")
Back to Journals » OncoTargets and Therapy » Volume 10
Suppression of microRNA-629 enhances sensitivity of cervical cancer cells to 1′S-1′-acetoxychavicol acetate via regulating RSU1
Authors Phuah NH, Azmi MN , Awang K, Nagoor NH
Received 18 July 2016
Accepted for publication 19 November 2016
Published 20 March 2017 Volume 2017:10 Pages 1695—1705
DOI https://doi.org/10.2147/OTT.S117492
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr XuYu Yang
Neoh Hun Phuah,1 Mohamad Nurul Azmi,2 Khalijah Awang,2 Noor Hasima Nagoor1,3
1Faculty of Science, Institute of Biological Science (Genetics and Molecular Biology), 2Faculty of Science, Department of Chemistry, Centre for Natural Product Research and Drug Discovery (CENAR), 3Centre for Research in Biotechnology for Agriculture (CEBAR), University of Malaya, Kuala Lumpur, Malaysia
Background: Cervical cancer is the fourth most frequent malignancy affecting women worldwide, but drug resistance and toxicities remain a major challenge in chemotherapy. The use of natural compounds is promising because they are less toxic and able to target multiple signaling pathways. The 1'S-1'-acetoxychavicol acetate (ACA), a natural compound isolated from wild ginger Alpinia conchigera, induced cytotoxicity on various cancer cells including cervical cancer. MicroRNAs (miRNAs) are short noncoding RNAs that regulate numerous biological processes, such as apoptosis and chemosensitivity. Past studies reported that miR-629 is upregulated in many cancers, and its expression was altered in ACA-treated cervical cancer cells. However, the role of miR-629 in regulating sensitivity toward ACA or other anticancer agents has not been reported. Hence, this study aims to investigate the role of miR-629 in regulating response toward ACA on cervical cancer cells.
Methods: The miR-629 expression following transfection with miR-629 hairpin inhibitor and hairpin inhibitor negative control was measured using quantitative real-time polymerase chain reaction (RT-qPCR). The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to investigate sensitivity toward ACA. Apoptosis was detected using Annexin V/propidium iodide and Caspase 3/7 assays. The gene target for miR-629 was identified using miRNA target prediction programs, luciferase reporter assay and Western blots. Gene overexpression studies were performed to evaluate its role in regulating response toward ACA.
Results: Transfection with miR-629 hairpin inhibitor downregulated its expression in both cervical cancer cell lines. Suppression of miR-629 increased sensitivity toward ACA by reducing cell proliferation and inducing apoptosis. Luciferase reporter assay confirmed RSU1 as a direct target of miR-629. Overexpression of miR-629 decreased RSU1 protein expression, while inhibition of miR-629 increased RSU1 protein expression. Overexpression of RSU1 augmented antiproliferative and apoptosis-inducing effects of ACA.
Conclusion: Our findings showed that combination of ACA with miR-629 and RSU1 may provide a potential strategy in treating cervical cancer.
Keywords: miR-629, 1'-acetoxychavicol acetate, cervical cancer, Ras suppressor-1, apoptosis, cell proliferation
Introduction
Cancer of the cervix is the fourth most common malignancy affecting women worldwide after breast cancer. In 2012, there were an estimated 527,600 new cases diagnosed and 265,700 mortalities worldwide, with the highest incidences happening in less developed countries.1 Chemotherapy is often used together with surgery and radiotherapy to treat cervical cancer in combinatorial approach. However, poor clinical outcomes and treatment failures still occur, especially in recurrent or advanced cervical cancer, due to the development of drug resistance and toxicities.2,3 Thus, it is necessary to identify new therapeutic strategies to improve overall response and survival outcome in cervical cancer patients.
Natural compounds have been used in cancer treatment for many years either as stand-alone or as adjuvant to improve therapeutic efficacy. They are able to target multiple signaling pathways and are less toxic compared to some conventional anticancer agents.4 Oncovin (vincristine), Velban (vinblastine), Hycamtin (topotecan) and Taxol (paclitaxel) are some examples of natural compounds derived from plants currently used in chemotherapy to treat various cancers.5 The 1′S-1′-acetoxychavicol acetate (ACA) is a natural compound isolated from the wild ginger, Alpinia conchigera. We have previously reported that it is able to induce cytotoxicity when used as stand-alone and potentiates the effects of cisplatin when used in combination on different cancer cells including cervical cancer.6,7
MicroRNAs (miRNAs) are short noncoding RNAs that regulate genes posttranscriptionally by suppressing translation or inducing messenger RNA (mRNA) degradation.8 Studies have shown that miRNAs are dysregulated in many cancers and they play crucial roles in regulating a wide variety of biological processes such as cell death, metastasis, cell proliferation and chemosensitivity.9–13 In a study carried out by a group of researchers to analyze miRNA expression profiles across multiple cancers using data sets obtained from The Cancer Genome Atlas (TCGA), miR-629 was found to be upregulated in various cancers, such as bladder, head and neck, lung, kidney, breast and uterine cancer.14 It was also reported that circulating miR-629 is upregulated in lung cancer patients and can potentially be used in diagnosis of lung and gastric cancer.15,16
In this study, we first showed that suppression of miR-629 in cervical cancer cells enhanced sensitivity toward ACA by reducing cell proliferation and inducing apoptosis. We next identified and confirmed RSU1 as a direct target of miR-629 and found that overexpression of miR-629 reduced RSU1 protein expression, whereas inhibition of miR-629 increased RSU1 protein expression. Furthermore, we demonstrated that overexpression of RSU1 augmented antiproliferative and apoptosis-inducing effects of ACA.
