Back to Journals » Drug Design, Development and Therapy » Volume 20
Swarm Intelligence in Drug Discovery Applications: Unlocking Deeper Insights on the Identification and Optimization of Potential Drug Candidates
Authors Gao Z, Ding P, Oguztuzun C ![]() , Xu R
, Xu R ![]()
Received 31 March 2026
Accepted for publication 18 June 2026
Published 23 June 2026 Volume 2026:20 561251
DOI https://doi.org/10.2147/DDDT.S561251
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Solomon Tadesse Zeleke
Zhenxiang Gao,1,* Pingjian Ding,2,* Cerag Oguztuzun,1,* Rong Xu1
1Center for Artificial Intelligence in Drug Discovery, School of Medicine, Case Western Reserve University, Cleveland, OH, 44106, USA; 2School of Computer Science, University of South China, Hengyang, 421001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Rong Xu, Center for Artificial Intelligence in Drug Discovery, School of Medicine, Case Western Reserve University, Cleveland, Ohio, 44106, USA, Email [email protected]
Abstract: Swarm-based analysis technology represents a class of computational approaches inspired by biological systems, such as bees or ants, to solve complex and high-dimensional problems through the collective behavior of interacting agents. This review provides an overview of swarm intelligence methods in drug discovery, covering foundational concepts, major algorithms, and representative applications in molecular docking, drug screening, de novo molecular design, and combinatorial chemical space exploration. We summarize classical swarm-based approaches and discuss recent hybrid frameworks integrating swarm intelligence with machine learning, deep learning, and large language model (LLM)-based multi-agent systems. In addition to highlighting their potential for adaptive search and multi-objective optimization, we critically examine current limitations, including scalability, convergence reliability, parameter sensitivity, and computational cost in high-dimensional biomedical settings. We further emphasize that many emerging frameworks, particularly LLM-enhanced and multi-agent swarm systems, remain at an early stage and have not yet been extensively validated in real-world drug discovery pipelines. Overall, swarm-based methods provide flexible and interpretable strategies for complex optimization tasks, while continued advances in data integration, benchmarking, and biologically informed modeling will be important for their broader application in drug discovery.
Keywords: swarm intelligence model, drug discovery, artificial intelligence, large language model
Introduction
Drug discovery is a complex, multi-stage process involving target identification, lead optimization, and rigorous safety and efficacy evaluations.1 This process requires navigating large and high-dimensional search spaces while balancing multiple, often competing objectives, such as biological activity, toxicity, pharmacokinetics, and synthetic feasibility.2 Despite substantial progress in experimental high-throughput technologies, structural biology, and data-driven predictive modeling, many candidates still fail during development, timelines frequently exceed a decade, and costs continue to rise.3 These persistent challenges continue to drive the development of computational strategies that can efficiently explore high-dimensional search spaces, generate novel molecular structures, and support multiple stages of the drug discovery pipeline.
Swarm intelligence (SI) represents a class of computational methods inspired by the collective behavior of decentralized and self-organized systems observed in nature, such as ant colonies, bird flocks, and bee swarms.4 Algorithms such as particle swarm optimization (PSO), ant colony optimization (ACO), and artificial bee colony (ABC) methods model how agents interact locally to produce coordinated global behavior.5 These approaches are typically population-based and rely on iterative updates driven by shared information, allowing them to explore complex search spaces without requiring gradient information or large amounts of labeled data.6 Their ability to handle discrete and continuous optimization problems, as well as to accommodate multiple objectives, has led to applications in biomedical domains.7 In recent years, several reviews have summarized the development of swarm intelligence algorithms and their applications in optimization problems. However, most prior surveys have either focused broadly on computational intelligence methods or discussed swarm-based techniques without emphasizing their translational role across the modern drug discovery pipeline.4–6
In drug discovery, swarm-based methods have been applied to a range of tasks, including molecular docking, drug screening, de novo molecular design, feature selection in quantitative structure-activity relationship (QSAR) and quantitative structure-property relationship (QSPR) modeling, and exploration of large combinatorial chemical spaces.8–11 These applications leverage the swarm intelligence to address the high dimensionality, multimodality, and uncertainty inherent in pharmaceutical optimization problems. In drug screening, adaptive and multi-swarm strategies help maintain diversity and avoid focusing too early on a small set of candidates.8 In de novo molecular design, multi-objective formulations allow different drug-relevant properties, such as activity, stability, drug-likeness, and synthesizability, to be optimized simultaneously.9 In QSAR and QSPR modeling, swarm methods support efficient selection of relevant features and parameter settings without exhaustive search.10 For large-scale combinatorial spaces, where the number of possible drug combinations grows rapidly, swarm methods improve tractability by decomposing the search into smaller interacting components through cooperative co-evolution, while constraint-aware strategies focus the search on feasible regions.11
Recent work has increasingly explored the integration of SI with artificial intelligence (AI) including machine learning (ML) and deep learning (DL), aiming to combine data-driven prediction with search-based optimization.12,13 Hybrid frameworks couple swarm algorithms with neural networks or learned representations to enable more efficient and smoother exploration of chemical space. In emerging directions, large language models are incorporated as components within swarm-like multi-agent systems.14 These models can incorporate information from diverse biomedical sources and provide structured guidance without task-specific retraining. Such integrations aim to mitigate limitations of standalone swarm methods while preserving advantages such as relatively low data requirements, transparent search processes, and natural support for multi-objective optimization.
