Back to Journals » International Journal of Nanomedicine » Volume 17
Supramolecular Hydrogel Based Post-Surgical Implant System for Hydrophobic Drug Delivery Against Glioma Recurrence
Authors Wanjale MV, Sunil Jaikumar V, Sivakumar KC, Ann Paul R, James J, Kumar GSV ![]()
Received 9 November 2021
Accepted for publication 15 March 2022
Published 16 May 2022 Volume 2022:17 Pages 2203—2224
DOI https://doi.org/10.2147/IJN.S348559
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Mrunal Vitthal Wanjale,1,2 Vishnu Sunil Jaikumar,3 KC Sivakumar,4 Riya Ann Paul,2,5 Jackson James,5 GS Vinod Kumar1
1Nano Drug Delivery Systems (NDDS), Cancer Biology Division, Rajiv Gandhi Centre for Biotechnology, Thycaud P.O, Thiruvananthapuram, Kerala, 695014, India; 2Research Scholar, Department of Biotechnology, Faculty of Applied Sciences & Technology, University of Kerala, Thiruvananthapuram, Kerala, 695581, India; 3Animal Research Facility, Rajiv Gandhi Centre for Biotechnology, Thycaud P.O, Thiruvananthapuram, Kerala, 695014, India; 4Distributed Information Sub-Centre (Bioinformatics Centre), Bio-Innovation Center (BIC), Rajiv Gandhi Centre for Biotechnology, Poojappura, Thiruvananthapuram, Kerala, 695014, India; 5Neuro-Stem Cell Biology Lab, Rajiv Gandhi Centre for Biotechnology, Thycaud P.O, Thiruvananthapuram, Kerala, 695014, India
Correspondence: GS Vinod Kumar, Tel +91 471 2781217, Fax +91 471 2348096, Email [email protected]
Purpose: The brain, protected by the cranium externally and the blood–brain barrier (BBB) internally, poses challenges in chemotherapy of aggressive brain tumors. Maximal tumor resection followed by radiation and chemotherapy is the standard treatment protocol; however, a substantial number of patients suffer from recurrence. Systemic circulation of drugs causes myelodysplasia and other side effects. To address these caveats, we report facile synthesis of a polyester-based supramolecular hydrogel as a brain biocompatible implant for in situ delivery of hydrophobic drugs.
Methods: Polycaprolactone-diol (PCL) was linked to polyethyleneglycol-diacid (PEG) via an ester bond. In silico modeling indicated micelle-based aggregation of PCL-PEG co-polymer to form a supramolecular hydrogel. Brain biocompatibility was checked in Sprague Dawley rat brain cortex with MRI, motor function test, and histology. Model hydrophobic drugs carmustine and curcumin entrapment propelled glioma cells into apoptosis-based death evaluated by in vitro cytotoxicity assays and Western blot. In vivo post-surgical xenograft glioma model was developed in NOD-SCID mice and evaluated for efficacy to restrict aggressive regrowth of tumors.
Results: 20% (w/v) PCL-PEG forms a soft hydrogel that can cover the uneven and large surface area of a tumor resection cavity and maintain brain density. The PCL-PEG hydrogel was biocompatible, and well-tolerated upon implantation in rat brain cortex, for a study period of 12 weeks. We report for the first time the combination of carmustine and curcumin entrapped as model hydrophobic drugs, increasing their bioavailability and yielding synergistic apoptotic effect on glioma cells. Further in vivo study indicated PCL-PEG hydrogel with a dual cargo of carmustine and curcumin restricted aggressive regrowth post-resection significantly compared with control and animals with intravenous drug treatment.
Conclusion: PCL-PEG soft gel-based implant is malleable compared with rigid wafers used as implants, thus providing larger surface area contact. This stable, biocompatible, supramolecular gel without external crosslinking can find wide applications by interchanging formulation of various hydrophobic drugs to ensure and increase site-specific delivery, avoiding systemic circulation.
Keywords: glioma, drug, delivery, brain, tumor, implant, surgery, hydrogel
Graphical Abstract:

Introduction
Intracranial implants for chemotherapeutic delivery had revolutionized glioma therapy once, since the conception of bis-chloroethyl nitrosourea (BCNU) releasing wafers in 1993.1,2 Polymeric implants can be delivered post-surgically in the tumor resection cavities, which helps in immediate drug release to tackle the relapse arising from the remnant glioma cells. A study by Choucair et al. reported that over 90% of glioblastoma multiforme (GBM) patients suffer from recurrence at the site of the primary tumor, with 5% of patients showing multiple lesions after treatment.3,4 In a prospective study of 222 randomised, placebo-controlled glioma patients, it was found that polymer implant-based chemotherapy directly to brain tumors at the time of surgery could be a safe and effective treatment for recurrent malignant gliomas.5 Many researchers have noted these advantages and worked upon implant-based systems to deliver several drugs such as paclitaxel, camptothecin, and temozolomide, in animal models.6–8
However, the current treatment strategy includes oral and IV modes of treatment that lead to systemic circulation of DNA alkylating chemotherapy drugs, potentially causing treatment-induced myelodysplasia.9,10 In addition, the drug has to be hydrophobic to pass through the blood–brain barrier (BBB) into the lipophilic environment of the brain. However hydrophobic drugs have low bioavailability.11 Also, chemotherapy is not advised if the patient is suffering from low blood cell count post-surgery and radiation, thus creating a lag that gives time for the aggressive tumor cells to diffuse and proliferate.2 Hence, in this study, we have focused on implant-based delivery to bridge the delay in the treatment and offer localized dispersion of the drug. The main advantage of using hydrogel instead of a wafer is the coverage of surface area achieved. It can be used to spread over the uneven edges formed after tumor resection and it can form direct contact with the residual tumor cells.
Polymer-based local delivery systems must have attributes such as high drug-loading capacity, low volume requirement, biocompatibility, biodegradability, good conformity, and efficiency in co-delivery of chemotherapy agents.12 Polyethyleneglycol-polycaprolactone based co-polymer combinations have been widely reported for biomedical and nanomedicine applications in the form of nanoparticles, micelles, and hydrogels.13 It is a biocompatible drug delivery system with reported applications in theranostics, nucleic acid, plasmid delivery, passive and targeted delivery of chemotherapeutic drugs, cytokines, growth factors, etc.14–16 Apart from its biocompatibility, the advantages of PCL-PEG co-polymeric systems include ease of synthesis, adaptability, and versatility offered by a change in hydrophilic (PEG) and hydrophobic (PCL) proportions. PCL and PEG have been approved by the Food and Drug Administration (FDA) for acute and chronic cytotoxicity and have been found safe for biomedical applications.17 In this paper, we have explored a facile way of open-ended co-polymer synthesis by using polycondensation reaction, thus minimising the use of solvents and other catalysts. We have synthesized a PCL-PEG co-polymer by ester bond using PCL diol and PEG diacid, which forms a stable hydrogel at 37°C without any crosslinker forming a supramolecular hydrogel. Our main focus is to develop a biodegradable carrier system in the form of a soft hydrogel, which is suitable for implanting in the brain without toxicity and further utilizing this hydrogel to encapsulate chemotherapeutic agents for glioma treatment.
Glioma is a highly heterogeneous tumor; the most commonly noted mutations are in the following genes: EGFR, p53, p16INK4a and PTEN, IDH1/2, PDGFR, and it varies upon the stages of tumor and from patient to patient.18,19 Hence it is very difficult to conquer this disease with a drug aiming at a single pathway, such as bevacizumab targeting VEGF-based angiogenesis or cilengitide-based EGFR targeting. Tumor cells tend to escape when targeted with a single moiety, and hence the drugs that have shown treatment efficiency are DNA alkylating agents such as BCNU and TMZ. Recent studies have reported the use of multiple concurrent chemotherapy agents and a combination of chemotherapy and biotherapeutics to address highly heterogeneous and aggressive glioma.20,21 In this study, we propose delivery of BCNU drug-loaded PCL-PEG hydrogel; supplemented with an adjuvant curcumin as an implant. Curcumin is the supermolecule that has anti-proliferative, anti-inflammatory, antibacterial, anti-angiogenic, anti-oxidant properties that have been investigated for decades in cancer therapy.22,23 As mentioned earlier, glioma can arise from changes in signaling pathways such as PI3K-AKT and RAS oncogenic pathway, p53 pathway of tumor suppressor, RB cell cycle control, and metabolic IDH1/2. To combat this versatility, curcumin acts by deregulation of these pathways such as interfering in ERK and PI-3K downstream signaling cascade of the EGFR, deregulation of PI-3K/AKT/mTOR and PI-3K/AKT NF- kB pathways which lead to decrease in the proliferation and survival of glioma cells.22,24 Zhuang et al. have also studied the potential of curcumin to promote autophagy and differentiation in glioma initiating cells in vitro, in vivo and in human glioma patients.25 Studies have also reported curcumin-based attenuation of chemotherapy and radiation-induced apoptosis in normal tissue, thus protecting normal cells from destructive effects on account of its anti-inflammatory properties.26 Although curcumin has tremendous potential as an anti-glioma molecule, its bioavailability remains an issue and requires drug delivery systems for its effective transport.27 Studies suggesting encapsulation of curcumin inside PLGA nanoparticles faces insufficient passage through the BBB, and thus cannot reduce a tumor, but only restrict its growth.28 In this study, we have explored curcumin as an adjuvant therapeutic molecule along with DNA alkylating BCNU as a primary drug. In our system, we propose to use minimal drug concentration, carmustine being a DNA alkylating agent, but restoring the treatment efficiency with addition of adjuvant curcumin, which can act on various pathways to curb proliferation of the remaining glioma cells.
Here, we have optimized the lowest concentration combinations of carmustine and curcumin that gave maximum cell cytotoxicity. As it has to be implanted in the brain for prolonged drug delivery, we checked its biocompatibility in Sprague Dawley rat brains. We have inserted polymer in the frontal cortex of healthy rats, and analysed its survival efficiency and motor function for 1 week, 6 weeks and 12 weeks. In vivo efficacy of the carrier system with drug and adjuvant was tested in SCID mice with U251-luciferase transfected cells induced glioma model. As per our hypothesis, our results suggest that even 0.1% of remnant tumor cells could lead to aggressive recurrence. With overlay of the proposed PCL-PEG carmustine-curcumin system in the post-surgical tumor cavity, we could see a reduction in tumor volume and regrowth.
Materials and Methods
Polycaprolactone diol (MW 2000), poly(ethylene glycol) diacid (MW 600), thiazolyl blue tetrazolium bromide (MTT), carmustine, curcumin, Monoclonal anti β actin antibody (A5316) were used as received from Sigma-Aldrich. Live dead assay kit and Apo-BrdU TUNEL Assay kit was purchased from Invitrogen. Cleaved Caspase 3 and cleaved PARP Antibody (Apoptosis sampler kit Cell signaling technologies #9664). DeadEnd Colorimetry TUNEL system (G7130) was from Promega. Cell culture inserts were purchased from Millipore (Cat No. MCSP24H48) and Hi media (TCP078).
