https://doi.org/10.2147/CMAR.S187597
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Jingjing Zhang,1,* Huayun Geng,2,* Ling Liu,1 Hao Zhang1
1Department of Hematology, Affiliated Hospital of Jining Medical University, Jining 272029, Shandong Province, China; 2Department of Hematology, Dongchangfu People’s Hospital of Liaocheng, Liaocheng 252000, Shandong Province, China
*These authors contributed equally to this work
Background/Aims: Cytotoxicity induced by reactive oxygen species (ROS) is critical for the effectiveness of chemotherapeutic drugs used in the treatment of acute myeloid leukemia (AML). This study aimed to investigate whether ROS contributes to cytotoxicity in AML cells when treated with homoharringtonine (HHT) and etoposide (ETP) in combination.
Methods: AML cell lines THP1 and HL60 and primary AML cells from patients were treated with HHT and ETP alone or in combination, and cell viability was determined by trypan blue exclusion test, and apoptosis was analyzed by annexin-V/propidium iodide double staining as well as Western blot for measuring expression of cleaved caspase-9 and cleaved caspase-3. Intracellular ROS level was detected by DCFH-DA fluorescence assay, and N-Acetyl-L-cysteine (NAC) was used to scavenge intracellular ROS. Retroviral infection was applied to mediate stable overexpression in AML cells.
Results: We show that HHT and ETP exhibit synergistic cytotoxicity in AML cell lines and primary AML cells in vitro, and meanwhile, HHT causes elevated ROS generation in ETP-treated AML cells. We next reveal that the elevated ROS is a critical factor for the synergistic cytotoxicity, since ROS scavenge by NAC remarkably diminishes this effect. Mechanistically, we demonstrate that HHT causes elevated ROS generation by disabling thioredoxin-mediated antioxidant defense. Finally, similar to HHT treatment, depletion of thioredoxin sensitizes AML to ETP treatment.
Conclusion: These results provide the foundation for augmenting the efficacy of ETP in treating AML with HHT, and also highlight the importance of targeting ROS in improving treatment outcome in AML.
Keywords: homoharringtonine, etoposide, synergistic cytotoxicity, acute myeloid leukemia, antioxidant defense, reactive oxygen species, thioredoxin
Introduction
Acute myeloid leukemia (AML) is a heterogeneous group of hematopoietic malignancies originating from transformed hematopoietic stem cells and myeloid progenitor cells. The standard induction chemotherapy for AML commonly consists of a deoxycytidine analog (Ara-C), an anthracyclin and etoposide (ETP).1,2 Although most patients achieve complete remission after initial treatment, due to frequent relapse and toxicity of futile chemotherapy, the long-term overall remission rate is only 30%–40%.3 The main cause of relapse is the persistence of leukemic stem cells that are refractory to cell death induced by chemotherapeutic drugs.4 In regard to the mechanism of action of chemotherapeutic drugs, including ETP, one of the first identified topoisomerase II inhibitors,5 in addition to inducing the inhibition of DNA synthesis and DNA double-strand breaks, the elevated generation of reactive oxygen species (ROS) has also been known as a critical mediator to induce cell death of AML.6–9 Therefore, exploiting ROS-induced cytotoxicity or abrogating the antioxidant capacity of resistant AML cells holds the promise to enhance the therapeutic benefits of induction chemotherapy.10
Homoharringtonine (HHT), a plant alkaloid derived from cephalotaxus fortune, has been widely used for the treatment of myelogenous leukemia for the past 30 years in China.11 In 2012, omacetaxine mepesuccinate, a highly purified HHT compound, was approved by the Food and Drug Administration for treating chronic myeloid leukemia (CML) after the treatment failure of tyrosine kinase inhibitors.12 Additionally, HHT also exhibits considerable therapeutic efficacy and serves as an adjunct for treating AML.2,13–15 Although previous studies have associated the inhibition of protein synthesis and cell apoptosis with the action of HHT, the anti-leukemic mechanisms of HHT are still largely unknown.16,17 Recently, one clinic study has shown that the induction chemotherapy of HHT in combination with cytarabine and ETP is effective when treating AML.18 Moreover, ETP-resistant human cancer cells exhibit sensitivity to HHT.19 These observations imply that these cancer cells have no obvious cross-resistance to HHT and ETP. In the current study, we report the ROS-dependent synergistic cytotoxicity of HHT and ETP in AML cells, in which the disrupted thioredoxin-mediated antioxidant defense plays an important role.
Materials and methods
Antibodies and reagents
The antibodies and reagents used in this study were purchased from the following sources: cleaved caspase-9 (ab25758; abcam, Cambridge, MA, USA), cleaved caspase-3 (C8487; Sigma-Aldrich Co., St Louis, MO, USA), PARP (9542; Cell Signaling, Danvers, MA, USA), thioredoxin (abcam, ab26320), GAPDH (10494–1-AP; Proteintech Group, Inc., Rosemont, IL, USA), goat anti-rabbit IgG-horseradish peroxidase (HRP) (ab6721; abcam), goat anti-mouse IgG-HRP (abcam, ab6789), HHT (H0635; Sigma-Aldrich Co.), ETP (E1383; Sigma-Aldrich Co.), 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Sigma-Aldrich Co., D6883), N-acetyl-l-cysteine (NAC) (Sigma-Aldrich Co.; A7250).