The role of swarm-based methods in drug discovery continues to evolve alongside advances in computational technologies. This review summarizes swarm-based analysis approaches in this domain, covering core algorithms, representative applications, recent methodological extensions, and existing challenges (Figure 1). The literature discussed in this review was identified through searches of major scientific databases, including PubMed, Web of Science, and Google Scholar, using keywords related to swarm intelligence, particle swarm optimization, ant colony optimization, molecular docking, virtual screening, de novo molecular design, QSAR/QSPR modeling, and multi-agent systems. Priority was given to representative studies describing major methodological developments, applications in drug discovery, and emerging computational frameworks. By integrating insights from the literature, we aim to characterize the potential and limitations of swarm-based approaches and to highlight promising directions for future research.
 |
Figure 1 Overview of swarm intelligence algorithms and their applications in drug discovery. The upper panel illustrates the major categories of swarm-based computational approaches, including Particle Swarm Optimization (PSO), Ant Colony Optimization (ACO), and Artificial Bee Colony (ABC), to more recent hybrid frameworks integrating machine learning and deep learning methods. The lower panel summarizes representative applications of swarm-based approaches in drug discovery, including molecular docking, drug screening, de novo molecular design, and drug combination discovery. |
Overview of Swarm Intelligence Algorithms
The development of swarm intelligence methods has evolved through several stages over the past three decades. Early work primarily focused on heuristic optimization strategies designed to solve combinatorial and continuous optimization problems. More recently, swarm-based algorithms have increasingly been integrated with broader AI frameworks, where they are integrated with machine learning, deep learning and large language models to enhance model performance and enable large-scale data-driven discovery.
The Classical Era: Heuristic Search
The early development of swarm intelligence algorithms was primarily centered on heuristic optimization strategies inspired by collective behaviors observed in biological systems (Figure 1). Among these, particle swarm optimization (PSO) is one of the most widely studied approaches, originally motivated by the coordinated movement of bird flocks during foraging.15 In PSO, candidate solutions are modeled as particles that move through the search space, guided by both their own past best positions and the best solution identified by the swarm.
Another foundational swarm-based algorithm is ant colony optimization (ACO), which is inspired by the foraging behavior of ants that deposit pheromone trails to guide others toward efficient paths between their nest and food sources.5 In ACO, artificial pheromone trails and probabilistic path selection are used to construct solutions, enabling the identification of high-quality paths in combinatorial optimization problems such as routing, scheduling, and graph traversal.
Building on these early developments, a variety of additional swarm-based algorithms have been proposed to tackle diverse optimization tasks. For instance, the artificial bee colony (ABC) algorithm models the foraging behavior of honeybee colonies, where employed, onlooker, and scout bees collectively balance exploration and exploitation.16 Similarly, the firefly algorithm (FA) was inspired by the attraction behavior of fireflies, in which candidate solutions are guided toward brighter peers that represent better objective values.17
While these methods have shown effectiveness in global optimization tasks, their performance is often problem-dependent and sensitive to parameter settings (Table 1). In biomedical applications, they have been explored for tasks such as feature selection, molecular docking optimization, and biomarker discovery. However, their scalability and robustness remain limited in high-dimensional and noisy settings commonly encountered in biological and clinical data, which has limited their widespread adoption in large-scale data-driven studies.
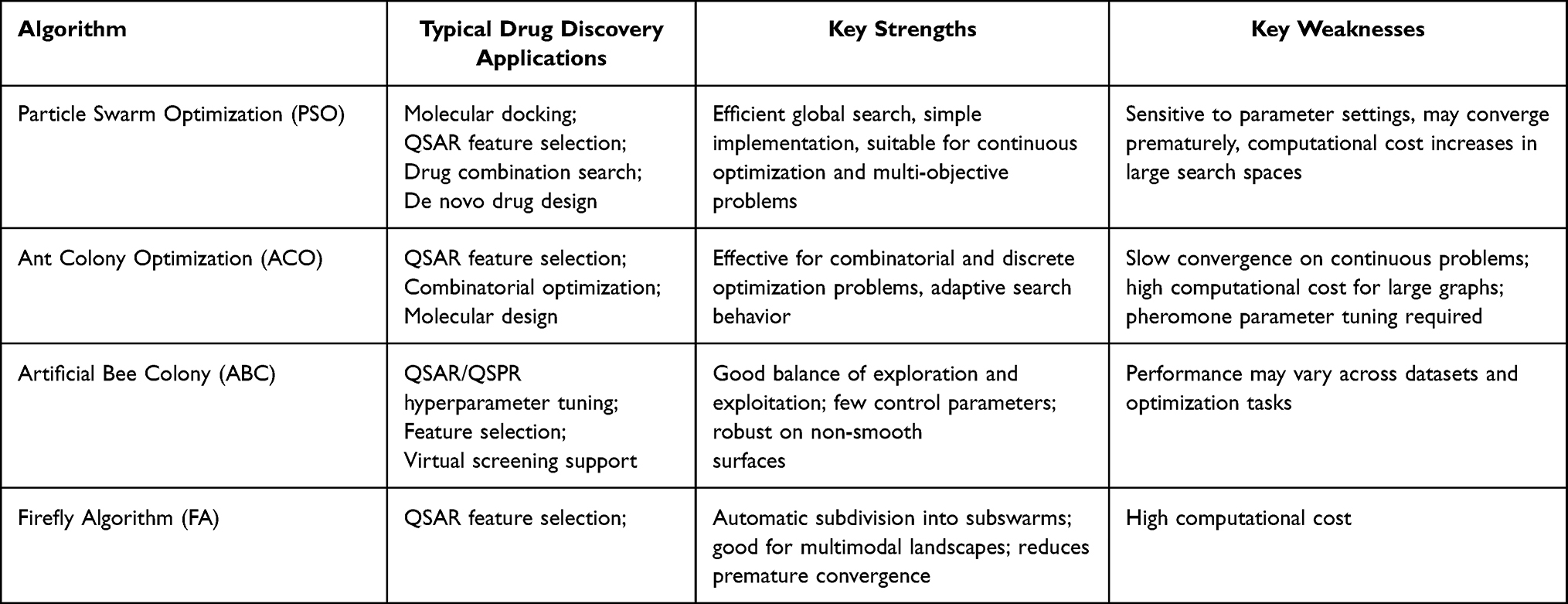 |
Table 1 Major Swarm Intelligence Approaches in Drug Discovery |
The Hybrid Era: Integrating with ML/DL
The development of SI methods has increasingly moved toward integration with both predictive and generative machine learning (ML) and deep learning (DL) frameworks (Figure 1). SI is commonly used as an optimization component within ML/DL pipelines rather than as a standalone method.18
One common use of hybrid SI-ML approaches is feature-selection and hyperparameter tuning. In biomedical applications, including QSAR modeling and classification tasks, methods such as PSO and ACO are used for feature selection, which can improve the performance of models such as SVM and random forests for drug screening.19 SI has been used in molecular docking and virtual screening, to optimize model parameters or search processes.8 In deep learning settings, SI is primarily used for tuning hyperparameters or network structures.20 Compared with grid search or random search, SI provides an efficient way to explore the parameter space. However, the computational cost can be high, especially when training large models. Hybrid approaches combining SI with generative ML/DL models have been explored for molecular design and optimization, where swarm-based search is applied in latent or graph-based chemical spaces to guide the generation of candidate molecules with desired properties.21 Overall, these integrations extend classical optimization strategies into data-intensive AI-driven settings.
