")
Back to Journals » International Journal of Nanomedicine » Volume 14
Synergism of cisplatin-oleanolic acid co-loaded calcium carbonate nanoparticles on hepatocellular carcinoma cells for enhanced apoptosis and reduced hepatotoxicity
Authors Khan MW, Zhao P, Khan A, Raza F, Raza SM , Sarfraz M, Chen Y, Li M, Yang T, Ma X, Xiang G
Received 3 December 2018
Accepted for publication 19 March 2019
Published 28 May 2019 Volume 2019:14 Pages 3753—3771
DOI https://doi.org/10.2147/IJN.S196651
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Muhammad Waseem Khan,1 Pengxuan Zhao,1 Asifullah Khan,2 Faisal Raza,2 Shahid Masood Raza,1 Muhammad Sarfraz,3 Yan Chen,1 Minsi Li,1 Tan Yang,1 Xiang Ma,1 Guangya Xiang1
1School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, People’s Republic of China; 2State Key Laboratory of Natural Medicines and Department of Pharmaceutics, China Pharmaceutical University, Nanjing 210009, People’s Republic of China; 3School of Basic Medical Sciences, Henan University, Kaifeng, Henan 475001/475004, People’s Republic of China
Background: Cisplatin (CDDP), a widely used chemotherapeutic agent against hepatocellular carcinoma (HCC), faces severe resistance and hepatotoxicity problems which can be alleviated through combination therapy.
Purpose: The objective of this study was to develop a pH-dependent calcium carbonate nano-delivery system for the combination therapy of CDDP with oleanolic acid (OA).
Methods: A microemulsion method was employed to generate lipid coated cisplatin/oleanolic acid calcium carbonate nanoparticles (CDDP/OA-LCC NPs), and the loading concentration of CDDP and OA was measured by atomic absorption spectroscopy and HPLC respectively.Transmission electron microscopy (TEM) was used to examine the nanoparticles morphology while its pH dependent release characteristics were investigated through in vitro release study. Cellular uptake was examined through a fluorescence microscopy. Apoptotic assays and western blot analysis were conducted to explore the synergistic apoptotic effect of OA on CDDP against HCC cells. The hepatoprotective of OA for CDDP was evaluated through H&E staining.
Results: TEM analysis revealed nanoparticles spherical shape with an average particle size of 206±15 nm, and the overall entrapment efficiency was 63.70%±3.9%. In vitro drug release study confirmed the pH-dependent property of the formulation, with the maximum CDDP release of 70%±4.6% at pH 5.5, in contrast to 28%±4.1% CDDP release at pH 7.4. Annexin V-FITC/PI assay and cell cycle analysis confirmed that CDDP and OA synergistically promoted greater HepG2 cells apoptosis for the CDDP/OA-LCC NPs as compared to their individual free drug solutions and NPs-treated groups. Western blot analysis also proved that CDDP/OA-LCC NPs induced the apoptosis by enhancing the proapoptotic protein expressions through downregulating P13K/AKT/mTOR pathway and upregulating p53 proapoptotic pathway. OA helped CDDP to overcome the resistance by downregulating the expression of proteins like XIAP, Bcl-2 via NF-κB pathway. OA also significantly alleviated CDDP-induced hepatotoxicity as evident from the decreased alanine transaminase, aspartate transaminase levels and histochemical evaluation. The possible mechanism may be related to the Nrf-2 induction via its antioxidant mechanism to maintain the redox balance and reduction in CYP2E1 activity which can lead to ROS-mediated oxidative stress.
Conclusion: These results suggest that CDDP/OA-LCC NPs have promising applications for co-delivering CDDP and OA to synergize their anti-tumor activity against HCC and to utilize OA’s protective effect against CDDP-induced hepatotoxicity.
Keywords: combination therapy, cisplatin, oleanolic acid, hepatocellular carcinoma, hepatotoxicity
Introduction
Hepatocellular carcinoma (HCC) is the most common form of primary hepatic carcinoma and considered to be the second most leading cause of cancer-related deaths worldwide.1 Major risk factors for HCC are liver cirrhosis, hepatitis B virus, hepatitis C virus, tobacco use, alcohol consumption, and aflatoxin exposure.2 Despite the latest advancements in the treatment techniques and early diagnosis, HCC is still a highly lethal disease.3 Hepatic resection and liver transplantation provide good treatment that offers a possible cure for HCC, though high risks of developing metastasis or recurrence have been noted in patients receiving liver resection.4 Sorafenib is currently one of a few FDA-approved HCC targeted therapeutic drugs since 2007. Since the recent adjuvant sorafenib related trial against HCC has been failed,5 the development and identification of effective therapeutic strategies are needed. Cisplatin (cis-diamminedichloroplatinium, CDDP), one of the most widely used chemotherapeutic drugs for a variety of tumors, exhibits anticancer activity via formation of CDDP-DNA adducts.6 However, its clinical use has been hampered by the intrinsic and adaptive resistances of the tumor cells along with severe toxic effects, which greatly subsides its efficacy.7,8 Therefore, an ideal therapeutic regimen is required to minimize the therapeutic resistance of tumor cells to CDDP-therapy and reduce its toxic effects.
Oleanolic acid (3β-hydroxyolean-12-en-28-oic acid, OA), the most abundant pentacyclic triterpenoid in the plant kingdom, is widely known for its important pharmacological potentials, such as hepato-protective9 and antitumor activities.10 OA exhibits anticancer activity through various pathways, including activation of AMP-activated Protein Kinase (AMPK) pathway,11 inhibition of P13K/AKT/mTOR/NF-κB signaling pathway12 and AMPK/mTOR pathway,13 upregulation of tumor protein (p53) mediated activation of mitochondrial apoptotic pathway,14 and blocking the release of anti-apoptotic inhibitor of apoptotic protein (IAP) family proteins.15 Recently, OA has been used in combination therapy to enhance the cancer cells apoptosis and reduce the severe side effects of doxorubicin.16
Combination therapy is often employed to improve the tumor prognosis and reduce the side effects.17 However, administering chemotherapeutic drugs in combination was challenged by the right dosage forms and drugs delivery to the target site at a suitable ratio in order to get the desired effects. Since the majority of anticancer drugs are low molecular-weight type compounds, they can be easily excreted via glomerular filtration or entrapped and metabolized by reticuloendothelial system.18 In these regards, the nanoscale drug delivery systems have demonstrated the potential for effective delivery of multiple chemotherapeutic drugs at the tumor site with enhanced drug circulation half-life, minimum free drug toxicity, and unspecific drug uptake.19 However, one of the major challenges in nanomedicine is the development of an effective matrix for targeted drugs delivery, as the release of active drug in appropriate concentration at the target site from the carrier is of utmost importance to overcome cisplatin resistance in cancer cells.20 Numerous stimuli-responsive nano-targeted delivery, particularly pH-sensitive response systems have been designed for the prompt and effective release of drugs at the tumor site to improve therapeutic effects.21 The pH-responsive materials dramatically alter their physicochemical properties at acidic tumor conditions, enabling a selective tumor targeting.22 Among them, an inorganic material delivery system calcium carbonate (CC) has been captivating much attention as it offers tremendous biocompatible and biodegradable properties, alongside its porous structure making it a perfect carrier for anticancer drugs.23 CC structure remains intact in the neutral environment toprotect the cargos from degradation and phagocytosis during circulation,24 while slightly acidic atmosphere prompts its dissolution for faster drug release with greater uptake,25 which makes it an ideal candidate for pH-sensitive sustained drug delivery systems.26 In the present study, lipid-coated cisplatin/oleanolic acid calcium carbonate nanoparticles (CDDP/OA-LCC NPs) have been developed. The NPs were optimized and their physicochemical properties were examined. In vitro cytotoxicity of NPs and their synergistic combination index (CI) analysis against HepG2 liver cancer cells were conducted. In vitro release profiles of the individual drugs in the NPs were also assessed alongside the toxicity evaluation study. Western blot analysis was performed to determine the beneficial effects of OA in NPs for CDDP-induced anti-tumor activity against HCC, and the protective effect against CDDP-induced hepatotoxicity was examined.
Materials and methods
Materials
CDDP (Pt, 65%) was purchased from Shanghai Macklin Biochemical Co., Ltd while OA from Aladdin Industrial Corporation, Shanghai (People’s Republic of China). 1,2-Dioleoyl-in-glycerol-3-phosphate (DOPA) and mono-methoxy polyethylene glycol 2000-distearoyl phosphatidylethanolamine (PEG-DSPE 2000) were purchased from Avanti Polar Lipids Inc (Alabaster, AL, USA). Dehydrogenated soya phosphatidylcholine (HSPC) from Shanghai Advanced Vehicle Technology Ltd. (Shanghai, People’s Republic of China) and cholesterol (CHOL) from Acros Organics (Geel, Belgium). Silver nitrate (AgNO3), calcium chloride, sodium carbonate, ethanol, chloroform, DMSO, methanol (AR grade), and Tween 80 (CP grade) were purchased from Sinopharm Chemical Reagent Co. (Shanghai, People’s Republic of China) while acetonitrile and methanol (high-performance liquid chromatography [HPLC] grade) from Thermo Fisher Scientific (Geel, Belgium). Dialysis bags were purchased from Shanghai Yaunye Biotechnology Co., Ltd. (People’s Republic of China) while trifluoroacetic acid (TFA) (AR) from Merck (Darmstadt, Germany). Cyclohexane, Igepal CO-520, coumarin-6 (C-6), DMEM, and PBS from Biosharp (Anhui, People’s Republic of China) while MTT was purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA); FBS was obtained from Zhejiang Tianhang Biological Technology Co., Ltd. (Hangzhou, People’s Republic of China). Annexin V-FITC/PI Kit was purchased from BestBio, while DAPI from KeyGen Biotech. Nanjing, People’s Republic of China. All the chemicals and solvents were of reagent grade and were used without any further purification unless specified.
Preparation of CDDP/OA-LCC NPs
Synthesis of Cis-[Pt (NH3)2(H2O) 2] (NO3)2 precursor
Cis-(Pt(NH3)2(H2O)2)](NO3)2 was prepared according to the previous literature.27 To a suspension of CDDP (60 mg, 0.20 mM) in double distilled water (1 mL), AgNO3 (66.2 mg, 0.39 mM) was added gently to generate a uniform mixture. The mixture was then heated at 60°C for 3 hrs followed by overnight stirring in a flask protected from the light with aluminum foil. Next, the mixture was centrifuged at 12,000 g for 15 mins to separate the AgCl precipitate, formed during the reaction, followed by the supernatant filtration through 0.2-mm syringe filter. SpectrAA-24OFS Atomic Absorption Spectrometer (Varin, USA) was used to determine the concentration of cis-[Pt(NH3)2(H2O)2](NO3)2 concentration in the final solution obtained.
