Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Surface modification of MPEG-b-PCL-based nanoparticles via oxidative self-polymerization of dopamine for malignant melanoma therapy
Authors Xiong W, Peng L, Chen H, Li Q
Received 19 December 2014
Accepted for publication 18 February 2015
Published 16 April 2015 Volume 2015:10(1) Pages 2985—2996
DOI https://doi.org/10.2147/IJN.S79605
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Wei Xiong,1,2 Lixia Peng,1,2 Hongbo Chen,3 Qin Li1,2
1Southern Medical University, Guangzhou, 2Department of Plastic Surgery, General Hospital of Guangzhou Military Command of PLA, Guangzhou, People’s Republic of China; 3Division of Life Sciences and Health, Graduate School at Shenzhen, Tsinghua University, Shenzhen, People’s Republic of China
Abstract: To enhance the therapeutic effects of chemotherapy on malignant melanoma, paclitaxel (PTX)-loaded methoxy poly(ethylene glycol)-b-poly(ε-caprolactone) nanoparticles (MPEG-b-PCL NPs) that had their surfaces modified with polydopamine (PTX-loaded MPEG-b-PCL NPs@PDA) were prepared as drug vehicles. The block copolymer MPEG-b-PCL was synthesized by ring-opening polymerization and characterized by proton nuclear magnetic resonance spectroscopy and gel permeation chromatography. The PTX-loaded NPs were prepared by a modified nanoprecipitation technique. The PTX-loaded NPs and PTX-loaded NPs@PDA were characterized in terms of size and size distribution, zeta potential, surface morphology, drug encapsulation efficiency, and drug release. Confocal laser scanning microscopy showed that coumarin-6-loaded NPs@PDA could be internalized by human melanoma cell line A875 cells. The cellular uptake efficiency of NPs was greatly enhanced after PDA modification. The antitumor efficacy of the PTX-loaded NPs@PDA was investigated in vitro by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and in vivo by a xenograft tumor model. The PTX-loaded NPs@PDA could significantly inhibit tumor growth compared to Taxol® and precursor PTX-loaded NPs. All the results suggested that the PTX-loaded MPEG-b-PCL NPs that had their surfaces modified with PDA are promising nanocarriers for malignant melanoma therapy.
Keywords: cancer nanotechnology, drug delivery, surface modification, polydopamine, malignant melanoma
Introduction
Malignant melanoma is the mainly aggressive form of skin cancer, causing the most number of skin cancer deaths.1 According to the National Cancer Institute database of the USA, melanoma is one of the most prevalent types of cancer, with >65,000 new cases and 8,650 deaths from malignant melanoma in the USA alone.2 During the past two decades, the incidence and the mortality rate of melanoma are increasing at a faster rate than for most other types of cancer.3 Primary melanoma is a good candidate for surgical therapy, but once metastasis occurs, the therapeutic effect of surgical removal therapy is very poor, although various chemotherapeutic drugs and radiotherapy may have been adopted. Hence, enhancing the clinical therapeutic efficacy of chemotherapy for malignant melanoma is very important. There are some reports in literature that paclitaxel (PTX) has significant effects in the treatment of malignant melanoma.1,4–6 However, the extremely poor water-soluble property, high toxicity, and low bioavailability of PTX have greatly hindered its therapeutic effects and limited its clinical application.7 Taxol® is one of the formulations of PTX that can be used in the therapy of malignant melanoma. Unfortunately, Cremophor-EL, which is an adjuvant used in Taxol®, causes serious side effects and leads to hypersensitivity, cardiotoxicity, and nephrotoxicity reactions in many patients. To reduce the side effects, many efforts have been devoted to replace the Cremophor-EL-based vehicle for PTX delivery.8
Recently, new strategies for malignant melanoma therapy based on anticancer drug-loaded nanocarrier formulations have emerged. Nanoparticles (NPs) represent promising drug vehicles, especially for specific delivery of anticancer agents to the tumor site, which could enhance the clinical effectiveness and reduce systemic toxic side effects.9–11 The NPs used as drug vehicles have many important advantages, such as high encapsulation efficiency (EE) and drug-loading capacity, minor drug leakage, sustained release, and excellent feasibility of routes of administration.12,13 Many chemotherapeutic drugs, such as docetaxel, puerarin, PTX, and doxorubicin, can be delivered by using NPs.14–16 The preparation techniques of polymeric NPs include nanoprecipitation, dialysis, solvent evaporation, and multiple emulsion preparation.13,17,18
Biodegradable copolymers have the potential to be developed as polymeric NPs for drug delivery. Poly(ε-caprolactone) (PCL) is used for applications in the field of drug delivery and tissue engineering. PCL has attracted considerable interest because of its excellent permeability to drugs, noncytotoxicity, thermal properties, and biocompatibility. However, it is well known that PCL is extremely hydrophobic and its degradation is too slow to meet the needs for drug delivery.19–21 Moreover, the PCL NPs could be rapidly removed in the liver and captured by the reticuloendothelial system (RES) when they are injected into the blood stream.22 A large number of approaches for prolonging the circulation of NPs in blood have been reported. In particular, introduction of the flexible, hydrophilic poly(ethylene glycol) (PEG) can help the NPs to escape uptake by the mononuclear phagocytic system (MPS) and RES, resulting in prolonged blood circulation time.22–24 Although PEG has many advantages such as biocompatibility, antigenicity, and lack of toxicity, as well as the fact that PEGylation increases circulation longevity, it has been reported that PEGylated NPs do not completely avoid cumulative uptake by MPS cells, and they cannot mostly eliminate complement system activation for their corresponding naked NPs.25,26 Kaneda et al27 reported that PEG showed a rather high urinary excretion and peripheral distribution volume. Besides, the molecular structure of PEG as a general polymeric modifier does also not readily allow the addition of new functions along its chain. To surmount these challenges, we use a simple and versatile surface modification method based on polydopamine (PDA).
PDA, a mimic of the specialized adhesive foot protein Mefp-5 (mytilus edulis foot protein-5) secreted by mussels, can modify almost all the drug-loaded polymeric NPs.28 PDA coating displays excellent biocompatibility and low cytotoxicity, making it a versatile platform for applications, including biomolecule immobilization, molecular surface imprinting for protein recognition, cells adhesion, and encapsulation.29–31 Furthermore, PDA is also a major pigment of naturally occurring eumelanin.32 PDA displays a lot of striking properties of naturally occurring eumelanin in terms of electrical, optical, and magnetic properties. Another valuable advantage of PDA lies in its chemical structure that possesses many functional groups such as amine, catechol, and imine. These functional groups could serve as the starting points for covalent modification with the targeting ligand for drug delivery. Due to these benefits, PDA has been rapidly changed into a wide range of applications across the biological, chemical, and nanomedicine fields. PDA coating prepared by oxidative self-polymerization of dopamine on solid or liquid templates showed negligible toxicity and exhibited promising applications for drug delivery.33,34
In this study, to enhance the therapeutic efficacy and minimize the side effects of malignant melanoma treatment, we prepared novel PTX-loaded methoxy poly(ethylene glycol)-b-poly(ε-caprolactone) NPs (PTX-loaded MPEG-b-PCL NPs) and used surface modification with PDA as drug carrier. The surface-modified NPs were characterized and the antitumor effect was evaluated both in vitro and in vivo. The results were closely compared with the current PTX clinical formulation Taxol®.