Sprague Dawley rats 6–8 weeks old were used for material biocompatibility study and NOD-SCID mice 4–6 weeks of age (18–20 g) were used for xenograft tumor study. Animals were maintained in Animal Research facility, Rajiv Gandhi Centre for Biotechnology (RGCB) and housed in cages with food and water ad libitum. Animal experiments were carried out in full accordance with the rules and regulations of the Government of India’s Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA). The Institutional Animal Ethics Committee (IAEC) of Rajiv Gandhi Centre for Biotechnology, gave their approval to all animal experiments under the protocol no IAEC/732/GSV/2019 for brain biocompatibility and IAEC/750/GSV/2019 for NOD-SCID mice xenograft-based drug delivery system efficacy study.
Synthesis of Polymer
PCL-PEG co-polymer was synthesized by a polycondensation reaction. Briefly, ester bond is synthesized in this process where poly (caprolactone) diol (MW 2000) and poly (ethylene glycol) diacid (MW 600) 1:1 (w/v) was heated to 115°C in oil bath under an inert nitrogen atmosphere for 14–16 h with constant stirring. The hydroxyl group of PCL diol reacts with the acid group of PEG diacid to eliminate water molecules and proceed to form a co-polymer having an ester bond. After heating, the co-polymer was cooled and dialysed for 48 h against water using Serva dialysis membrane (MW 3500 cut-off) to remove unreacted monomers and further lyophilized.
Characterization of Polymer
Fourier Transformed Infra-Red Spectroscopy (FT-IR)
FT-IR spectroscopy was performed on Spectrum 65 (Perkin Elmer). Spectra were recorded between 4000 and 600 cm−1 wave number range. Dried samples were mixed with KBr and compressed into pellets for making measurements.
Differential Scanning Calorimetry
Differential Scanning Calorimetry (DSC 6000, PerkinElmer, USA) was employed for determining the nature of the synthesized PCL-PEG co-polymer. The accurately weighed sample was sealed in an aluminium pan and was scanned while heating between −30 and 150°C at a rising rate of 10°C/min. Nitrogen at 20 mL/min was purged into the system.
1H-Nmr
The formation of co-polymer was confirmed through Proton nuclear magnetic resonance (1H−NMR) spectroscopy using Bruker 500MHz spectrometer (Bruker BioSpin, Billerica, MA). CDCl3 was used as a solvent.
Gel Permeation Chromatography
Gel Permeation Chromatography (GPC) (Agilent 110 HPLC, USA) was used to determine the macromolecular weight and distribution of PCL-PEG co-polymer. The sample was freshly dissolved in distilled tetrahydrofuran (THF) at a concentration of 5 mg/mL. THF was eluted at a rate of 1.0 mL/min through the Waters Styragel HT column. The external and column temperature was kept at 35°C. The molecular weights of samples were calculated based on polystyrene standard samples with a known narrow molecular weight distribution.
Rheology
Amplitude sweeps and frequency sweep study was performed on PCL-PEG supramolecular hydrogel using Anton Paar Modular Compact MCR-102 Rheometer. Cone plate (CP25-2) was used with a slope of 5pt/dec. The sample was placed on the clean sample holder and the cone plate was adjusted to a length of 22µm. Amplitude sweep was done at variable strain (0.01–100%) and frequency sweep at constant strain at 0.1% with variable angular frequency (100–0.1 rads/s) at 37°C.
Scanning Electron Microscopy
SEM analysis of the lyophilized PCL-PEG hydrogel was performed on Nova NanoSEM 450, SE mode at 5.00 kV. We scanned for surface morphology at 400x, 1000x and 15,000x. Further 15,000x image was scanned with Image j software to show surface layers.
Drug Release Study
To form a gel, 200 mg of PCL-PEG co-polymer was weighed and 800 µL PBS was added to it at room temperature. The tube was inverted several times to mix the polymer into the solution uniformly. It was observed that excessive vortex leads to precipitation of polymer, which further fails to form a sturdy gel. After mixing the solution was heated at 50°C for 3 min with intermittent mixing and then cooled to 20°C gradually using Thermomixer. Once the solution had cooled to 20°C, 200 µL of drug mixture was added to the tube and mixed uniformly and then incubated at 37°C to form a stable gel.
Release of carmustine and curcumin from PCL-PEG hydrogel in PBS was studied up to 360 h at 37°C. Concentration of carmustine and curcumin was estimated by C18 HPLC column with methanol: acetonitrile: water: acetic acid (41:36:23:1) as mobile phase. Carmustine and curcumin absorbance was acquired at 262 nm and 425 nm, respectively.
In silico Modeling – Mechanism of Gelation
In order to further understand aggregation behavior of PCL-PEG polymer, we performed in silico modeling studies. In molecular dynamic (MD) simulations, initial systems with 100 molecules were enclosed and minimized for two possible PCL-PEG chains: PEG-PCL-PEG-PCL-PEG and PCL-PEG-PCL-PEG-PCL using an explicit TIP3P water model, solvated using a cubic box of water measuring 70 Å on each side. A series of simulations looked at the effect of temperature on the micelle aggregation of the polymers. For three different temperatures, the polymer systems were subjected to free molecular dynamics lasting 50 ns each, with periodic boundary conditions and full particle-mesh Ewald electrostatics using Gromacs 4.5 and PRODRG was used to generate a topology of co-polymer chains to be further used with GROMOS force field.29–31 To facilitate micelle formation, 32 additional SDS molecules were added with random placement of the structure, thus increasing the detergent concentration. In the case of each system, the molecular configuration was taken from the equilibration performed under NVT for 10 ns and then followed by NPT for 10 ns was subjected to MD production run at three different temperatures of 50°C, 20°C, and 37°C.
Cell Culture Study
Neuro 2a cells (RRID: CVCL_6268) were procured from National Centre for Cell Sciences (NCCS), Pune, India. U251 cells were obtained from Cell repository, Rajiv Gandhi Centre for Biotechnology. Cells were maintained in prescribed standard medium (unless stated otherwise) consisting of Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) Gibco (Life Technologies AG, Basel, Switzerland) and 1% Anti-anti cocktail (Gibco). For Neuro 2a cells above media was supplemented with 0.1% sodium pyruvate (Gibco), 0.1% non-essential amino acids (NEAA) (Gibco), 0.1% Glutamax (Gibco) under humidified atmosphere at 37°C with 5% CO2.
Biocompatibility Study – Neuro Excitotoxicity
Neuro excitotoxicity was studied according to Lee et al. with modification.32 Briefly, Neuro 2a cells were used to study the excitotoxicity of the polymer before in vivo cranial implantation. (100 µL) PCL-PEG hydrogel was incubated with media at 37°C for 24, 48, 72, 96 and 120 h. For each time point, a separate media control tube was kept to neutralise any death caused due to depletion of nutrients. Glutamate was used as a positive control for neuro excitotoxicity and was measured by MTT assay.
In vivo Studies; Biocompatibility
Biocompatibility Study by the Intracranial Implant of Hydrogel
The procedures described by He et al. and Hou et al. were followed for stereotactic rat brain implantation.33,34 Briefly, 6–8 weeks old Sprague Dawley Rats (220–250 g) were used as an animal model for compatibility testing of the biodegradable polymeric hydrogel. Animals were anesthetized with intraperitoneal injection of ketamine (75 mg/kg) xylazine (4 mg/kg). Animals were kept on heating blankets to maintain body temperature. After being entirely anesthetized, the head and neck were shaved and thoroughly cleaned with 70% ethanol and prepared for surgery. Under aseptic conditions, a midline incision was made to mark Bregma on the skull. A drill hole was made in the cranium marked stereotactically, penetrating into the cortical tissue in the left frontal region at 2 mm forward from bregma and 2 mm on left and 3mm deep. 20 µL of hydrogel was injected into the brain using a Hamilton syringe. The syringe was held in place for 30 s and then slowly removed. The cranium was sealed with dental plaster to avoid any infection and sutured. Meloxicam (0.2 mg/kg) was administered as an analgesic. Animals were housed in appropriate cages with ad libitum food and water supply and checked for any sign of distress, weight loss and loss of motor activity due to surgery or inflammation due to implant.
After 1 week, 6 weeks and 12 weeks (n = 5 for each time point of control and PCL-PEG implant), separate groups of animals were deeply anesthetized and 0.9% saline solution was perfused through their heart followed by a fixative containing 4% paraformaldehyde. The region of the frontal lobe containing gel implant was resected and fixed in 4% paraformaldehyde for 24 h followed by 30% sucrose for 72 hfor cryopreservation. OCT embedded cryosections were taken and processed for histological evaluation by hematoxylin and eosin staining. Inflammation and scar formation was studied by immunofluorescence using IBA1 (Abcam, ab178847[EPR16589]) and GFAP (Invitrogen (GA5) eFluor 660, e Bioscience) antibodies.
Motor Function Test: Wire Hang Test
A non-invasive and easy method to determine the proper functioning of the motor cortex (implant region) and its function was tested following the protocol of wire hang test described by Paylor et al.35 Briefly, the rat was placed on a wire cage lid, and the lid was gently waved in the air so that the animal gripped the wire. The lid was then turned upside down, approximately 30–38 cm above a surface of bedding material. Latency to fall onto the bedding material was recorded with a 60 s cut-off time.
Live Animal MRI Scanning
MRI images were taken using a 7T animal MRI station (70/30 USR Bruker Biospec, USA) at Amritha University, Kochi. T2-weighted MRI scan of the brain was done for 1 week and 12 weeks post-implantation and control animals under anesthesia to check for edema and confirm the presence of material in the implanted site in the different time periods.
In vitro Cytotoxicity assays
MTT Assay Combination Index
MTT reduction assay was performed to assess cell viability. Briefly, U251 cells were seeded (5×103 cells/well) in a 96-well culture plate and allowed to adhere overnight. Cells were treated with formulations of carmustine (100–750 µM), curcumin (10–70 µM) and combination of carmustine and curcumin for 72 h. Thereafter, 100 µL of MTT at a concentration of 5 µg in PBS (pH 7.4) was added to each well and incubated for 4 h at 37°C. The unreacted medium was discarded and formazan purple crystals were solubilised in 200 µL isopropyl alcohol, and absorbance was measured at 570 nm.
Cell viability (%) related to control wells containing cell culture medium without treatment was calculated by [A] test/[A] control×100, where [A] test is the absorbance of the test sample and [A]control is the absorbance of the control sample. Cytotoxicity (%) is 100−cell viability (%).
Combination Index: (CI) = (D)1 /(Dχ)1 + (D)2/(Dχ)2
(Dχ)1 and (Dχ)2 represented IC50 concentrations of each drug alone
(D)1 and (D)2 represented IC50 concentrations of drugs in combination
CI < 1, = 1 and > 1 indicated synergism, additivity, and antagonism, respectively.