Materials and methods
Cell culture and ACA
Human cervical cancer cells Ca Ski and SiHa were procured from American Type Cell Culture (ATCC, Manassas, VA, USA) and cultured in Roswell Park Memorial Institute (RPMI)-1640 (HyClone, Logan, UT, USA) and DMEM (HyClone) supplemented with 10% (v/v) fetal bovine serum (HyClone), respectively. All cells were maintained in humidified incubator containing 5% CO2 at 37°C. The ACA was provided by the Centre for Natural Product Research and Drug Discovery (CENAR), Department of Chemistry, University of Malaya, Malaysia.
Transfection
For miRNA transfection, cells were transfected with 100 nM of miRIDIAN microRNA human hsa-miR-629 mimic or miRIDIAN microRNA human hsa-miR-629 hairpin inhibitor (Thermo Fisher Scientific, Waltham, MA, USA), using DharmaFECT 1 Transfection Reagent (Thermo Fisher Scientific) for 24 h according to the manufacturer’s protocol. miRIDIAN microRNA Mimic Negative Control #1 and miRIDIAN microRNA Hairpin Inhibitor Negative Control #1 (Thermo Fisher Scientific) were used as negative controls.
For plasmid transfection, cells were transfected with 50 ng of pCMV6-RSU1 (Origene Technologies, Rockville, MD, USA) using DharmaFECT 1 Transfection Reagent (Thermo Fisher Scientific) for 12 h according to manufacturer’s protocol. The empty vector pCMV6 (Origene Technologies) was used as negative control.
Quantitative real-time polymerase chain reaction (RT-qPCR)
The total RNA was extracted using miRNeasy Mini Kit (Qiagen, Venlo, the Netherlands) and reverse-transcribed into cDNA with TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol using Veriti 96-well Thermal Cycler (Applied Biosystems). The TaqMan MicroRNA Assays (Applied Biosystems) were used to quantified miR-629 expression according to the manufacturer’s protocol using a Bio-Rad CFX96™ Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) and analyzed by Bio-Rad CFX Manager v2.1 (Bio-Rad Laboratories). The U6 small nuclear RNA was used to normalize RNA input. Fold changes were calculated using the 2−ΔΔCt method and presented as normalized fold expression.
Cell proliferation assay
Transfected cells were treated with ACA for 12 h (plasmid transfection) or 48 h (miRNA transfection). A total of 30 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Merck, Kenilworth, NJ, USA) (5.0 mg/mL) was added into each well and incubated in the dark at 37°C for 1 h. Media containing excess MTT reagent were then aspirated and 200 μL dimethyl sulfoxide (Merck) was added to dissolve the purple formazan precipitates. Results were obtained using microtiter plate reader (Tecan, Männedorf, Zürich, Switzerland), which detects absorbance wavelength at 570 nm.
Annexin V/propidium iodide (PI) assay
Transfected cells were treated with ACA (20 μM for Ca Ski and 30 μM for SiHa) for 12 h (plasmid transfection) or 48 h (miRNA transfection). Apoptosis assays were carried out using BD Pharmingen™ Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s protocol. Results were obtained using BD FACSCanto II flow cytometer (BD Biosciences) and analyzed with BD FACSDiva software (BD Biosciences).
Caspase 3/7 assay
Transfected cells were treated with ACA (20 μM for Ca Ski and 30 μM for SiHa) for 6 h (plasmid transfection) or 12 h (miRNA transfection). The activities of caspases 3 and 7 were then measured with Caspase-Glo® 3/7 Assay (Promega, Fitchburg, WI, USA) according to the manufacturer’s protocol using GloMax®-Multi Jr (Promega).
Bioinformatic analyses of miR-629′s putative targets
miRNA target prediction programs TargetScan v7.1 and miRanda were used to identify potential targets of miR-629. The predicted targets were then subjected to gene annotation enrichment analysis using Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8.
Vector construction
The total RNA was first converted into cDNA using RevertAid First Strand cDNA (Thermo Fisher Scientific). The 3′-UTR regions of RSU1 containing the predicted binding site were then amplified from the cDNA and inserted into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega) to construct the luciferase reporter plasmids. Mutations in the predicted binding site for RSU1 were performed using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA, USA) according to the manufacturer’s protocol. The sequences of the constructs were then verified by sequencing.
Luciferase assay
Cells were cotransfected with 40 ng of 3′-UTR reporter constructs containing wild-type or mutated binding sites and 100 nM of miR-629 mimic or negative control using DharmaFECT 1 Transfection Reagent (Thermo Fisher Scientific). Firefly and Renilla luciferase activities were assayed 48 h after transfection with Dual-Glo® Luciferase Assay System (Promega) according to the manufacturer’s protocol using GloMax-Multi Jr (Promega). The firefly luciferase activity was normalized to Renilla activity, which was used as an internal control.