Materials and methods
Materials
Stannous octoate (Sn(Oct)2), ε-caprolactone (ε-CL), methoxy PEG (MPEG, molecular weight Mn=2,000 Da), d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), and dopamine hydrochloride were purchased from Sigma-Aldrich (St Louis, MO, USA). PTX was purchased from Melone Pharmaceutical Co, Ltd (Dalian, People’s Republic of China). Acetonitrile and methanol were purchased from EM Science (ChromAR, high-performance liquid chromatography (HPLC) grade, Mallinckrodt Baker, NJ, USA). All other chemicals were of the highest quality commercially available and were used as received. Human melanoma cell line A875 cells were provided by Shanghai Maisha Biotech (Shanghai, People’s Republic of China).
Synthesis of block copolymer MPEG-b-PCL
Copolymer MPEG-b-PCL was synthesized from ε-CL and MPEG in the presence of Sn(Oct)2 as a catalyst by ring-opening copolymerization, as presented in Figure 1. Briefly, ε-CL (9.1 g, 80 mmol), MPEG (2.0 g, 1 mmol), and Sn(Oct)2 (0.1 mol% of monomer ε-CL) were added in a glass tube, which was connected to a vacuum system. Then an exhausting–refilling process with nitrogen was repeated three times. The tube was sealed and heated to 140°C in oil bath for 24 hours. After the reaction tube was cooled to room temperature, the resulting product was dissolved in dichloromethane, then precipitated in excess cold anhydrous ether, and washed with methanol. The final product was designated MPEG-b-PCL and vacuum dried at 40°C for 24 hours.
 | Figure 1 Synthesis scheme of block copolymer MPEG-b-PCL. |
Characterization of copolymer MPEG-b-PCL
Proton nuclear magnetic resonance (1H-NMR; Bruker AMX500) was used to confirm the structure of the prepared copolymer MPEG-b-PCL; CDCl3 was used as a solvent. Gel permeation chromatography (GPC, Waters 150C) was used to obtain the molecular weight and molecular weight distribution, which were estimated using polystyrene standards. The eluent was tetrahydrofuran with 1 mL/min flow rate.
Formulation of PTX-loaded MPEG-b-PCL NPs and PTX-loaded MPEG-b-PCL NPs@PDA
PTX-loaded MPEG-b-PCL NPs were prepared by a modified nanoprecipitation method.13,35 Briefly, 20 mg anticancer drug PTX powder and 100 mg of copolymer MPEG-b-PCL were dissolved in 10 mL of acetone by vortexing and sonication. This mixture was added dropwise into 100 mL of 0.03% TPGS aqueous solution under stirring. The resulting suspension was then stirred uncovered overnight to remove acetone fully. The NP suspension was centrifuged at 15,000 rpm for 20 minutes and then washed three times to remove the emulsifier TPGS and unencapsulated drug PTX. Finally, the dispersed solution was lyophilized 2 days for further surface modification. Fluorescent coumarin-6 (C6)-loaded MPEG-b-PCL NPs were prepared by the same procedure, except that PTX was replaced by C6.
Surface modification of NPs was carried out by the oxidative self-polymerization of dopamine.29,36 In brief, 50 mg lyophilized NPs were dispersed ultrasonically in 50 mL Tris-HCl buffer (pH 8.5, 10 mM) and then 20 mg dopamine hydrochloride was added. The mixture was magnetically stirred for 6 hours in the dark. The oxidative self-polymerized solid PDA-coated PTX-loaded MPEG-b-PCL NPs (designated PTX-loaded MPEG-b-PCL NPs@PDA) were centrifuged (15,000 rpm, 10 minutes) and washed with water to remove the unpolymerized dopamine. PTX-loaded MPEG-b-PCL NPs@PDA was lyophilized 2 days for further use.
Characterization of NPs
Size and zeta potential
The particle size and size distribution were measured by Malvern Mastersizer 2000 (Malvern Instruments Ltd, Malvern, UK). Before measurement, the freshly prepared particles were diluted and the concentration was 1 mg/mL. The measurements were performed in triplicate.
Surface morphology
The surface morphology of NPs was examined by field-emission scanning electron microscopy (FESEM) using a JEOL JSM-6700F system operated at a 5.0 kV accelerating voltage. To prepare samples for FESEM, the particles were fixed on the stub by a double-sided sticky tape and then coated with platinum layer by JFC-1300 automatic fine platinum coater (JEOL, Tokyo, Japan) for 60 seconds.
Drug loading and encapsulation efficiency
The drug loading content (LC) and EE of NPs were obtained by HPLC (LC 1200; Agilent Technologies, Santa Clara, CA, USA) according to the reported procedures.37,38 In summary, NPs (5 mg) were dissolved in 1 mL dichloromethane under vigorous vortexing. This solution was transferred to 5 mL of mobile phase consisting of deionized water and acetonitrile (50:50, v/v). A nitrogen stream was introduced for about 15 minutes to evaporate the dichloromethane, and then a clear solution was obtained for HPLC analysis. A reverse-phase C18 column (250 mm ×4.6 mm; Agilent Technologies) was used. The flow rate of mobile phase was 1 mL/min. The column effluent was detected at λmax of 227 nm with an ultraviolet-visible detector. The drug EE was calculated as the ratio between the amount of PTX encapsulated in the NPs and that fed into the process. The data were obtained as the average of three measurements.
In vitro drug release
The dialysis method was used to analyze the drug release in vitro.39 Briefly, NPs (5 mg) were dispersed in 2 mL of phosphate-buffered saline (PBS, pH =7.4). The suspension was put into a regenerated cellulose dialysis bag (molecular weight cutoff =3,500 Da; Spectra/Por 6, Spectrum Laboratories, Inc., CA, USA). The closed bag was then put into a centrifuge tube and immersed into 15 mL of release medium PBS. The tube was transferred into an orbital water bath and shaken at 120 rpm at 37°C. An aliquot (10 mL) of release medium was taken at various time intervals for HPLC analysis and replaced with fresh PBS solution. The analysis method was similar to the measurement of LC and EE. Each batch of experiments was performed three times. The accumulative release of drug from PTX-loaded NPs and PTX-loaded NPs@PDA was plotted against time.