Applications of Swarm-Based Methods in Drug Discovery
Molecular Docking
Molecular docking is a computational technique used to predict how a ligand binds to a target macromolecule, typically a protein, and to estimate the strength of their interaction.22,23 Its goal is to identify the most appropriate binding pose of a ligand within a protein’s binding site by searching the conformational space and evaluating each pose using a scoring function. The process of docking flexible ligand with rigid receptor can be depicted into three stages:24,25 1) translation: the ligand is moved in the three-dimensional Cartesian space to position it within or near the target protein’s binding site. 2) orientation: the ligand is rotated as a rigid body to explore different spatial orientations within the binding site. 3) conformation adjustment: the internal flexibility of the ligand is considered by adjusting the torsion angles of its rotatable bonds. As a result, docking programs output one or more low-energy binding conformations, which can be used for virtual screening, binding mode analysis, and structure-based drug design.24 In practice, molecular docking involves two core components: the search algorithm and the scoring function.26 The search algorithm explores possible ligand postures by sampling translation, orientation, and internal conformational degrees of freedom (eg, torsion angles of rotatable bonds). The scoring function evaluates each candidate poses by estimating the binding affinity based on intermolecular interactions such as van der Waals forces, electrostatics, and hydrogen bonding.27
In molecular docking, swarm intelligence methods are employed as search strategies to explore the ligand-target conformational space, while the scoring function evaluates the quality of each searched pose. Most docking software packages adopt flexible ligand docking under the assumption that the binding site is already known, requiring prior identification of the region containing potential binding sites.28 To enhance docking accuracy and efficiency, various SI-based strategies have been developed. A multi-swarm competitive algorithm29 improves docking for highly flexible ligands by combining competitive feedback between swarms with adaptive gradient-based local refinement. A parallel master-slave multi-swarm particle swarm optimization (PSO) strategy30 facilitates information exchange and fully parallel execution, resulting in improved docking accuracy and virtual screening performance compared with Vina-based methods. AutoDock Vina itself employs an empirical, knowledge-based scoring function to approximate protein-ligand binding free energy and combines stochastic global sampling with iterative local optimization using a quasi-Newton search method. Moldina,31 built on AutoDock Vina, integrates PSO to enable simultaneous multi-ligand docking, achieving comparable accuracy while reducing computational time by several hundred-fold. RDPSOVina32 replaces Vina’s original search strategy with a random drift particle swarm optimization algorithm while retaining the same scoring function, enhancing global exploration via stochastic drift and mutation mechanisms. For DNA-ligand docking, specialized SI frameworks have been developed. For example, FCMGSO33 integrates Fuzzy C-Means Clustering with Glowworm Swarm Optimization to provide an efficient and accurate approach for DNA-ligand mapping. These examples illustrate how integrating SI with established docking frameworks can improve both the efficiency and accuracy of ligand-target interaction predictions.
In some cases, it is necessary to dock a ligand across the entire surface of a protein without prior knowledge of the binding site to either identify potential binding sites or reproduce the crystallographic docking pose. This approach is referred to as blind docking.26,34 The key distinction between blind docking and conventional docking is that the search algorithms must explore the entire protein surface to identify candidate binding sites.35 For example, a hybrid docking algorithm34 combines quantum particle swarm optimization with a diversity-guided attractor strategy and a modified Solis-Wets local search to enhance search quality. This framework supports both conventional and blind docking by enabling global-scale exploration. LightDock36 is an open-source docking platform for protein-protein, protein-peptide, and protein-DNA interactions. It uses glowworm swarm optimization to globally sample the interaction energy landscape, capturing multiple binding modes through swarm-based clustering and score-based ranking. These approaches demonstrate how SI can facilitate comprehensive, global searches in complex docking scenarios where the binding site is unknown.
Swarm-based docking methods are particularly useful for complex docking tasks involving flexible ligands or large conformational spaces. Compared with conventional local search approaches, swarm-based methods can improve global exploration by maintaining multiple candidate solutions during optimization. Hybrid methods that combine swarm optimization with established docking platforms, such as AutoDock Vina, suggest that swarm-based strategies are effective when integrated with efficient scoring functions and local refinement procedures. However, some swarm-based methods improve docking accuracy at the cost of increased computational complexity or longer optimization times, indicating a trade-off between search performance and computational efficiency.