AML cell lines and clinical primary AML cells
Human AML cell lines HL60 and THP1 were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA) and maintained in RPMI-1640 medium supplemented with 10% FBS at 37°C in a humidified incubator containing 5% CO2. For obtaining clinical primary AML cells, three patients diagnosed with de novo AMLs were recruited into the present study. The peripheral blood mononuclear cells were purified from peripheral blood by Ficoll-Hypaque (Sigma-Aldrich Co.) density gradient centrifugation, and bone marrow aspirates containing leukemic blast cells from these patients were obtained as previously described.4 This study was conducted in accordance with the Declaration of Helsinki. Signed informed consent was obtained from each patient. The study design was approved by the Ethics Committee of Affiliated Hospital of Jining Medical University.
Cell treatment and cell viability determination
HL60, THP1, and primary AML cells were treated with HHT (10 or 20 nM) or ETP (1 or 2 µM) alone or with graded concentrations of drugs at fixed ratios (HHT/ETP, 10 nM/1 µM or 20 nM/2 µM) in the presence or absence of NAC for 48 hours. These two concentrations of 10/1 and 20/2 (nM/µM, HHT/ETP) used in this study were chosen according to our pilot study, in which they showed considerable cytotoxicity among these AML cells. Control group was cultured in medium containing the same volume of vehicle. Each treatment was set with six replicates per assay, and each experiment was performed in three repeats. After treatment, the cell viability was assessed by trypan blue exclusion according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA, 15250061). The trypan blue positive cells were considered non-viable. The cell viability was calculated as the number of viable cells divided by the total number of cells within the grids on the hemacytometer. The formula used is as follows: % viable cells = (1.00 – [Number of blue cells ÷ Number of total cells]) ×100. The results were represented as relative to control group (%).
Apoptosis analysis
The apoptosis of HL60, THP1, and primary AML cells was determined with the Annexin V-FITC Apoptosis Detection Kit (APOAF; Sigma-Aldrich Co.) following the manufacturer’s instructions. Briefly, cells were collected and resuspended in binding buffer after washing with PBS and 0.5 µL fluorochrome-conjugated Annexin V were added into the100 µL cell suspension to incubate for 20 minutes at room temperature in the dark, and then 4 µL propidium iodide were added into the cell suspension prior to flow cytometry analysis. The percentage of apoptotic cells was determined by flow cytometry analysis (FACSCalibur Flow Cytometer; BD Biosciences, San Jose, CA, USA). Data were analyzed using the Flowjo software (Treestar Inc., Ashland, OR, USA).
Western blot analysis
Cells were washed with cold PBS for three times and harvested in tubes. For protein extraction, cells were lysed using RIPA Lysis Buffer System (sc-24948; Santa Cruz Biotechnology Inc., Dallas, TX, USA) supplemented with 1% (v/v) protease inhibitor Cocktail (Promega Corporation, Fitchburg, WI, USA) on ice for 20 minutes. The tubes were flicked every 5 minutes. The whole cell lysates were then centrifuged at 12,000×g for 10 minutes at 4°C. The supernatants containing protein samples were collected and quantified using BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Protein samples were then denatured in 5× loading buffer at 100°C for 5 minutes. Western blot was performed as described previously.20 In brief, equal amount of total proteins were loaded and resolved by SDS-PAGE. After electrophoresis, proteins were transferred onto polyvinylidene fluoride (PVDF) membrane with 0.2 µm pore size (EMD Millipore, Billerica, MA, USA). PVDF membranes blotted with proteins were washed with TBS supplemented with 0.1% Tween (TBST) and blocked with 5% skim milk (BD) in TBST solution for 1 hour at room temperature. Following block, PVDF membranes were incubated overnight with primary antibodies diluted in blocking buffer at 4°C. PVDF membranes were washed with TBST for three times and incubated with secondary antibodies conjugated with HRP for 1 hour at room temperature. After washing, PVDF membranes were incubated with enhanced chemiluminescence Plus Western Blotting Substrate (32132; Thermo Fisher Scientific) to detect protein bands with the platform of ImageQuant-LAS-4000 instrument (GE Healthcare, Chicago, IL, USA). The intensity of protein bands was analyzed by ImageJ software.
Intracellular ROS measurement
After treatment, cells were washed with PBS for three times and harvested in tubes. For measuring ROS level, cells were treated with 10 µM H2DCFH-DA for 30 minutes in the dark, followed by two times washing with PBS. The reduced H2DCF-DA is oxidized and converted into fluorescent DCF-DA by intracellular ROS. The fluorescent signals were quantified by flow cytometry analysis (FACSCalibur Flow Cytometer; BD Biosciences). Roughly 10,000 cells were analyzed per sample. Data were analyzed using the Flowjo software (Treestar).
Retroviral infections
Retroviral constructs expressing human thioredoxin were constructed by inserting human thioredoxin cDNA into the pMIG retroviral vector. The pMIG retroviral vectors encoding shRNA against thioredoxin were also constructed. Viruses were generated in HEK293T cells by transfecting with viral constructs together with gag-pol and env (VSVG) package vectors using Lipofectamine 2000 (Invitrogen) according to manufacturers’ instructions. Meanwhile, viruses containing pMIG retroviral empty vectors were generated. Viral supernatants were harvested 48 hours after transfection and filtered through 0.45 mM strainer. HL60 cell line was infected with retroviral solution for 24 hours. After cell expansion, the stably pMIG-infected cells were isolated using flow cytometry analysis according to green fluorescent protein expression.
Statistical analysis
All data are expressed as means ± SD. The statistical analyses were performed by unpaired Student’s t-test with Prism 6 software. Differences are considered significant when P-values are <0.05.