Preparation of CDDP/OA-LCC NPs
CDDP-LCC NPs were prepared in two steps. First, the CDDP-CC cores were prepared followed by outer lipid coating. The CDDP-CC cores were developed through water-in-oil microemulsion method in accordance with the previously reported literature with slight modifications.28,29 Two water-in-oil microemulsions were prepared; 1) calcium emulsion: briefly, 300 µL CaCl2 aqueous solution (500 mM) was dispersed in 15 mL oil phase (cyclohexane/Igepal CO-520) (71:29, v/v) to generate a well-dispersed water-in-oil reverse micro-emulsion. 2) Carbonate emulsion: the carbonate part was prepared by dispersing 300 µL of sodium carbonate (250 mM) aqueous solution in a separate 15 mL oil phase (cyclohexane/Igepal CO-520) (71:29, v/v). Cisplatin prodrug solution (250 µL, 2 mg/mL) and dioleoylphosphatydicacid (DOPA) (200 µL, 20 mg/mL) (as an inner leaflet lipid) in chloroform were also added to the carbonate phase. The two oil phases were mixed together after a separate mixing for 20 mins. After mixing the two microemulsions for 30 mins, 30 mL of absolute ethanol was added to break the micro-emulsion system followed by centrifugation at 12,000 g for 30 mins to remove the surfactants, cyclohexane and to collect the pellets. The pellets were washed 2–3 times with absolute ethanol to remove any residual of DOPA and cyclohexane. Finally, after the extensive washing, the pellets were collected in chloroform (10 mL) and stored in a glass vial for further modifications.
To prepare the lipid-coated cisplatin-calcium carbonate NPs (CDDP-LCC NPs), HSPC: CHOL: DSPE-PEG-2000 at a molar ratio of 11:1:1 mM and the CDDP/CC core solution (1 mL) prepared in the first step were dispersed in chloroform (5 mL). Later, the chloroform was removed under reduced pressure using rotary evaporator. Finally, the thin film that formed on the inner wall of the vial was shattered to NPs by adding phosphate buffer saline (pH 7.4) or H2O (1 mL) under brief sonication.
To prepare oleanolic acid-lipid coated calcium carbonate NPs (OA-LCC NPs), the same procedure was adopted with the only difference that blank CC NPs were used rather than CDDP incorporated CC cores.
To prepare CDDP/OA-LCC NPs, CDDP-CC cores solution (1 mL) and OA solution (695 µL 4 mg/mL, ethanol) were dispersed in chloroform (5 mL). After the chloroform was removed by rotary evaporation, residual lipids were dispersed in PBS (pH 7.4) or H2O (1 mL) to generate CDDP/OA-LCC NPs.
The same procedure was applied to formulate the fluorescent coumarin-6 loaded CDDP-LCC NPs with OA being replaced by coumarin-6.
Characterization of NPs
Particle size, polydispersity index, and zeta potential measurement
Physicochemical characteristics of the NPs were determined using a Zeta PALS instrument (Brookhaven Instruments, Austin, TX, USA). Dynamic light scattering (DLS) was employed to determine the NPs diameter and polydispersity index. The samples were prepared by diluting the NP formulation with PBS (pH 7.4) to a count of 300–500 and sonicated for 5 mins before the readings being taken at 25±1°C. All the readings were taken in triplicate.
Morphology
Morphological examination of CDDP/OA-LCC NPs was done through a JEOL 100CX transmission electron microscope (TEM; Tokyo, Japan). Briefly, a drop of freshly prepared NPs diluted with double distilled water was deposited onto a 300-mesh copper grid coated with carbon (Ted Pella, Inc., Redding, CA, USA) and allowed to dry at room temperature. Thereafter, a drop of 0.2% phosphotungstic acid was used as a negative stain. The excess solution was removed with a dry filter paper and was placed for a total dryness in a dust-free zone before being observed under the TEM.
Entrapment efficiency
Platinum contents in the NPs were measured by atomic absorption spectroscopy (AAS) (SpectrAA-24OFS Atomic Absorption Spectrometer, Varin, USA). Samples were digested first in nitric acid (70%) and then diluted to a final acid content of 2% in water. Platinum concentration was determined using 195Pt isotope. OA loading was determined by high-performance liquid chromatography (HPLC) (Agilent Infinity 1220 LC System., Germany) analysis against the standard curve using Chemstation software (Agilent) for data acquisition and analysis. The chromatographic columns used were: Agilent Zorbax SB-C18 (2.1 mm, 50 mm) and Sepax Technologies Sapphire C18 analytical column (4.6 mm×250 mm, 5 µm). Mobile phase was composed of 0.1% TFA in water and a mixture of acetonitrile: methanol (17:1) at a ratio of 10:90. The elute time was 15 mins with the column temperature maintained at 30°C.30 The flow rate was set at 1.3 mL/min, with the detection wavelength of 210 nm. The following equation was used to determine the entrapment efficiency.
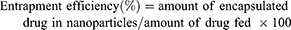
Cell line
HepG2 cells obtained from the China Center for Type Culture Collection at Wuhan University (Wuhan, People's Republic of China) were cultured with high glucose DMEM medium supplemented with 10% heat-inactivated, FBS, 100 U/mL of penicillin and 100 mg/mL of streptomycin at 37°C in an atmosphere of 5% CO2 and 95% air (RH) in the incubators.
Animals
Kunming mice were obtained from the Animal Care Facility Centre of Huazhong University of Science and Technology, Wuhan, People's Republic of China. They were kept at animal care center with food and water provided at libitum with 12 hrs light/dark cycle. All the animal studies were conducted following the guidelines and with the approval of the University Animal Experimentation Ethics Committee, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People's Republic of China (S 850).
In vitro cell viability assay (free drugs) and combinationa index (CI) analysis
The relative cytotoxicity of the free CDDP (CDDP-sol), OA solution (OA-sol), and their combinations toward the HepG2 cells were evaluated by measuring the cell viability through methyl thiazolyl tetrazolium (MTT) assay. Briefly, cells were overnight cultured in 96-well plates at a density of 5×103 cells per well in 100 µL of complete culture medium. Subsequently, the culture medium was removed and cells were treated with CDDP-sol and OA-sol in a 200 µL complete culture medium. The cells were then subjected to MTT assay after being incubated for another 24, 48, or 72 hrs. The control was a complete culture medium having untreated cells.31 To determine their synergistic CI ratios, HepG2 cells were treated with various molar ratios of CDDP-sol and OA-sol at a series of dilutions in complete culture medium for 72 hrs. Each concentration was tested in five wells and the data were presented in mean±SD. Relative cell viability was determined by comparing the absorbance of the treated cells to untreated control wells (containing only cell culture medium) through a microplate reader (Multiskan MK3: Thermo Fisher Scientific, Atlanta, GA, USA) at 490 nm. The IC50 was determined using Compusyn Software (Version 1.0, Combo-Syn Inc., USA).
CI analysis
CI analysis of the in vitro free drug solution combination was evaluated using Compusyn software based on Chou and Talalay method.32 Briefly, for each value of fraction affected (Fa), the CI values for CDDP and OA combinations were calculated according to the following equation:
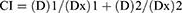
where (D)1 and (D)2 are the concentrations of drug 1 and drug 2, respectively, in the combination resulting in Fa ×100% growth inhibition, while (Dx)2 and (Dx)2 are the concentrations of drugs alone resulting in Fa ×100% growth inhibition. CI values for drug combinations were plotted as a function of Fa. CI values <1, CI=1 and CI>1 represent synergistic, additive, and antagonistic effect, respectively.
In vitro drug release study
In vitro release profiles of CDDP and OA from CDDP/OA-LCC NPs were examined through dialysis diffusion bag technique.33 The incubation medium, PBS was used at different pH conditions (ie, pH =5.5 and 7.4). Freeze-dried NPs suspended in 2 mL of 0.1 M PBS containing 0.1 mg/mL of CDDP, 20 mg/ml of OA (freeze-dried) were added into the pre-swelled dialysis bags with 12-kDa molecular weight cutoff and dialyzed against 60 mL of 0.1 mol/L PBS buffer (pH=5.5 and 7.4) with stirring speed of 200 rpm at 37°C for 72 hrs. At each time point, 2 mL sample was collected from the incubation medium and immediately replenished by an equal volume of fresh medium. CDDP and OA concentrations were then determined by AAS and HPLC, respectively, at specific time points. The collected samples having the released drug were split equally into two parts before being freeze-dried; one portion was used to quantify the released platinum; the other part was used to determine the released OA from the NPs. The released drug concentration was expressed as a percentage of total drugs in the NPs and plotted as a function of time. Experiments were performed in triplicates and the results were presented as a mean±SD of the three individual experiments. Statistical analysis for both CDDP and OA release was conducted via a Student's two-tailed t-test.
In vitro cytotoxicity of NPs
The relative cytotoxic effect of the developed NPs against HepG2 cell was determined through MTT assay. Briefly, cells were seeded onto a 96-well plate at a density of 5×103 cells/well for 24 hrs. After proper confluence, the cells were exposed to various concentrations of CDDP-LCC NPs, OA-LCC NPs, and their various combination ratios for 72 hrs in a fresh DMEM medium. During each group experiment, a complete culture medium without drug was taken as a control. After 72-hr incubation, the proportion of the viable cells against each formulation were determined using the MTT assay following the user’s manual for each group.
Cellular uptake study
Coumarin-6, a fluorescent dye widely used as a substitute for hydrophobic drugs in cellular uptake studies,34 was used to examine the efficiency of NPs uptake by the tumor cells. HepG2 cells were seeded onto the 6-well plates at a density of 5×104 cell/well for 24 hrs. The cells were then exposed to medium containing coumarin-6 (12.5 µg/mL) co-loaded CDDP-LCC NPs for different time points. Later, the cells were washed with cold PBS to remove free NPs along with cellular uptake blockage and were then fixed by 70% ethanol for 20 mins. Cells were washed twice with PBS again followed by nuclei staining with DAPI (5 µg/ml) for 10 min. Free and excess DAPI was removed through PBS washing. Finally, the DAPI-stained cells were observed and photographed using a fluorescence microscope (Olympus 1X71 microscope, Japan).