QSAR/QSPR Modelling for Drug Screening
QSAR models quantify the relationship between molecular structures and their biological activities, while QSPR predicts physicochemical properties.37,38 QSAR typically involves calculating molecular descriptors, selecting the most relevant features, building a predictive model using statistical or machine learning methods, and applying the model to predict activities of new compounds and guide molecular design.
In practice, QSAR modeling can be decomposed into two core components: molecular descriptors which capture diverse molecular features, and the predictive model, which maps these features to target biological activity or physicochemical property. SI methods are widely employed within this framework as optimization tools. They are used not only for selecting the most relevant descriptors but also for tuning model hyperparameters, such as kernel settings, regularization coefficients, or other model-specific parameters. By simultaneously optimizing descriptor subsets and model hyperparameters, SI-based strategies enable QSAR models to capture key factors governing molecular activity or properties, thereby improving predictive performance and generalization.
The integration of SI into QSAR modeling can be broadly categorized into feature selection-oriented and predictive model selection-oriented methods. In feature selection, SI algorithms optimize high-dimensional descriptor spaces to identify subsets most relevant to biological activity or properties, improving model interpretability, generalization, and reducing redundancy. Examples include a hybrid Sinh Cosh Optimizer with the Arithmetic Optimization Algorithm for indoloquinoline derivatives,39 a chaotic-map-enhanced golden jackal optimizer for high-dimensional QSAR datasets,40 and binary swarm-based methods such as hybrid whale optimization for drug-induced liver injury prediction.41 Other SI applications, including firefly algorithm-SVR frameworks42 and modified binary PSO for cholinesterase inhibitor QSAR,43 further demonstrate the effectiveness of SI-driven feature selection in enhancing predictive accuracy and model robustness. In predictive model selection, SI algorithms optimize QSAR model structure and parameters, including model type, hyperparameters, and architecture. For example, a coati optimizer with elite opposite-based learning improved kernel ridge regression hyperparameter selection, enhancing classification and regression performance across seven chemical datasets.44
Overall, these studies demonstrate that SI provides a versatile and effective framework for both feature and predictive model optimization in QSAR modeling. By efficiently navigating high-dimensional descriptor spaces and guiding model configuration, SI-driven approaches enhance predictive accuracy, robustness, and generalization, highlighting their significant potential for accelerating data-driven drug discovery. However, the effectiveness of SI-based QSAR approaches often depends on dataset quality, descriptor design, and optimization settings. In some studies, the performance improvements over conventional optimization methods are relatively small and vary across datasets and prediction tasks. In addition, optimization can become computationally expensive when large descriptor sets, or complex predictive models are involved. In practice, SI-based methods appear to be most useful for high-dimensional QSAR problems where traditional search strategies become inefficient or difficult to scale.
Drug Combination Discovery
Identifying effective drug combinations is important for improving therapeutic efficacy, overcoming resistance, and reducing adverse effects.45 However, the vast combinatorial search space of possible drug pairs presents substantial challenges for traditional experimental and computational screening methods.46
Swarm-based approaches have been applied to explore large, high-dimensional search spaces for identifying candidate drug combinations. For example, in breast cancer therapy, PSO was used to infer model parameters and predict combinations of inhibitors targeting the MAPK and PI3K/AKT pathways.47 In addition, Kang et al48 further combined PSO with genetic algorithms to iteratively search for effective drug combinations involving imatinib in leukemia.
The performance of these approaches, however, depends strongly on the design of the objective function, the underlying biological model, and the quality of experimental feedback. Future work should focus on improving the scalability, robustness, and translational relevance of swarm-based drug combination discovery methods. For example, integrating swarm-based optimization with graph neural networks, reinforcement learning, or patient-specific data may improve the ability to optimize efficacy, toxicity, and resistance profiles simultaneously. In addition, closer collaboration between computational and experimental researchers will be important for validating predicted drug combinations through in vitro and in vivo studies and for improving the translation of computational predictions into clinically relevant therapeutic strategies.
De-Novo Drug Design
De novo drug design creates novel molecules with desired properties by exploring chemical space.49 SI has been employed to iteratively refine candidate molecules within this vast search space. Early applications adapted swarm algorithms such as PSO to combinatorial molecular design. For example, Hartenfeller et al50 developed the COLIBREE framework, which applies PSO to fragment-based de novo design around predefined scaffolds, using building blocks derived from pseudo-retrosynthetic fragmentation and similarity-based scoring functions. Subsequent work extended swarm-based methods to multi-objective optimization, addressing trade-offs in drug discovery, such as potency, toxicity, and solubility. For instance, Reutlinger et al51 applied ant colony optimization (ACO) to polypharmacology-guided design, where fragment selection is influenced by predicted activities across multiple targets.
Recent work has explored the integration of SI with learned molecular representations, allowing optimization in continuous latent spaces. For example, Winter et al52 combined PSO with a variational autoencoder-derived representation, enabling gradient-free optimization of multiple molecular properties, including predicted binding affinity to targets such as EGFR and BACE1, as well as properties related to drug-likeness. Similarly, ChemMORT53 integrates PSO with learned molecular representations to optimize multiple ADMET-related properties during molecule generation. In this framework, candidate molecules are iteratively refined based on predicted property scores to identify structures that balance efficacy, safety, and physicochemical constraints.
Beyond latent representation-based approaches, alternative SI frameworks operate directly on molecular structures. For instance, Devi et al54 introduced MOBifi, a multi-objective biofilm-inspired algorithm that models process such as attachment, maturation, and dispersal through chemotaxis-like movements and Pareto-based selection. In single-objective settings, Liu et al11 proposed SIB-SOMO, a swarm-based method that works directly on discrete molecular graphs without relying on latent representations, iteratively modifying molecules through substructure exchange, mutation, and random perturbations.