Results
HHT and ETP exhibit synergistic cytotoxicity in AML cells
HHT and ETP are cytotoxic reagents for AML cells.21,22 To test whether HHT and ETP have synergistic cytotoxicity in AML cells, the chemosensitive AML model cell lines (THP1 and HL60) were treated with HHT and ETP alone or in combination (10/1 and 20/2, nM/µM) for 48 hours. As expected, the trypan blue exclusion assay showed a dose-dependent decease of cell viability by treatment of HHT and ETP alone (Figure 1A). More importantly, the combination treatment exhibited a stark synergistic effect on reducing the cell viability of both THP1 and HL60 cells (Figure 1A), suggesting synergistic cytotoxicity in AML cell lines. Analysis of apoptosis using annexin-V/propidium iodide double staining found that the combination treatment of HHT and ETP enhanced their apoptosis-inducing effect on HL60 cells (Figure 1B). Moreover, their synergistic effect on apoptosis was further confirmed by the increased expressions of cleaved caspase-9 and cleaved caspase-3, as shown by Western blotting analysis (Figure 1C). Thus, these data demonstrate that HHT and ETP display synergistic cytotoxicity in AML cell lines.
 | Figure 1 Synergistic cytotoxicity of HHT and ETP in AML cells. Notes: (A) THP1 and HL60 cells were treated with vehicle control (Ctrl), HHT, ETP, or in combination as indicated for 48 hours. Cell viability was determined via trypan blue exclusion assay. Results relative to Ctrl are shown (%). (B, C) HL60 cells were treated as in (A). (B) The percentage of apoptotic cells was determined by flow cytometry analysis using annexin-V/propidium iodide double staining. The statistical analysis is shown. (C) The protein expressions of cleaved caspase-9 and cleaved caspase-3 were measured by Western blot analysis. PARP was used as a loading control. (D) Primary AML cells from three patients (AML-1, AML-2, and AML-3) were treated as indicated for 48 hours. Cell viability was determined via trypan blue exclusion assay. Results relative to Ctrl are shown (%). (E, F) Primary AML cells (AML-2) were treated as in (D). (E) The percentage of apoptotic cells was determined by flow cytometry analysis using Annexin-V/propidium iodide double staining. The statistical analysis is shown. (F) The protein expressions of cleaved caspase-9 and cleaved caspase-3 were measured by Western blot analysis. PARP was used as a loading control. Data were obtained from at least three independent experiments and analyzed by Student’s t-test. Data are expressed as mean ± SD. **P<0.01. Abbreviations: AML, acute myeloid leukemia; ETP, etoposide; HHT, homoharringtonine. |
To broaden this finding, we utilized primary AML cells from newly diagnosed patients with AML (denoted as AML-1, AML-2, and AML-3). Similar to AML cell lines, HHT and ETP manifested synergistic cytotoxicity in these primary AML cells, as shown by enhanced decrease of cell viability (Figure 1D). Since AML-2 displayed the optimal synergistic cytotoxicity effect among three clinical samples, we therefore focused on utilizing it for further analyses. The synergistic cytotoxicity of HHT and ETP in AML-2 cells was further validated by the enhanced apoptosis (Figure 1E, F). Thus, these results from in vitro cell lines and clinical samples illustrate that HHT and ETP exhibit synergistic cytotoxicity in AML cells.
HHT elevates ROS level in ETP-treated AML cells
The generation of ROS is a key element for ETP cytotoxicity, and studies have shown that the oncogenic Akt,23 PKCδ,24 and curcumin25 enhance ETP cytotoxicity through ROS-mediated damage. Intriguingly, we found that the combination treatment of HHT and ETP synergistically elevated ROS generation in HL60 (Figure 2A) and THP1 (Figure 2B) cell lines, as well as in primary AML cells, as measured by a fluorescence assay (DCFH-DA; Figure 2C). Consistent with a previous report that leukemic cells respond to HHT-specific cellular stress via an ROS-independent apoptotic pathway,26 we noticed that HHT treatment alone had no obvious effect on ROS level in these AML cells, and different from HHT treatment, ETP alone caused an elevated ROS generation (Figure 2A–C). Together, these observations suggest that the induced elevation of ROS level in AML cells treated with the combination of HHT and ETP is due to the promotive role of HHT in amplifying the ROS-inducing effect of ETP.