Cellular uptake of NPs
Human melanoma cell line A875 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum and antibiotics. A875 cells, at the initial density of 1×105 cells per well, were plated in 96-well plates. The culture was kept in humidified atmosphere containing 95% air and 5% CO2 at 37°C. The cells were incubated with 100 μg/mL C6-loaded NPs and C6-loaded NPs@PDA at 37°C for 2 hours, washed with cold PBS three times, and then fixed with 4% paraformaldehyde for 20 minutes. Thereafter, cells were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Fluka, Buchs, Switzerland) for 10 minutes and washed twice with PBS. The cells were observed by a confocal laser scanning microscope (Olympus Fluoview FV-1000, Olympus Optical Co, Tokyo, Japan) with imaging software. The images of the cells were captured with differential interference contrast channel; the images of C6-loaded NPs and the nuclei of DAPI-stained cells were recorded with following channels: blue channel (DAPI) excited at 340 nm and green channel (C6) excited at 488 nm.10 For quantitative analysis, the A875 cells at an initial density of 1×104 cells per well were plated in 96-well plates and left overnight. The cells were equilibrated with Hank’s buffered salt solution at 37°C for 1 hour before C6-loaded NPs and C6-loaded NPs@PDA were added at concentrations of 50 μg/mL, 100 μg/mL, and 200 μg/mL. After incubation for 2 hours, the medium was removed and the wells were washed three times with 50 μL cold PBS solution. Then, 50 μL 0.5% Triton X-100 in 0.2 mol/L sodium hydroxide was added into each sample well to lyse the cells.13
In vitro cytotoxicity of PTX-loaded NPs and PTX-loaded NPs@PDA
A875 cells were seeded in 96-well plates at the density of 5×103 cells per well and incubated 24 hours to allow cell adherence. The cells were incubated with the PTX-loaded MPEG-b-PCL NPs, PTX-loaded MPEG-b-PCL NPs@PDA suspension, and Taxol® at equivalent drug concentrations ranging from 0.25 μg/mL to 25 μg/mL for 12 hours, 24 hours, and 48 hours. The PTX-free MPEG-b-PCL NPs@PDA had the same amount of NPs. Before harvest, the culture media were replaced with fresh media containing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; 5 mg/mL) and incubated for additional 4 hours. MTT was aspirated off and dimethyl sulfoxide was added to dissolve the formazan crystals. Absorbance was measured at 570 nm using a microplate reader (Bio-Rad Model 680, UK). Untreated cells were taken as control with 100% viability, and cells without addition of MTT were used as blank to calibrate the spectrophotometer to zero absorbance.40
In vivo antitumor efficacy and side effect analysis
Animal experiments were approved by the Administrative Committee on Animal Research (Southern Medical University, Guangzhou, People’s Republic of China). The mice were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences. The tumor growth inhibitory activities of PTX-loaded MPEG-b-PCL NPs@PDA were evaluated with A875 cell xenograft-bearing nude mice model. Human melanoma A875 cells (2×106 cells per mouse) were inoculated subcutaneously to mice at the right axilla. After inoculation of A875 cells, the tumor growth in each mouse was closely observed. The tumor volume was monitored with a caliper every 2 days until the 20th day. Tumor volume can be calculated from the following formula: V = d2 × D/2, where d and D are the shortest and the longest diameters of the tumor in millimeters, respectively.13,41 When the tumor volume reached around 100 mm3 (designated as the 0 day), treatments were carried out. The mice were randomly divided into four groups (each group has five animals; n=5). Physiological saline was used in the control experiment. PTX (Taxol®; 10 mg/kg), PTX-loaded NPs, and PTX-loaded NPs@PDA in PBS were injected via the caudal vein on days 0, 4, 8, 12, and 16. Animals were closely observed for clinical signs and behavior changes. Furthermore, to estimate the side effects of the PTX-loaded NPs and the NPs having surface modification with PDA (NPs@PDA), body weight of the animals was recorded during the treatment period.
Statistical methodology
All results are reported as the mean ± standard error of the mean of three independent experiments. Student’s t-test statistical analysis was used, and probability (P) less than 0.05 was considered statistically significant.
Results and discussion
Characterization of block copolymer MPEG-b-PCL
The biodegradable block copolymer MPEG-b-PCL was prepared by the ring-opening polymerization of monomer ε-CL using MPEG as the initiator and Sn(Oct)2 as the catalyst in bulk at 140°C for 24 hours. To confirm the formation of copolymer MPEG-b-PCL, 1H-NMR in CDCl3 was used, as shown in Figure 2, with following results: peak a (δ=4.06 parts per million [ppm], CL repeating unit: –CO–CH2–CH2–CH2–CH2–CH2–O–), b (δ=2.32 ppm, CL repeating unit: –CO–CH2–CH2–CH2–CH2–CH2–O–), c (δ=1.62–1.67 ppm, CL repeating unit: –CO–CH2–CH2–CH2–CH2–CH2–O–), and d (δ=1.38 ppm, CL repeating unit: –CO–CH2–CH2–CH2–CH2–CH2–O–). Peak e is the solvent CDCl3. The peak f at 3.65 ppm was assigned to the (–CH2–CH2) protons of the initiator MPEG. Similar results were reported by Wan et al42 and Knop et al.43 The molecular weight of MPEG-b-PCL was calculated from the peak areas integral ratio of 4.06 ppm and 3.65 ppm; the molecular weight Mn of the copolymer MPEG-b-PCL was determined by 1H-NMR as 12,150 Da. The molecular weight and polydispersity index (PDI) of copolymer MPEG-b-PCL, which were evaluated by GPC, are 11,893 Da and 1.19, respectively. As shown, the molecular weights detected from GPC and 1H-NMR could confirm each other.