Apoptosis by Activation of Caspase 3 Cleavage by Immunostaining
1x104 U251 cells were seeded in 8-well chamber slides. Cells were treated with carmustine (150 µM), curcumin (20µM) and combination of both (150µM+20µM). An equivalent amount of ethanol required to make the concentration of drug was added in the control well. After 48 h incubation, media with drug was removed and washed thrice with PBS and fixed with ice-cold 4% paraformaldehyde at 4°C for 15 min. After washing with PBS, post fixing cells were blocked using 10% normal goat serum plus Triton X for 45 min at room temperature. Cells were incubated with Cleaved Caspase 3 primary antibody (Apoptosis sampler kit Cell signaling technologies #9664) overnight at 4°C. Cells were washed well with PBS and incubated with anti-rabbit secondary antibody tagged with Alexa fluor 488 (Abcam 150077) for 1 h. At the end of incubation, nucleus was stained with Hoechst 33342 for 5 min. Finally, the cells were washed 3 times with PBS and imaged with Leica confocal microscope.
Apoptotic Index was calculated by the formula:
Apoptotic Index = (Cleaved caspase 3 positive cells/Total number of cells) x 100.
Tunel Assay
APO-BrdU Tunel assay kit (Invitrogen A23210) was used to analyse DNA fragmentation as a marker of apoptosis caused due to drug released from the hydrogel. Briefly, U251 cells 3×105 were seeded in 6-well plates. Drug-loaded PCL-PEG hydrogel (100 µL) was suspended in cell culture insert above cells with media. Cells were trypsinized and processed after 24, 48, and 72 h post-treatment. Cells were fixed with paraformaldehyde, and DNA double-strand nick was labelled with BrdU DNA labelling solution. Cytotoxicity was analysed by FACS.
Live Dead Assay
Live dead assay was performed following the protocol detailed in the kit by Thermo Scientific. Briefly, U251 cells were cultured overnight in 6-well plate (3x105 per well). Cells were treated with PCL-PEG hydrogel loaded with carmustine and curcumin, which was suspended in a transmembrane cell culture insert for 24, 48, and 72 h.36
In-Vivo Study
Evaluation of Anti-Tumor Efficacy
4–6 weeks old SCID mice (male/female) were subcutaneously injected with 5×106 cells/0.2 mL of U251 glioma cells in the right flank of each mouse. These U251 cells were electroporated with pGL 4.51 luc plasmid expressing luciferase enzyme under SV40 promoter. After injection, the tumor growth dimensions and body weight was monitored physically. After the tumor reaches a palpable size (300–400 mm3), animals were randomly divided into the following groups under isoflurane anesthesia: Control, PCL-PEG carrier alone, intravenous administration of drug (carmustine and curcumin) and drug-loaded PCL-PEG gel. In all animals, 98–99% tumor was resected. Control sham-treatment animals were sutured immediately; 100–125 µL of carmustine and Ccurcumin loaded PCL-PEG hydrogel was applied at the resection site of implant treated animals; the intravenous drug group was given the same concentration of carmustine and curcumin combination as that of the implant through tail vein injection 3 times at 3 day intervals. The polymer was pre-sterilised by UV radiation and hydrogel was made aseptically and applied at the tumor resection site in the carrier alone group. Animals were sutured and kept under observation for 6 weeks. Live images of animals were recorded before and after the surgery and implant using luciferin-based bioluminescence imaging IVIS Xenogen, Perkin Elmer, USA. Live images of the tumor progression/death were recorded. At the end of treatment, all the animals were sacrificed by CO2 asphyxiation and vital organs such as the brain, liver, kidney, lungs, heart, spleen, along with residual tumor were collected for histological studies. Paraffin-embedded tumor and organ sections were stained with hematoxylin and eosin. Tumor tissues were also further stained for immunofluorescence with Ki67 and GFAP (Invitrogen (GA5) eFluor 660, e Bioscience) and imaged with confocal microscope. Paraffin-embedded tumor tissue sections were processed and stained with DeadEnd colorimetry TUNEL assay following the prescribed protocol and developed with DAB and chromogen in dark for 30 min. Slides were mounted using DPX and visualized at 10x under a light microscope.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 6. Student’s t-test was used to compare two samples (*P <0.05, **P <0.01, ***P <0.001) to find statistical variations. For multiple comparisons between three or more groups, one-tailed analysis of variance ANOVA was used. Data were presented as mean value ± S.D. (standard deviation).
Results and Discussion
PCL-PEG Co-polymer Synthesis and Characterization
PCL-PEG co-polymer was synthesized by polycondensation followed by post-dialysis yield was 76%. Use of any catalyst such as stannous octate was avoided as this co-polymer-based hydrogel was to be implanted in the brain and we wanted to avoid use of any harsh chemicals that could cause inflammation or side effects of the material. Hence a facile approach of polycondensation employed by heat and the monomers used in the form of diol and diacid provide the necessary OH and COOH groups for ester bond synthesis (Figure 1A). FTIR, H1NMR, DSC and GPC analysis were used to characterize the synthesized co-polymer.
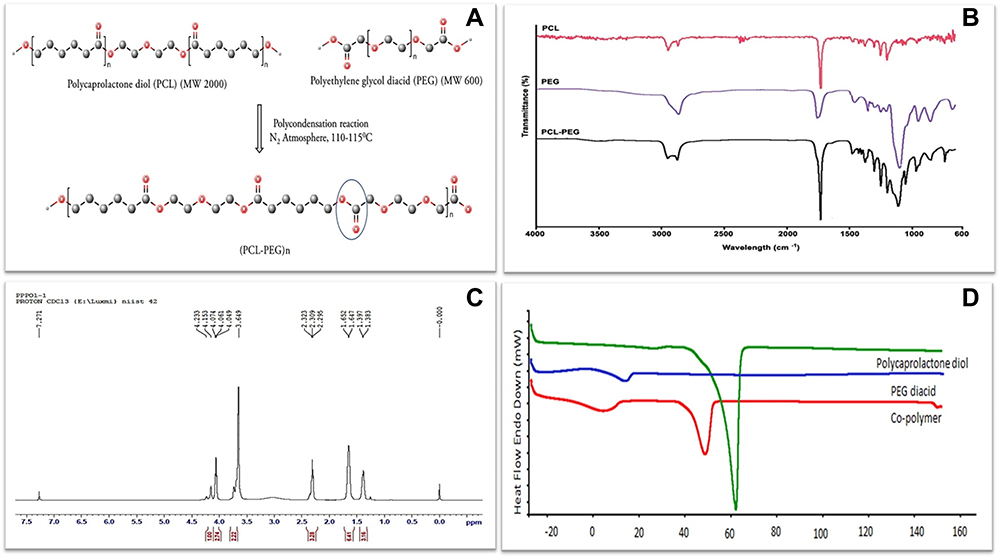 |
Figure 1 (A) Schematic representation of PCL-PEG co-polymer synthesis by polycondensation reaction of polycaprolactone diol and polyethylene glycol diacid. Characterization of co-polymer (B) FTIR spectrum of PCL diol, PEG diacid and PCL-PEG co-polymer with sharp peak at 1722 cm−1 representing ester bond. (C) H1 NMR spectrum of co-polymer. (D) DSC thermogram of PCL diol, PEG diacid and PCL-PEG co-polymer. |
FTIR spectrum analysis (Figure 1B) shows absorption bands at 1102 cm−1 and 1242–1294 cm−1 was attributed to the characteristic C–O–C stretching vibrations of the repeated –(OCH2CH2) units of PEG and the –COO bands stretching vibrations respectively and a strong C-O stretching band appeared at 1722 cm−1 which was attributed to the ester bond. For further confirmation, H1NMR spectra recorded a sharp peak at 3.64 ppm which is attributed to methylene protons of –(CH2CH2O) in PEG units in co-polymer and peaks at 1.38, 1.64, 2.32, and 4.06 are attributed to methylene protons of –(CH2), –(OCCH2), and –(CH2OOC) in PCL blocks. The weak peak at 4.20 ppm was attributed to methylene protons of -O–CH2–CH2–in the PEG end block that linked with PCL blocks (Figure 1C).
DSC results (Figure 1D) shows the DSC thermogram corresponds to polycaprolactone, PEG and PCL-PEG co-polymer. The endothermic peak at 62°C indicating PCL, endothermic peak at 18°C indicating PEG, and endothermic peak at 52°C indicating the co-polymer was observed. The visible shift of endothermic peak observed in co-polymer indicates the shifting of crystalline nature of co-polymer. The co-polymer has the capability to form in situ gelation behavior. In lower critical solution temperature, it can behave as a solid due to the interaction between hydrophobic PCL core and polar environment. While above lower critical solution temperature, it behaves as a gel.
The gelation process of the PCL-PEG co-polymer, although simple, requires meticulous control over temperature and time. We compared gelation between synthesized co-polymer PCL-PEG and equal quantity of monomer PCL diol and PEG diacid blend. After processing at sequential temperature and time, the monomer blend did not form a hydrogel. It formed a stable hydrogel only in co-polymer state (Figure 2A). 200 mg/mL of PCL-PEG co-polymer (20% w/v) formed a stable hydrogel with 10% tolerance to ethanol (Figure 2B and C). Here ethanol was used as a solubilizing agent for hydrophobic drugs. An increase in ethanol concentration led to precipitation of the polymer in the gelation step. Also, with lower concentration of polymer the tolerance level of ethanol reduced which led to an undesirable decrease in drug entrapment.