Western blot
Proteins were extracted using radioimmunoprecipitation assay buffer containing 1× Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) and quantified using BCA Protein Assay Kit (Pierce, Waltham, MA, USA) according to manufacturer’s protocol. Total cell lysates were fractionated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electroblot onto nitrocellulose membranes. Nonspecific binding sites of membranes were blocked for 1 h with 5% (w/v) nonfat milk in 1× TBST (1× Tris buffered saline [TBS] with 0.1% [v/v] Tween-20). Membranes were then incubated at 4°C overnight with primary antibodies against RSU1 (1:1,000; Abcam, Cambridge, UK) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1,000; Cell Signaling Technology, Danvers, MA, USA), washed with 1× TBST and followed by incubation with secondary immunoglobulin G (IgG) horseradish peroxidase (HRP)-linked antibody and antibiotin HRP-linked antibody (1:1,000; Cell Signaling Technology). Membranes were then washed with 1× TBST and 1× TBS, and bands were visualized using WesternBright Quantum (Advansta, Menlo Park, CA, USA) on the Fusion FX7 system (Vilber Lourmat, Torcy, Marne La Vallée, France) and quantified using the ImageJ Analyst software (NIH, Bethesda, MD, USA), with band intensities normalized to GAPDH, which was used as loading control.
Statistical analysis
All experiments were carried out in triplicates and presented as mean values ± standard error mean. Mann–Whitney U-test was used to evaluate the statistical significance of results, whereby P-value <0.05 was considered as statistically significant.
Results
ACA downregulates miR-629 expression and suppression of miR-629 enhances sensitivity toward ACA
To study the response of miR-629 toward ACA, the expression of miR-629 was determined using RT-qPCR following treatment with ACA. Figure 1A shows that miR-629 is downregulated following treatment with ACA in both Ca Ski and SiHa cells. To explore the role of miR-629, we transfected Ca Ski and SiHa cells with miR-629 hairpin inhibitor and hairpin inhibitor negative control. Figure 1B shows successful inhibition of miR-629 expression level in both cell lines after transfection. To investigate the effects of ACA when miR-629 is downregulated, MTT cell viability assays were carried out. Results showed that transfection with miR-629 hairpin inhibitor enhanced sensitivity toward ACA, as shown by the decrease in the half maximal inhibitory concentration (IC50) values for ACA in Ca Ski (Figure 1C) and SiHa (Figure 1D) cells. However, overexpression of miR-629 did not cause significant differences in sensitivity toward ACA (Figure S1).
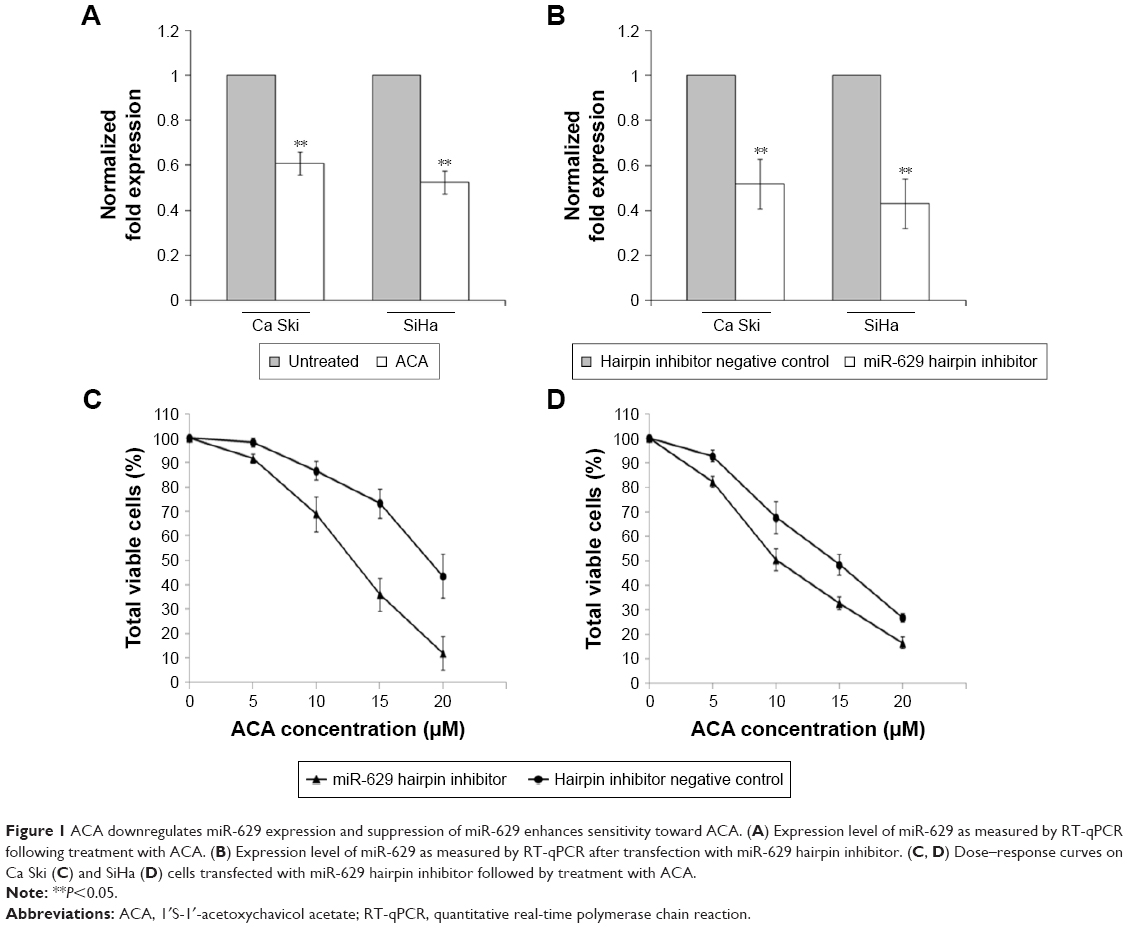 | Figure 1 ACA downregulates miR-629 expression and suppression of miR-629 enhances sensitivity toward ACA. (A) Expression level of miR-629 as measured by RT-qPCR following treatment with ACA. (B) Expression level of miR-629 as measured by RT-qPCR after transfection with miR-629 hairpin inhibitor. (C, D) Dose–response curves on Ca Ski (C) and SiHa (D) cells transfected with miR-629 hairpin inhibitor followed by treatment with ACA. |
Suppression of miR-629 increases sensitivity toward ACA by inducing apoptosis
To assess if combination of miR-629 hairpin inhibitor and ACA modulates apoptosis, both Annexin V/PI and Caspase 3/7 assays were employed. Results showed that suppression of miR-629 promoted induction of apoptosis when cells were treated with ACA, as shown by the increased apoptosis level in Figure 2A and higher Caspase 3/7 activity in Figure 2B. These results indicated that suppression of miR-629 increases sensitivity toward ACA by inducing apoptosis. On the other hand, no obvious differences in apoptosis were detected when miR-629 overexpressing cells were treated with ACA (Figure S2).