Cell apoptosis assay
Cell apoptosis assay was conducted in two ways: 1) Hoechst 33258 staining method was employed to determine the qualitative apoptosis of HepG2 cells, seeded onto a 12-well plate (106/well). After an overnight stay, cells were treated with the culture medium containing different formulations (sol/NP) for 48 hrs. The control group was a complete cell culture medium well. Later, the cells were washed with PBS twice and then fixed with 4% paraformaldehyde (500 µL/well) for 30 mins. After fixation, the cells were washed with PBS again and then stained with Hoechst 33258 (500 µL, 5 µg/mL) at 37°C in the dark for 10 mins. Finally, the cells were washed with PBS and then examined by a fluorescence microscope (IX71; Olympus, Japan) and photographed.35 2) Cell apoptosis was also assessed through an Annexin V-fluorescein isothiocyanate/propidium iodide (Annexin V-FITC/PI) double staining assay. HepG2 cells were cultured in 6-well plates at a density of 2×105 per well. After proper confluence, the cells were exposed to different formulations in a fresh medium for 48 hrs. Later, the cells were collected using EDTA-free pancreatin and then stained with the detection kit as per instructions. Early apoptotic cells with intact cell membranes but exposed phosphatidylserine bound to Annexin-V-FITC while necrotic cells were stained by PI. A complete cell culture medium well was taken as a control. The percentage of the live, apoptotic and necrotic cells were analyzed by flow cytometry36 and through a computer station running Cell-Quest software (BD Biosciences, San Jose, CA, USA).
Cell cycle study
Cell cycle analysis was conducted to determine the cell cycle progress after the drug exposure. Briefly, HepG2 cells (2×105 cells/well) cultured in a 6-well plate were treated with both free drug solutions and their NPs for a defined incubation time. In each plate, wells with untreated cells taken as a control were also included. After 48 hrs, the cells were washed with cold PBS, harvested (floating and adherent cells) and washed with PBS again through centrifugation, later fixed with 70% ethanol at 4°C for overnight. On the day of analysis, ethanol was removed from the cells by centrifugation (5 mins, 37°C, 2,000 rpm), followed by PBS washing and then processed with RNAase (20 µg/mL) at 37°C for 30 mins before being stained with PI (50 µg/mL) for 30 mins at 4°C in the darkness. Flow cytometry (BD Biosciences, San Jose, CA, USA) was conducted to determine the distribution of the cells where proportion of the cells in G0/G1, S, G2/M phases; presented as DNA histogram while apoptotic cells with hypodiploid DNA content; determined through quantifying sub-G1 peak in cell cycle pattern, were determined through FlowJo-V10/Cell Quest acquisition software (BD Biosciences). For each experiment, 10,000 events per sample were analyzed.
Hepatoprotective effect of OA against CDDP induced injury in mice
To study the protective effect of OA on CDDP-induced liver injury in mice, 49 mice were randomly divided into seven groups (seven mice per group). Group 1 (control group): served as untreated control received normal saline; Group 2 (corn oil group): received corn oil orally at a daily dose of 5 mL/kg body weight (b.wt.) intraperitoneally (i.p.) for 7 days which was used a vehicle for OA in the study. Group 3 (OA-sol group): received OA-sol injection at a daily dose of 20 mg/kg b.wt. i.p. and a single i.v. CDDP-sol injection at a dose of 7.5 mg/kg b.wt, 1 hr after the OA-Sol injection (second day). Group 4 (OA-LCC NPs group) received the same treatments of OA and CDDP as a third group but in NPs form. Group 5 (CDDP-sol group) and Group 6 (CDDP-LCC NPs group) were given a single i.v. injection of CDDP-sol and CDDP-LCC NPs at a dose of 7.5 mg/kg b.wt. on the second day of study. Group 7 (combined NPs group): received a single i.v. injection of CDDP/OA-LCC NPs (CDDP, 7.5 mg: OA, 20 mg/kg) on the second day of the experiment. At the end of the study period, all the groups were subjected to fasting for an overnight but allowed to drink water at libitum. Later, the mice were euthanized and the blood samples were collected and centrifuged (3,000 rpm×5 mins, 4°C) for serum collection to conduct various biochemical tests. For histological examination, livers were dissected out after animals sacrifice, washed with saline and then dried between filter papers. Later, they were fixed in 10% formalin for histological analysis.
Western blot analysis
HepG2 cells treated with various formulations (Drug-sol/NPs) or DMEM (as a control) were washed with cold PBS and then lysed with RIPA lysis buffer (Beyotime, People's Republic of China). Cell lysates were quantified for proteins concentration with BCA protein assay kit (Wuhan Service biotechnology CO., LTD). Afterward, protein loading buffer was added to the protein at a ratio of 5:1, mixed at 95°C for 5 mins to denature the proteins and stored at −20°C. Equal amounts of proteins were loaded onto a 10–12% SDS-PAGE gels and thereafter electro-transferred to PVDF membranes for each treatment. Later, the membranes were blocked with 5% non-fat dry milk in Tris-buffered saline (10 mM Tris, 150 mM NaCl) for 1 hr and then incubated with primary monoclonal antibodies against p53 (1:1,000, Proteintech, USA), BAX (1:1,000, Proteintech, USA), BCL-2 (1:1,000, Proteintech, USA), BAD (1:1,000, Proteintech, USA), cyto-c (1:1,000, Proteintech, USA), caspase-3 (1:1,000, Proteintech, USA), NF-κB (1:1,000, Proteintech, USA), XIAP (1:1,000, Proteintech, USA), and β-actin (1:1,000, Proteintech, USA) overnight at 4°C. Later, the membranes were washed three times with TBST (10 mM Tris, 150 millimole NaCl and 0.1% Tween-20) and then incubated with an anti-rabbit or anti-mouse secondary antibodies (diluted, 1:10,000) conjugated with horseradish peroxidase, at room temperature for 1 hr. Finally, the protein bands were rinsed with TBST thrice for 5 mins each and later an ECL-western blot system was used for qualitative analysis of the protein bands via densitometry using chemiluminescent reagent according to the manufactures guidelines (Cell Signaling Technology, Beverly, Perkin Elmer, Waltham, MA, USA).
Statistical analysis
Data values are expressed as mean±SD. Statistical significances were determined with one-way ANOVA followed by Dunnett’s multiple comparison test using GraphPad prism Version 5 software (GraphPad Software, USA) with p-values <0.05 considered to be statistically significant.
Results and discussion
In vitro cell viability and synergistic effect analysis (free drugs)
MTT assay was conducted to determine the in vitro cytotoxicity effect of CDDP and OA (free drug solutions) against HepG2 cells for 24 , 48, and 72 hrs time period. As shown in Figure S1A and B, the cytotoxic effect was concentration and time-dependent for both drugs. The cell viability for each drug was considerably less in 72 hrs than in 48 and 24 hrs. Because of the greatest cytotoxic effects, 72 hrs time point was chosen to determine the synergistic combination ratios of CDDP and OA against HepG2 cells.
In order to find out an appropriate synergistic combination ratio for free CDDP and free OA drug solutions against HepG2 cells, the viability of the cells for 72 hrs against various combined molar ratios were also determined through MTT assay. Both CDDP and OA were used at a non-fixed ratios as shown in Figure 1A. The synergistic inhibitory effect was evaluated using the median-effect method and CI analysis. A total of 10 non-fixed ratios of free CDDP and OA solution molar ratios were evaluated to determine the best possible synergistic combination ratios. As shown in Figure 1A and B, CI values fall in synergistic range (<1) at a non-fixed ratio when OA molar concentration was kept constant at 35 µM while CDDP was used at a different molar concentration, ie, 2, 1.5, 1, 0.5, and 0.25 µM. On the other hand, when CDDP was kept constant at 0.25 µM while OA was applied at different molar concentration, ie, 274, 219, 137, 110, and 82 µM, the cytotoxic effect was prominent but the CI values were >1, clearly showing the antagonistic effect. Of all the synergistic ratios, 2 µM vs 35 µM concentration ratio (representing CDDP and OA, respectively) was selected the best possible synergistic ratio because of the narrowest synergism range for the two drugs against HepG2 cells. Therefore, this synergistic combination ratio, ie, 2:35 (CDDP:OA) was used as an optimized drug loading ratio in the final NPs formulation.
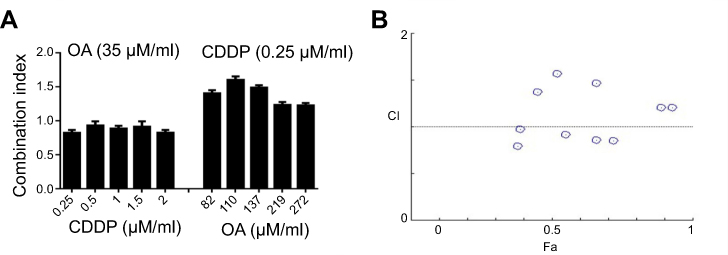 | Figure 1 Cytotoxicity assay of free CDDP and free OA against HepG2 cells. (A) Combination indices at non-fixed ratios (72 h). (B) Corresponding CI vs Fa plot at various non-fixed ratios generated through Compusyn Software. Data presented as mean±SD, n=5. Abbreviations: CDDP, cisplatin; OA, oleanolic acid; CI, combination index; Fa, fraction affected. |
Experimental design, preparation, and characterization of CDDP/OA-LCC NPs
Experimental design
This CIS/OA-LCC NPs system was composed of CDDP (D1), OA (D2), lipids (HSPC: CHOL: DSPE-PEG-2000), CaCl2, Na2CO3, and surfactant (DOPA). Amounts of all the composites were optimized in a preliminary study using a hit and trial method where the composites on the properties of the developed system were determined in terms of particle size, zeta potential, and entrapment efficiency shown in Table 1. The amounts of D1 and D2 were kept constant throughout the experiment. The lipids concentration were optimized by changing the lipid amounts while keeping the other two variables constant as indicated in Table 1 (F1, F2, and F3). A combination of lipids were used to develop this system as mixed lipid matrix yields particles with smaller size, lower crystallinity, and greater entrapment efficacy.37 A slight increase in particle size was observed for F2 when the lipids concentration were increased, however, the entrapment efficiency was significantly enhanced (56.11%) as compared to F1 and F3. This could be attributed to the lipid solubility of the drugs. Therefore, lipids concentrations of F2 were selected for further study. Next, the ratio of CaCl2: Na2CO3 was optimized by varying their formulation ratios to 1:1, 1:2, and 2:1 as mentioned in Table 1 (F4, F5, and F6). A significantly enhanced entrapment efficiency (58.91%) was noted as the ratio of CaCl2:Na2CO3 was changed to 1:2. This could be attributed to the decreased Ca2+:CO32− ratios resulted in increased particle size as reported by Chen.38 Finally, the amount of surfactant was investigated at three different ranges (180, 200, and 150 µL) mentioned in Table 1 (F7, F8, and F9), respectively. The entrapment efficiency significantly increased to 63.70% for F8, since the surfactant in this system acted as a steric stabilizer, resulting in high drug entrapment.39 Moreover, the decrease in particle size at high surfactant concentrations might be related to an effective reduction in interfacial tension between aqueous and lipid phases. Formulation F8 (Table 1) was selected for further studies based on its smaller particle size (217±16 nm), suitable zeta potential (−23.7±2 mV), and enhanced entrapment efficiency (63.70%±3.9%).