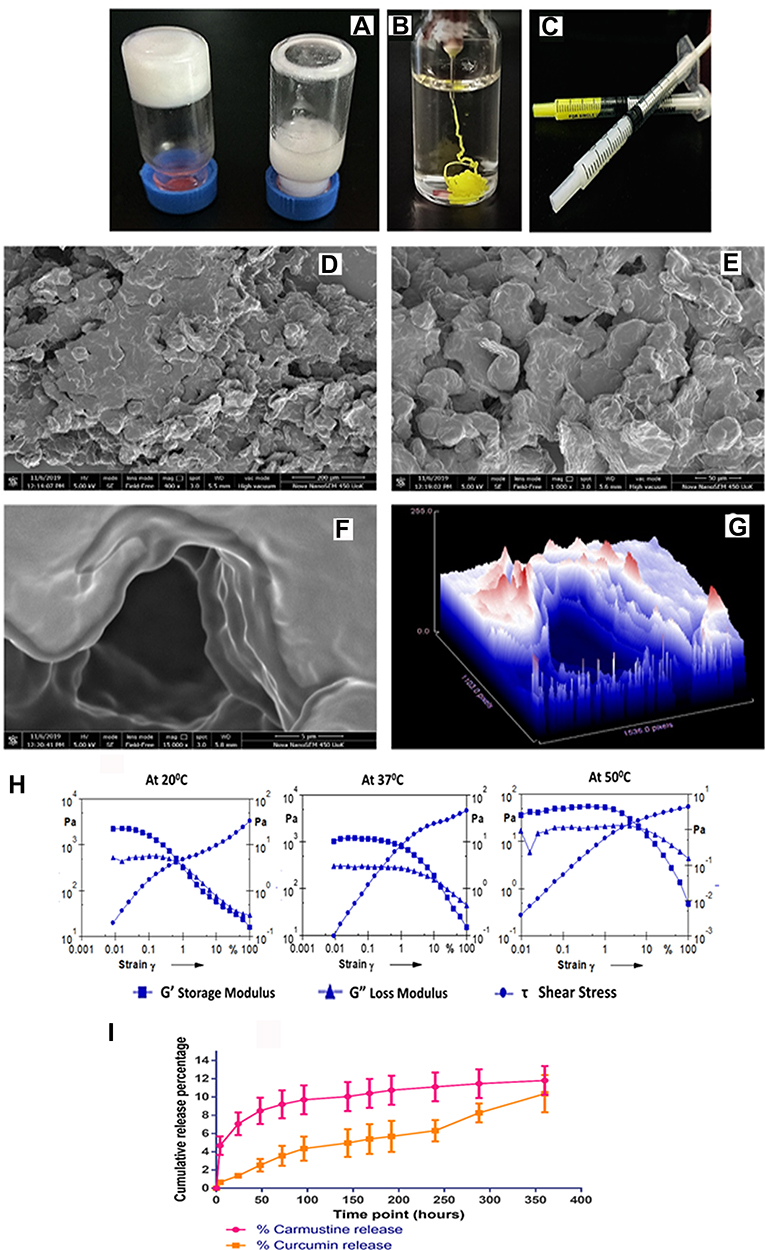 |
Figure 2 (A) 20% (w/v) PCL-PEG co-polymer forming hydrogel vs 20% (w/v) monomer PCL diol and PEG diacid blend, that does not form hydrogel. (B) Drug entrapped hydrogel holds its stable gel nature upon injection in excess PBS. (C) PCL-PEG hydrogel can be filled in syringes and ready for application for ease of administration. (D and E) Scanning electron micrograph of surface topology of PCL-PEG (400x) and 1000x, respectively. (F and G) 15,000x magnification of the surface shows layering of polymer in the gel and surface topography graph by Image J analysis of the same. (H) Rheology graph of amplitude sweep at 20°C , 37°C and 50°C with varying strain (0.001–100%). (I) Release study of carmustine and curcumin from the PCL-PEG hydrogel at 37°C in PBS for 360 h. |
Surface topology of PCL-PEG supramolecular hydrogel was analysed with a scanning electron microscope after lyophilising hydrogel. The surface was not smooth and upon magnification we can see a layering pattern in the lyophilized gel (Figure 2D-G). We believe these to be the pockets for drug entrapment which facilitates slow release upon gradual swelling and degradation by esterases.
The stability and viscosity of the supramolecular hydrogel was studied using rheometry. G’ storage modulus and G” loss modulus were determined in an amplitude sweep study corresponding to gradual increase in amplitude of strain exerted on the gel at three different temperatures (Figure 2H). It was observed that G’ was higher than G” which indicates stable gel and less fluidity. At physiological temperature, 37°C, the gel could sustain higher strain indicating stability compared with gel at 20°C and 50°C. At lower temperature, the G’ was higher than G” but could not withstand strain more than 1%. While at 50°C both the storage and loss modulus were lower than at 37°C, yet G’ was still higher than G”. Thus PCL-PEG hydrogel could sustain a difference in strain and temperature and was most stable at physiological temperature of 37°C. In an interesting study by Rowland et al., rheology of resected human glioma tissues was studied at 20°C with 1% strain and reported storage modulus values that range from 140–620 Pa. According to the authors having a lower G’ than resected tissue helps in reinforcing good contact with the brain surface in the surgical cavity.37 In our study 20% (w/v) PCL-PEG hydrogel at 20°C with 1% strain has G’ of 323 Pa which falls within the tissue range. Hence following resection, the surrounding brain tissue refolds gel which will be able to reshape and stay in touch providing a larger surface area for drug dissipation.
Study of cumulative release behaviour of carmustine and curcumin from PCL-PEG hydrogel for 360 h in PBS revealed slow and steady release of the active molecules from the gel (Figure 2I). As carmustine and curcumin are not crosslinked to the polymer, we can see a slow release based on swelling of the hydrogel in vitro considering the affinity of hydrophobic carmustine and curcumin to the hydrophobic matrix of PCL-PEG hydrogel. In vivo, due to the presence of esterase the release rate of active molecules from the gel could be higher.
The gel permeation chromatography of PCL-PEG co-polymer, indicates the molecular weight and mass distribution. The single peak (6384) indicates the mono distribution of the molecular weight of co-polymer and the absence of individual chains with low polydispersity. The results show that the weight and number average molecular weight of co-polymer are 6527 and 2488 (Figure S1).
PCL-PEG co-polymer has been synthesized previously by ring opening polymerization using initiators and catalyst to form block polymer which forms hydrogel.38,39 The gelation mechanism in this system is by micelle formation followed by packaging driven by hydrophobic forces of polycaprolactone.40 In our study, we have preferred elementary method for co-polymer synthesis to avoid involvement of any chemical initiators or solvents which could prove toxic to the brain. Hence to predict the mechanism of gelation and contemplate if similar micelle formation takes place in the PCL-PEG co-polymer chain we have synthesized, we performed insilico modeling of two possible PCL-PEG chains with reference to our GPC data. The GPC molecular peak was observed at 6384, so based on the molecular weight of the monomers we predicted possibility of presence of two PCL-PEG chains: PEG-PCL-PEG-PCL-PEG and PCL-PEG-PCL-PEG-PCL. Also, according to our gelation experiment, we subjected these PCL-PEG chains to sequential temperature which were required to form hydrogel: 50°C, 20°C, and 37°C. The characterization of micelle formation is provided by a total of 150 ns of free molecular dynamics. The following observations were found to be consistent with the micelle formation in the MD simulations of PEG-PCL-PEG-PCL-PEG and PCL-PEG-PCL-PEG-PCL. The first 50ns of MD simulation with a temperature of 50°C shows higher fluctuations in the RMSD for both polymers and starts to decrease after that, indicating the systems begin to gain reliable configuration (Figure 3A and C). The RMSD displacement decreases over time because the polymers with higher separation begin to take the closest approach to each other. At the start of the simulation, the PCL headgroup surrounded by water was initially unstable, as indicated by higher RMSD fluctuations. A higher peak RMSD of ~6 Å in the initial phase of MD simulations is equivalent to bending the hydrophobic head groups in each polymeric unit and further reduces the separation in the next 50 ns to 20°C, as the occupancy of micelle hydrocarbon into the interior occurs over a longer simulation time. The last 50 ns at 37°C showed a stable structure for the rest of the simulation with RMSD ~1.8 Å from their initial structure. The rate of micelle formation remained the same for both polymers subjected to free molecular dynamics. The integrity of the micelle was maintained throughout the simulation, and there was significant change in radius in the final phase of the simulation. As shown in Figure 3B and D, the resulting structure exhibits a micelle almost by the aggregation of ~20 polymers. In the course of the simulation, the radius of micelle PCL-PEG-PCL-PEG-PCL was slightly higher with 36.8 Å compared with 28.5 Å in the case of PEG-PCL-PEG-PCL-PEG, but remained stable. Notably, the aggregation of PCL-PEG units with PEG-PCL-PEG-PCL-PEG resulted in a −619 kJ/mol in the energy compared with −946 kJ/mol for aggregation of PCL-PEG-PCL-PEG-PCL (Figure S2). Thus, we could see PCL-PEG co-polymer aggregating and experimentally we could cast a stable hydrogel without using any crosslinker, attributing these characteristics to the supramolecular nature of PCL-PEG co-polymer hydrogel.
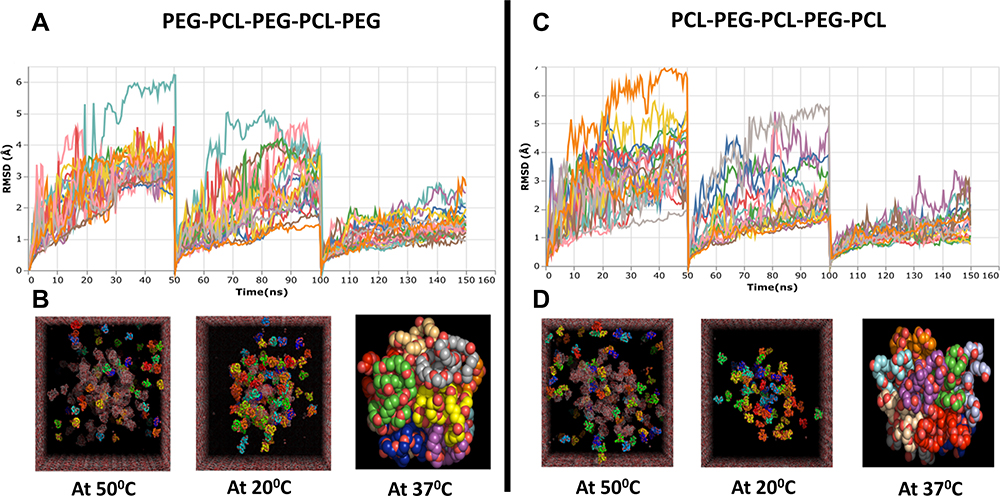 |
Figure 3 In silico modeling of two possible combination of PCL-PEG co-polymer chain PEG-PCL-PEG-PCL-PEG and PCL-PEG-PCL-PEG-PCL using Molecular Dynamics (MD) simulation. (A and C) Root means square deviation (RMSD) graph of total simulation time of 150 ns at 50°C, 20°C and 37°C for 50 ns each. (B and D) Representation of simulation of PCL-PEG molecules (ball chain) in cubic box with water molecules (mesh) at 50°C, 20°C, and 37°C (isolated final stable micelle formed from ~20 polymer molecules. |
Upon differential light scattering (DLS) study of the hydrogel we found micro-micellar structures of cumulant result diameter of 597 nm size (Figure S3). Based on this and in silico study we propose the gelation mechanism of the PCL-PEG co-polymer to be multimolecular micelle which aggregates with an increase in temperature and concentration. The hydrophobic blocks of PCL form the core while PEG forms the flanking bridge, which could also form a flexible loop that remains hydrated to form a hydrogel. After initial solubilization of PCL-PEG co-polymer at 50°C during the process of cooling, at 20°C micelle organization could take place further stabilizing at 37°C.
Excitotoxicity
Excitotoxicity leads to excessive activation of neuronal receptors of amino acids and can cause neuronal killing as well. Certain compounds such as glutamate, capsaicin, acetylcholine and solvents such as dichloromethane have been recognized to cause excitotoxicity.41 As a brain implant, to assess neuro-compatibility of PCL-PEG hydrogel, Neuro2a were used to evaluate excitotoxicity of the polymer initially following the model of glutamate excitotoxicity.42 Normalizing the viability of cells with incubated media at each time point, no significant death or loss of viability was observed. However, there was a reduction of viability at 24–72 h which increased later at 96 and 120 h time points (Figure 4A). With the PCL-PEG implant having a long degradation time frame it was considered suitable for further in vivo implant experiments as the viability was restored. Comparing viability at 96 and 120 h to positive control glutamate (0.5 mM and 5 mM), it was found that cells were significantly more viable than the glutamate.