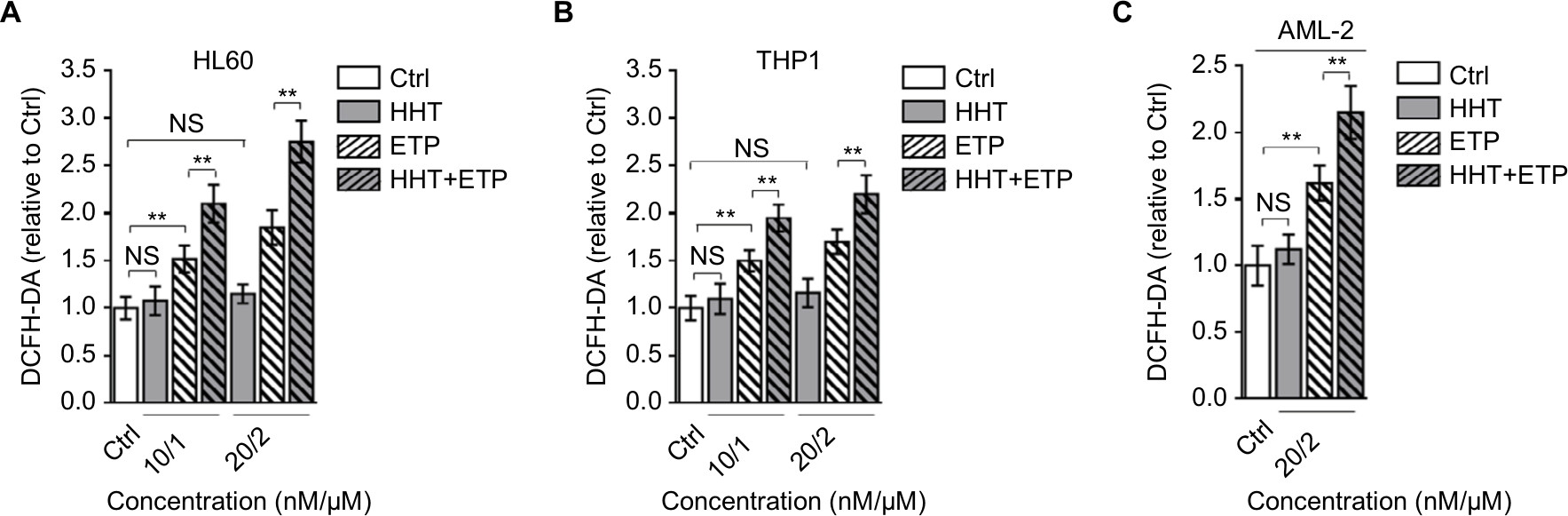 | Figure 2 HHT causes elevated ROS generation in AML cells treated with ETP. Notes: HL60 cells (A) and THP1 cells (B) were treated with vehicle control (Ctrl), HHT, ETP, or in combination as indicated for 48 hours. The intracellular ROS level was detected by flow cytometry analysis using DCFH-DA. Results relative to Ctrl are shown. (C) The primary AML cells (AML-2) were treated as indicated for 48 hours. The intracellular ROS level was detected by flow cytometry analysis using DCFH-DA. Results relative to Ctrl are shown. Data were obtained from at least three independent experiments and analyzed by Student’s t-test. Data are expressed as mean ± SD. **P<0.01. Abbreviations: AML, acute myeloid leukemia; DCFH-DA, dichlorofluorescein diacetate; ETP, etoposide; HHT, homoharringtonine; NS, not significant; ROS, reactive oxygen species. |
Elevated ROS generation is critical for synergistic cytotoxicity of HHT and ETP in AML cells
The excessive generation of ROS results in oxidative stress and induces cell death via caspase activation.27 To examine whether the elevated ROS generation contributes to the synergistic cytotoxicity of HHT and ETP in AML cells, we employed NAC, an effective scavenger of intracellular ROS,28 to treat AML cells. The elimination of ROS by NAC was highly effective in AML cells treated with ETP or in combination with HHT (Figure 3A). Along with the eliminated ROS generation, the enhanced decrease of cell viability by the combination treatment of HHT and ETP was substantially recovered (Figure 3B). Of note, ROS elimination did not completely erase the synergistic cytotoxicity, suggesting that other mechanisms may exist. In any case, these data indicate that the promoted elevation of ROS generation by HHT is a critical contributor to their synergistic cytotoxicity. In agreement with the result of cell viability, NAC addition largely reversed the increased expressions of cleaved caspase-9 and cleaved caspase-3 in HL60 cells treated with the combination of ETP and HHT (Figure 3C). Except for HL60 cells, similar results were obtained in primary AML cells when ROS was eliminated by NAC treatment (Figure 3D–F). Collectively, these findings show that HHT-elevated ROS generation plays a critical role in the synergistic cytotoxicity of HHT and ETP in AML cells.
 | Figure 3 ROS scavenge diminishes HHT-augmented ETP cytotoxicity. Notes: (A–C) HL60 cells were treated with vehicle control (Ctrl), HHT, ETP, or in combination in the presence or absence of NAC as indicated for 48 hours. (A) The intracellular ROS level was detected by flow cytometry analysis using DCFH-DA. Results relative to Ctrl are shown. (B) Cell viability was determined via trypan blue exclusion assay. Results relative to Ctrl are shown (%). (C) The protein expressions of cleaved caspase-9 and cleaved caspase-3 were measured by Western blot analysis. PARP was used as a loading control. (D–F) Primary AML cells (AML-2) were treated as in (A). The intracellular ROS level (D), cell viability (E), and protein expressions of cleaved caspase-9 and cleaved caspase-3 (F) were determined as in (A–C). Data were obtained from at least three independent experiments and analyzed by Student’s t-test. Data are expressed as mean ± SD. **P<0.01. Abbreviations: AML, acute myeloid leukemia; DCFH-DA, dichlorofluorescein diacetate; ETP, etoposide; HHT, homoharringtonine; NAC, N-acetyl-l-cysteine; NS, not significant; ROS, reactive oxygen species. |
HHT causes elevated ROS generation by reducing thioredoxin
The oxidative stress derives from an imbalance between ROS generation and antioxidant defenses. There are plenty of antioxidant molecules acting to detoxify excessive ROS, including thioredoxins, glutaredoxins, peroxiredoxins, and others.29 HHT has been shown to inhibit protein synthesis by preventing the binding of aminoacyl-tRNAs to peptidyl-transferase A-site cleft in the ribosome.30 Besides, one proteomic study has reported that HHT treatment reduces thioredoxin level in K562 cells.31 To gain insight into the mechanism by which HHT promotes ETP-induced ROS generation in AML cells, we investigated the possible role of thioredoxin involved in this scenario. We found that ETP treatment alone induced thioredoxin expression in AML cell lines and primary AML cells; however, this was inhibited when combinatorially treated with HHT (Figure 4A), implying that HHT may inhibit thioredoxin-mediated antioxidant defense against ETP-induced ROS generation. To test this possibility, we overexpressed thioredoxin in HL60 cells to overcome the decreasing effect of HHT on its expression (Figure 4B). Astonishingly, when thioredoxin was restored in HHT-treated HL60 cells, the elevated ROS generation was totally recovered to the basal level (Figure 4C), mimicking the treatment of NAC (Figure 3A). This suggests that the reduced level of thioredoxin by HHT accounts for the promoted generation of ROS in ETP-treated AML cells. Functionally, thioredoxin restoration in HHT-treated HL60 cells remarkably recovered the decreased cell viability by combinatory treatment of HHT and ETP (Figure 4D), therefore establishing a causal link between suppressed thioredoxin-mediated antioxidant defense and the synergistic cytotoxicity of HHT and ETP in AML cells.