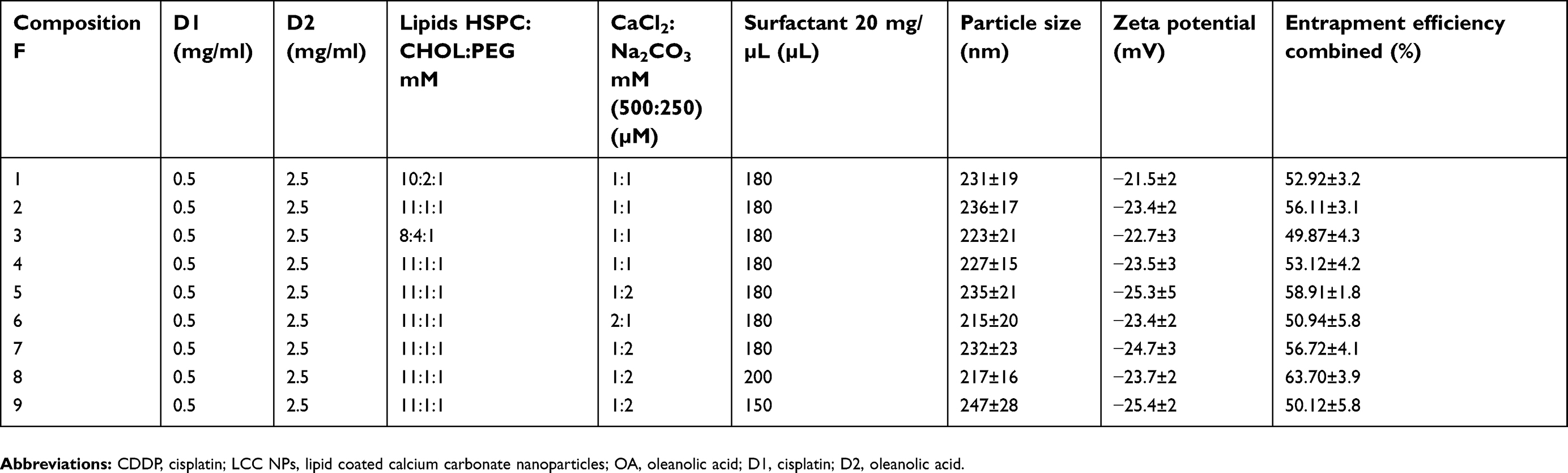 | Table 1 Experimental design followed for the formulation of CDDP/OA-LCC NPs |
Preparation and characterization
Microemulsion method, involving a nanoprecipitation process, was conducted to generate CDDP-CC cores using DOPA as a pre-coating agent for the nano-sized cores. The schematic illustration of the preparatory method is presented in Figure 2A. DOPA, which strongly reacts with platinum cations40 at the interface between carbonate and calcium ion generating stable precipitates, allows the control over the nano-precipitate size and prevents the possible aggregation during the centrifugal separation step. The DOPA coating was extensively washed with chloroform to remove free DOPA and cyclohexane on the CDDP-CC cores. DOPA-coated CDDP-CC cores were then obtained through centrifugation. The composition of the outer leaflet lipids were carefully chosen as they can greatly affect the pharmacokinetics and tissue distribution of the final NPs.41 Thus, an outer leaflet lipid layer of HSPC, CHOL, and DSPE-PEG-2000 (11:1:1) in chloroform was coated onto the cores to generate stable NPs. OA was also loaded onto the NPs at this time point. The hydrated CDDP/OA-LCC NPs were then purified through centrifugation to remove the unbound drug and free liposomes. Drug loading was 76%±5% and 50%±7% (w) for CDDP and OA, respectively. The average NP diameter was 217±20 nm via DLS measurement while zeta potential was −23.7±2. The average polydispersity index was 0.187, indicating the narrow and uniform dispersion of the NPs. TEM analysis (Figure 2B) confirmed that NPs were spherical in shape with a dark CC core and an average diameter of 206 nm, smaller than the size results obtained through DLS technique. The reason should be the NPs dehydration happened during the TEM sample preparation process and that of DLS sensitivity to the interference of large particles. Overall, these results indicated that the prepared NPs were of uniform size, within the acceptable size range, well dispersed in aqueous solution and with a high drug loading.
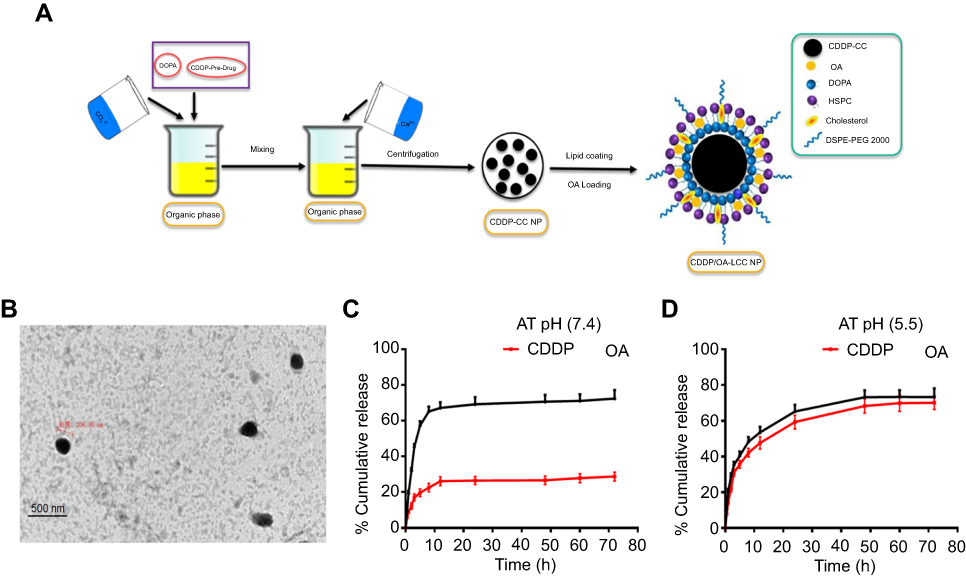 | Figure 2 (A) Schematic illustration of the formulation of lipid-coated cisplatin/oleanolic acid co-loaded calcium carbonate nanoparticles (CDDP/OA-LCC NPs); (B) TEM images of CDDP/OA-LCC NPs, scale bar: 500 nm; (C) In vitro cumulative release profiles of CDDP and OA from the CDDP/OA-LCC NPs in PBS (72 h) at pH 7.4; (D) In vitro cumulative release profiles of CDDP and OA from the CDDP/OA-LCC NPs in PBS (72 h) at pH 5.5. Abbreviations: CDDP, cisplatin; DOPA, dioleoyl-in-glycerol-3-phosphate; OA, oleanolic acid; PEG-DSPE 2000, mono-methoxy polyethylene glycol 2000-distearoyl phosphatidylethanolamine; LCC, lipid coated calcium carbonate; NP, nanoparticles; HSPC, dehydrogenated soya phosphatidylcholine; PBS, phosphate buffer saline. |
In vitro release study
The drug release behavior of CDDP/OA-LCC NPs was investigated in PBS solution (pH 7.4, pH 5.5) at 37°C. Both the pH conditions were applied since the microenvironment in the tumor is more acidic than in the normal tissue.21 A sustained drug release profiles were noted for both the drugs from the NPs after an initial burst release at pH 7.4 and pH 5.5 as indicated in Figure 2C and D. It was noted that the platinum release from the NPs in PBS solution at acidic range (pH =5.5) was considerably higher than its release at neutral PBS (pH 7.4). The cumulative Pt release in the acidic medium (pH 5.5) was 70%±4.6%, while its release at neutral medium (pH 7.4) was 28%±4.1% from the NPs after 72 hrs. The statistical calculations (p<0.001) also clearly depicted the highly significant difference between the CDDP release profiles at different pH conditions.
The difference in the Pt release profiles might be due to the fact that Pt was encapsulated inside the CC cores which remained stable at neutral pH, so limited amount of Pt (28%) released from the NPs. CC cores collapsed rapidly at low pH ranges resulting in the fast and easy release of encapsulated drugs,25 which was clearly evident from our data as large amount of Pt was released at acidic pH conditions. This pH dependence drug delivery system provided opportunities for tumor-targeted delivery because the pH level drops to 5 when the endocytotic vesicles delivered from the cytoplasm (pH 7.4).21 Contrary to Pt release profiles, there was no obvious difference noted in the OA release profile in both neutral and acidic medium shown in Figure 2C and D. An initial burst release was observed in both, ie, acidic and basic medium followed by a sustained release. The overall OA release was found to be 75%±4.8% at pH 5.5 while 72%±2.1% at pH 7.4 without any significant difference (p=0.8300).
This in vitro drug release data analysis clearly defines that CDDP/OA-LCC NPs with a pH-sensitive drug delivery vehicle will be able to delay the drug release from containers in the bloodstream (7.4) and target the cancer cells to achieve high selectivity.
In vitro cytotoxicity of NPs
MTT assay of the prepared NPs against HepG2 cell lines was conducted for CDDP-LCC, OA-LCC, and CDDP/OA-LCC NPs. The cytotoxic effect of CDDP-LCC NPs and OA-LCC NPs against HepG2 cells indicated in Figure 3A and B, respectively, were slightly higher than their corresponding-free drugs solutions effect. Moreover, the cytotoxic effect was considerably greater when CDDP/OA-LCC NPs were applied, indicating their synergistic apoptotic effect at this ratio (1:10) as indicated in Figure 3C.
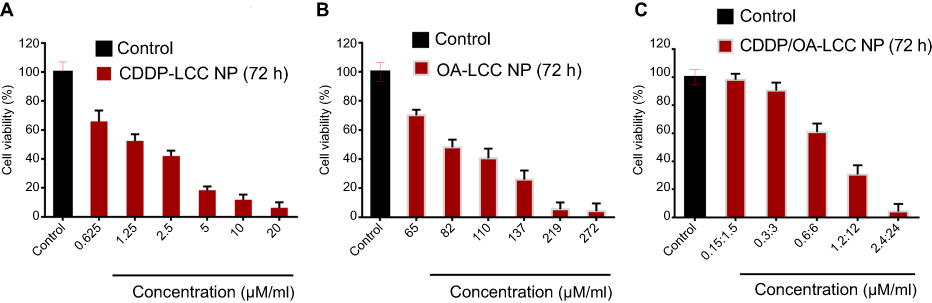 | Figure 3 Cytotoxicity assay of the nanoparticles against HepG2 cells (72 h). (A) CDDP-LCC NPs MTT assay; (B) OA-LCC NPs MTT assay; (C) CDDP/OA-LCC NPs MTT assay with fixed ratios. Data presented as mean±SD, n=5. Abbreviation: CDDP/OA-LCC NPs, CDDP and OA-lipid coated calcium carbonate nanoparticles. |
Cell uptake study
Cellular uptake and internalization efficiency of drug loaded NPs determine the fate of therapeutic outcomes since small molecular drugs entrapped in the NPs can penetrate the cells via endocytosis rather than passive diffusion42 which makes the cellular uptake an important factor as far as the cytotoxic effect is concerned. C-6 loaded CDDP-LCC NPs were used, with the results extrapolated for both CDDP and OA cell uptake. The fluorescence images of HepG2 cells incubated with C-6 loaded NPs are shown in Figure 4. As we can see that, at early time points (1 hr, 2 hrs), there was not any significant C-6 fluorescence surrounding the blue stained DAPI nuclei, however at time points (8 hrs, 24 hrs), we observed more C-6 loaded NPs circumvented alongside the cell nuclei, depicting the localization of C-6 NPs in the cytoplasm. Thus, we can extrapolate these results in both CDDP and OA in the CDDP/OA-LCC NPs that they could interact with the cells in an efficient way and so the internalized NPs could deliver their cargo to the target sites effectively.