The effectiveness of these approaches depends strongly on the molecular representation, scoring function, and optimization objectives used during the search process. Latent-space methods rely on the quality of learned molecular representations, while graph-based methods depend on chemically meaningful mutation and substructure operations. In practice, swarm-based approaches appear particularly useful for multi-objective optimization problems where exhaustive exploration of chemical space is difficult. Future work may further improve these methods through integration with reaction prediction, synthetic accessibility estimation, and experimentally guided molecular constraints.
Hybrid Large Language Model and Swarm Technologies in Drug Discovery
Recent work has begun to explore the integration of large language models (LLMs) with swarm-based search strategies in drug discovery. In these approaches, LLMs provide structured knowledge, reasoning, or candidate generation, while swarm-inspired mechanisms iteratively refine hypotheses or molecular designs. This hybrid approach generally takes two forms: augmenting traditional swarm algorithms using LLMs, and designing multi-agent systems in which LLM-driven agents operate as cooperative “swarms” for hypothesis generation and molecular optimization tasks.
LLM-Enhanced Swarm Intelligence
LLMs have recently been incorporated into swarm optimization frameworks to guide updates, propose candidate solutions, or modify search operators. For example, Shinohara et al55 introduced Language Model Particle Swarm Optimization (LMPSO), in which particle updates are generated by an LLM conditioned on the current position, personal best, and global best solutions. This formulation allows the method to produce structured outputs, such as sequences or expressions, within a PSO-inspired framework. The approach has been evaluated on combinatorial optimization tasks, including the traveling salesman problem and symbolic regression, demonstrating feasibility in structured search spaces. Although not yet applied to drug discovery, its ability to handle discrete and structured representations suggests potential for molecular graph or sequence optimization in de novo drug design.
More broadly, the incorporation of LLMs into swarm optimization frameworks offers a promising direction for de novo drug discovery, particularly in tasks that require reasoning over chemical knowledge, multi-step design constraints, or optimization in discrete molecular spaces. By conditioning LLM-driven updates on domain-specific prompts, such as reaction rules, pharmacophore constraints, or ADMET filters, these hybrid approaches can generate chemically plausible modifications guided by relevant biochemical constraints. Furthermore, the capacity of LLMs to interact with external tools, including database queries, predictive models, and synthesizability assessments, enables the incorporation of additional constraints into the search process without requiring model retraining.
Multi-Agent LLM Swarms and Agentic Frameworks
Multi-agent systems powered by LLMs consist of multiple LLM instances functioning as distinct agents, each assigned specific roles, tools, or subtasks. These agents communicate through defined protocols and iteratively refine solutions based on shared information. Such systems can benefit from principles drawn from SI, such as parallel exploration and distributed coordination, enabling more effective search over complex solution spaces than single-model approaches. In drug discovery, this paradigm can be applied to tasks such as hypothesis generation, target validation, and early-stage lead optimization, where multiple types of evidence and reasoning steps need to be integrated.
A representative example is PharmaSwarm,56 a multi-agent framework designed for hypothesis-driven drug discovery. In this system, specialized agents perform tasks such as genomic data analysis, knowledge graph querying, pathway analysis, and binding affinity estimation. An evaluator agent ranks candidate hypotheses based on criteria including plausibility and predicted efficacy, and the system iteratively refines proposed targets and compounds through feedback across agents. This design enables the integration of heterogeneous data sources within a single workflow while maintaining traceability of intermediate steps.
More broadly, multi-agent LLM systems provide a flexible way to coordinate complex drug discovery processes that involve multiple interdependent tasks. Their swarm-like behavior arises from the interaction of agents that explore different aspects of the problem space and update shared hypotheses over time.
Overall, LLM-enhanced swarm intelligence and multi-agent systems represent a promising direction for drug discovery platforms. By combining population-based search with agent-based reasoning and access to biomedical knowledge, these approaches may support more flexible exploration of complex search spaces and assist in identifying candidate therapeutic strategies. However, most current studies remain at an early stage, and their practical impact in real-world drug discovery settings has yet to be fully established.
Discussion
Drug discovery is a multi-dimensional, multi-step process involving complex search and optimization. This review has highlighted the potential of swam-based intelligence algorithms in this domain. Swarm-based methods provide a set of population-based optimization strategies that are well suited for a range of drug discovery tasks. Compared with purely data-driven approaches, these methods are less dependent on large, labeled datasets and can be used in settings where data are limited or heterogeneous. Moreover, their search process is relatively transparent, allowing the evolution of candidate solutions to be tracked and individual updates to be examined. Swarm algorithms also naturally support multi-objective optimization, enabling the simultaneous consideration of factors such as biological activity, toxicity, and physicochemical properties. Their distributed search mechanisms provide robustness in the presence of noisy or uncertain objective evaluations, and their flexible structure allows them to be adapted to diverse challenges across the drug discovery pipeline.
Challenges and Limitations
While swarm-based methods are effective for exploring large chemical spaces and handling multi-objective trade-offs, they also have several inherent limitations. A key challenge is scalability: as the search space grows or as evaluations involve computationally intensive steps such as docking or molecular simulations, the overall cost can increase substantially.
Another limitation is the heuristic nature of swarm algorithms. Swarm algorithms rely on stochastic search and do not guarantee convergence to a global optimum, particularly in high-dimensional or discrete chemical spaces. In de novo design, this can result in molecules that are chemically valid but show limited novelty or require further assessment of synthetic feasibility, and in some cases the generated structures may be variations of known scaffolds.
In addition, drug discovery often involves balancing multiple objectives, such as activity, toxicity, and solubility. While swarm-based methods can generate diverse trade-off solutions, selecting the most relevant candidates typically requires domain expertise or further downstream analysis. As the number of objectives increases, the optimization process becomes more complex, which may reduce efficiency in certain settings.