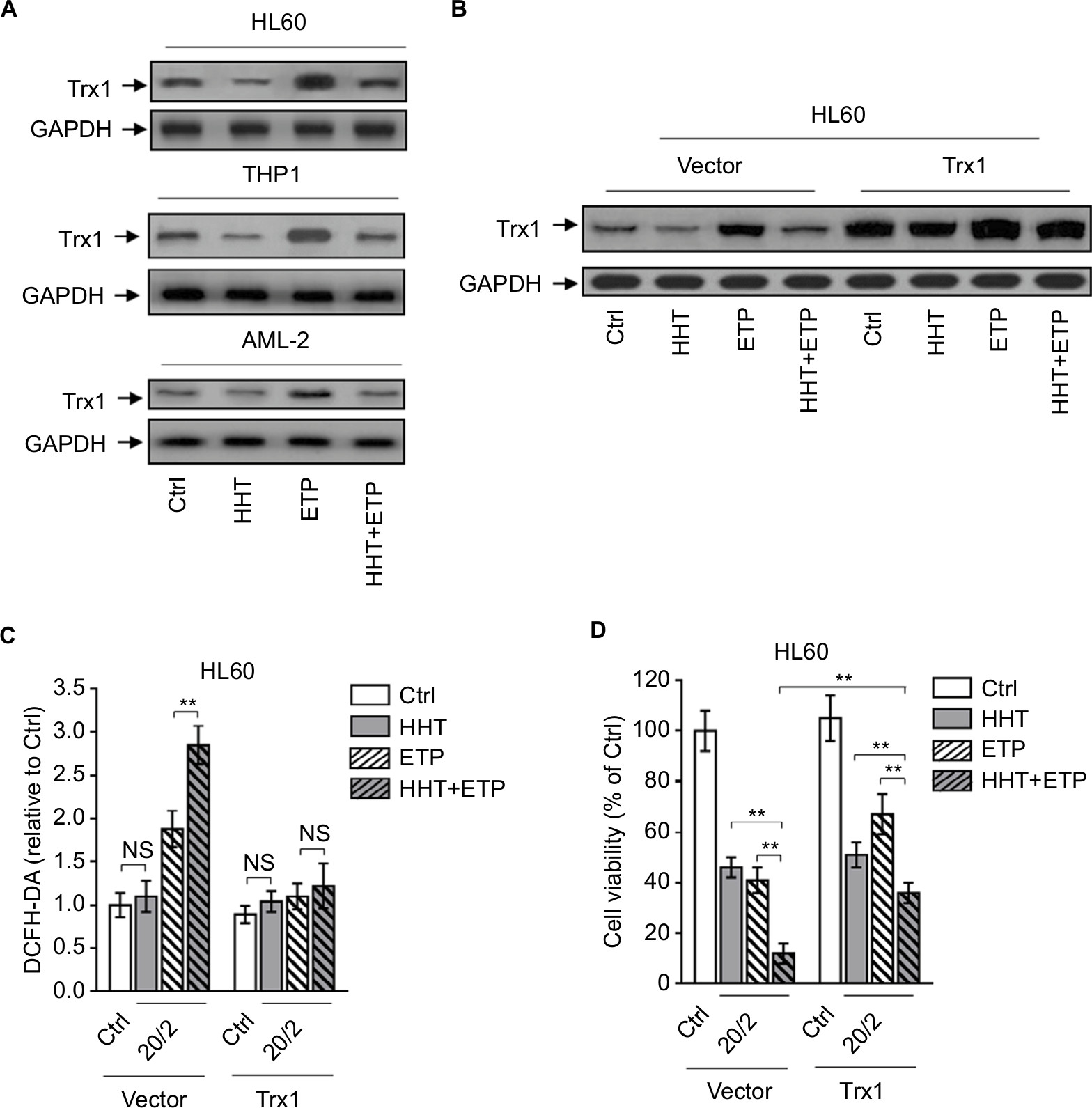 | Figure 4 HHT causes elevated ROS generation by disabling thioredoxin-mediated antioxidant defense. Notes: (A) HL60, THP1, and primary AML cells (AML-2) were treated with vehicle control (Ctrl), HHT, ETP, or in combination as indicated for 48 hours. The protein expression of Trx1 was determined by Western blot. GAPDH was used as a loading control. The representative images (left) and the quantification of band intensity (right) are shown. (B–D) HL60 cells stably overexpressing vector or Trx1 were treated as in (A). (B) The protein expression of Trx1 was determined by Western blot. GAPDH was used as a loading control. The representative images (upper) and the quantification of band intensity (lower) are shown. (C) The intracellular ROS level was detected by flow cytometry analysis using DCFH-DA. Results relative to Ctrl are shown. (D) Cell viability was determined via trypan blue exclusion assay. Results relative to Ctrl are shown (%). Data were obtained from at least three independent experiments and analyzed by Student’s t-test. Data are expressed as mean ± SD. **P<0.01. Abbreviations: AML, acute myeloid leukemia; DCFH-DA, dichlorofluorescein diacetate; ETP, etoposide; HHT, homoharringtonine; NS, not significant; ROS, reactive oxygen species. |
Disabling thioredoxin-mediated antioxidant defense sensitizes AML to ETP
To further reinforce the importance of thioredoxin-mediated antioxidant defense in protecting against oxidative insults caused by ETP-induced ROS generation, we depleted thioredoxin in HL60 cells treated with or without ETP (Figure 5A). Phenocopying HHT treatment, thioredoxin depletion increased the ETP-induced ROS generation (Figure 5B) and resulted in enhanced decrease of cell viability (Figure 5C). Taken together, these lines of evidence indicate that the thioredoxin-mediated antioxidant defense is important for detoxifying ETP-induced ROS generation, and that HHT reduces thioredoxin, thereby disrupting this antioxidant defense and augmenting the oxidative insults, which could at least partly explain the synergistic cytotoxicity of HHT and ETP in AML cells (Figure 5D).