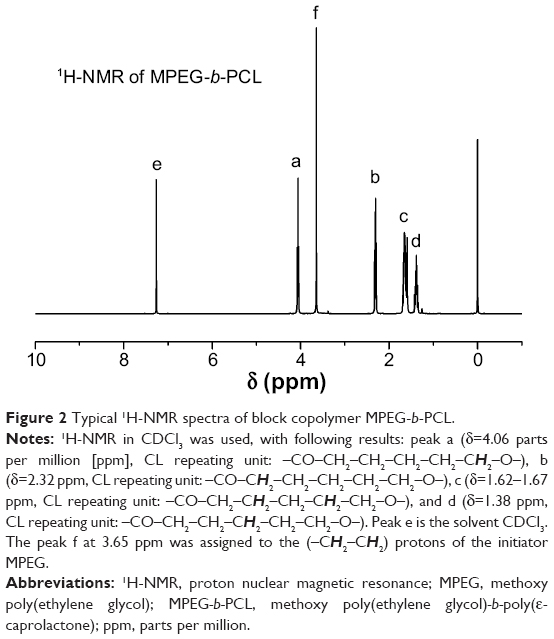 | Figure 2 Typical 1H-NMR spectra of block copolymer MPEG-b-PCL. |
Preparation and characterization of NPs
PTX-loaded MPEG-b-PCL NPs were prepared by a modified nanoprecipitation method; acetone was selected as an acceptable solvent. Nanoprecipitation provides a mild, facile, and low-energy input method for the formulation of polymeric NPs. The preparation technique of NPs is shown in Figure 3. The copolymer MPEG-b-PCL and drug PTX could be fully dissolved in acetone to generate a homogeneous and clear solution. This mixture phase was injected into a stirred aqueous solution containing 0.03% TPGS as a surfactant. Copolymer deposition on the interface between the water and the organic solvent acetone, caused by fast diffusion of the solvent, leads to the instantaneous formation of an NP suspension.13,44 The surface modification of NPs was carried out by the oxidative self-polymerization of dopamine in Tris-HCl buffer (pH 8.5). Polymerized dopamine is known to bind tightly on solid surfaces via covalent and noncovalent interactions, forming a durable layer that serves as an intermediate for ligand incorporation.45
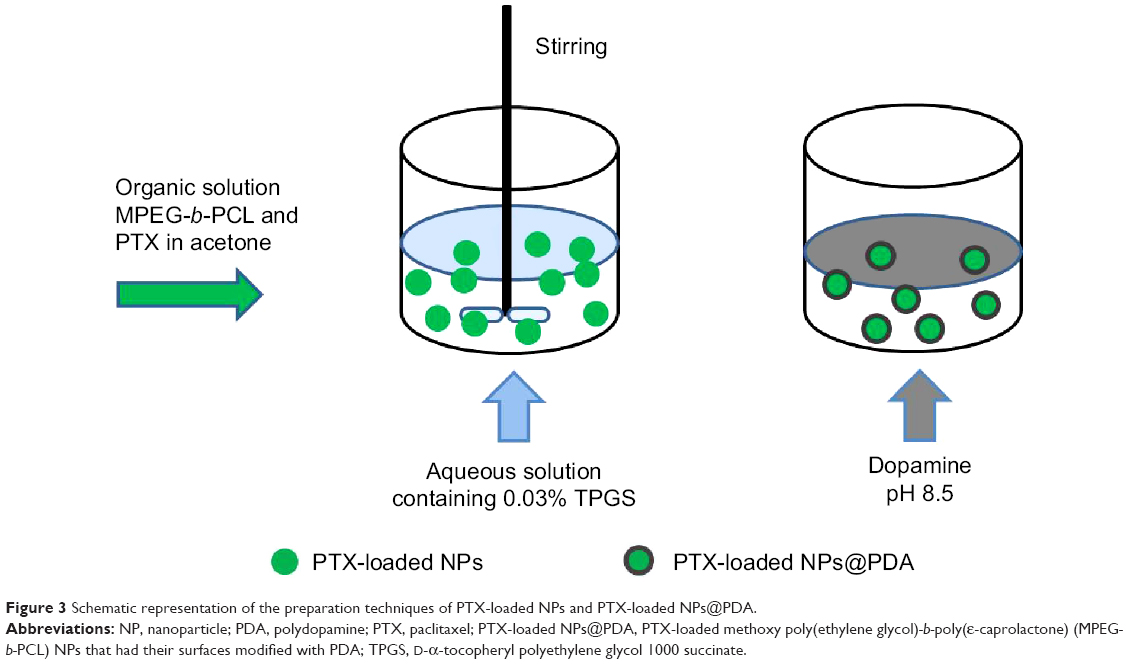 | Figure 3 Schematic representation of the preparation techniques of PTX-loaded NPs and PTX-loaded NPs@PDA. |
The size and size distribution of the PTX-loaded MPEG-b-PCL NPs and PTX-loaded MPEG-b-PCL NPs@PDA in this study are displayed in Table 1. Physicochemical characteristics, such as surface properties and particle size, play an important role in the drug release, cellular uptake, and cytotoxicity of these NPs, as well as their in vivo biodistribution and pharmacokinetics. Thus, the physicochemical properties of NPs affect the clinical application of the anticancer drug.38,46,47 The size of NPs was 120–140 nm in diameter, which may result in highest cellular uptake by endocytosis and inhibit elimination by the RES, as well as promoting accumulation in tumor vasculature because of the enhanced permeability and retention effect on passive tumor targeting.48,49 The size of the PTX-loaded MPEG-b-PCL NPs@PDA was larger than that of the nonmodified PTX-loaded MPEG-b-PCL NPs; this could be ascribed to the coating of PDA on PTX-loaded NPs. The PDIs of PTX-loaded NPs and PTX-loaded NPs@PDA were 0.136 and 0.121, respectively. The PDI is rather narrow, which is advantageous for cancer therapy. The size distribution of the PTX-loaded MPEG-b-PCL NPs@PDA obtained from dynamic light scattering (DLS) is presented in Figure 4A. As can be seen from Table 1, PTX-loaded NPs and PTX-loaded NPs@PDA have similar drug LC and EE values. Furthermore, the drug LC of PTX in NPs@PDA could reach about 16.8%, providing satisfactorily efficient drug delivery system.
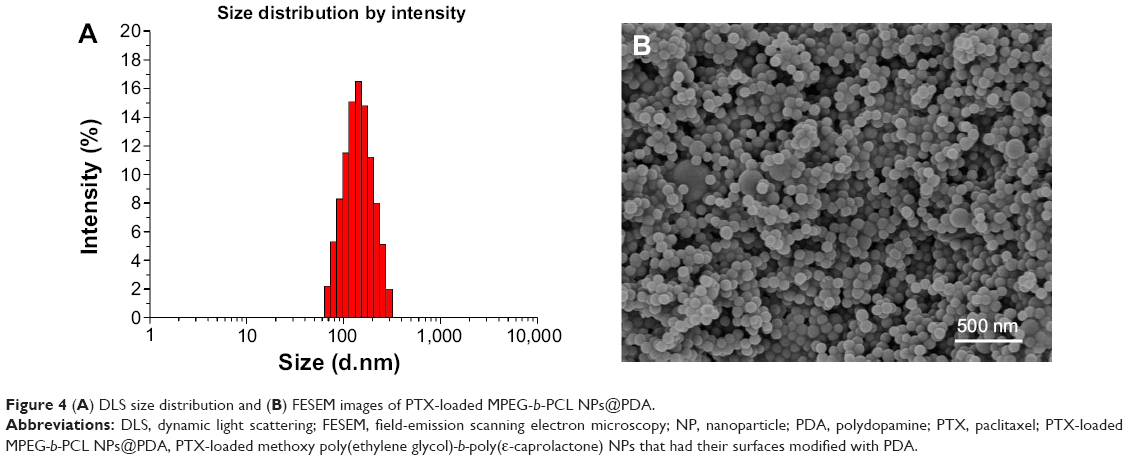 | Figure 4 (A) DLS size distribution and (B) FESEM images of PTX-loaded MPEG-b-PCL NPs@PDA. |
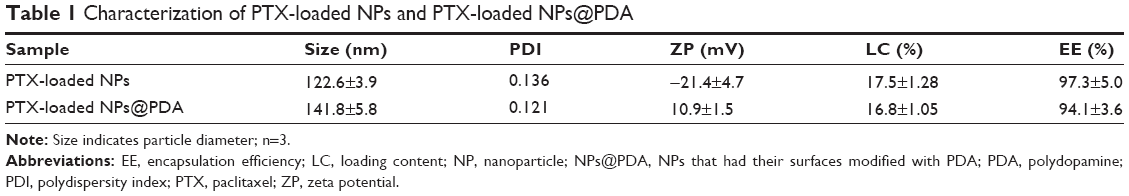 | Table 1 Characterization of PTX-loaded NPs and PTX-loaded NPs@PDA |
The zeta potential also reflects the stability of NPs in vivo through electrostatic repulsion. As shown in Table 1, the zeta potentials of PTX-loaded NPs and PTX-loaded NPs@PDA range from -21.4 mV to 10.9 mV. The negative surface charge of the PTX-loaded NPs resulted from the ionized carboxyl groups of PCL segments. Zeta potential of PTX-loaded NPs@PDA was positive, which could enhance mucosal uptake due to the anionic nature of the mucous layer.50
The surface morphology of the PTX-loaded MPEG-b-PCL NPs@PDA was observed by FESEM. Figure 4B shows the FESEM image for NPs@PDA. The NPs seemed to be about 100 nm in size and had smooth surface within the resolution level used. However, the particle size evaluated from FESEM was smaller than that from DLS analysis. This could be attributed to the fact that dry NPs were prone to shrinking and collapsing.13,51
The in vitro drug release profiles of the PTX-loaded NPs and PTX-loaded NPs@PDA in the first 30 days are shown in Figure 5. As can be seen from Figure 5, similar drug release curves of PTX-loaded NPs and PTX-loaded NPs@PDA were observed at the same time. The results suggested that the chemotherapeutic drug PTX of PDA-modified NPs was almost totally preserved during the preparation procedure. The NPs showed steady, continuous release patterns, with initial burst releases. The drug release from PTX-loaded NPs@PDA was found to be 53.6% and 88.2% of the encapsulated drug in the first 5 days and after 30 days, respectively. The PTX release behaviors could be ascribed to the following three important factors. First, the drug was hardly entrapped in the polymeric core and just located beneath the periphery of NPs. Second, the drug was dispersed amorphously or in the solid solution state in the NPs, which accelerated drug release because amorphous PTX dissolves much faster than its crystalline form.52 Third, sustained release mainly resulted from the diffusion of drug that was well encapsulated in the hydrophobic inner shell of NPs.