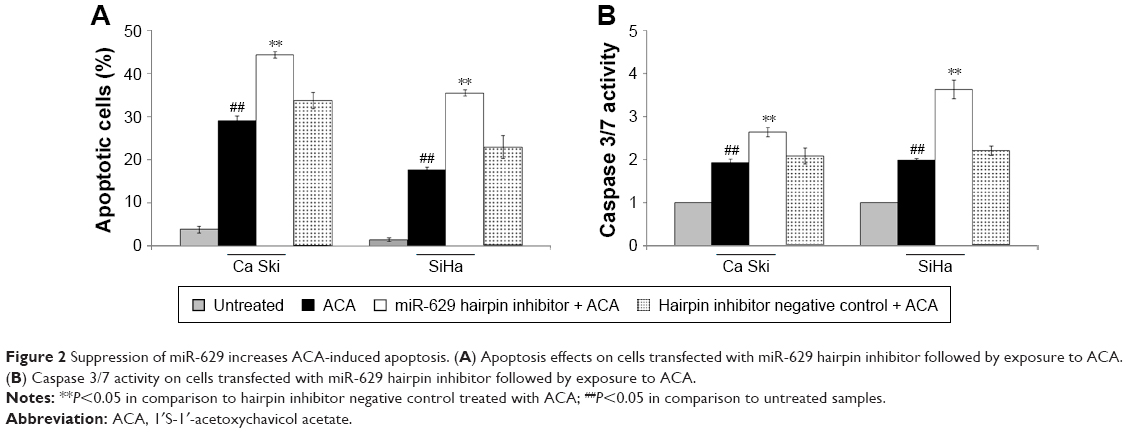 | Figure 2 Suppression of miR-629 increases ACA-induced apoptosis. (A) Apoptosis effects on cells transfected with miR-629 hairpin inhibitor followed by exposure to ACA. (B) Caspase 3/7 activity on cells transfected with miR-629 hairpin inhibitor followed by exposure to ACA. |
MiR-629 directly targets the 3′UTR of RSU1
The hypothetical signaling network depicting the interaction between miR-629 and its putative targets revealed that miR-629′s putative targets are involved in regulating cell proliferation and apoptosis (Figure 3). RSU1, which was chosen based on scores, novelty and function, was identified as a potential target by TargetScan v7.1 and miRanda (Figure 4A). To study the response of RSU1 toward ACA, the expression of RSU1 was determined using Western blot following treatment with ACA. Figure 4B shows that RSU1 is upregulated following treatment with ACA in both Ca Ski and SiHa cells. To confirm the direct interaction, luciferase reporter assay was performed. As shown in Figure 4C, luciferase activity was significantly decreased when cells were cotransfected with miR-629 mimic and luciferase reporter vector containing the wild-type sequence. However, no obvious differences were observed when the binding site is mutated. This confirmed that RSU1 is a direct target of miR-629. To assess if miR-629 can regulate the expression of RSU1, Western blots were carried out. Figure 4D shows that miR-629 mimic significantly reduced the expression of RSU1. Conversely, the expression of RSU1 was significantly increased when miR-629 is suppressed. Collectively, these results revealed that miR-629 directly targets RSU1.