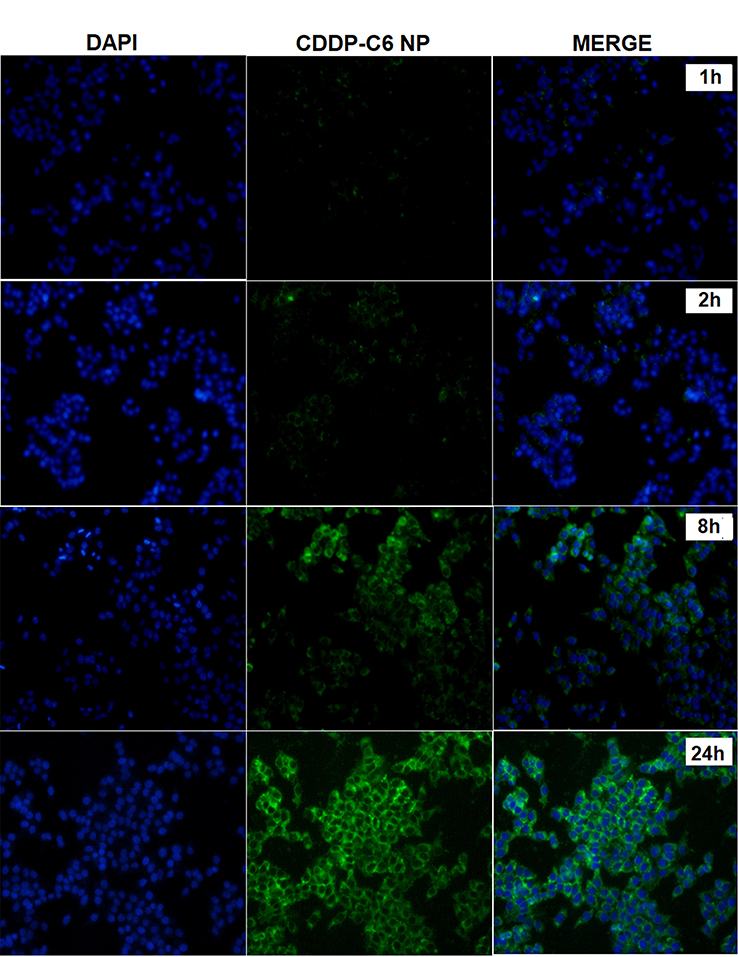 | Figure 4 Fluorescence microscopic images of the HepG2 cells treated with C-6 labeled CDDP-LCC NPs. Cells nuclei stained blue by DAPI, green fluorescence distributed in the cytoplasm from C-6 labeled nanoparticles, and finally merging of two images. Abbreviations: CDDP-LCC NPs, CDDP lipid coated calcium carbonate nanoparticles; C-6, coumarin-6. |
Cell apoptosis assay
To investigate the synergistic effect arising from the co-delivery of CDDP and OA NPs, apoptotic assay on HepG2 cells via Hoechst 33258 staining and Annexin V-FITC/PI staining methods were conducted. Hoechst 33258 nuclear staining was performed to determine the apoptosis by examining the nuclear morphological changes after the drug exposure. As shown in Figure S2A–F, the cells treated with free drugs solutions (CDDP, OA) or their corresponding individual NPs had smaller nuclei, much brighter staining with condensed chromatin than the control group. On the other hand, the cells treated with combined NPs (CDDP/OA-LCC NPs) had the highest apoptotic proportion, with the highest proportion of bright staining, nucleus shrinkage, condensation and fragmentation, which are the typical signs for cell apoptosis.43
Annex V-FITC/PI staining assay was also conducted to determine the cellular apoptotic level after the drug exposure shown in Figure 5A. The apoptotic cells proportion (the early and late apoptosis) for CDDP/OA-LCC NPs were 41%, which were higher than those of CDDP-LCC NPs (34%) and OA-LCC NPs (24%). On the other hand, the apoptotic effect of both free drugs solutions was lower than their corresponding NPs groups. These results indicated that CDDP/OA-LCC NPs have much higher apoptotic effect against HepG2 cells than the individual CDDP NPs, OA NPs and their free drug solutions at the same concentrations, elaborating beneficial synergistic behavior of OA with CDDP in CDDP/OA-LCC NPs against HepG2 cancer cells.
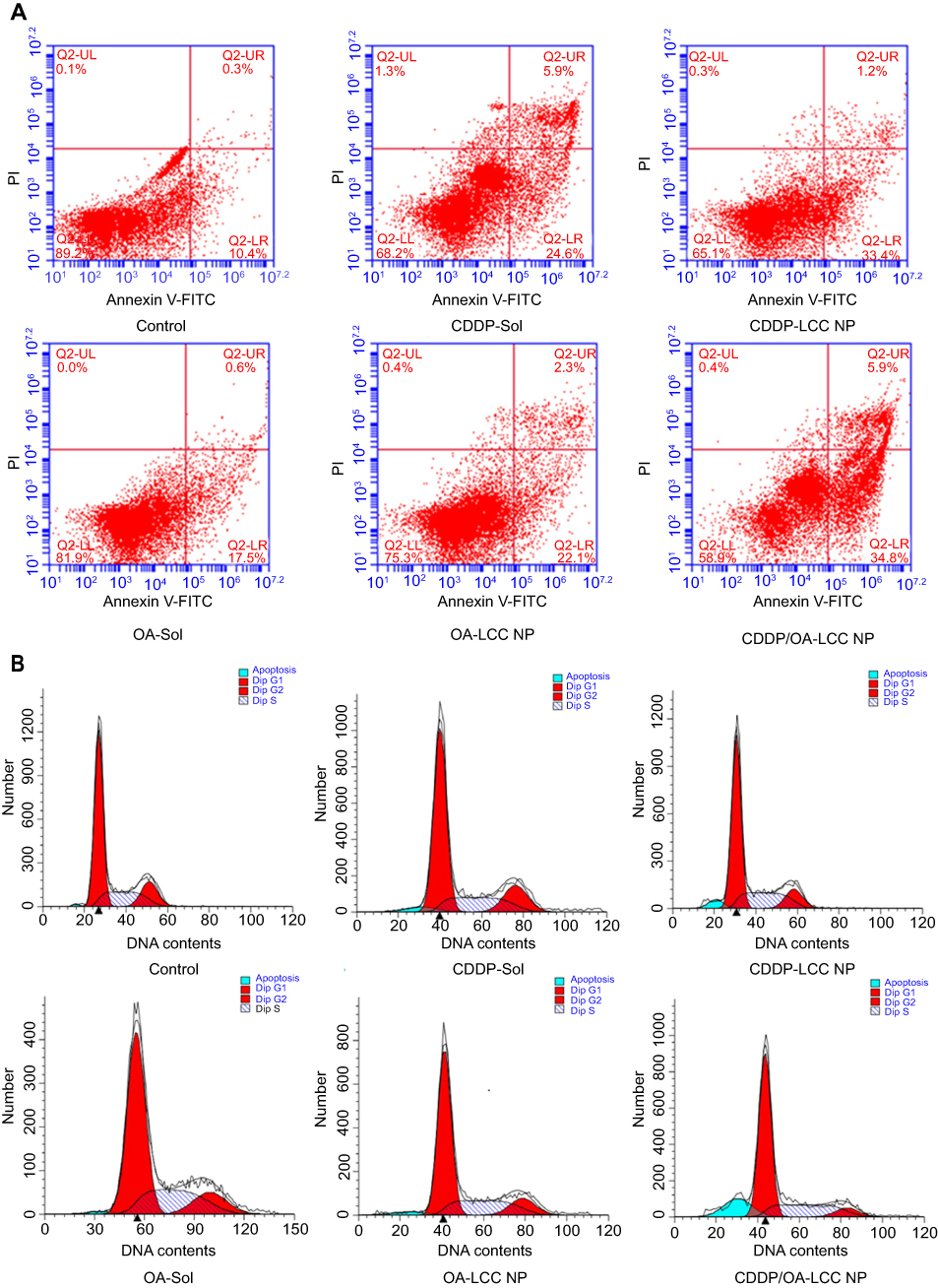 | Figure 5 CDDP/OA-LCC NPs induce apoptosis in HepG2 cells. (A) Annexin V-FITC/PI double staining apoptosis assay results; (B) cell cycle analysis of HepG2 cancer cells. Abbreviations: CDDP, cisplatin; OA, oleanolic acid; LCC, lipid coated calcium carbonate; NP, nanoparticles, UR, upper right; UL, upper left; LL, lower left; LR, lower right; Q2, quadrant. |
Cell cycle study
The cell cycle distribution of the HepG2 cells incubated with CDDP-sol/NPs, OA-sol/NPs, and CDDP/OA-LCC NPs is mentioned in Figure 5B. Less arrested cells in G0/G1 phases and sub-G1 region (apoptotic region) were observed for OA treated groups (sol/NPs) in contrast to CDDP- treated groups (sol/NPs). However, the cells treated with CDDP/OA-LCC NPs have greater number of cells in the G0/G1 phase, the apoptotic region (sub-G1) but having less proportion of cells in the G2/M phase in contrast to the rest of the treated groups, indicating their highest apoptotic effect as compared to the rest of treated groups.
OA attenuates CDDP-induced hepatotoxicity in mice
The potential hepatoprotective effect of OA against CDDP-induced liver injuries in mice was evaluated through serum biochemical markers evaluation, ie, ALT and AST and histological liver tissue screening. Serum biochemical markers are regarded as the most sensitive detective indicator of hepatic impairment by WHO.44 In the present study, as shown in Figure 6H, mice groups which received a single CDDP i.p. injection both in free drug sol/NPs forms, respectively (Group 5 and Group 6) caused a significant increase in ALT and AST levels, suggesting severe liver dysfunction. For the groups, who received OA both in free drug solution and NPs form, respectively (Group 3 and Group 4), markers levels showed no significant difference from the untreated control groups (Group 1 and Group 2). However, in CDDP/OA-LCC NPs-treated group (Group 7), the serum marker levels were lower than the levels for both Group 5 and Group 6 indicating the hepatoprotective effect of OA in our optimized combined NPs of CDDP and OA (CDDP/OA-LCC NPs).