 | Figure 5 Depletion of thioredoxin sensitizes AML to ETP treatment. Notes: (A–C) HL60 cells transfected with siCtrl or siTrx1 were treated with vehicle control (Ctrl) or 2 µM ETP for 48 hours. (A) The protein expression of Trx1 was determined by Western blot. GAPDH was used as a loading control. The representative images (left) and the quantification of band intensity (right) are shown. (B) The intracellular ROS level was detected by flow cytometry analysis using DCFH-DA. Results relative to Ctrl are shown. (C) Cell viability was determined via trypan blue exclusion assay. Results relative to Ctrl are shown (%). Data were obtained from at least three independent experiments and analyzed by Student’s t-test. Data are presented as mean ± SD. **P<0.01. (D) Brief schematic graph of a model for the role of HHT in augmenting ETP cytotoxicity in AML cells through targeting thioredoxin-mediated ROS elimination. Abbreviations: AML, acute myeloid leukemia; DCFH-DA, dichlorofluorescein diacetate; ETP, etoposide; HHT, homoharringtonine; NS, not significant; ROS, reactive oxygen species. |
Discussion
To date, for the front-line treatment of AML, the orthodox chemotherapeutic drugs remain the standard therapy, including the options of Ara-C, anthracyclin, and ETP. However, the treatment efficacy of chemotherapeutic drugs still requires improvement, since the frequently emerging drug resistance induces disease relapses that eventually result in a relatively poor long-term overall remission rate.32 Recent studies have suggested that the increased generation of ROS and an altered redox status observed in cancer cells can be exploited for therapeutic benefits.33 High dosage of chemotherapy used in AML treatment is often accompanied by ROS-induced cytotoxicity, whereas, the upregulation of antioxidant capacity in adaptation to oxidative stress can confer drug resistance. Therefore, abrogation of this drug-resistant mechanism could have significant therapeutic implications.34 In the present study, we show that HHT, a plant alkaloid with antileukemic activity, has synergistic cytotoxicity with ETP in AML cell lines and primary AML cells. Further mechanistic evidence demonstrates that HHT causes reduction of thioredoxin, an important element in eliminating intracellular excessive ROS, thus disrupting this antioxidant defense, which leads to promoted ROS generation and exaggerated ETP-induced oxidative insults. By using ROS scavenger NAC, we prove that this mechanism contributes to their synergistic cytotoxicity in vitro. The finding that thioredoxin knockdown enhances the sensitivity of AML to ETP further validates the important role of thioredoxin-mediated antioxidant defense in detoxifying the oxidative insults induced by ETP treatment. Thus, our study connects the synergistic cytotoxicity of HHT and ETP in AML cells to HHT-disrupted antioxidant capacity in adaptation to oxidative stress aroused by ETP, and also provides insights into a novel mechanism that underlies the antileukemic activity of HHT.
HHT is known to function as a translation inhibitor by preventing substrates from binding to the receptor site on ribosome subunit, thereby blocking chain elongation and inhibiting protein synthesis.35,36 The function of HHT in inhibiting protein synthesis is important for its apoptosis-inducing effect on AML cells, and HHT is even a more potent inhibitor of protein synthesis than cyclohexane.37 In this study, we found that in AML cell lines, HL60 and THP1, and primary AML cells, HHT treatment decreased thioredoxin expression. Coincidently, it has been reported in a comparative proteomic study that in K562 CML cells, thioredoxin is also decreased by HHT treatment.31 Although direct evidence is unavailable, we speculate that the reduction of thioredoxin upon HHT treatment is at least partially caused by inhibition of thioredoxin synthesis. Solid evidence such as from [3H]Leucine incorporation assay is preferably needed to demonstrate this issue. In addition to this mechanism that could explain the reduction of thioredoxin, the regulation by DJ-1 may also be involved in this process. DJ-1 is an antioxidant protein that induces thioredoxin expression via Nrf2-mediated transcriptional induction, and when DJ-1 is lost, the expression of thioredoxin is accordingly downregulated.38,39 DJ-1 has also been reported to be decreased by HHT treatment.31 Therefore, the reduction of thioredoxin in response to HHT treatment may be due to inhibited protein synthesis and deficiency of positive regulation by DJ-1.