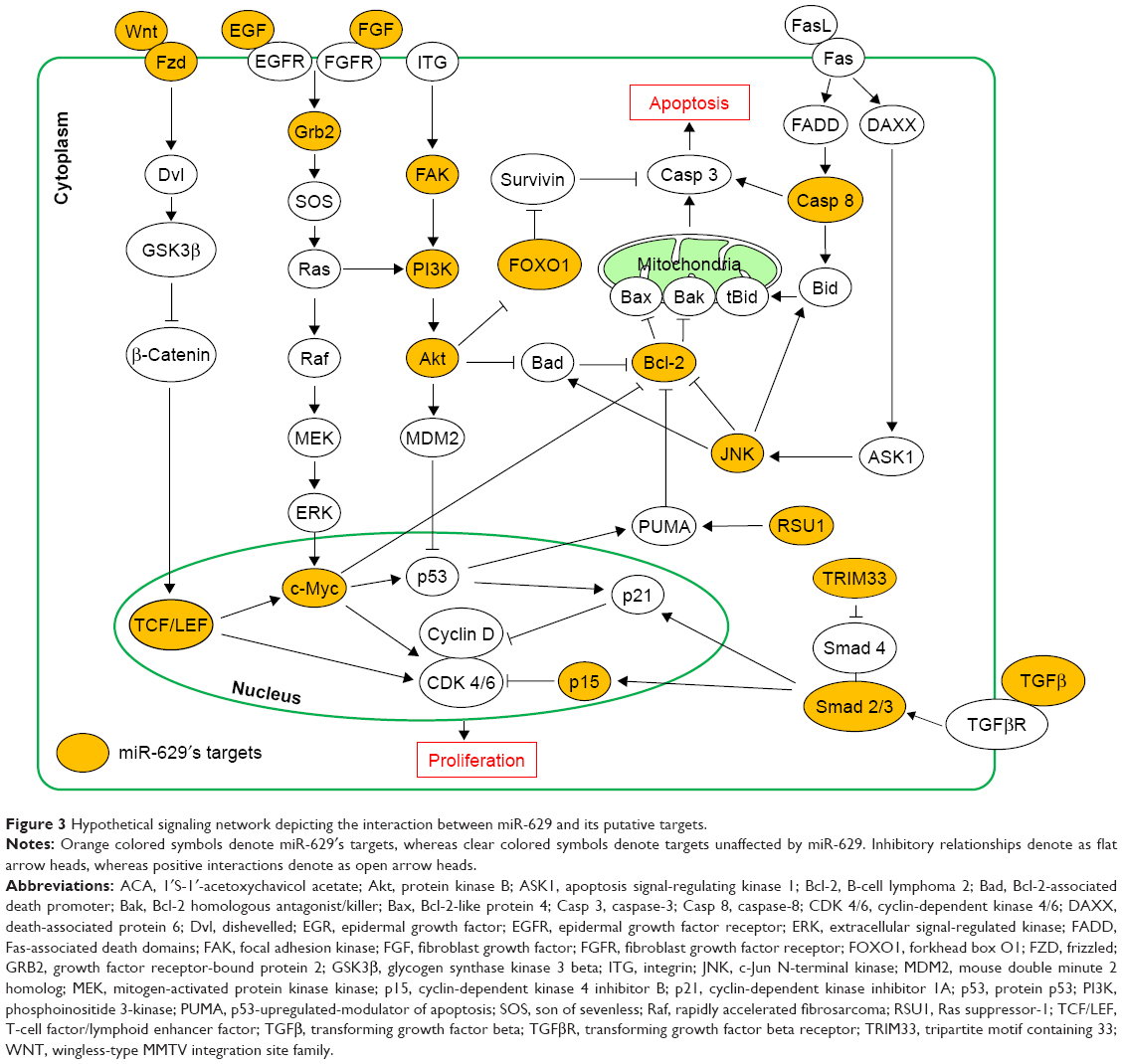 | Figure 3 Hypothetical signaling network depicting the interaction between miR-629 and its putative targets. |
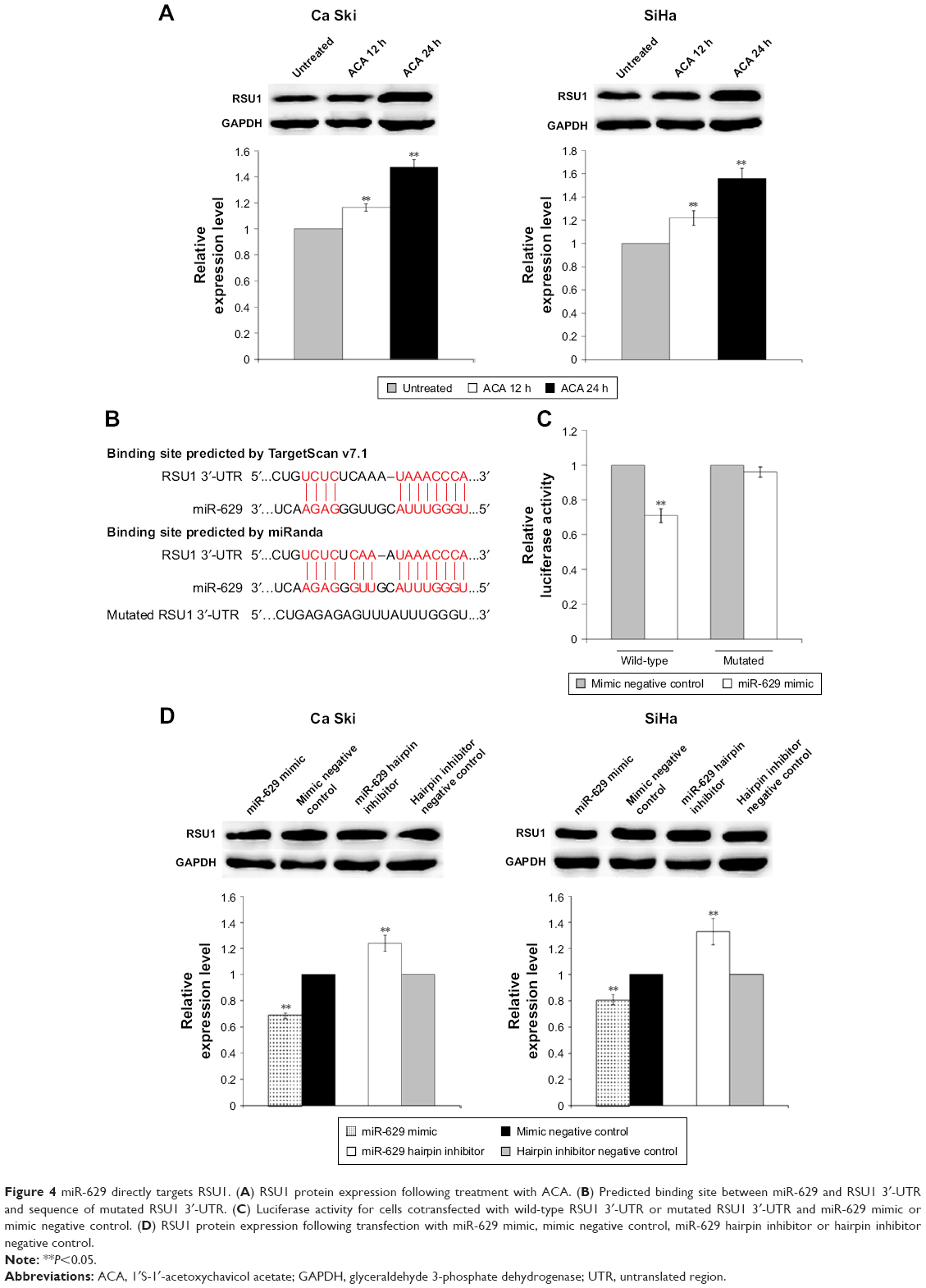 | Figure 4 miR-629 directly targets RSU1. (A) RSU1 protein expression following treatment with ACA. (B) Predicted binding site between miR-629 and RSU1 3′-UTR and sequence of mutated RSU1 3′-UTR. (C) Luciferase activity for cells cotransfected with wild-type RSU1 3′-UTR or mutated RSU1 3′-UTR and miR-629 mimic or mimic negative control. (D) RSU1 protein expression following transfection with miR-629 mimic, mimic negative control, miR-629 hairpin inhibitor or hairpin inhibitor negative control. |
Overexpression of RSU1 increases RSU1 protein expression level and reduces cell viability
To elucidate the role of RSU1 in regulating response toward ACA, RSU1 was overexpressed using pCMV6-XL5 vector containing RSU1 sequence (pCMV6-RSU1) while empty vector lacking RSU1 sequence (pCMV6) served as negative control. As seen in Figure 5A, an increase in the RSU1 protein expression level was observed in both cell lines after transfection with