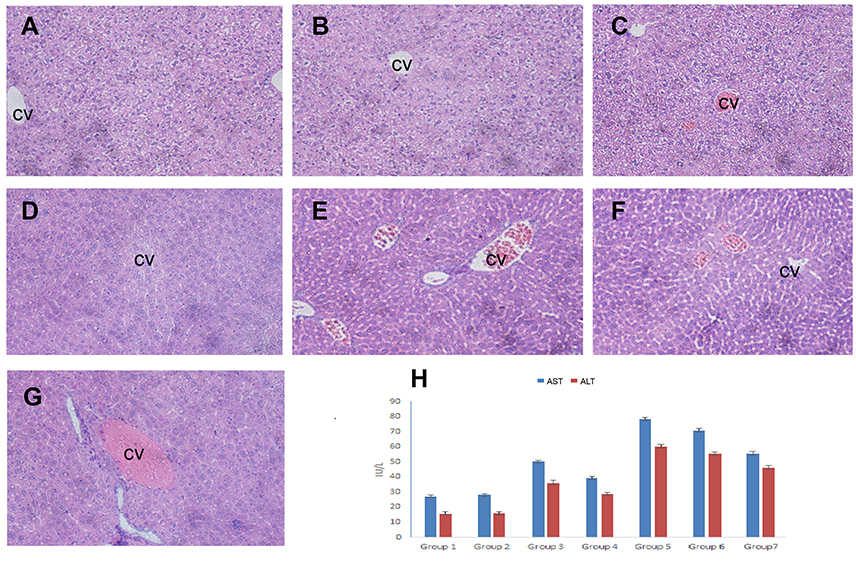 | Figure 6 OA alleviates cisplatin-induced hepatotoxicity. Representative sections from mice liver tissue stained by H & E (200× magnification). (A) Control (NS): normal histological structure of the hepatic lobule. (B) Corn oil group: normal hepatic histology with few hydrophobic degenerations of hepatocytes and normal central vein. (C) OA sol+CDDP sol group: mild hydrophobic degeneration of hepatocytes and mild congested central vein. (D) OA-LCC NP and CDDP-LCC NP group: moderate hepatocytes vacuolization. (E, F). CDDP-sol and CDDP-LCC NPs injection group’s, respectively, hepatic necrosis, severe toxicity, congestion and dilatation of the central vein. (G). CDDP/OA-LCC NPs injection group: less toxicity, less vacuolization; (H) oleanolic acid pre-treatment improved liver function and inhibited cisplatin-induced aberrations in ALT, AST levels. Abbreviations: ALS, Alanine Aminotransferase; AST, aspartate aminotransferase; CDDP, cisplatin; OA, oleanolic acid; LCC, lipid coated calcium carbonate; NP, nanoparticles; CV, central vein. |
Histological screening of the liver tissue also confirmed the hepatoprotective effect of the OA in the CDDP-treated mice. As shown in Figure 6A–G, the mice groups (E and F) given a single CDDP injection in free drug solution and NPs form (7.5 mg/kg) respectively, has shown greater toxicity as evident by the hepatic necrosis, vacuolization and dilatation and congestion of the central vein. The mice groups (C and D) showed a less toxicity, vacuolization, and hydrophobicity as compared to group (E and F), suggesting OA greater hepatoprotective effect against CDDP-induced hepatic toxicity because of its repeated doses. In group G, mice which were given a single CDDP/OA-LCC NPs injection shown slight morphological changes, but it was significantly better than the CDDP-treated groups (E and F). CDDP being one of the most potent anticancer drugs has several dose-limiting reported toxicities, with hepatotoxicity being one of them.45 The exact mechanisms of CDDP-induced hepatotoxicity are not fully known, however, oxidative stress plays a key role.46 CDDP induces increased NF-κB activation leading to oxidative stress inflammation which plays an important physiological role in CDDP-induced hepatotoxicity through various intercalating pathways. NF-κB signaling regulates various inflammatory genes, such as iNOS, TNF-α, IL-6, and IL-1β, with the primary action of NF-κB in liver dysfunction is to facilitate pro-inflammatory cytokines, chemokines and adhesion molecules release which leads to liver regeneration impairment resulting in CDDP- induced hepatotoxicity. CDDP-induced hepatotoxicity has also been reported to be enhanced by the high expression of Cytochrome P450 2E1 (CYP2E1),47 mainly expressed in the liver. CYP2E1 elevates oxidative stress, a key factor in CDDP-induced hepatic injury.
OA counteracts CDDP-induced liver injury and plays its hepatoprotective role by minimizing the factors that leads to that condition as illustrated in Figure S3. Owing to this beneficial aspect, OA has been used in People’s Republic of C,hina for liver disorders such as viral hepatitis for ages, though the exact mechanism of OA hepatoprotective action still remains to be fully decoded.48 It has been reported that OA activates and enhances nuclear accumulation of nuclear factor erythroid 2-related factor 2 (Nrf-2) through signaling pathways mediated through the P13/AKT pathway.49 Nrf-2, a nuclear transcriptional regulating factor of detoxifying and antioxidant enzymes through interaction with antioxidant response element. Antioxidant and protective enzymes induced via Nrf-2 through OA application countering the toxic effects of ROS imbalance include superoxide dismutase, heme oxygenase-1, and glutathione reductase. OA also increases the amount of hepatic GSH level and GSH: GSSG ratio.50 GSH, a primary non-enzymatic antioxidant protector, scavenges ROS effectively, detoxifies xenobiotics and their metabolites while GSH: GSSG ratio is taken as cellular redox balance indicator.
OA significantly inhibits the metabolizing activity of CYPs51 including the inhibition of CYP2E1 expression, as OA hepatoprotective effect against chemicals, eg, ethanol, carbon tetrachloride and acetaminophen inducing hepatic injury has been reported,52 since those chemicals are primarily metabolized mainly by CYP2E1 leading to the production of toxic metabolites and ultimately oxidative stress, we can relate the hepatoprotective effect of OA to the inhibition or reduction of CYP2E1 isoforms metabolic activities.
Western blot analysis
Combination therapy with CDDP and OA synergistically enhanced the HepG2 cell apoptosis in CDDP/OA-LCC treatment
The lack of sensitivity to CDDP-based chemotherapy was probably resulted from the cross-competition for transcriptional coactivators and suppression of chemotherapy-induced p53 stabilization and activation. OA in the CDDP/OA-LCC NPs significantly enhanced the sensitivity of HepG2 cells toward CDDP and ultimately enhanced the apoptosis through a number of pathways, as clearly indicated in Figure 7, with the greater intensity of pro-apoptotic proteins and a lesser concentration of anti-apoptotic protein bands in the OA treated groups.
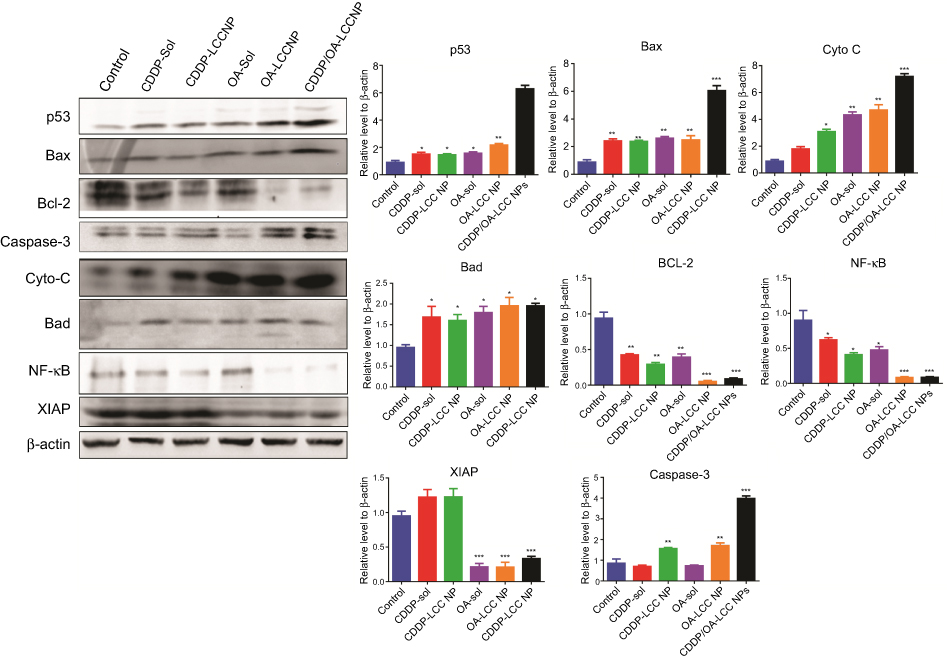 | Figure 7 Western blot analysis of protein levels (p53, Bax, Bad, Cyto-C, caspase-3, NF-κB, Bcl-2, and XIAP) after treating HepG2 cells with CDDP-Sol, CDDP-LCC NP, OA-Sol, OA-LCC NP, CDDP/OA-LCC NP in vitro. β-actin was used as a loading control. Quantification of protein level using Image J. Data presented as mean±SD (n=3). Abbreviations: CDDP, cisplatin; OA, oleanolic acid; LCC, lipid coated calcium carbonate; NP, nanoparticles. |
Activated P13K/Akt/mTOR pathway attenuates the antitumor effects of chemotherapeutics leading to cancer cells survival and its proliferation,53 which plays a pivotal role in cancer cells resistance toward cisplatin and reducing its antitumor efficacy.54 As revealed in Figure 7, OA increases the apoptosis through Bad overexpression by suppressing the mTOR pathway activation via AMPK pathway,11 which inhibits mTOR phosphorylation to suppress protein synthesis in cancer cells.55 OA also significantly enhances the antitumor activity via p53-induced caspase-mediated pro-apoptotic signaling pathway.56 As shown in Figure 7, p53 pathway proapoptotic proteins, such as p53, Bax, cytochrome C, and caspase-3 were upregulated, while the anti-apoptotic Bcl-2 expression was decreased. P53, a major regulator in CDDP-induced cancer cell apoptosis57 via a transcription-dependent or independent pathway. Increased Bax can rupture the outer mitochondrial membrane to induce the cytochrome C release to activate the generation of caspase-3 and ultimately induces extrinsic apoptosis, while downregulated Bcl-2 leads to the intrinsic apoptosis through reduced formation of Bcl-2/Bax heterodimers which promotes the cytochrome C release for the initiation of apoptosis through caspase activation.58 OA exerts an AMPK-dependent antitumor activity to increase p53 nuclear accumulation and apoptosis.59
OA also greatly enhances the apoptosis by deactivating the NF-κB pathway.12 Nuclear factor-Kb (NF-κB), on activation, enhances the apoptotic potential of chemotherapeutic agents, presenting its crucial role in the development of resistance to cisplatin in cancer cells.60
Activated NF-κB results in chemotherapy resistance probably through the cross-competition for transcriptional coactivators and suppression of chemotherapy-induced p53 stabilization.61 NF-κB promotes cell survival by expressing anti-apoptotic proteins BCL-2 family proteins, or by directly blocking caspase activation through increasing the expression of IAPs, especially X-linked inhibitor of apoptotic protein (XIAP).62 XIAP, is highly expressed in the tumor samples causing poor prognosis and tumor recurrence. In our western blot analysis as shown in Figure 7, OA downgraded the expressions of NF-κB targeted genes, ie, BCL-2 and XIAP as compared to CDDP-treated groups alone. The basic mechanism is that OA downgrades PI3K/AKT/mTOR/NF-κB phosphorylation,12 resulted in the down expression of anti-apoptotic proteins like Bcl-2 and XIAP and ultimately enhanced HepG2 cells apoptosis for CDDP/OA-LCC NPs.