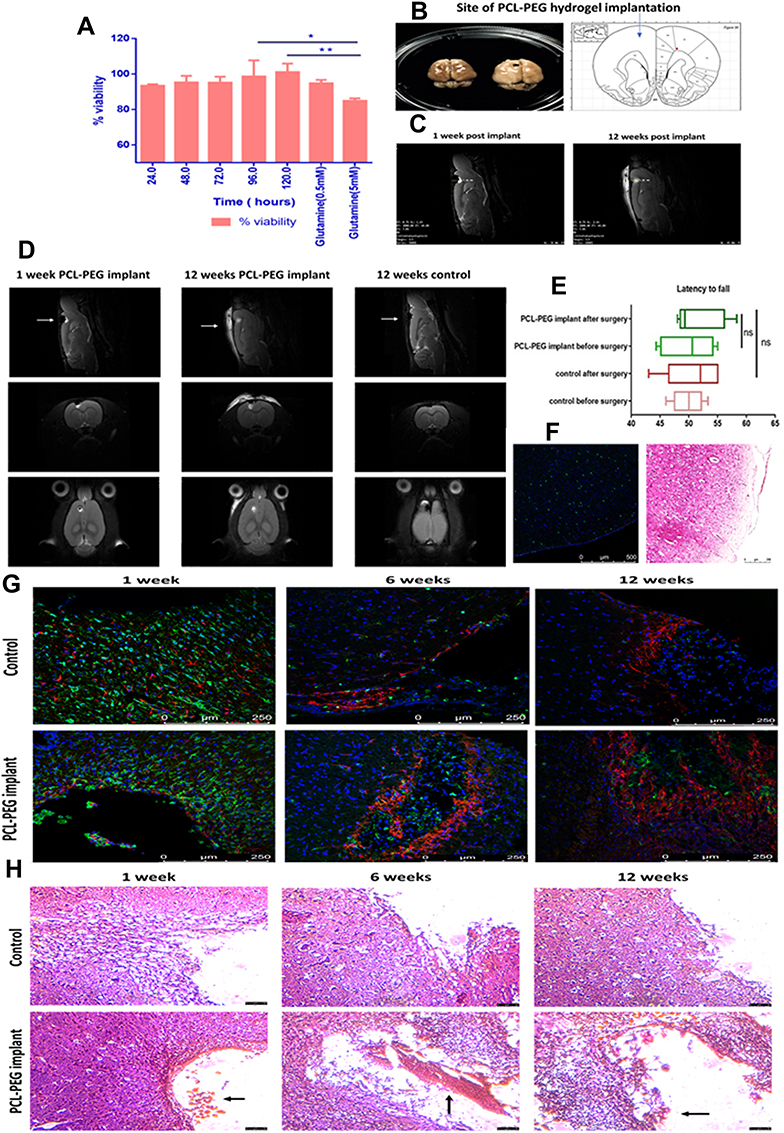 |
Figure 4 (A) Neuro-excitotoxicity study of PCL-PEG co-polymer enriched media (24–120 h) with glutamate (5 mM) as positive control analysed by MTT assay for viability post 24 h incubation (*P< 0.05, **P<0.01). In vivo biocompatibility study. (B) Rat brain(left), with implant in cortex (right) and site of implant of hydrogel 3 mm deep in cortex rat brain atlas (extreme right). (C) MRI scan at 3 mm depth PCL-PEG hydrogel implant at 1 week post-surgery and 12 weeks post-surgery (hydrogel intact in brain, swollen and partially degraded). (D) MRI scan of rat brain implanted with polymer (at 1 week and 12 weeks) and control animal with PBS implant (12 weeks). (E) Motor cortex function test comparison of rats with and without PCL-PEG implant hydrogel analysed by latency to fall and active movements before and after implant surgery (6 weeks post implant). (F) Immunofluorescence and hematoxylin and eosin staining of right hemisphere of rat brain cortex (control for inflammation caused due to surgical intervention in brain). (G) Immunostaining of rat brain tissues control and PCL-PEG implant at 1 week, 6 weeks and 12 weeks time point stained with Iba 1 antibody (green) GFAP antibody (red) and Hoechst (blue). IBA 1 positive cells are indicator of microglial activation as a part of inflammatory response. (H) Hematoxylin and eosin staining for histopathological analysis of rat brain tissues of control and PCL-PEG implanted animals at 1 week, 6 weeks and 12 weeks time point. Arrows indicate implant at the site of surgery. |
In-Vivo Biocompatibility
Intracranial Implantation of PCL-PEG Hydrogel System
Intracranial implants need to be studied primarily for their biocompatibility which depends on many factors such as the site of implant, material composition, size, viscosity, sterility, contact area, by-products after degradation; and mainly defined by cell-cell and cell-polymer interaction.43,44 Inflammatory reactions due to implantation or any intervention should be as minimum as possible for the implant to be a successful candidate for the drug delivery system. Inflammatory reactions can be of two types primarily: Acute phase response, the first step in response due to any injury, implant or infection. Immune cell infiltration along with cytokine production and activation of macrophages marks this phase. In the brain, we can see the activation of Iba 1 positive macrophages.45 In Chronic phase response, the proliferation of fibroblasts and macrophages can be observed along with collagen deposition. If inflammation persists, adverse effects such as edema, pain and intense granuloma, and infection can be seen.46 In this study, PCL-PEG hydrogel was implanted in Sprague Dawley rat brain cortex using stereotaxis apparatus. Animals were observed for any adverse effects, weight and motor function test and overall coordination, which could be affected if chronic inflammation persists.
The PCL-PEG hydrogel was implanted in the left-brain cortex at a depth of 3mm (Figure 4B and C). During the T2-weighted MRI imaging of rat brain implanted with PCL-PEG hydrogel, we could find hydrogel as white contrast present in the brain cavity created surgically at 1 week and 12 week time point compared with control animal brain imaged at 12 weeks time point. We also observed the hydrogel swollen and partially degraded at 12 weeks compared with 1 week implanted animals (Figure 4D). We did not find significant fluid accumulation or signs of chronic inflammation upon implanting at 1 week and up to 12 weeks post-implantation. PCL-PEG hydrogel was implanted 2 mm further from Bregma, 2 mm to the left and 3 mm deep in the cortex region of the rat brain which is responsible for motor function. To observe any significant changes in motor function of the animal due to inflammation in this region, a simple wire hang test was done before surgery and after implantation. We did not find any significant change in latency to the animal falling off before and after 6 weeks of the surgery (Figure 4E). Immunofluorescence and hematoxylin and eosin staining of right hemisphere of rat brain cortex (control for inflammation caused due to surgical intervention in brain) are shown in Figure 4F. The hydrogel was stable and well-tolerated in the Sprague Dawley rat brain.
Cryosections of Sprague Dawley rat brain implanted with PCL-PEG hydrogel and control were stained with Iba 1 marker for microglial activation as an inflammatory marker and astrocytes that migrate to the site of surgery as a part of healing are labeled with GFAP antibody. We could see severe infiltration of Iba 1 cells in 1 week group in both control and implanted animals (Figure 4G). The inflammation was acute in nature and subsided over the next 2 time points of 6 weeks and 12 weeks. Iba 1 cells were high in 1-week animals and reduced in 12 weeks group whereas GFAP cells increased from 1 week to 12 weeks, signifying healing. Hoechst 33342 was used to stain the nucleus of cells and we can see cells localizing in the implant region, supporting that the hydrogel is biocompatible and supports wound healing without causing chronic inflammation. The right hemisphere of the brain was used as an internal control in animals.
Hematoxylin and Eosin Staining of Sprague Dawley Rat Brain
Rat brain cryosections were stained with hematoxylin and eosin. A number of inflammatory cells which infiltrated in the surgical implant region were counted. PBS was injected into control animals. Infiltrating inflammatory cells such as neutrophils, leukocytes along with activated microglial cells were counted after staining. We could see a surge in inflammatory cells in animals sacrificed at 1 week time point in both control and PCL-PEG hydrogel implanted animals, attributing the inflammation mainly to the surgical intervention (Figure S4). Later in animals with PCL-PEG hydrogel implant, number of inflammatory cells reduced significantly at 6 weeks interval however it was still more than the control animals with PBS. At 12 weeks interval, the inflammation had substantially reduced compared with the first week and 6 weeks implant and compared with the control animals (Figure 4H).
PCL-PEG Hydrogel –Drug Delivery Implant System
Drug Toxicity Evaluation by the Synergistic Method
As the PCL-PEG hydrogel was found to be biocompatible, well-tolerated, and could form a stable hydrogel, which remained intact even after passing through a syringe, the next step was to entrap a drug to complete it as a drug delivery system. Carmustine is the prescribed drug for glioma treatment, but it is a DNA alkylating drug and not only specific for glioma cells. In the commercially available wafer system, each wafer is impregnated with 7.7 mg of carmustine. Our aim was to reduce the amount of drug with a chemosensitizer to enhance its efficacy in tumor recurrence suppression. Hence to reduce the amount of carmustine to be entrapped and make it cost-effective, we have tried a combination of curcumin as an adjuvant along with it.
U251 glioma cells were treated with carmustine (100–750 µM) and curcumin (10–70 µM) concentrations individually for 72 h and processed with MTT. Media with PBS and ethanol was used as control (carmustine stock was prepared in ethanol) to normalize data and calculate cytotoxicity of the drugs. IC50 of carmustine was calculated as 293.8 µM and that of curcumin was 27.23 µM (Figure 5A and B). PCL-PEG supramolecular hydrogel acts as a carrier of hydrophobic drugs and thus can entrap both the drug carmustine and adjuvant curcumin. Thus, U251 cells were treated with a combination of the two with the least concentration (i.e. below its IC50) and which had the highest cytotoxicity in MTT assay was selected. Supplementing carmustine with curcumin caused higher cytotoxicity and helped as an adjuvant (Figure 5C). For individual drugs, cytotoxicity was found to be dose-dependent and increased upon an increase in concentration. However, in the increased combination, curcumin concentration helped to increase cytotoxicity and at lower concentration of carmustine i.e., 150 µM with supplementation of curcumin (10, 20, and 50 µM), significantly higher toxicity was achieved than the free drug. Thus, we chose the least concentration of each carmustine and curcumin i.e., below IC50, which was cytotoxic above 75%. The combination index was calculated and found that CI at IC50 of the combination carmustine 150 µM and curcumin 20 µM was 0.649 (less than 1) i.e., the combination of drug is synergistic.