pCMV6-RSU1. To investigate the effects of RSU1 overexpression and ACA on cell viability, MTT cell viability assay was performed. Results showed a significant reduction in cell viability when RSU1 is overexpressed and treated with ACA (Figure 5B), indicating that RSU1 enhanced sensitivity toward ACA.
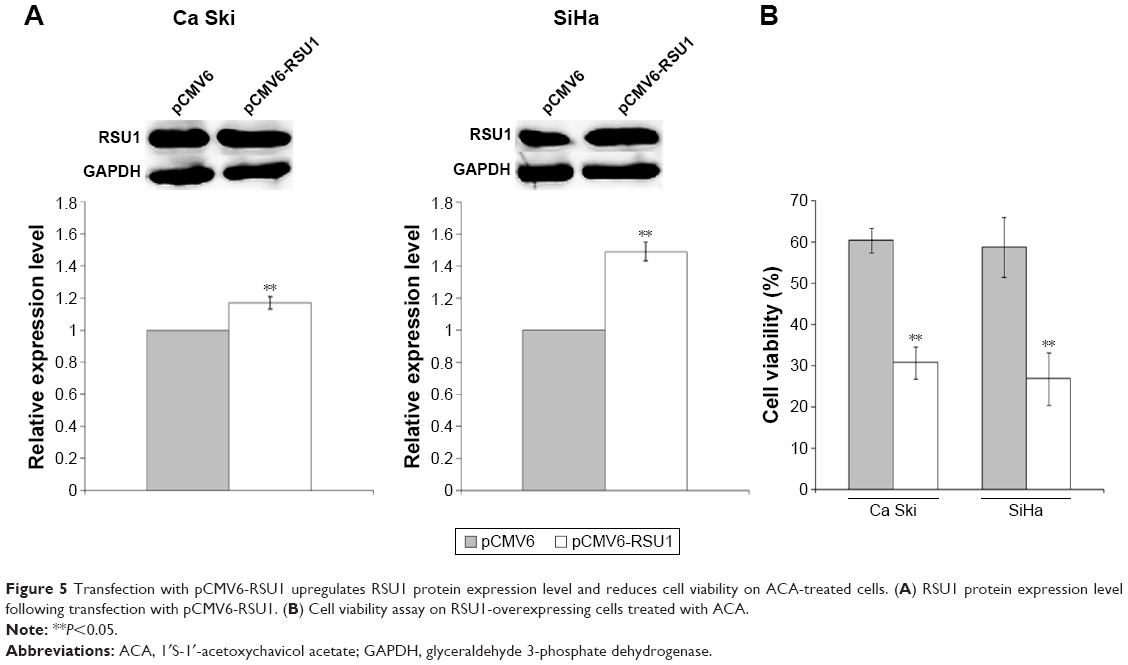 | Figure 5 Transfection with pCMV6-RSU1 upregulates RSU1 protein expression level and reduces cell viability on ACA-treated cells. (A) RSU1 protein expression level following transfection with pCMV6-RSU1. (B) Cell viability assay on RSU1-overexpressing cells treated with ACA. |
RSU1 overexpression increases ACA-induced apoptosis
To evaluate if the inhibitory effects of RSU1 overexpression in combination with ACA were mediated by apoptosis, both Annexin V/PI and Caspase 3/7 assays were used. As shown in Figure 6A, overexpression of RSU1 increased apoptosis level when treated with ACA. Likewise, a marked increase in Caspase 3/7 activity was observed on RSU1-overexpressing cells treated with ACA (Figure 6B). These results indicated that overexpression of RSU1 increases ACA-induced apoptosis.