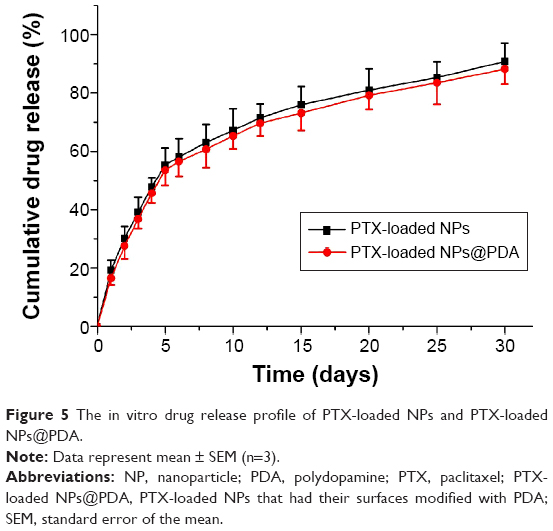 | Figure 5 The in vitro drug release profile of PTX-loaded NPs and PTX-loaded NPs@PDA. |
Cellular uptake of C6-loaded NPs and NPs@PDA
Generally speaking, the therapeutic effects of the drug-loaded NPs depend on internalization and sustained release of the NPs by the cancer cells. C6, a fluorescence molecule, has been widely used as a probe for marking NPs in cellular uptake investigation.53,54 C6 was used for replacing the PTX in the NPs to observe and analyze cellular uptake of drug-loaded NPs. Figure 6 presented confocal laser scanning microscopy images of the melanoma cell line A875 cells after 24-hour incubation with the C6-loaded NPs and C6-loaded NPs@PDA suspension in DMEM at 100 μg/mL NP concentration. The images were obtained from (A) enhanced green fluorescent protein channel (green), (B) DAPI channel (blue), and (C) the merger of the two channels. The C6-loaded NPs and C6-loaded NPs@PDA (green) were densely dispersed around the blue nuclei (stained by DAPI), implying that the fluorescent NPs had been internalized into the A875 cells. Furthermore, the fluorescence of C6-loaded NPs@PDA in the cytoplasm was much brighter than that of C6-loaded NPs. Thus, NPs could be more efficiently internalized into the A875 cell to deliver drug after surface modification with PDA. The results could be attributed to the facilitated internalization of cationic NPs (modified with PDA) by the negatively charged cell membranes.34
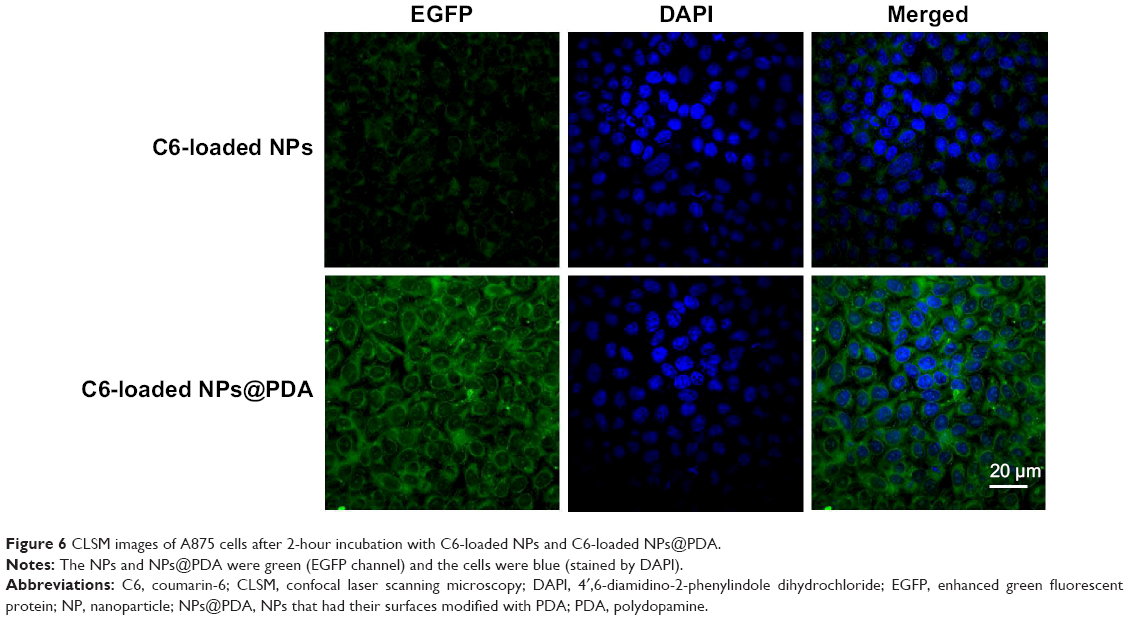 | Figure 6 CLSM images of A875 cells after 2-hour incubation with C6-loaded NPs and C6-loaded NPs@PDA. |
The cellular uptake efficiency of the C6-loaded NPs and C6-loaded NPs@PDA was evaluated and is shown in Figure 7. As shown in Figure 7, the cellular uptake efficiency of both C6-loaded NPs and NPs@PDA decreased with increase in the incubated NP concentration from 50 μg/mL to 200 μg/mL. The cellular uptake efficiency of C6-loaded NPs@PDA was 1.87-, 1.90-, and 2.24-fold higher than that of C6-loaded NPs at the incubated NP concentrations of 50 μg/mL, 100 μg/mL, and 200 μg/mL, respectively. Therefore, we strongly believe that the surface PDA could enhance the cellular uptake efficiency of NPs, which is very useful for malignant melanoma therapy.
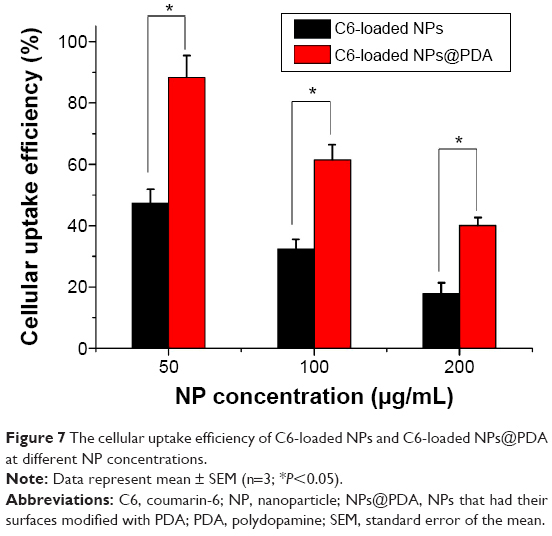 | Figure 7 The cellular uptake efficiency of C6-loaded NPs and C6-loaded NPs@PDA at different NP concentrations. |
In vitro cell viability of DTX-loaded NPs
Human melanoma cell line A875 cells were used to investigate the in vitro cytotoxicity of PTX-loaded NPs@PDA and to prove that the drug that was encapsulated in the NPs remained bioactive.13 According to the plasma levels of PTX achievable in human beings, the cells were treated with PTX-loaded MPEG-b-PCL NPs, NPs@PDA, and Taxol® at equivalent PTX concentrations of 0.25 μg/mL, 2.5 μg/mL, and 25 μg/mL for 12 hours, 24 hours, and 48 hours. The percentage of cell viability was determined by the MTT method (Figure 8). As shown in Figure 8, we can arrive at four conclusions. First, the cytotoxicity of the three PTX formulations increased with increasing drug dose and incubation time. Second, the cytotoxicity of the three formulations is very similar in the first 12 hours of incubation. However, after 24 hours and 48 hours of incubation, the PTX-loaded NPs@PDA showed significantly higher cytotoxicity than Taxol®. Third, the surface-modified PTX-loaded NPs@PDA are most destructive to A875 cells. Fourth, the synthesized copolymer MPEG-b-PCL and PDA seemed to be nontoxic in cell culture because no significant cytotoxicity was found for the drug-free NPs at various concentrations.