Recent studies integrating large language models (LLMs) and multi-agent systems with swarm-based optimization represent an emerging direction in computational drug discovery. However, many current studies remain proof-of-concept reports or early methodological explorations rather than extensively validated applications in pharmaceutical research. Their practical value in medicinal chemistry and translational drug discovery therefore remains uncertain. Several limitations require further investigation, including hallucination effects, reproducibility, interpretability, and the computational cost of complex multi-agent optimization workflows.
Furthermore, benchmarking standards remain insufficiently standardized, with many studies relying on different datasets, scoring metrics, and experimental settings, limiting reproducibility and direct comparison across methods. Comparative evaluations between swarm-based approaches and alternative optimization strategies, such as gradient-based optimization and evolutionary algorithms, also remain relatively limited in many application domains. Several unresolved issues therefore persist, including parameter sensitivity, convergence stability, scalability in high-dimensional chemical spaces, and the limited translational validation of computationally generated candidates in experimental and clinical settings.
Future Perspectives
Swarm-based methods have demonstrated utility across multiple stages of drug discovery, yet several areas warrant further development. Scalability remains a key consideration, particularly as chemical space and biomedical data continue to expand, requiring more efficient optimization in high-dimensional settings. Another important direction is the integration of heterogeneous data sources, including chemical structures, omics data, clinical records, and literature, to better capture complex biological relationships. In such context, maintaining interpretability of the search process is important, especially for applications requiring mechanistic insight. Recent developments involving LMMs and multi-agent systems provide additional opportunities, as these models can offer structured guidance, such as candidate modifications or biologically informed suggestions, within swarm-based frameworks. Finally, systematic benchmarking across tasks, datasets, and methodological settings remains necessary to more clearly define the strengths and limitations of these approaches in real-world drug discovery pipelines. Future swarm-based frameworks may also benefit from tighter integration with experimental validation pipelines, including high-throughput screening, patient-derived systems, and real-world clinical data. Improving the ability of computationally prioritized candidates to translate into experimentally reproducible and clinically relevant outcomes remains an important challenge. Continued efforts toward standardized benchmarking, translational validation, and closer collaboration between computational researchers and experimental scientists will be important for advancing the practical utility and clinical relevance of swarm-based approaches in modern drug discovery.
In conclusion, swarm-based methods provide a flexible framework for exploring complex search spaces in drug discovery, with applications spanning molecular docking, virtual screening, de novo design, and QSAR/QSPR modeling. Despite challenges related to scalability, convergence, and sensitivity to problem formulation, their ability to operate under limited data and support interpretable, multi-objective optimization suggests they will remain a valuable component in both research and applied drug discovery.
Ethical Considerations
This study did not involve human participants, animals, or identifiable personal data.
Funding
This work received no funding support.
Disclosure
The authors declare no competing interests in this work.
References
1. Palanki R, Bose SK. Drug discovery and development. In: Translational Surgery. Academic Press; 2023:35–11.
2. Dhudum R, Ganeshpurkar A, Pawar A. Revolutionizing drug discovery: a comprehensive review of AI applications. Drugs Drug Candidates. 2024;3(1):148–171.
3. Khalil AS, Jaenisch R, Mooney DJ. Engineered tissues and strategies to overcome challenges in drug development. Adv Drug Delivery Rev. 2020;158:116–139.
4. Chao WANG, Zhang S, Tianhang MA, Yuetong XIAO, Chen MZ, Lei WANG. Swarm intelligence: a survey of model classification and applications. Chin J Aeronaut. 2025;38(3):102982.
5. Priyadarshi R, Kumar RR. Evolution of swarm intelligence: a systematic review of particle swarm and ant colony optimization approaches in modern research. Arch Comput Meth Eng. 2025;32(6):3609–3650.
6. Altshuler Y. Recent developments in the theory and applicability of swarm research. Appl Swarm Intell. 2024;2024:1–31.
7. Zendehbad SA, Rad EM, Bajestani SS. Swarm intelligence in biomedical engineering. Intell Based Med. 2025;12:100308. doi:10.1016/j.ibmed.2025.100308
8. Mostafa AA, Alhossary AA, Salem SA, Mohamed AE. GBO-kNN a new framework for enhancing the performance of ligand-based virtual screening for drug discovery. Expert Syst Appl. 2022;197:116723. doi:10.1016/j.eswa.2022.116723
9. Mahapatra B. Swarm Intelligence and Evolutionary Algorithms for Drug Design and Development. In: Swarm Intelligence and Evolutionary Algorithms in Healthcare and Drug Development. Chapman and Hall/CRC; 2019:117–140.
10. Nicolaou CA, Brown N, Pattichis CS. Molecular optimization using computational multi-objective methods. Current Opinion Drug Discovery Dev. 2007;10(3):316.
11. Liu HP, Phoa FKH, Dutta S. Molecule discovery and optimization via evolutionary swarm intelligence. Scient Rep. 2024;14(1):24510. doi:10.1038/s41598-024-75515-w
12. Wang J, Di Y, Rui X. Research and application of machine learning method based on swarm intelligence optimization. J Comput Meth Sci Eng. 2019;19(1_suppl):179–187. doi:10.3233/JCM-191025
13. Pawan YN, Prakash KB, Chowdhury S, Hu YC. Particle swarm optimization performance improvement using deep learning techniques. Multimedia Tools Appl. 2022;81(19):27949–27968. doi:10.1007/s11042-022-12966-1
14. Jimenez-Romero C, Yegenoglu A, Blum C. Multi-agent systems powered by large language models: applications in swarm intelligence. Front Artificial Intell. 2025;8:1593017. doi:10.3389/frai.2025.1593017
15. Abualigah L, Sheikhan A, Ikotun AM, et al. Particle swarm optimization algorithm: review and applications. Metaheuristic Optim Algorithms. 2024;2024:1–14.