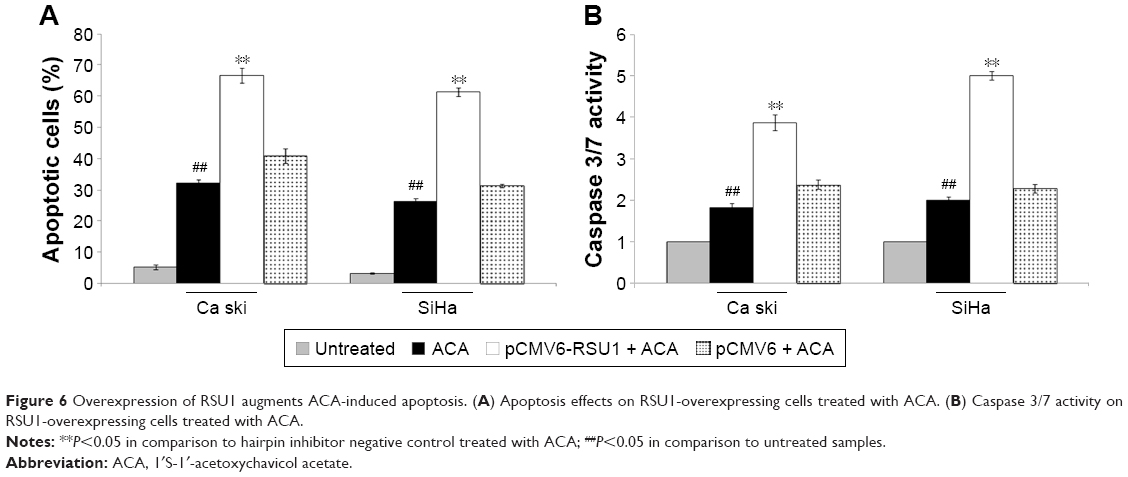 | Figure 6 Overexpression of RSU1 augments ACA-induced apoptosis. (A) Apoptosis effects on RSU1-overexpressing cells treated with ACA. (B) Caspase 3/7 activity on RSU1-overexpressing cells treated with ACA. |
Discussion
Many studies have demonstrated that miRNAs play an important role in cancer initiation and progression.17 Interestingly, a single miRNA is able to target different genes, enabling them to simultaneously regulate multiple signaling pathways.18 Different studies have reported that anticancer agents such as plant-derived natural compounds can modulate miRNA expression, and alterations in these miRNA expression can affect their anticancer activities.19 Studies have shown that cancer is a multistage process caused by gene alterations in more than one signaling pathway.20 Therefore, the use of combination therapy involving natural compounds and miRNAs targeting could possibly help to improve therapeutic efficacy due to their pleiotropic effects.
Although miR-629 is overexpressed in many cancers, there is only a few published work on the role of miR-629 in cancer.14 Past studies have demonstrated that miR-629 plays an important role in hepatocellular carcinogenesis by regulating HNF4α-miRNA inflammatory feedback circuit and increases lung cancer risk by targeting tumor suppressor NBS1 gene.21,22 It was also revealed that miR-629 is upregulated in clear cell renal cell carcinoma and accelerates cell motility and invasion by targeting tripartite motif-containing 33 (TRIMM33), a tumor suppressor.23 We have also previously reported that miR-629 expression is dysregulated following treatment with ACA in cervical cancer cells.7 However, the mechanistic role of miR-629 in regulating response toward anticancer agents has not been reported. Both HPV-16 positive Ca Ski and SiHa cells were used in this study because it was previously reported that HPV-positive cervical cancer cell lines exhibited high expression of miR-629, partly due to the continuous expression of E6/E7 in these cells.24 Our results showed that downregulation of miR-629 enhanced sensitivity toward ACA by reducing cell proliferation and increasing apoptosis, indicating that miR-629 could also play a role in regulating response toward anticancer agents.
In addition, we also demonstrated that miR-629 directly binds to RSU1, a tumor suppressor capable of suppressing Ras-dependent oncogenic transformation, and inhibits anchorage independent growth.25–27 A study by Donthamsetty et al showed that ectopic expression of RSU1 suppressed cell proliferation and migration in hepatocellular cancer cells. It was suggested in the same study that RSU1 could play a role in regulating liver size and tumorigenesis in mice together with particularly interesting new cysteine-histidine-rich protein (PINCH).28 More recently, it was reported that RSU1 is able to hinder proliferation by inhibiting prosurvival PINCH-1 and promote apoptosis by activating proapoptotic p53-upregulated modulator of apoptosis (PUMA) in breast cancer cells.29 Nevertheless, its role in regulating response toward anticancer agents has also not been reported. In this study, we demonstrated that overexpression of RSU1 augments antiproliferative and apoptosis-inducing effects of ACA, indicating that this tumor suppressor could also play a role in regulating sensitivity toward anticancer agents. Apart from RSU1, however, we could not rule out the possibility that other targets of miR-629 could also mediate sensitivity toward ACA, as there are many other targets which remain unvalidated.