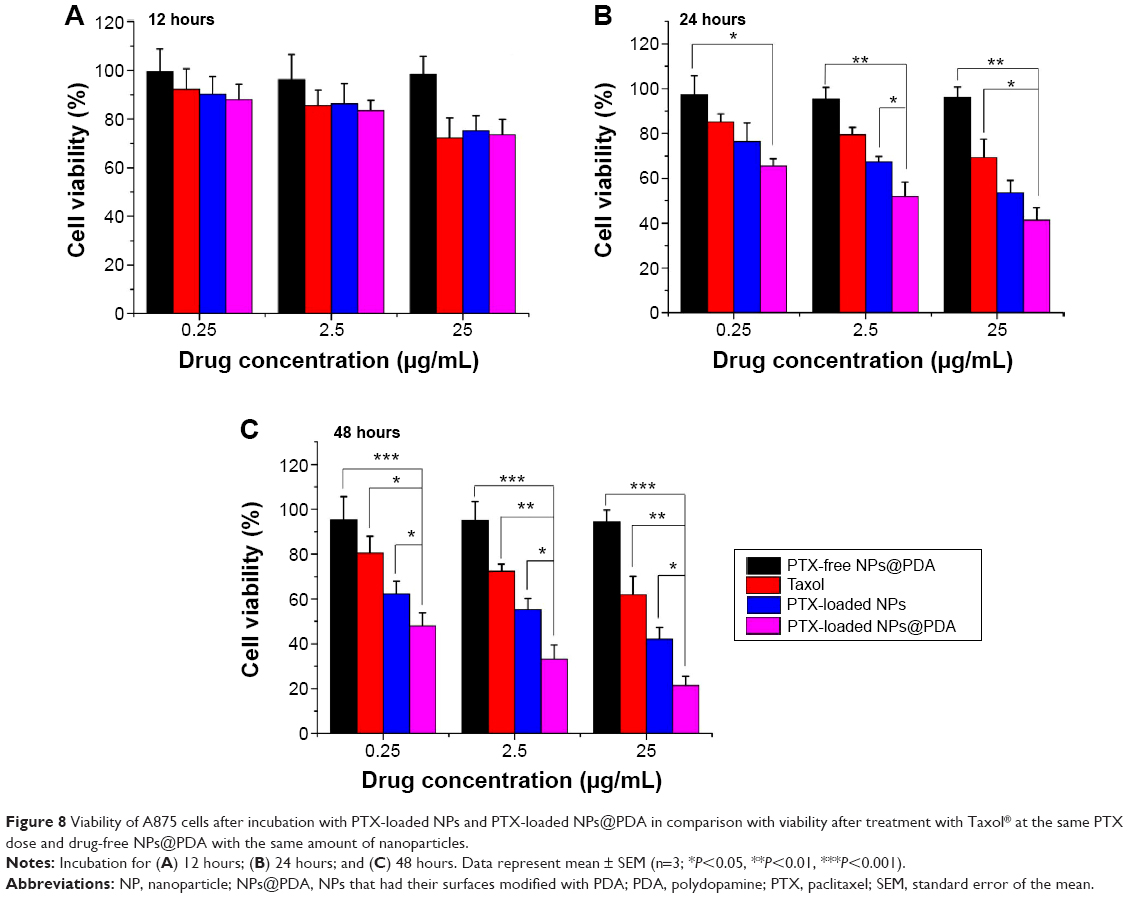 | Figure 8 Viability of A875 cells after incubation with PTX-loaded NPs and PTX-loaded NPs@PDA in comparison with viability after treatment with Taxol® at the same PTX dose and drug-free NPs@PDA with the same amount of nanoparticles. |
After 12 hours of incubation at a drug concentration of 2.5 μg/mL, the cell viability was 85.61% for Taxol®, 86.32% for PTX-loaded NPs, and 83.45% for PTX-loaded NPs@PDA. However, after 24 hours and 48 hours of incubation, the cytotoxicity of PTX-loaded NPs@PDA against A875 cells was, respectively, 27.48% and 39.17% higher than that of Taxol®. The PTX-loaded NPs@PDA showed better cytotoxicity efficacy against A875 cells than PTX-loaded NPs and commercial Taxol®; this could be attributed to the special physicochemical properties of PDA-modified PTX-loaded NPs. The PDA can improve the cellular uptake efficiency of PTX-loaded NPs due to its surface charge and biocompatibility.32,55
In vivo antitumor efficacy and side effect analysis
Due to the satisfactory in vitro cytotoxicity against melanoma cells A875, PTX-loaded MPEG-b-PCL NPs@PDA can be used as drug vehicles for treating melanoma. The antitumor efficacy of the PTX-loaded NPs and the NPs having surfaces modified with PDA was investigated in human melanoma cell line A875 cell-bearing nude mice; the clinical drug Taxol® and physiological saline were used for comparison. The drug was injected into the mice every 4 days for five consecutive cycles, and the tumor volumes and weights of the mice were recorded every 2 days.
Figure 9A shows the tumor growths of the mice after intravenous injection with PTX-loaded NPs, PTX-loaded NPs@PDA, Taxol®, and physiological saline after 20 days’ treatment. The tumor size of the saline group increased obviously. However, tumor growths in the groups treated with Taxol®, PTX-loaded NPs, and PTX-loaded NPs @PDA were significantly inhibited. The melanoma tumor inhibitory effects were in the following order: PTX-loaded NPs@PDA > PTX-loaded NPs > Taxol® > saline. The results of the antitumor efficacy experiment demonstrated that PTX-loaded NPs having surfaces modified with PDA could be released from the NPs effectively and that the released PTX maintained its bioactivity for melanoma treatment.