Conclusion
In summary, a pH-sensitive, CC NPs, co-loading cisplatin and oleanolic acid (CDDP/OA-LCC NPs) against HCC were prepared using a microemulsion method. NPs were optimized based on their particle size and entrapment efficiency. Various studies were conducted to determine their safety, efficacy, and suitability in handling the HCC. In vitro release study confirmed the pH sensitivity of the delivery vehicle as greater amount of drug was released at acidic pH as compared to basic pH medium, making it an ideal carrier for chemotherapeutics. In vitro cell viability studies confirmed CDDP and OA synergistic apoptotic effect against HCC in the CDDP/OA-LCC NPs, as dual-loaded NPs showed enhanced cancer cell growth inhibition than either of the single drug NPs as well as their individual free drugs solution. Toxicity evaluation study confirmed the safety profile of this combination therapy as OA in the dual-loaded NP system greatly reduced the cisplatin-induced hepatotoxicity. These results demonstrated that CDDP/OA-LCC NPs is an ideal approach in dealing with HCC alongside keeping the cisplatin toxicity parameters in check.
Acknowledgments
This research was supported by National Natural Science Foundation of China (NSFC) (No. 81673368), the Fundamental Research Fund for the Central Universities of Huazhong University of Science and Technology (HUST) (2016YXMS140, 2016YXMS147, 2016JCTD109, and 2014TS090).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Choo SP, Tan WL, Goh BK, Tai WM, Zhu AX. Comparison of hepatocellular carcinoma in Eastern versus Western populations. Cancer. 2016;122(22):3430–3446. doi:10.1002/cncr.30237
2. Abdel-Hamid NM, Abass SA, Mohamed AA, Hamid DM. Herbal management of hepatocellular carcinoma through cutting the pathways of the common risk factors. Biomed Pharmacother. 2018;107:1246–1258. doi:10.1016/j.biopha.2018.08.104
3. Bertuccio P, Turati F, Carioli G, et al. Global trends and predictions in hepatocellular carcinoma mortality. J Hepatol. 2017;67(2):302–309. doi:10.1016/j.jhep.2017.03.011
4. Menahem B, Lubrano J, Duvoux C, et al. Liver transplantation versus liver resection for hepatocellular carcinoma in intention to treat: an attempt to perform an ideal meta‐analysis. Liver Transplant. 2017;23(6):836–844. doi:10.1002/lt.24758
5. Bruix J, Takayama T, Mazzaferro V, et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015;16(13):1344–1354. doi:10.1016/S1470-2045(15)00198-9
6. Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332(2):237–248. doi:10.1016/j.canlet.2012.01.007
7. Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869. doi:10.1038/onc.2011.627
8. Al-Malki AL, Sayed AAR. Thymoquinone attenuates cisplatin-induced hepatotoxicity via nuclear factor kappa-β. BMC Complement Altern Med. 2014;14(1):282. doi:10.1186/1472-6882-14-282
9. Yu Z, Sun W, Peng W, Yu R, Li G, Jiang T. Pharmacokinetics in vitro and in vivo of two novel prodrugs of oleanolic acid in rats and its hepatoprotective effects against liver injury induced by CCl4. Mol Pharm. 2016;13(5):1699–1710. doi:10.1021/acs.molpharmaceut.6b00129
10. Liese J, Abhari BA, Fulda S. Smac mimetic and oleanolic acid synergize to induce cell death in human hepatocellular carcinoma cells. Cancer Lett. 2015;365(1):47–56. doi:10.1016/j.canlet.2015.04.018
11. Liu J, Zheng L, Wu N, et al. Oleanolic acid induces metabolic adaptation in cancer cells by activating the AMP-activated protein kinase pathway. J Agric Food Chem. 2014;62(24):5528–5537. doi:10.1021/jf500622p
12. Wu J, Yang C, Guo C, et al. SZC015, a synthetic oleanolic acid derivative, induces both apoptosis and autophagy in MCF-7 breast cancer cells. Chem Biol Interact. 2016;244:94–104. doi:10.1016/j.cbi.2015.11.013
13. Nie H, Wang Y, Qin Y, Gong XG. Oleanolic acid induces autophagic death in human gastric cancer cells in vitro and in vivo. Cell Biol Int. 2016;40(7):770–778. doi:10.1002/cbin.10612
14. Wang X, Bai H, Zhang X, et al. Inhibitory effect of oleanolic acid on hepatocellular carcinoma via ERK–p53-mediated cell cycle arrest and mitochondrial-dependent apoptosis. Carcinogenesis. 2013;34(6):1323–1330. doi:10.1093/carcin/bgt058
15. Shyu M-H, Kao T-C, Yen G-C. Oleanolic acid and ursolic acid induce apoptosis in HuH7 human hepatocellular carcinoma cells through a mitochondrial-dependent pathway and downregulation of XIAP. J Agric Food Chem. 2010;58(10):6110–6118. doi:10.1021/jf100574j
16. Sarfraz M, Afzal A, Raza SM, et al. Liposomal co-delivered oleanolic acid attenuates doxorubicin-induced multi-organ toxicity in hepatocellular carcinoma. Oncotarget. 2017;8(29):47136. doi:10.18632/oncotarget.17559
17. Khan IU, Khan RU, Asif H, et al. Co-delivery strategies to overcome multidrug resistance in ovarian cancer. Int J Pharm. 2017. doi:10.1016/j.ijpharm.2017.09.060
18. Liao J, Zheng H, Fei Z, et al. Tumor-targeting and pH-responsive nanoparticles from hyaluronic acid for the enhanced delivery of doxorubicin. Int J Biol Macromol. 2018;113:737–747. doi:10.1016/j.ijbiomac.2018.03.004
19. Li R, Liu B, Gao J. The application of nanoparticles in diagnosis and theranostics of gastric cancer. Cancer Lett. 2017;386:123–130. doi:10.1016/j.canlet.2016.10.032
20. Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. Polyvalent oligonucleotide gold nanoparticle conjugates as delivery vehicles for platinum (IV) warheads. J Am Chem Soc. 2009;131(41):14652–14653. doi:10.1021/ja9071282
21. Wang Z, Deng X, Ding J, Zhou W, Zheng X, Tang G. Mechanisms of drug release in pH-sensitive micelles for tumour targeted drug delivery system: a review. Int J Pharm. 2018;535(1–2):253–260. doi:10.1016/j.ijpharm.2017.11.003
22. Kim SK, Park H, Lee JM, Na K, Lee ES. pH‐responsive starch microparticles for a tumor‐targeting implant. Polym Adv Technol. 2018;29(5):1372–1376. doi:10.1002/pat.v29.5
23. Guragain S, Torad NL, Alghamdi YG, et al. Synthesis of nanoporous calcium carbonate spheres using double hydrophilic block copolymer poly (acrylic acid-bN-isopropylacrylamide). Mater Lett. 2018;230:143–147. doi:10.1016/j.matlet.2018.07.060
24. Dong Z, Feng L, Zhu W, et al. CaCO3 nanoparticles as an ultra-sensitive tumor-pH-responsive nanoplatform enabling real-time drug release monitoring and cancer combination therapy. Biomaterials. 2016;110:60–70. doi:10.1016/j.biomaterials.2016.09.025
25. Gong M-Q, Wu J-L, Chen B, Zhuo R-X, Cheng S-X. Self-assembled polymer/inorganic hybrid nanovesicles for multiple drug delivery to overcome drug resistance in cancer chemotherapy. Langmuir. 2015;31(18):5115–5122. doi:10.1021/acs.langmuir.5b00542
26. Zhao P, Li M, Wang Y, et al. Enhancing anti-tumor efficiency in hepatocellular carcinoma through the autophagy inhibition by miR-375/sorafenib in lipid-coated calcium carbonate nanoparticles. Acta Biomater. 2018;72:248–255. doi:10.1016/j.actbio.2018.03.022
27. Guo S, Wang Y, Miao L, et al. Lipid-coated cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy. ACS Nano. 2013;7(11):9896–9904. doi:10.1021/nn403606m
28. Maleki Dizaj S, Barzegar-Jalali M, Zarrintan MH, Adibkia K, Lotfipour F. Calcium carbonate nanoparticles as cancer drug delivery system. Expert Opin Drug Deliv. 2015;12(10):1649–1660. doi:10.1517/17425247.2015.1049530
29. Kim SK, Foote MB, Huang L. Targeted delivery of EV peptide to tumor cell cytoplasm using lipid coated calcium carbonate nanoparticles. Cancer Lett. 2013;334(2):311–318. doi:10.1016/j.canlet.2012.07.011
30. Cai S, Shi C-H, Zhang X, et al. Self-microemulsifying drug-delivery system for improved oral bioavailability of 20 (S)-25-methoxyl-dammarane-3β, 12β, 20-triol: preparation and evaluation. Int J Nanomedicine. 2014;9:913.
31. Amna T, Hassan MS, Yang J, et al. Virgin olive oil blended polyurethane micro/nanofibers ornamented with copper oxide nanocrystals for biomedical applications. Int J Nanomedicine. 2014;9:891. doi:10.2147/IJN.S54113
32. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2)440–446. CAN-0009-1947.