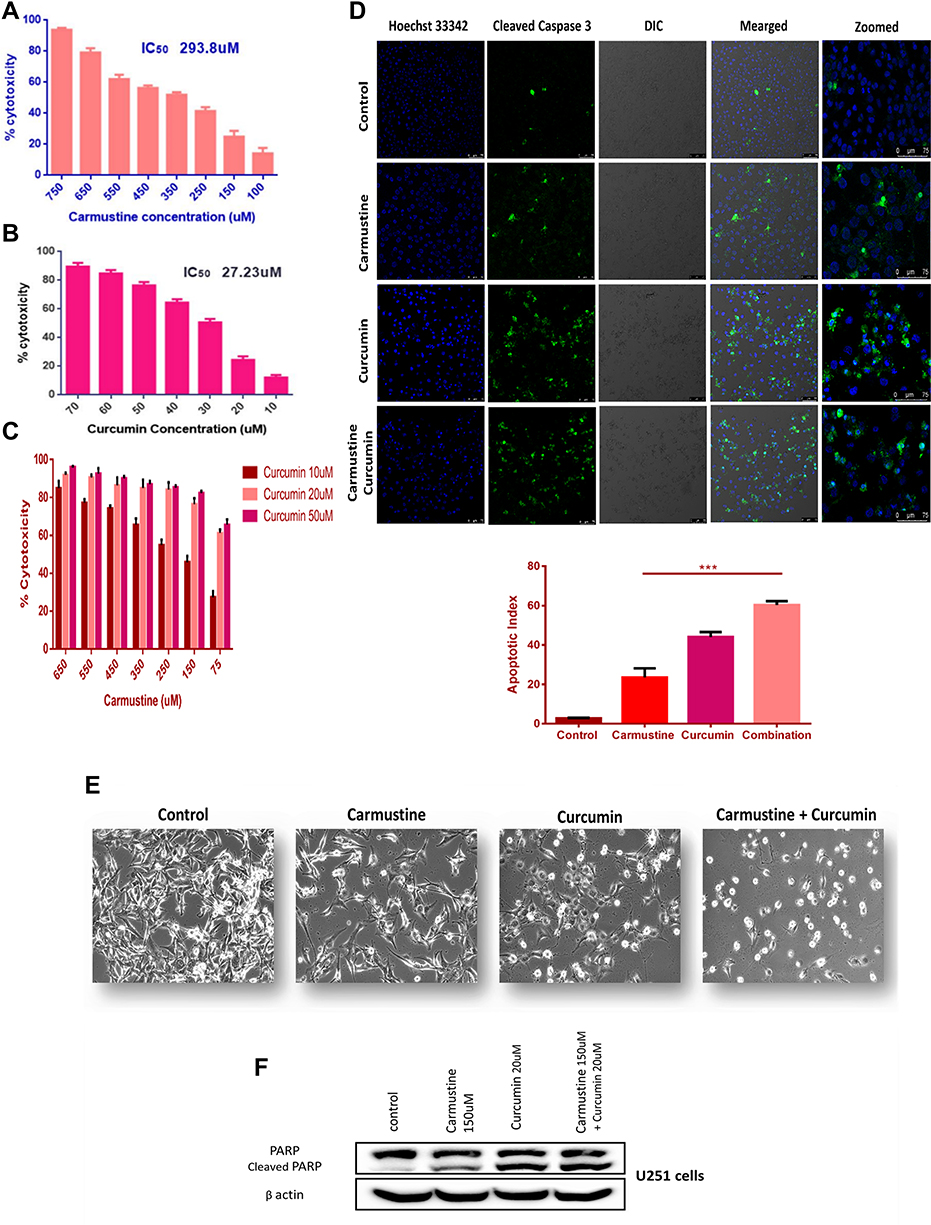 |
Figure 5 In vitro cytotoxicity analysis of (A) carmustine, (B) curcumin, (C) carmustine and curcumin combination on U251 glioma cells at the end of 72 h, cytotoxicity was assessed using MTT assay (n = 3). Based on Combination Index calculation carmustine and curcumin work synergistically. (D) Detection of caspase 3 cleavage by immunofluorescence. U251 glioma cells treated with carmustine (150 μM), Curcumin (20 μL) and combination of both for 48 h followed by staining with antibody against cleaved caspase 3 (Green- FITC) detected in cytoplasm, nucleus (Blue-Hoechst 33342). Combination Apoptotic index was significantly higher compared with individual carmustine (***P <0.0005) indicating positive impact of curcumin addition to increase cytotoxicity of carmustine. (E) Morphological changes in U251 cells treated with carmustine (150 μM), Curcumin (20 uM) and combination of both for 48 h (Brightfield image). (F) Cleavage of nuclear protein PARP, hallmark of apoptosis in U251 cells upon treatment with carmustine (150 μM), Curcumin (20μM) and combination of both for 48 h. Full length PARP (116 kDa), cleaved PARP (89 kDa) B actin (42 kDa). |
Immunocytochemistry Study: Caspase 3 Cleavage
As MTT results showed improved cytotoxicity upon carmustine-curcumin combination treatment, this led us to investigate further if the cells are undergoing apoptosis or the combination is only able to inhibit proliferation. Further, curcumin has an established role in time-dependent Caspase 3 activation via Apaf1 upregulation.47 Previous reports also suggest curcumin can induce receptor-mediated and mitochondria-mediated proteolytic mechanisms and induce apoptosis in T98G glioma cells.48 Hence, to check if the combination is able to induce apoptosis via the known executioner Caspase 3 activation, U251 cells treated with carmustine, curcumin and combination for 48 h, and stained with cleaved caspase 3 antibody by immunofluorescence-based imaging. Control cells have smaller number of cells with cleaved Caspase 3 and upon treatment with both the drugs; we can see an increase in the caspase 3 activation and cleavage. As curcumin is known to have autofluorescence, U251 cells treated with curcumin and stained with secondary antibody alone was used as control for immunostaining (Figure S5). Apoptotic index calculation showed a significant increase and influence of curcumin supplementation (Figure 5D).
Western Blotting: PARP Cleavage
Glioma cells, upon treatment with carmustine and curcumin for 48 h showed a change in morphology (Figure 5E). In comparison to the control cells, which have fibroblast-like morphology, drug combination-treated cells showed decreasing trend in viability with adverse changes in morphology such as rounding and shrinkage of cells along with blebbing of the plasma membrane observed under brightfield microscope. For further confirmation of apoptosis, we have performed immunofluorescence staining of Caspase 3 cleavage, which is a hallmark of cell priming towards apoptosis. Increased Caspase 3 along with other caspases is known to execute the cleavage of nuclear protein PARP.49 Further, with Western blotting, we have shown cleavage of PARP enzyme, thus confirming the apoptotic fate of the glioma cells upon effective treatment with a combination of carmustine and curcumin. Here again, an increase in PARP cleavage was observed when glioma cells were treated with a combination of carmustine and curcumin compared with the drug carmustine alone (Figure 5F). Treating cells with drug below its IC50 concentration, we could achieve 90% cytotoxicity by the addition of curcumin to the carmustine treatment regime. Also, the combination was not only able to inhibit the growth of glioma cells but also induce apoptosis.
Entrapment of Drug
Based on the in vitro cytotoxicity assays, the combination of concentration of carmustine and curcumin, which was lower than individual IC50 concentration, was entrapped in the hydrogel. It also helped in controlling the amount of ethanol that could be tolerated in PCL-PEG 20% gel without precipitating the polymer. As PCL-PEG was a slow drug-releasing gel, the ratio of carmustine and curcumin concentration was maintained and 50 times of 150 µM and 20 µM i.e., 7.5 mM and 1 mM respectively, was entrapped in 20% w/v of PCL-PEG hydrogel. The polymer was heated at 50°C and upon cooling at 20°C drug mixture was added to the polymer and allowed to form a stable gel at 37°C (Figure 2B and C). Further assays were done using this drug entrapped gel. Entrapment efficiency was 98.3% estimated by precipitating the polymer in excessive ethanol. Ethanol also solubilizes drugs carmustine and curcumin and the concentration entrapped was calculated with UV spectrophotometer.
PCL-PEG Efficacy to Deliver Drug in vitro: Live Dead Assay
Schematic representation of the experimental set-up to test in vitro efficacy of drug PCL-PEG hydrogel is shown in Figure 6A. Carmustine and curcumin-loaded PCL-PEG hydrogel was suspended over U251 cells in cell culture insert and cytotoxicity was assessed by a live dead assay using FACS. The live cells produced esterase, which cleaved calcein AM to calcein that emits green fluorescence. On the other hand, dead cells had lost membrane integrity and were thus labelled with ethidium homodimer, which is live cell impermeant (Figure S6). Upon FACS analysis we observed time-dependent increase in cytotoxicity from 19.8% in 24 h incubation to 84.6% in 72 h when carmustine and curcumin co-entrapped PCL-PEG hydrogel was suspended over cells in inserts (Figure 6B). As the gel is immersed in media above cells, carmustine and curcumin are released slowly into the media leading to significant time-dependent percentage cytotoxicity (Figure S7).
 |
Figure 6 (A) Schematic representation of experimental set up to test in vitro efficacy of drug PCL-PEG hydrogel. (B) Calcein-AM and ethidium homodimer based Live dead analysis of U251 cells treated with carmustine and curcumin loaded PCL-PEG hydrogel for 24 h, 48 h, 72 h and PCL-PEG carrier alone as control. (C) Double stranded DNA breaks analysed by extent of incorporation of BrdU -TUNEL assay, in U251 cells treated with carmustine and curcumin loaded PCL-PEG hydrogel conditioned media. Increase in apoptosis with increase in time-dependent drug release. |
TUNEL Assay
Carmustine crosslinking with DNA leads to blocking of DNA and RNA synthesis, blocks protein synthesis which results ultimately in apoptosis. Along with this curcumin also blocks cell proliferation by acting on various pathways and leads to apoptosis in glioma cells. One of the hallmarks, DNA double-stranded nicks, is formed when nucleases are activated in the process of apoptosis, a controlled cell destruction process. U251 glioma cells were treated with drug released in media 24–72 h. Post-treatment BrdU were incorporated to the double stranded DNA nicks. Anti BrdU antibody tagged with Alexa 488 was used to detect the BrdU incorporated in treated cells. With an increase in time, carmustine and curcumin entrapped in the hydrogel were released to the conditioned media and we could see an increase in apoptotic cell population from 6.3% at 24 h to 95.3% in 72 h (Figure 6C).