16. Wang Y, Jiao J, Liu J, Xiao R. A labor division artificial bee colony algorithm based on behavioral development. Inform Sci. 2022;606:152–172. doi:10.1016/j.ins.2022.05.065
17. Shaban AA, Almufti SM, Asaad RR, Ali RI. Firefly algorithm: overview, applications, and modifications. J Image Process Intell Remote Sens. 2025;5(02):1.
18. Shankar MS, Sivarajan S, Carie A, Anamalamudi S, Sobin CC. Machine Learning and Deep Learning with Swarm Algorithms. In: Swarm Intelligence. CRC Press:74–95
19. Salhi A, Alshamrani R, Althbiti A, Ismail A, Abd-ElRahman M, Hassan BM. Optimizing high dimensional data classification with a hybrid AI driven feature selection framework and machine learning schema. Scient Rep. 2025;15(1):35038. doi:10.1038/s41598-025-08699-4
20. Abdulsaed E, Alabbas M, Khudeyer R. Hyperparameter optimization for convolutional neural networks using the salp swarm algorithm. Informatica. 2023;47(9). doi:10.31449/inf.v47i9.5148
21. Meyers J, Fabian B, Brown N. De novo molecular design and generative models. Drug Discovery Today. 2021;26(11):2707–2715. doi:10.1016/j.drudis.2021.05.019
22. Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: a review. Biophys Rev. 2017;9(2):91–102. doi:10.1007/s12551-016-0247-1
23. Gackowski M, Madriwala B, Koba M. In silico design, docking simulation, and ANN-QSAR model for predicting the anticoagulant activity of thiourea isosteviol compounds as FXa inhibitors. Chem Papers. 2023;77(11):7027–7044. doi:10.1007/s11696-023-02994-y
24. Paggi JM, Pandit A, Dror RO. The art and science of molecular docking. Ann Rev Biochem. 2024;93(1):389–410. doi:10.1146/annurev-biochem-030222-120000
25. Gao Z, Ding P, Xu R. IUPHAR review-data-driven computational drug repurposing approaches for opioid use disorder. Pharmacol Res. 2024;199:106960. doi:10.1016/j.phrs.2023.106960
26. de Angelo RM, de Sousa DS, da Silva AP, Chiari LP, da Silva AB, Honorio KM. Molecular docking: state-of-the-art scoring functions and search algorithms. In: Computer-Aided and Machine Learning-Driven Drug Design: From Theory to Applications. Cham: Springer Nature Switzerland; 2025:163–198.
27. Liu J, Wang R. Classification of current scoring functions. J Chem Inform Model. 2015;55(3):475–482. doi:10.1021/ci500731a
28. Shin WH, Kim JK, Kim DS, Seok C. GalaxyDock2: protein-ligand docking using beta-complex and global optimization. J Comput Chem. 2013;34(30):2647–2656. doi:10.1002/jcc.23438
29. Zhou J, Yang Z, He Y, Ji J, Lin Q, Li J. A novel molecular docking program based on a multi-swarm competitive algorithm. Swarm Evol Comput. 2023;78:101292. doi:10.1016/j.swevo.2023.101292
30. Tripathi A, Suri K, Murugan NA, Murugan NA. Assessing the accuracy of binding pose prediction for kinase proteins and 7-azaindole inhibitors: a study with AutoDock4, Vina, DOCK 6, and GNINA 1.0. RSC Adv. 2025;15(55):47051–47065. doi:10.1039/D5RA05526A
31. Halfar R, Damborský J, Marques SM, Martinovič J. Moldina: a fast and accurate search algorithm for simultaneous docking of multiple ligands. J Cheminform. 2025;17(1):61. doi:10.1186/s13321-025-01005-4
32. Li J, Li C, Sun J, Palade V. RDPSOVina: the random drift particle swarm optimization for protein-ligand docking. J Comp-Aided Mol Design. 2022;36(6):415–425. doi:10.1007/s10822-022-00455-4
33. Kiruba Nesamalar E, SatheeshKumar J, Amudha T. Efficient DNA-ligand interaction framework using fuzzy C-means clustering based glowworm swarm optimization (FCMGSO) method. J Biomol Struct Dynam. 2023;41(13):6249–6261. doi:10.1080/07391102.2022.2105958
34. Li C, Sun J, Li LW, Wu X, Palade V. An effective swarm intelligence optimization algorithm for flexible ligand docking. IEEE/ACM Transactions Comput Biol Bioinform. 2021;19(5):2672–2684. doi:10.1109/TCBB.2021.3103777
35. Bálint M, Horváth I, Mészáros N, Hetényi C. Towards unraveling the histone code by fragment blind docking. Int J Mol Sci. 2019;20(2):422. doi:10.3390/ijms20020422
36. Jiménez-García B, Roel-Touris J, Barradas-Bautista D. The LightDock Server: artificial Intelligence-powered modeling of macromolecular interactions. Nucleic Acids Res. 2023;51(W1):W298–W304. doi:10.1093/nar/gkad327
37. Khan AU, Khan AU. Descriptors and their selection methods in QSAR analysis: paradigm for drug design. Drug Discovery Today. 2016;21(8):1291–1302. doi:10.1016/j.drudis.2016.06.013
38. Mądra-Gackowska K, Baumgart S, Jędrzejewski M, Studzińska R, Szeleszczuk Ł, Gackowski M. Computational QSAR study of novel 2-aminothiazol-4 (5H)-one derivatives as 11β-HSD1 inhibitors. J Comp-Aided Mol Design. 2025;39(1):67. doi:10.1007/s10822-025-00648-7
39. Ibrahim RA, Aly MAS, Moemen YS, El Sayed IET, Abd Elaziz M, Khalil HA. Boosting Sinh Cosh Optimizer and arithmetic optimization algorithm for improved prediction of biological activities for indoloquinoline derivatives. Chemosphere. 2024;359:142362. doi:10.1016/j.chemosphere.2024.142362