33. Petrović S, Tačić A, Savić S, Nikolić V, Nikolić L, Savić S. Sulfanilamide in solution and liposome vesicles; in vitro release and UV-stability studies. Saudi Pharm J. 2017;25(8):1194–1200. doi:10.1016/j.jsps.2017.09.003
34. Wu H, Zhong Q, Zhong R, et al. Preparation and antitumor evaluation of self-assembling oleanolic acid-loaded Pluronic P105/D-α-tocopheryl polyethylene glycol succinate mixed micelles for non-small-cell lung cancer treatment. Int J Nanomedicine. 2016;11:6337. doi:10.2147/IJN.S119839
35. Cheng Y, Zhao P, Wu S, et al. Cisplatin and curcumin co-loaded nano-liposomes for the treatment of hepatocellular carcinoma. Int J Pharm. 2018;545(1–2):261–273. doi:10.1016/j.ijpharm.2018.05.007
36. Awasthi BP, Kathuria M, Pant G, Kumari N, Mitra K. Plumbagin, a plant-derived naphthoquinone metabolite induces mitochondria mediated apoptosis-like cell death in Leishmania donovani: an ultrastructural and physiological study. Apoptosis. 2016;21(8):941–953. doi:10.1007/s10495-016-1259-9
37. ud Din F, Mustapha O, Kim DW, et al. Novel dual-reverse thermosensitive solid lipid nanoparticle-loaded hydrogel for rectal administration of flurbiprofen with improved bioavailability and reduced initial burst effect. Eur J Pharm Biopharm. 2015;94:64–72.
38. Chen S, Li F, Zhuo R-X, Cheng S-X. Efficient non-viral gene delivery mediated by nanostructured calcium carbonate in solution-based transfection and solid-phase transfection. Mol Biosyst. 2011;7(10):2841–2847. doi:10.1039/c1mb05147d
39. Bnyan R, Khan I, Ehtezazi T, et al. Surfactant effects on lipid-based vesicles properties. J Pharm Sci. 2018;107(5):1237–1246. doi:10.1016/j.xphs.2018.01.005
40. Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, Barthélémy P. Nucleoside–lipid-based nanoparticles for cisplatin delivery. ACS Nano. 2011;5(11):8649–8655. doi:10.1021/nn202291k
41. Zhang J, Chen Y, Li X, Liang X, Luo X. The influence of different long-circulating materials on the pharmacokinetics of liposomal vincristine sulfate. Int J Nanomedicine. 2016;11:4187. doi:10.2147/IJN.S109547
42. Rosen H, Abribat T. The rise and rise of drug delivery. Nat Rev Drug Discov. 2005;4(5):381. doi:10.1038/nrd1876
43. Yan W, Ma X, Zhao X. Baicalein induces apoptosis and autophagy of breast cancer cells via inhibiting Pi3K/aKT pathway in vivo and vitro. Drug Des Devel Ther. 2018;12:3961–3972. doi:10.2147/DDDT.S181939
44. Yang N, Jiang Y, Zhang H, et al. Active targeting docetaxel-PLA nanoparticles eradicate circulating lung cancer stem-like cells and inhibit liver metastasis. Mol Pharm. 2014;12(1):232–239. doi:10.1021/mp500568z
45. Omar HA, Mohamed WR, Arafa E-SA, et al. Hesperidin alleviates cisplatin-induced hepatotoxicity in rats without inhibiting its antitumor activity. Pharmacol Rep. 2016;68(2):349–356. doi:10.1016/j.pharep.2015.09.007
46. Bentli R, Parlakpinar H, Polat A, Samdanci E, Sarihan ME, Sagir M. Molsidomine prevents cisplatin-induced hepatotoxicity. Arch Med Res. 2013;44(7):521–528. doi:10.1016/j.arcmed.2013.09.013
47. Lu Y, Cederbaum AI. Cisplatin-induced hepatotoxicity is enhanced by elevated expression of cytochrome P450 2E1. Toxicol Sci. 2005;89(2):515–523. doi:10.1093/toxsci/kfj031
48. Wang X, Ye X-L, Liu R, et al. Antioxidant activities of oleanolic acid in vitro: possible role of Nrf2 and MAP kinases. Chem Biol Interact. 2010;184(3):328–337. doi:10.1016/j.cbi.2010.01.034
49. Chai J, Du X, Chen S, et al. Oral administration of oleanolic acid, isolated from Swertia mussotii Franch, attenuates liver injury, inflammation, and cholestasis in bile duct-ligated rats. Int J Clin Exp Med. 2015;8(2):1691.
50. Liu C, Huang X, Li Y, et al. The anti-portal hypertension effect of oleanolic acid in CCl4-induced cirrhosis rats. J Chin Med Mater. 2012;35(6):930–935.
51. Kim K-A, Lee J-S, Park H-J, et al. Inhibition of cytochrome P450 activities by oleanolic acid and ursolic acid in human liver microsomes. Life Sci. 2004;74(22):2769–2779. doi:10.1016/j.lfs.2003.10.020
52. Liu J, Wang X, Liu R, et al. Oleanolic acid co-administration alleviates ethanol-induced hepatic injury via Nrf-2 and ethanol-metabolizing modulating in rats. Chem Biol Interact. 2014;221:88–98. doi:10.1016/j.cbi.2014.07.017
53. Shackelford DB, Shaw RJ. The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563. doi:10.1038/nrc2676
54. Hahne JC. Platinum-resistance and AKT over-expression in ovarian cancer. Int J Gynecol Clin Pract. 2015; 2:14–21.
55. Sikka A, Kaur M, Agarwal C, Deep G, Agarwal R. Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle. 2012;11(7):1374–1382. doi:10.4161/cc.19798
56. Kim G-J, Jo H-J, Lee K-J, Choi JW, An JH. Oleanolic acid induces p53-dependent apoptosis via the ERK/JNK/AKT pathway in cancer cell lines in prostatic cancer xenografts in mice. Oncotarget. 2018;9(41):26370. doi:10.18632/oncotarget.25316
57. Herůdková J, Paruch K, Khirsariya P, et al. Chk1 inhibitor SCH900776 effectively potentiates the cytotoxic effects of platinum-based chemotherapeutic drugs in human colon cancer cells. Neoplasia. 2017;19(10):830–841. doi:10.1016/j.neo.2017.08.002
58. Safta TB, Ziani L, Favre L, et al. Granzyme B–activated p53 interacts with Bcl-2 to promote cytotoxic lymphocyte–mediated apoptosis. J Immunol. 2015;194(1):418–428. doi:10.4049/jimmunol.1401978
59. Fox MM, Phoenix KN, Kopsiaftis SG, Claffey KP. AMP-activated protein kinase α 2 isoform suppression in primary breast cancer alters AMPK growth control and apoptotic signaling. Genes Cancer. 2013;4(1–2):3–14. doi:10.1177/1947601913486346
60. Godwin P, Baird A-M, Heavey S, Barr M, O’Byrne K, Gately KA. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front Oncol. 2013;3:120. doi:10.3389/fonc.2013.00120
61. Yu L, Li L, Medeiros LJ, Young KH. NF-κB signaling pathway and its potential as a target for therapy in lymphoid neoplasms. Blood Rev. 2017;31(2):77–92. doi:10.1016/j.blre.2016.10.001
62. Song W, Tang Z, Shen N, et al. Combining disulfiram and poly (l-glutamic acid)-cisplatin conjugates for combating cisplatin resistance. J Control Release. 2016;231:94–102. doi:10.1016/j.jconrel.2016.02.039
Supplementary materials
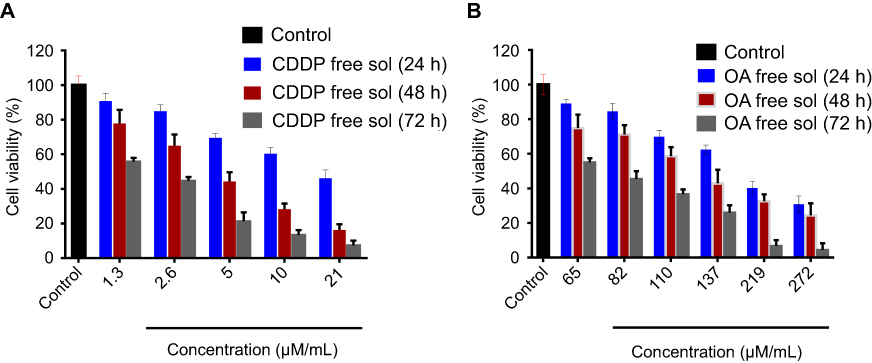 | Figure S1 Cytotoxicity assay of free CDDP and free OA against HepG2 cells. (A) MTT assay results of free CDDP solution against HepG2 cells for 24, 48, and 72 h; (B) MTT assay results of free OA solution against HepG2 cells for 24, 48, and 72 h. Data presented as mean±SD (n=5).Abbreviations: CDDP, cisplatin; OA, oleanolic acid. |
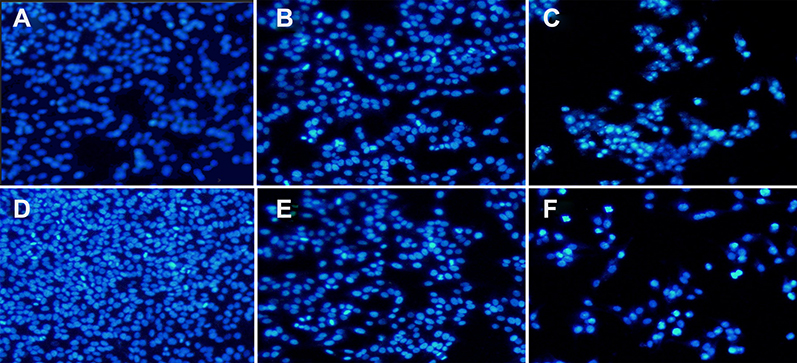 | Figure S2 CDDP/OA-LCC NPs induces apoptosis in HepG2 cells. Hoechst 33,258 nuclear staining cell apoptosis images. (A) Control; (B) CDDP-Sol; (C) CDDP-LCC NPs; (D) OA-Sol; (E) OA-LCC NPs; (F) CDDP/OA-LCC NPs. Abbreviations: CDDP, cisplatin; OA, oleanolic acid; LCC, lipid coated calcium carbonate; NP, nanoparticles. |
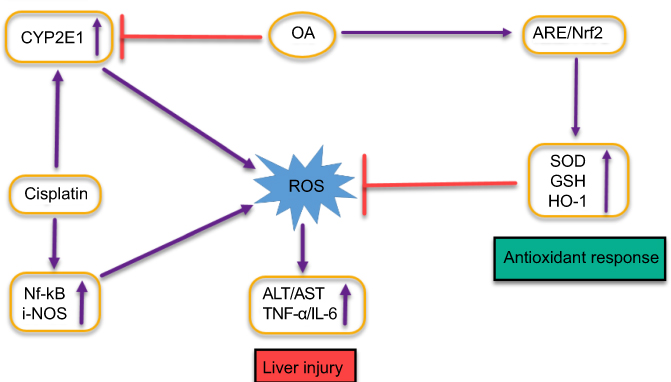 | Figure S3 Graphical representation depicting the proposed protective mechanisms of oleanolic acid (OA) co-administration on liver injury induced by cisplatin. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.