The elevated ROS generation by HHT in ETP-treated AML cells could be attributed to the decreased expression of thioredoxin, since the restoration of thioredoxin by enforced overexpression can completely eliminate the excessive ROS, even though DJ-1 is also decreased. The irrelevance of DJ-1 observed in this condition may be associated with the mechanism that the antioxidant function exerted by DJ-1 is dependent on increasing Trx1 expression.38 Given the important role of Trx1 in eliminating the excessive ROS induced by HHT, it could be deduced that DJ-1/Nrf2 may also be associated with this process. It is worth noting that HHT treatment alone could not cause ROS elevation in AML cells. This suggests that oxidative insults are not responsible for its antileukemic activity when HHT is administrated alone. However, this seems paradoxical, because the basal level of thioredoxin is decreased by HHT (Figure 4A). We guess that when AML cells are not subjected to oxidative stress environment, the function of thioredoxin may be in surplus, and the ROS level can achieve homeostasis status by the compensation from other antioxidant machineries, such as glutathione- and Nrf2-mediated responses.40 Whereas, when AML cells are exposed to ROS-inducing stimuli, like ETP, the oxidative stress would overwhelm the antioxidant ability of cells when thioredoxin is insufficient. This is much possible, since we observed that HHT only elevated ROS generation in ETP-treated AML cells. Moreover, the possibility that other antioxidant molecules, such as glutaredoxins and peroxiredoxins, may be involved in the synergistic effect of HHT and ETP in leukemia cannot be ruled out either, elucidating this issue could help us to fully understand the mechanism by which the antioxidant defense affects the antileukemic activity of HHT and ETP.
The synergistic cytotoxicity of HHT and ETP in AML cells is evidenced by enhanced decrease of cell viability and apoptosis. The synergistic effect is diminished when ROS is scavenged by NAC, proving that the elevated ROS is an important contributor that accounts for their synergistic cytotoxicity. On the other hand, the partial rescue effect of NAC also suggests that other mechanisms may exist. Fully addressing this question may help us to advance our understanding of the regulation of synergistic cytotoxicity of HHT and ETP in AML cells. It is well known that the excessive ROS causes damage to cellular components, such as proteins, nucleic acids, lipids, membranes, and organelles, which leads to the activation of main pathways of apoptotic cell death mediated by mitochondria, death receptors, and the endoplasmic reticulum.41 As shown by our evidence, the cleaved caspase-9 and cleaved caspase-3 are increased, indicating the activation of apoptotic protease cascade. However, how exactly the HHT-elevated ROS in ETP-treated AML cells causes apoptosis and whether other forms of cell death are simultaneously induced need further investigations. On the other hand, one caveat is that the examined sample size of primary and cell lines of AML is limited, and more evidence from studies with larger sample size is warranted to consolidate our findings in the future.
At last, the enhanced cytotoxicity of ETP in AML cells by the disruption of thioredoxin-mediated antioxidant defense through either HHT treatment or direct depletion of thioredoxin may highlight the importance of targeting this protective mechanism in AML cells in optimizing the therapeutic efficacy or overcoming the drug resistance to ETP. Additionally, our study provides a foundation of using HHT as an adjuvant to ETP for treating AML or even ETP-resistant AML.
Acknowledgment
This study was supported by Natural Science Foundation of Shandong Province, China (No. ZR2017LH029), PhD Research Foundation of Affiliated Hospital of Jining Medical University, China (No. 2016-BS-001), Medicine and Health Science Technology Development Program of Shandong Province, China (No. 2017WS514), Scientific Research Supported Foundation for the Young Teacher of Jining Medical University, China (No. JY2017FS008), and Project of Scientific Research Program of Affiliated Hospital of Jining Medical University (No. MP-2016-007).
Disclosure
The authors report no conflicts of interest in this work.
References
Pastore D, Specchia G, Carluccio P, et al. FLAG-IDA in the treatment of refractory/relapsed acute myeloid leukemia: single-center experience. Ann Hematol. 2003;82(4):231–235. | ||
Lam SS, Ho ES, He BL, et al. Homoharringtonine (omacetaxine mepesuccinate) as an adjunct for FLT3-ITD acute myeloid leukemia. Sci Transl Med. 2016;8(359):359ra129. | ||
Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29(5):487–494. | ||
Vergez F, Green AS, Tamburini J, et al. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica. 2011;96(12):1792–1798. | ||
Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34(10):1514–1521. | ||
Matés JM, Segura JA, Alonso FJ, Márquez J. Oxidative stress in apoptosis and cancer: an update. Arch Toxicol. 2012;86(11):1649–1665. | ||
Zhang J, Lei W, Chen X, Wang S, Qian W. Oxidative stress response induced by chemotherapy in leukemia treatment. Mol Clin Oncol. 2018;8(3):391–399. | ||
Yokomizo A, Ono M, Nanri H, et al. Cellular levels of thioredoxin associated with drug sensitivity to cisplatin, mitomycin C, doxorubicin, and etoposide. Cancer Res. 1995;55(19):4293–4296. | ||