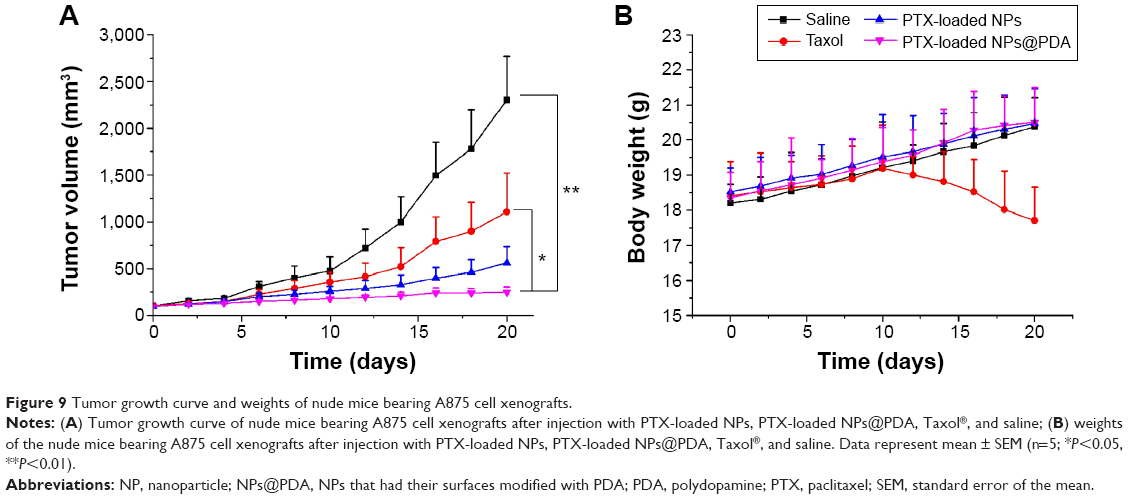 | Figure 9 Tumor growth curve and weights of nude mice bearing A875 cell xenografts. |
Minimizing the side effects of drug-loaded NPs is one of the crucial concerns while developing novel drug delivery vehicles.38 Safety profiles of PTX formulations were evaluated by analyzing the changes in the body weights of animals. Figure 9B shows the body weight variations of mice in all groups; body weight loss was only found at the end of Taxol® therapy. However, the body weight of the groups injected with PTX-loaded NPs and PTX-loaded NPs@PDA grew steadily. Meanwhile, the mice treated with PTX-loaded NPs@PDA remained healthy and vigorous throughout the therapy period, but the mice treated with Taxol® showed weakened vitality. These observations demonstrated that the side effects in mice injected with PTX-loaded NPs@PDA were fewer than those after administration of commercial Taxol®. In other words, the PTX-loaded MPEG-b-PCL NPs having surface modification with PDA could retain the pharmacological activity of PTX and they significantly inhibited tumor growth compared to the effect of Taxol® at the same dose.
Conclusion
In this study, a novel type of PTX-loaded MPEG-b-PCL NPs, which possessed surface modification with PDA, were prepared for malignant melanoma treatment. The PTX-loaded NPs were manufactured by a modified nanoprecipitation method and subsequent surface modification with PDA. The hydrodynamic size of PTX-loaded NPs@PDA is about 140 nm and it has smooth surface. The PTX-loaded NPs@PDA and precursor PTX-loaded NPs had similar drug LC, EE, and drug release profiles. The drug-loaded NPs@PDA possessed higher cellular uptake activity and showed greater cytotoxicity than precursor PTX-loaded NPs. The in vitro and in vivo antitumor effects of the PTX-loaded NPs@PDA were obviously superior to those of PTX-loaded NPs and Taxol®. In short, the novel PTX-loaded NPs@PDA is capable of eliciting high therapeutic effects and low side effects during malignant melanoma treatment.
Acknowledgments
This research was supported by the Guangdong Natural Science Foundation (2014A030313758), Doctoral Fund of Ministry of Education of China (20120002120020), and Science, Technology & Innovation Commission of Shenzhen Municipality (Numbers JCYJ20120616213411826 and JCYJ20140417115840285).
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang W, Shi Y, Chen Y, Hao J, Sha X, Fang X. The potential of pluronic polymeric micelles encapsulated with paclitaxel for the treatment of melanoma using subcutaneous and pulmonary metastatic mice models. Biomaterials. 2011;32:5934–5944. | ||
Agarwala SS, O’Day SJ. Current and future adjuvant immunotherapies for melanoma: blockade of cytotoxic T-lymphocyte antigen-4 as a novel approach. Cancer Treat Rev. 2011;37:133–142. | ||
Ding BY, Wu X, Fan W, et al. Anti-DR5 monoclonal antibody-mediated DTIC-loaded nanoparticles combining chemotherapy and immunotherapy for malignant melanoma: target formulation development and in vitro anticancer activity. Int J Nanomedicine. 2011;6:1991–2005. | ||
Gogas H, Bafaloukos D, Bedikian AY. The role of taxanes in the treatment of metastatic melanoma. Melanoma Res. 2004;14:415–420. | ||
Bedikian AY, Plager C, Papadopoulos N, Eton O, Ellerhorst J, Smith T. Phase II evaluation of paclitaxel by short intravenous infusion in metastatic melanoma. Melanoma Res. 2004;14:63–66. | ||
He W, Ma X, Yang X, Zhao Y, Qiu J, Hang H. A role for the arginine methylation of Rad9 in checkpoint control and cellular sensitivity to DNA damage. Nucleic Acids Res. 2011;39:4719–4727. | ||
Videira M, Almeida AJ, Fabra A. Preclinical evaluation of a pulmonary delivered paclitaxel-loaded lipid nanocarrier antitumor effect. Nanomedicine. 2012;8:1208–1215. | ||
Xu ZH, Gu WW, Huang J, et al. In vitro and in vivo evaluation of actively targetable nanoparticles for paclitaxel delivery. Int J Pharm. 2005;288:361–368. | ||
Xiao X, Zeng X, Zhang X, et al. Effects of Caryota mitis profilin-loaded PLGA nanoparticles in a murine model of allergic asthma. Int J Nanomedicine. 2013;8:4553–4562. | ||
Zhu H, Chen H, Zeng X, et al. Co-delivery of chemotherapeutic drugs with vitamin E TPGS by porous PLGA nanoparticles for enhanced chemotherapy against multi-drug resistance. Biomaterials. 2014;35:2391–2400. | ||
Dianzani C, Zara GP, Maina G, et al. Drug delivery nanoparticles in skin cancers. Biomed Res Int. 2014;2014:895986. | ||
Cho KJ, Wang X, Nie SM, Chen Z, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–1316. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34:6058–6067. | ||
Tao W, Zeng X, Liu T, et al. Docetaxel-loaded nanoparticles based on star-shaped mannitol-core PLGA-TPGS diblock copolymer for breast cancer therapy. Acta Biomater. 2013;9:8910–8920. | ||
Zeng X, Chen H, Zheng Y, et al. Enhanced adsorption of puerarin onto a novel hydrophilic and polar modified post-crosslinked resin from aqueous solution. J Colloid Interface Sci. 2012;385:166–173. | ||
Huang P, Song H, Wang W, et al. Integrin-targeted zwitterionic polymeric nanoparticles with acid-induced disassembly property for enhanced drug accumulation and release in tumor. Biomacromolecules. 2014;15:3128–3138. | ||
Barba AA, Dalmoro A, d’Amore M, Lamberti G. In vitro dissolution of pH sensitive microparticles for colon-specific drug delivery. Pharm Dev Technol. 2013;18:1399–1406. | ||
Barba AA, Dalmoro A, d’Amore M, Vascello C, Lamberti G. Biocompatible nano-micro-particles by solvent evaporation from multiple emulsions technique. J Mater Sci. 2014;49:5160–5170. | ||
Tao W, Zeng XW, Zhang JX, et al. Synthesis of cholic acid-core poly(epsilon-caprolactone-ran-lactide)-b-poly(ethylene glycol) 1000 random copolymer as a chemotherapeutic nanocarrier for liver cancer treatment. Biomater Sci. 2014;2:1262–1274. | ||
Lale SV, Aswathy RG, Aravind A, Kumar DS, Koul V. AS1411 aptamer and folic acid functionalized pH-responsive ATRP fabricated pPEGMA-PCL-pPEGMA polymeric nanoparticles for targeted drug delivery in cancer therapy. Biomacromolecules. 2014;15:1737–1752. | ||
Tamboli V, Mishra GP, Mitra AK. Novel pentablock copolymer (PLA-PCL-PEG-PCL-PLA)-based nanoparticles for controlled drug delivery: effect of copolymer compositions on the crystallinity of copolymers and in vitro drug release profile from nanoparticles. Colloid Polym Sci. 2013;291:1235–1245. | ||