Evaluation of Anti-Tumor Efficacy: In vivo SCID Mice Xenograft Study
The efficacy of the PCL-PEG hydrogel loaded with carmustine and curcumin, to help in drug delivery in situ and reduction of tumor recurrence was studied in NOD-SCID mice implanted with U251-Luc transfected cells. Schematic experimental representation of post-surgical treatment in xenograft Glioma model in NOD-SCID mice is represented in Figure 7A. Tumor of size 300–400mm3 was observed within 6–8 weeks of injection of 5×106 cells. Unbiased maximum surgical resection was performed removing 98–99% of the tumor. Maximum resection of the tumor could be done when the tumor is subcutaneous, but upon attachment to muscles, tumor was found to be more aggressive and maximum 98% resection of tumor was possible. The remnant U251 glioma cells could be visualised by Luciferin based bioluminescence (Figure 7B). NOD-SCID mice with U251 Luc cells tumors were divided into 4 groups (n = 5): (a) Control group where animals were sutured back after surgical resection without any implant, (b) PCL-PEG carrier alone (no drug cargo), (c) Intravenous drug injection: carmustine and curcumin equivalent to the implanted system were given through tail vein injection 3 doses at intervals of 3 days, and (d) Drug loaded PCL-PEG system: carmustine and curcumin loaded PCL-PEG system. In group three (IV drug) it was observed that the tumor regrowth was slow until 2 weeks post-surgery, coinciding with the drug treatment; however later, the tumor volume increased. This combination managed to restrict tumor regrowth compared with control and carrier alone. In the fourth group with carmustine and curcumin-loaded PCL-PEG hydrogel, significant tumor reduction was observed compared with control. Also, there was a significant reduction of tumor volume in hydrogel implanted animals compared with IV drug-treated animals (Figure 7C). Even though complete ablation of tumor could not be achieved with the 100–125 µL hydrogel implant treatment, increasing the hydrogel amount or replacing other potent drug can be investigated to achieve the desired restriction. However, in our study with PCL-PEG hydrogel loaded with carmustine and curcumin significant reduction of aggressive tumor regrowth was observed (Figure 7D). Post- surgery, weight of animals was monitored weekly. Initially there was a slight drop in the weight for 1 week and all animals showed recovery since then and weight was moreover maintained (Figure 7E). Along with it Kaplan–Meier survival analysis showed 100% animals survived till the 6-week end point, of the drug-loaded PCL-PEG compared with 60% of tail vein treated and 40% of aggressively growing control animals as the animals in control and carrier group had to be euthanized after reaching ethical end point of tumor volume (Figure 7F).
 |
Figure 7 In vivo efficacy analysis of PCL-PEG carmustine and curcumin loaded hydrogel to inhibit aggressive tumor recurrence. (A) Schematic experimental representation of post-surgical treatment in xenograft glioma model in NOD-SCID mice. (B) Representative gradual luciferin bioluminescence based tumor volume monitoring post- surgery. (C) Tumor size at end point of study − 6 weeks post-surgical resection in control, PCL-PEG carrier, intravenous drug and PCL-PEG hydrogel with carmustine and curcumin. (D) Tumor volume progression after surgical resection up to 6 weeks of study (*P <0.05. ****P <0.0001). (E) Weight of animals through the post-surgery treatment period. (F) Kaplan−Meier Survival percent analysis for the 6 weeks post-surgery treatment period. |
Hematoxylin and eosin staining of paraffin-fixed tissues shows tumor cells have elongated undifferentiated nuclei and are multinucleated cells. Apoptotic patches are seen in the interior region of the tumor (Figure 8A). Pyknotic nuclei which are shrunken and rounded are seen in the apoptotic region. The zone of apoptosis is seen in IV drug-treated animals and PCL-PEG hydrogel with carmustine and curcumin. However, this zone of apoptosis is not only restricted to the interior core of the tumor in PCL-PEG hydrogel with drug-treated animals and extends almost to the boundary of the tumor. No obvious evidence of organ damage was observed in the brain, heart, liver, spleen, lung, or kidneys of PCL-PEG hydrogel treatment groups compared with control groups based on histological inspection by hematoxylin and eosin staining. Splenomegaly was observed in all groups; most in control animals and least in implant-treated animals (Figure S8A and B). Previous reports suggest increase in ratio of circulating neutrophils to granulocytes in glioma along with secretory molecules such as granulocyte colony stimulating factors (G-CSF and GM-CSF) which are known to cause splenomegaly.50 Hence in the control group which has aggressive regrowth post-surgery most splenomegaly cases were seen compared with drug-loaded PCL-PEG hydrogel. 100–125 µL of PCL-PEG drug implant was well tolerated and reduced systemic circulation of drug thereby safeguarding vital organs from continuous exposure to chemotherapeutic drugs.
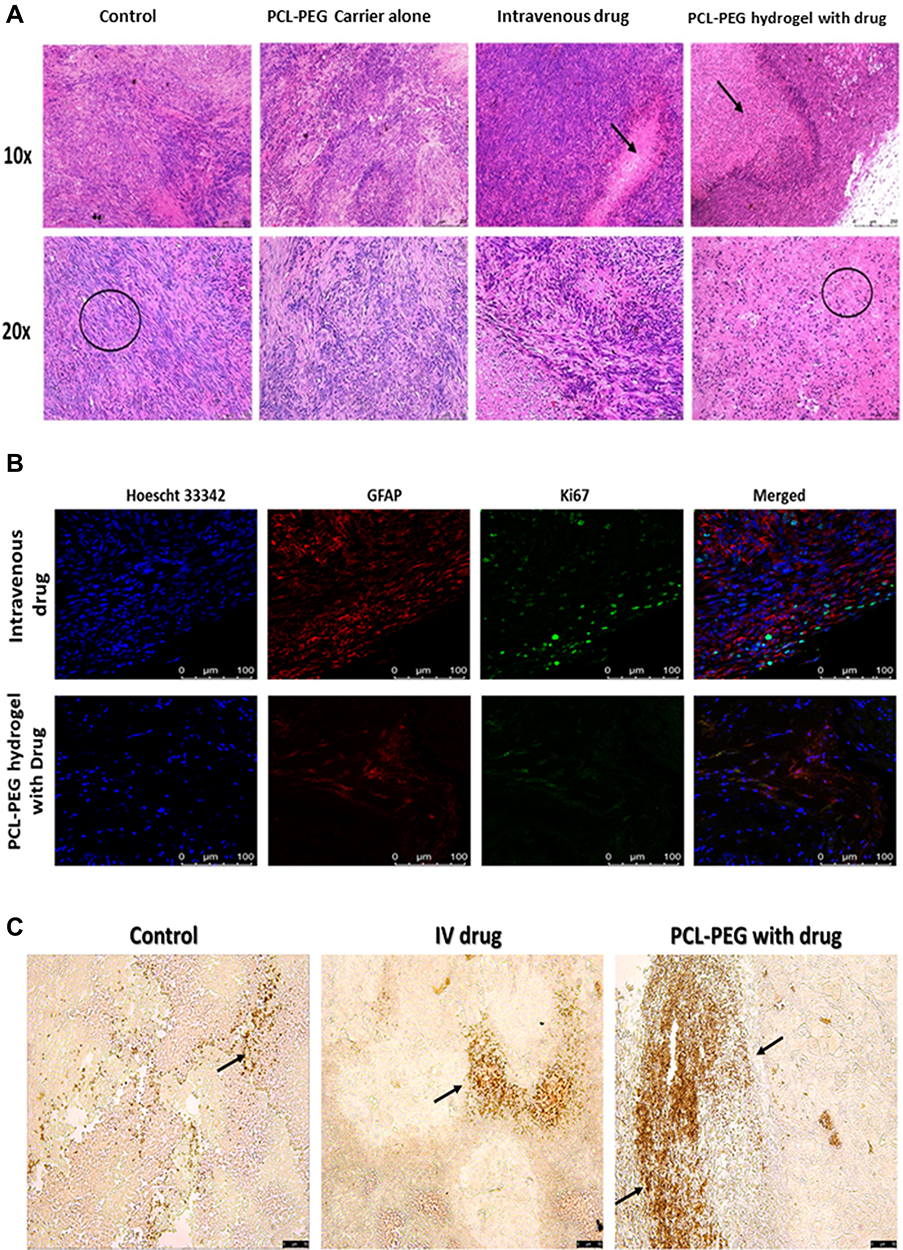 |
Figure 8 (A) Histopathological evaluation of paraffin-embedded tumor tissue by hematoxylin and eosin staining, after 6 weeks of treatment. (B) Confocal image-based evaluation by immunofluorescence of proliferation and prognostic marker Ki67 (Alexa 488), counter-stained with GFAP (eFluor 660) positive U251 cells to differentiate mice fibroblasts from human glioma cells and Hoechst 33342 to stain nucleus. (C) Brightfield image-based evaluation of tumor tissue sections stained with TUNEL assay, arrows indicate apoptotic cells with double stranded DNA breaks. |
Immunohistofluorescence Study with Ki67 Cell Proliferation Marker
Ki67 (or MIB-1) is a nonhistone nuclear protein, a proliferation marker expressed in cells undergoing mitosis, while it is absent in cells in G0 phase.51 According to WHO classification, lower Ki67 staining index has been correlated to low-grade glioma and thus better prognosis in patients.19,52 Here, we have counterstained paraffin-embedded tumor tissue sections with Hoechst 33342 staining the nucleus, GFAP stains U251 glioma cells, so that we can differentiate only the glioma cells from other tissue cells, that are proliferating. We have found a significant difference in the number of Ki67 positive glioma cells in group four drug-treated animals compared with animals post surgically implanted with carmustine and curcumin entrapped gel (Figure 8B). Although the studies continue to ascertain the use of Ki67 as a marker for prognosis in patients, it clearly indicates a reduction in the actively proliferating glioma cells. Thus, based on hematoxylin and eosin and Ki67 staining, the post-surgical implant of the drug-loaded hydrogel has reduced proliferation and caused apoptosis in the aggressively recurring glioma.
Tissue sections stained with TUNEL assay showed the presence of apoptotic cells with free 3ʹhydroxyl termini, a hallmark of DNA breaks formed during apoptosis (Figure 8C). In the animals treated with PCL-PEG hydrogel with carmustine and curcumin, large apoptotic patches were observed compared with IV drug-treated animals. Very few sparsely distributed apoptotic cells were observed in the control animal tumor. In our in vitro study, we have validated that a combination of carmustine and curcumin treatment of glioma cells primes them towards apoptosis, observed by caspase 3 activation and PARP cleavage which is in agreement with the TUNEL assay-stained in vivo xenograft study data.
Conclusions
This study is successful in developing an injectable and implantable brain biocompatible hydrogel efficient in carrying model hydrophobic drugs. A facile esterification synthesis is used for the co-polymer synthesis to avoid drastic chemicals for brain implant preparation. PCL-PEG hydrogel serves as a long-term implant and was studied to be intact for 12 weeks, confirmed by MRI. The MD simulations demonstrate that it is possible to simulate the formation of micelles evaluated by controlled temperatures through all-atom dynamics. It forms hydrogel with PBS without crosslinking, and it is very efficient in entrapping hydrophobic drugs such as carmustine and curcumin. The main advantage of using this supramolecular biodegradable hydrogel compared with currently existing wafers is the malleability and ability to cover a larger and uneven surface area exposed post-surgical resection of tumor. By using curcumin as an adjuvant, we were able to make this implant cost-effective with increasing glioma cell apoptosis and treatment efficacy. Thus, by implanting the carmustine-curcumin loaded PCL-PEG hydrogel, we can avoid systemic circulation of chemotherapeutic drugs, avoiding side effects and loss of drug due to metabolic processing. Although the tumor regrowth was significantly less in the treated group compared with control, increasing the drug concentration or amount of hydrogel loaded could further increase efficiency thus potentially increasing progression-free survival in patients. PCL-PEG supramolecular hydrogel provides future avenues for entrapping hydrophobic drugs efficiently and can be used for future drug discovery for better efficacy.
Ethics Approval
We certify that all applicable institutional and governmental regulations concerning the ethical use of animals were followed during the course of this research. The experiments were approved by Institutional Animal Ethics Committee (IAEC) of Rajiv Gandhi Centre for Biotechnology.