Mäntymaa P, Siitonen T, Guttorm T, et al. Induction of mitochondrial manganese superoxide dismutase confers resistance to apoptosis in acute myeloblastic leukaemia cells exposed to etoposide. Br J Haematol. 2000;108(3):574–581. | ||
Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. | ||
Lu S, Wang J. Homoharringtonine and omacetaxine for myeloid hematological malignancies. J Hematol Oncol. 2014;7(1):2. | ||
Kantarjian HM, O’Brien S, Cortes J. Homoharringtonine/omacetaxine mepesuccinate: the long and winding road to food and drug administration approval. Clin Lymphoma Myeloma Leuk. 2013;13(5):530–533. | ||
Kantarjian H, O’Brien S, Jabbour E, Barnes G, Pathak A, Cortes J. Effectiveness of homoharringtonine (Omacetaxine mepesuccinate) for treatment of acute myeloid leukemia: a meta-analysis of Chinese studies. Clin Lymphoma Myeloma Leuk. 2015;15(1):13–21. | ||
Wang L, Jin J. The mechanisms of synergistically cytotoxicity induced by homoharringtonine and aclarubicin in acute myeloid leukemia cells. Blood. 2012;120(21):4313–4313. | ||
Yu W, Mao L, Qian J, et al. Homoharringtonine in combination with cytarabine and aclarubicin in the treatment of refractory/relapsed acute myeloid leukemia: a single-center experience. Ann Hematol. 2013;92(8):1091–1100. | ||
Chen J, Mu Q, Li X, et al. Homoharringtonine targets Smad3 and TGF-β pathway to inhibit the proliferation of acute myeloid leukemia cells. Oncotarget. 2017;8(25):40318–40326. | ||
Tang R, Faussat AM, Majdak P, et al. Semisynthetic homoharringtonine induces apoptosis via inhibition of protein synthesis and triggers rapid myeloid cell leukemia-1 down-regulation in myeloid leukemia cells. Mol Cancer Ther. 2006;5(3):723–731. | ||
He J, Li L, Zhu J. Homoharringtonine in combination with cytarabine and etoposide for induction therapy in patients with de novo acute myelogenous leukemia. Blood. 2013;122(21):5006–5006. | ||
Wilkoff LJ, Dulmadge EA, Vasanthakumar G, Donahue JP. Etoposide-resistant human colon and lung adenocarcinoma cell lines exhibit sensitivity to homoharringtonine. Cancer Chemother Pharmacol. 1993;33(2):149–153. | ||
Zhang T, Shen S, Zhu Z, et al. Homoharringtonine binds to and increases myosin-9 in myeloid leukaemia. Br J Pharmacol. 2016;173(1):212–221. | ||
Jin J, Wang JX, Chen FF, et al. Homoharringtonine-based induction regimens for patients with de-novo acute myeloid leukaemia: a multicentre, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2013;14(7):599–608. | ||
Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354–365. | ||
Oh SY, Sohn YW, Park JW, et al. Selective cell death of oncogenic Akt-transduced brain cancer cells by etoposide through reactive oxygen species mediated damage. Mol Cancer Ther. 2007;6(8):2178–2187. | ||
Marengo B, De Ciucis C, Ricciarelli R, et al. PKCδ sensitizes neuroblastoma cells to L-buthionine-sulfoximine and etoposide inducing reactive oxygen species overproduction and DNA damage. PLoS One. 2011;6(2):e14661. | ||
Papież MA, Krzyściak W, Szade K, et al. Curcumin enhances the cytogenotoxic effect of etoposide in leukemia cells through induction of reactive oxygen species. Drug Des Devel Ther. 2016;10:557–570. | ||
Cai Z, Lin M, Wuchter C, et al. Apoptotic response to homoharringtonine in human wt p53 leukemic cells is independent of reactive oxygen species generation and implicates Bax translocation, mitochondrial cytochrome c release and caspase activation. Leukemia. 2001;15(4):567–574. | ||
Ryter SW, Kim HP, Hoetzel A, et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9(1):49–89. | ||
Kaur A, Webster MR, Marchbank K, et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature. 2016;532(7598):250–254. | ||
Raninga PV, Trapani GD, Tonissen KF. Cross talk between two antioxidant systems, thioredoxin and DJ-1: consequences for cancer. Oncoscience. 2014;1(1):95–110. | ||
Gürel G, Blaha G, Moore PB, Steitz TA. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2009;389(1):146–156. | ||
Zhou X, Xu N, Li R, et al. A comparative proteomic study of Homoharringtonine-induced apoptosis in leukemia K562 cells. Leuk Lymphoma. 2015;56(7):2162–2169. | ||
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. | ||
Reczek CR, Chandel NS. The two faces of reactive oxygen species in cancer. Annu Rev Cancer Biol. 2017;1(1):79–98. | ||
Zhou Z, Song J, Nie L, Chen X. Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chem Soc Rev. 2016;45(23):6597–6626. | ||
Fresno M, Jiménez A, Vázquez D. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem. 1977;72(2):323–330. | ||
Tujebajeva RM, Graifer DM, Karpova GG, Ajtkhozhina NA. Alkaloid homoharringtonine inhibits polypeptide chain elongation on human ribosomes on the step of peptide bond formation. FEBS Lett. 1989;257(2):254–256. | ||
Chen R, Guo L, Chen Y, Jiang Y, Wierda WG, Plunkett W. Homoharringtonine reduced Mcl-1 expression and induced apoptosis in chronic lymphocytic leukemia. Blood. 2011;117(1):156–164. | ||
Im JY, Lee KW, Woo JM, Junn E, Mouradian MM. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum Mol Genet. 2012;21(13):3013–3024. | ||
Im JY, Lee KW, Junn E, Mouradian MM. DJ-1 protects against oxidative damage by regulating the thioredoxin/ASK1 complex. Neurosci Res. 2010;67(3):203–208. | ||
Pekovic-Vaughan V, Gibbs J, Yoshitane H, et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 2014;28(6):548–560. | ||
Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863(12):2977–2992. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.