He W, Zhao Y, Zhang C, et al. Rad9 plays an important role in DNA mismatch repair through physical interaction with MLH1. Nucleic Acids Res. 2008;36:6406–6417. | ||
Zhang WP, Sun J, Liu Y, et al. PEG-stabilized bilayer nanodisks as carriers for doxorubicin delivery. Mol Pharm. 2014;11:3279–3290. | ||
Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: focus on cancer. Int J Nanomedicine. 2014;9:467–483. | ||
Madani F, Bessodes M, Lakrouf A, Vauthier C, Scherman D, Chaumeil JC. PEGylation of microspheres for therapeutic embolization: preparation, characterization and biological performance evaluation. Biomaterials. 2007;28:1198–1208. | ||
Zhu ZS, Li Y, Li XL, et al. Paclitaxel-loaded poly(N-vinylpyrrolidone)-b-poly(epsilon-caprolactone) nanoparticles: preparation and antitumor activity in vivo. J Control Release. 2010;142:438–446. | ||
Kaneda Y, Tsutsumi Y, Yoshioka Y, et al. The use of PVP as a polymeric carrier to improve the plasma half-life of drugs. Biomaterials. 2004;25:3259–3266. | ||
Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 2007;318:426–430. | ||
Zheng QS, Lin TR, Wu HY, et al. Mussel-inspired polydopamine coated mesoporous silica nanoparticles as pH-sensitive nanocarriers for controlled release. Int J Pharm. 2014;463:22–26. | ||
Li F, Meng JQ, Ye JF, Yang B, Tian Q, Deng CH. Surface modification of PES ultrafiltration membrane by polydopamine coating and poly(ethylene glycol) grafting: morphology, stability, and anti-fouling. Desalination. 2014;344:422–430. | ||
Yang K, Lee JS, Kim J, et al. Polydopamine-mediated surface modification of scaffold materials for human neural stem cell engineering. Biomaterials. 2012;33:8186–8187. | ||
Liu YL, Ai KL, Lu LH. Polydopamine and its derivative materials: synthesis and promising applications in energy, environmental, and biomedical fields. Chem Rev. 2014;114:5057–5115. | ||
Postma A, Yan Y, Wang YJ, Zelikin AN, Tjipto E, Caruso F. Self-polymerization of dopamine as a versatile and robust technique to prepare polymer capsules. Chem Mater. 2009;21:3042–3044. | ||
Cheng FF, Zhang JJ, Xu F, Hu LH, Abdel-Halim ES, Zhu JJ. pH-sensitive polydopamine nanocapsules for cell imaging and drug delivery based on folate receptor targeting. J Biomed Nanotechnol. 2013;9:1155–1163. | ||
Wang Z, Zeng X, Ma Y, et al. Antitumor efficiency of D-alpha-tocopheryl polyethylene glycol 1000 succinate-b-poly(epsilon-caprolactone-ran-lactide) nanoparticle-based delivery of docetaxel in mice bearing cervical cancer. J Biomed Nanotechnol. 2014;10:1509–1519. | ||
Park J, Brust TF, Lee HJ, Lee SC, Watts VJ, Yeo Y. Polydopamine-based simple and versatile surface modification of polymeric nano drug carriers. ACS Nano. 2014;8:3347–3356. | ||
Zhang YQ, Tang L, Sun LL, et al. A novel paclitaxel-loaded poly(epsilon-caprolactone)/Poloxamer 188 blend nanoparticle overcoming multidrug resistance for cancer treatment. Acta Biomater. 2010;6:2045–2052. | ||
Zhao TJ, Chen HZ, Dong YC, et al. Paclitaxel-loaded poly(glycolide-co-epsilon-caprolactone)-b-D-alpha-tocopheryl polyethylene glycol 2000 succinate nanoparticles for lung cancer therapy. Int J Nanomedicine. 2013;8:1947–1957. | ||
Ma YD, Huang LQ, Song CX, Zeng XW, Liu G, Mei L. Nanoparticle formulation of poly(epsilon-caprolactone-co-lactide)-D-alpha-tocopheryl polyethylene glycol 1000 succinate random copolymer for cervical cancer treatment. Polymer. 2010;51:5952–5959. | ||
Zhang X, Dong Y, Zeng X, et al. The effect of autophagy inhibitors on drug delivery using biodegradable polymer nanoparticles in cancer treatment. Biomaterials. 2014;35:1932–1943. | ||
Zhang X, Zeng X, Liang X, et al. The chemotherapeutic potential of PEG-b-PLGA copolymer micelles that combine chloroquine as autophagy inhibitor and docetaxel as an anti-cancer drug. Biomaterials. 2014;35:9144–9154. | ||
Wan YM, Gan ZH, Li ZB. Effects of the surface charge on the stability of PEG-b-PCL micelles: simulation of the interactions between charged micelles and plasma components. Polym Chem. 2014;5:1720–1727. | ||
Knop K, Stumpf S, Schubert US. Drugs as matrix to detect their own drug delivery system of PEG-b-PCL block copolymers in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2013;27:2201–2212. | ||
Dalmoro A, Lamberti G, Titomanlio G, Barba AA, d’Amore M. Enteric micro-particles for targeted oral drug delivery. AAPS Pharm Sci Tech. 2010;11:1500–1507. | ||
Lee H, Scherer NF, Messersmith PB. Single-molecule mechanics of mussel adhesion. Proc Natl Acad Sci U S A. 2006;103:12999–13003. | ||
Zeng X, Tao W, Wang Z, et al. Docetaxel-loaded nanoparticles of dendritic amphiphilic block copolymer H40-PLA-b-TPGS for cancer treatment. Part Part Syst Charact. 2015;32:112–122. | ||
Yan F, Zhang C, Zheng Y, et al. The effect of poloxamer 188 on nanoparticle morphology, size, cancer cell uptake, and cytotoxicity. Nanomedicine. 2010;6:170–178. | ||
Rakhmatullin RM, Kurkin IN, Pavlov VV, Semashko VV. EPR, optical, and dielectric spectroscopy of Er-doped cerium dioxide nanoparticles. Phys Status Solidi B. 2014;251:1545–1551. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63:170–183. | ||
Hariharan S, Bhardwaj V, Bala I, Sitterberg J, Bakowsky U, Ravi Kumar MN. Design of estradiol loaded PLGA nanoparticulate formulations: a potential oral delivery system for hormone therapy. Pharm Res. 2006;23:184–195. | ||
Liu TT, Seiffert S, Thiele J, Abate AR, Weitz DA, Richtering W. Non-coalescence of oppositely charged droplets in pH-sensitive emulsions. Proc Natl Acad Sci U S A. 2012;109:384–389. | ||
Zhang LY, Sun MJ, Guo R, et al. Chitosan surface-modified hydroxycamptothecin loaded nanoparticles with enhanced transport across Caco-2 cell monolayer. J Nanosci Nanotechnol. 2006;6:2912–2920. | ||
Ma YD, Zheng Y, Zeng XW, et al. Novel docetaxel-loaded nanoparticles based on PCL-Tween 80 copolymer for cancer treatment. Int J Nanomedicine. 2011;6:2679–2688. | ||
Ma WX, Chen MS, Kaushal S, et al. PLGA nanoparticle-mediated delivery of tumor antigenic peptides elicits effective immune responses. Int J Nanomedicine. 2012;7:1475–1487. | ||
Gullotti E, Park J, Yeo Y. Polydopamine-based surface modification for the development of peritumorally activatable nanoparticles. Pharm Res. 2013;30:1956–1967. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.