Acknowledgments
This work was supported by the Department of Biotechnology (DBT, India) and PhD fellowship from INSPIRE, Department of Science and Technology (DST, India). We are thankful to Sree Chitra Tirunal Institute for Medical Sciences and Technology (SCTIMST) Thiruvananthapuram for GPC analysis and National Institute for Interdisciplinary Science (NIIST) Thiruvananthapuram for Rheology work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Tamargo RJ, Myseros JS, Epstein JI, Yang MB, Chasin M, Brem H. Interstitial Chemotherapy of the 9L Gliosarcoma: controlled Release Polymers for Drug Delivery in the Brain. Cancer Res. 1993;1:45.
2. Nagpal S. The Role of BCNU Polymer Wafers (Gliadel) in the Treatment of Malignant Glioma. Neurosurg Clin. 2012;23(2):289–295. doi:10.1016/J.NEC.2012.01.004
3. Choucair AK, Levin VA, Gutin PH, et al. Development of multiple lesions during radiation therapy and chemotherapy in patients with gliomas. J Neurosurg. 1986;65(5):654–658. doi:10.3171/JNS.1986.65.5.0654
4. Loeffler J, Alexander E, Hochberg FH, et al. Clinical patterns of failure following stereotactic interstitial irradiation for malignant gliomas. Int J Radiat Oncol Biol Phys. 1990;(1):758. doi:10.1016/0360-3016(90)90358-Q
5. Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. Lancet. 1995;345(8956):1008–1012. doi:10.1016/S0140-6736(95)90755-6
6. Walter KA, Cahan MA, Gur A, et al. Interstitial Taxol Delivered from a Biodegradable Polymer Implant against Experimental Malignant Glioma1. Cancer Res. 1994;1:53.
7. Storm PB, Moriarity JL, Tyler B, Burger PC, Brem H, Weingart J. Polymer delivery of camptothecin against 9L gliosarcoma: release, distribution, and efficacy. J Neurooncol. 2002;2:43. doi:10.1023/A:1015003232713
8. Vellimana AK, Recinos VR, Hwang L, et al. Combination of paclitaxel thermal gel depot with temozolomide and radiotherapy significantly prolongs survival in an experimental rodent glioma model. J Neurooncol. 2013;2:5443. doi:10.1007/s11060-012-1014-1
9. Su YW, Chang MC, Chiang MF, Hsieh RK. Treatment-related myelodysplastic syndrome after temozolomide for recurrent high-grade glioma. J Neuro-Oncology. 2005;71(3):315–318. doi:10.1007/S11060-004-2028-0
10. Natelson EA, Pyatt D. Temozolomide-induced myelodysplasia. Adv Hematol. 2010;2010:45. doi:10.1155/2010/760402
11. Asyikin Binti Abdul Aziz Z, Ahmad A, Hamidah Mohd-Setapar S, et al. Recent Advances in Drug Delivery of Polymeric Nano-Micelles. Current Drug Metabo. 2017;18(1):16
12. Tseng YY, Chen TY, Liu SJ. Role of Polymeric Local Drug Delivery in Multimodal Treatment of Malignant Glioma: a Review. Int J Nanomedicine. 2021;16:4597. doi:10.2147/IJN.S309937
13. Grossen P, Witzigmann D, Sieber S, Huwyler J. PEG-PCL-based nanomedicines: a biodegradable drug delivery system and its application. J Control Release. 2017;260:46–60. doi:10.1016/j.jconrel.2017.05.028
14. Mei JH, Ma LG, Qian ZY, et al. One-step preparation of poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) nanoparticles for plasmid DNA delivery. J Biomed Mater Res. 2008;86(4):979–986. doi:10.1002/jbm.a.31704
15. Wang P, Wang H, Ma K, et al. Novel cytokine-loaded PCL-PEG scaffold composites for spinal cord injury repair. RSC Adv. 2020;10(11):6306–6314. doi:10.1039/c9ra10385f
16. Khodaverdi E, Delroba K, Mohammadpour F, et al. In-vitro Release Evaluation of Growth Hormone from an Injectable In-Situ Forming Gel Using PCL-PEG-PCL Thermosensitive Triblock. Curr Drug Deliv. 2020;17(2):174–183. doi:10.2174/1567201817666200120120105
17. Singh S, Alrobaian MM, Molugulu N, Agrawal N, Numan A, Kesharwani P. Pyramid-Shaped PEG-PCL-PEG Polymeric-Based Model Systems for Site-Specific Drug Delivery of Vancomycin with Enhance Antibacterial Efficacy. ACS Omega. 2020;5(21):11935–11945. doi:10.1021/ACSOMEGA.9B04064
18. Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19(4):764–772. doi:10.1158/1078-0432.CCR-12-3002
19. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–820. doi:10.1007/s00401-016-1545-1
20. Tseng YY, Huang YC, Yang TC, et al. Concurrent Chemotherapy of Malignant Glioma in Rats by Using Multidrug-Loaded Biodegradable Nanofibrous Membranes. Sci Reports. 2016;6(1):1–10. doi:10.1038/srep30630
21. Smith SJ, Tyler BM, Gould T, et al. Overall Survival in Malignant Glioma Is Significantly Prolonged by Neurosurgical Delivery of Etoposide and Temozolomide from a Thermo-Responsive Biodegradable Paste. Clin Cancer Res. 2019;25(16):5094–5106. doi:10.1158/1078-0432.CCR-18-3850
22. Vasudevan SM, Ashwanikumar N, Kumar GSV. Peptide decorated glycolipid nanomicelles for drug delivery across the blood–brain barrier (BBB). Biomater Sci. 2019;7(10):4017–4021.
23. Anto RJ, Mukhopadhyay A, Denning K, Aggarwal BB. Curcumin (diferuloylmethane) induces apoptosis through activation of caspase-8, Bid cleavage and cytochrome c release: its suppression by ectopic expression of Bcl-2 and Bcl-xl. Carcinogenesis. 2002;23(1):143–150.
24. Zanotto-Filho A, Braganhol E, Edelweiss MI, et al. The curry spice curcumin selectively inhibits cancer cells growth in vitro and in preclinical model of glioblastoma. J Nutr Biochem. 2012;23(6):591–601.
25. Zhuang W, Long L, Zheng B, et al. Curcumin promotes differentiation of glioma-initiating cells by inducing autophagy. Cancer Sci. 2012;103(4):684–690.
26. Mortezaee K, Salehi E, Mirtavoos-mahyari H, et al. Mechanisms of apoptosis modulation by curcumin: implications for cancer therapy. J Cell Physiol. 2019;234(8):12537–12550. doi:10.1002/JCP.28122
27. Shabaninejad Z, Pourhanifeh MH, Movahedpour A, et al. Therapeutic potentials of curcumin in the treatment of glioblastoma. Eur J Med Chem. 2020;188:112040.
28. Orunoğlu M, Kaffashi A, Pehlivan SB, et al. Effects of curcumin-loaded PLGA nanoparticles on the RG2 rat glioma model. Mater Sci Eng C. 2017;78:32–38.
29. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1998;103(19):8577.
30. Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25.
31. Schüttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Cryst. 2004;D60:1355–1363.
32. Lee SM, Yang EJ, Choi SM, Kim SH, Baek MG, Jiang JH. Effects of bee venom on glutamate-induced toxicity in neuronal and glial cells. Evid Based Complement Alternat Med. 2012;2012:356.
33. He J, Wang XM, Spector M, Cui FZ. Scaffolds for central nervous system tissue engineering. Front Mater Sci. 2012;6(1):1–25.
34. Hou S, Xu Q, Tian W, et al. The repair of brain lesion by implantation of hyaluronic acid hydrogels modified with laminin. J Neurosci Methods. 2005;148(1):60–70.
35. Paylor R, Nguyen M, Crawley JN, Patrick J, Beaudet A, Orr-Urtreger A. α7 nicotinic receptor subunits are not necessary for hippocampal- dependent learning or sensorimotor gating: a behavioral characterization of Acra7-deficient mice. Learn Mem. 1998;5(4–5):302–316.
36. Zhao H, Feng H, Liu J, et al. Dual-functional guanosine-based hydrogel integrating localized delivery and anticancer activities for cancer therapy. Biomaterials. 2020;230:119598.
37. Rowland MJ, Parkins CC, McAbee JH, et al. An adherent tissue-inspired hydrogel delivery vehicle utilised in primary human glioma models. Biomaterials. 2018;179:199–208.
38. Lu C, Guo S, Zhang Y, Yin M. Synthesis and aggregation behavior of four types of different shaped PCL-PEG block copolymers. Polym Int. 2006;55(6):694–700.
39. Asadi N, Annabi N, Mostafavi E, et al. Synthesis, characterization and in vitro evaluation of magnetic nanoparticles modified with PCL–PEG–PCL for controlled delivery of 5FU. Artif Cells Nanomed Biotechnol. 2018;46:938–945.
40. Liu CB, Gong CY, Huang MJ, et al. Thermoreversible gel-sol behavior of biodegradable PCL-PEG-PCL triblock copolymer in aqueous solutions. J Biomed Mater Res B Appl Biomater. 2008;84(1):165–175.
41. Liao R, Pon J, Chungyoun M, Nance E. Enzymatic protection and biocompatibility screening of enzyme-loaded polymeric nanoparticles for neurotherapeutic applications. Biomaterials. 2020;257:120238.
42. Grebenik EA, Surin AM, Bardakova KN, et al. Chitosan-g-oligo(L,L-lactide) copolymer hydrogel for nervous tissue regeneration in glutamate excitotoxicity: in vitro feasibility evaluation. Biomed Mater. 2020;15(1):015011.
43. Fournier E, Passirani C, Montero-Menei CN, Benoit JP. Biocompatibility of implantable synthetic polymeric drug carriers: focus on brain biocompatibility. Biomaterials. 2003;24(19):3311–3331.
44. Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47(1):65–81.
45. Fourniols T, Randolph LD, Staub A, et al. Temozolomide-loaded photopolymerizable PEG-DMA-based hydrogel for the treatment of glioblastoma. J Control Release. 2015;210:95–104.
46. Tang L, Eaton JW. Inflammatory responses to biomaterials. Am J Clin Pathol. 1995;103(4):466–471.
47. Gogada R, Amadori M, Zhang H, et al. Curcumin induces Apaf-1-dependent, p21-mediated caspase activation and apoptosis. Cell Cycle. 2011;10(23):4128.
48. Karmakar S, Banik NL, Patel SJ, Ray SK. Curcumin activated both receptor-mediated and mitochondria-mediated proteolytic pathways for apoptosis in human glioblastoma T98G cells. Neurosci Lett. 2006;407(1):53–58.
49. Chaitanya GV, Alexander JS, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31.
50. Kast RE, Hill QA, Wion D, et al. Glioblastoma-synthesized G-CSF and GM-CSF contribute to growth and immunosuppression: potential therapeutic benefit from dapsone, fenofibrate, and ribavirin. Tumour Biol. 2017;39:5.
51. Alkhaibary A, Alassiri AH, AlSufiani F, Alharbi MA. Ki-67 labeling index in glioblastoma; does it really matter? Hematol Oncol Stem Cell Ther. 2019;12(2):82–88.
52. Nielsen LAG, Bangsø JA, Lindahl KH, et al. Evaluation of the proliferation marker Ki-67 in gliomas: interobserver variability and digital quantification. Diagn Pathol. 2018;13(1):38.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.