40. Alharthi AM, Kadir DH, Al-Fakih AM, Algamal ZY, Al-Thanoon NA, Qasim MK. Improving golden jackel optimization algorithm: an application of chemical data classification.Chemomet Intell Lab Syst. 2024;250:105149. doi:10.1016/j.chemolab.2024.105149
41. Zhou R, Zhang Y, He K. A novel hybrid binary whale optimization algorithm with chameleon hunting mechanism for wrapper feature selection in QSAR classification model: a drug-induced liver injury case study. Expert Syst Appl. 2023;234:121015. doi:10.1016/j.eswa.2023.121015
42. Fouad MA, Serag A, Tolba EH, El-Shal MA, El Kerdawy AM. QSRR modeling of the chromatographic retention behavior of some quinolone and sulfonamide antibacterial agents using firefly algorithm coupled to support vector machine. BMC Chem. 2022;16(1):85. doi:10.1186/s13065-022-00874-2
43. Shamsi E, Rahati A, Dehghanian E. A modified binary particle swarm optimization with a machine learning algorithm and molecular docking for QSAR modelling of cholinesterase inhibitors. SAR QSAR Environ Res. 2021;32(9):745–767. doi:10.1080/1062936X.2021.1971761
44. Mahmood SW, Basheer GT, Algamal ZY. Quantitative structure-activity relationship modeling based on improving kernel ridge regression. J Chemomet. 2025;39(5):e70027. doi:10.1002/cem.70027
45. He B, Lu C, Zheng G, et al. Combination therapeutics in complex diseases. J Cell Mol Med. 2016;20(12):2231–2240. doi:10.1111/jcmm.12930
46. Motohashi S, Katsuta E, Ban D. Advances and challenges in drug screening for cancer therapy: a comprehensive review. Bioengineering. 2025;12(12):1315. doi:10.3390/bioengineering12121315
47. Iadevaia S, Lu Y, Morales FC, Mills GB, Ram PT. Identification of optimal drug combinations targeting cellular networks: integrating phospho-proteomics and computational network analysis. Cancer Res. 2010;70(17):6704–6714. doi:10.1158/0008-5472.CAN-10-0460
48. Kang Y, Hodges A, Ong E, Roberts W, Piermarocchi C, Paternostro G. Identification of drug combinations containing imatinib for treatment of BCR-ABL+ leukemias. PLoS One. 2014;9(7):e102221. doi:10.1371/journal.pone.0102221
49. Hartenfeller M, Schneider G. Enabling future drug discovery by de novo design. Wiley Interdiscip Rev. 2011;1(5):742–759.
50. Hartenfeller M, Proschak E, Schüller A, Schneider G. Concept of combinatorial de novo design of drug-like molecules by particle swarm optimization. Chem Biol Drug Design. 2008;72(1):16–26. doi:10.1111/j.1747-0285.2008.00672.x
51. Reutlinger M, Rodrigues T, Schneider P, Schneider G. Multi-objective molecular de novo design by adaptive fragment prioritization. Angewandte Chem Int Ed. 2014;53(16):4244–4248. doi:10.1002/anie.201310864
52. Winter R, Montanari F, Steffen A, Briem H, Noé F, Clevert DA. Efficient multi-objective molecular optimization in a continuous latent space. Chem Sci. 2019;10(34):8016–8024. doi:10.1039/C9SC01928F
53. Yi JC, Yang ZY, Zhao WT, et al. ChemMORT: an automatic ADMET optimization platform using deep learning and multi-objective particle swarm optimization. Brief Bioinform. 2024;25(2):bbae008. doi:10.1093/bib/bbae008
54. Devi RV, Sathya SS, Coumar MS. Multi-objective biofilm algorithm (MOBifi) for de novo drug design with special focus to anti-diabetic drugs. Appl Soft Comput. 2020;96:106655. doi:10.1016/j.asoc.2020.106655
55. Shinohara Y, Xu J, Li T, Iba H. Large language models as particle swarm optimizers. 2025. arXiv preprint arXiv:2504.09247.
56. Song K, Trotter A, Chen JY. LLM agent swarm for hypothesis-driven drug discovery. 2025. arXiv preprint arXiv:2504.17967.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Application of Artificial Intelligence Generated Content in Medical Examinations
Li R, Wu T
Advances in Medical Education and Practice 2025, 16:331-339
Published Date: 25 February 2025
Generative AI/LLMs for Plain Language Medical Information for Patients, Caregivers and General Public: Opportunities, Risks and Ethics
Pal A, Wangmo T, Bharadia T, Ahmed-Richards M, Bhanderi MB, Kachhadiya R, Allemann SS, Elger BS
Patient Preference and Adherence 2025, 19:2227-2249
Published Date: 31 July 2025
Assessing DeepSeek-R1 for Clinical Decision Support in Multidisciplinary Laboratory Medicine
Li Q, Zhan L, Cai X
Journal of Multidisciplinary Healthcare 2025, 18:4979-4988
Published Date: 12 August 2025
AI-Driven Large Language Models in Health Consultations for HIV Patients
Zhao CY, Song C, Yang T, Huang AC, Qiang HB, Gong CM, Chen JS, Zhu QD
Journal of Multidisciplinary Healthcare 2025, 18:5187-5198
Published Date: 25 August 2025
Assessing the Diagnostic Capabilities of ChatGPT-4 Omni in Grading Diabetic Retinopathy Fundoscopy Using Color Fundus Photographs
Chetla N, Samayamanthula SS, Chang JH, Leigh AY, Akosman S, Tandon M, Hage TR, Cusick M
Clinical Ophthalmology 2025, 19:3103-3112
Published Date: 31 August 2025