Conclusion
We demonstrated that suppression of miR-629 enhanced sensitivity toward ACA by reducing cell proliferation and inducing apoptosis. We also identified RSU1 as a direct target of miR-629 and showed that RSU1 expression is regulated by miR-629. Furthermore, we revealed that overexpression of RSU1 augmented antiproliferative and apoptosis-inducing effects of ACA. To the best of our knowledge, this is the first study which demonstrated that miR-629 and RSU1 play a role in regulating response toward anticancer agents. Taken together, our findings showed that combination of ACA with miR-629 and RSU1 may provide a potential strategy in treating cervical cancer.
Acknowledgment
This study was supported by University of Malaya Research Grant (UMRG) (RP001B-13BIO) and RU015-2016.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Crafton SM, Salani R. Beyond chemotherapy: an overview and review of targeted therapy in cervical cancer. Clin Ther. 2016;38(3):449–458. | ||
Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. | ||
Millimouno FM, Dong J, Yang L, Li J, Li X. Targeting apoptosis pathways in cancer and perspectives with natural compounds from mother nature. Cancer Prev Res (Phila). 2014;7(11):1081–1107. | ||
Cragg GM, Newman DJ. Plants as a source of anti-cancer agents. J Ethnopharmacol. 2005;100(1–2):72–79. | ||
Awang K, Azmi MN, Aun LI, Aziz AN, Ibrahim H, Nagoor NH. The apoptotic effect of 1′S-1′-acetoxychavicol acetate from Alpinia conchigera on human cancer cells. Molecules. 2010;15(11):8048–8059. | ||
Phuah NH, In LL, Azmi MN, Ibrahim H, Awang K, Nagoor NH. Alterations of microRNA expression patterns in human cervical carcinoma cells (Ca Ski) toward 1′S-1′-acetoxychavicol acetate and cisplatin. Reprod Sci. 2013;20(5):567–578. | ||
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. | ||
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–866. | ||
Othman N, In LL, Harikrishna JA, Hasima N. Bcl-xL silencing induces alterations in hsa-miR-608 expression and subsequent cell death in A549 and SK-LU1 human lung adenocarcinoma cells. PLoS One. 2013;8(12):e81735. | ||
Ho CS, Yap SH, Phuah NH, In LL, Hasima N. MicroRNAs associated with tumour migration, invasion and angiogenic properties in A549 and SK-Lu1 human lung adenocarcinoma cells. Lung Cancer. 2014;83(2):154–162. | ||
Liu J, Wang H, Wang Y, et al. Repression of the miR-93-enhanced sensitivity of bladder carcinoma to chemotherapy involves the regulation of LASS2. Onco Targets Ther. 2016;9:1813–1822. | ||
Yan B, Guo Q, Nan XX, et al. Micro-ribonucleic acid 29b inhibits cell proliferation and invasion and enhances cell apoptosis and chemotherapy effects of cisplatin via targeting of DNMT3b and AKT3 in prostate cancer. Onco Targets Ther. 2015;8:557–565. | ||
Liu Z, Zhang J, Yuan X, et al. Detecting pan-cancer conserved microRNA modules from microRNA expression profiles across multiple cancers. Mol Biosyst. 2015;11(8):2227–2237. | ||
Cazzoli R, Buttitta F, Di Nicola M, et al. microRNAs derived from circulating exosomes as noninvasive biomarkers for screening and diagnosing lung cancer. J Thorac Oncol. 2013;8(9):1156–1162. | ||
Shin VY, Ng EK, Chan VW, Kwong A, Chu KM. A three-miRNA signature as promising non-invasive diagnostic marker for gastric cancer. Mol Cancer. 2015;14:202. | ||
Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10(6):389–402. | ||
Wu S, Huang S, Ding J, et al. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene. 2010;29(15):2302–2308. | ||
Phuah NH, Nagoor NH. Regulation of microRNAs by natural agents: new strategies in cancer therapies. Biomed Res Int. 2014;2014:804510. | ||
Sugimura T, Terada M, Yokota J, Hirohashi S, Wakabayashi K. Multiple genetic alterations in human carcinogenesis. Environ Health Perspect. 1992;98:5–12. | ||
Hatziapostolou M, Polytarchou C, Aggelidou E, et al. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell. 2011;147(6):1233–1247. | ||
Yang L, Li Y, Cheng M, et al. A functional polymorphism at microRNA-629-binding site in the 3′-untranslated region of NBS1 gene confers an increased risk of lung cancer in Southern and Eastern Chinese population. Carcinogenesis. 2012;33(2):338–347. | ||
Jingushi K, Ueda Y, Kitae K, et al. miR-629 targets TRIM33 to promote TGFbeta/Smad signaling and metastatic phenotypes in ccRCC. Mol Cancer Res. 2015;13(3):565–574. | ||
Honegger A, Schilling D, Bastian S, et al. Dependence of intracellular and exosomal microRNAs on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS Pathog. 2015;11(3):e1004712. | ||
Cutler ML, Bassin RH, Zanoni L, Talbot N. Isolation of rsp-1, a novel cDNA capable of suppressing v-Ras transformation. Mol Cell Biol. 1992;12(9):3750–3756. | ||
Tsuda T, Marinetti MR, Masuelli L, Cutler ML. The Ras suppressor RSU-1 localizes to 10p13 and its expression in the U251 glioblastoma cell line correlates with a decrease in growth rate and tumorigenic potential. Oncogene. 1995;11(2):397–403. | ||
Vasaturo F, Dougherty GW, Cutler ML. Ectopic expression of Rsu-1 results in elevation of p21CIP and inhibits anchorage-independent growth of MCF7 breast cancer cells. Breast Cancer Res Treat. 2000;61(1):69–78. | ||
Donthamsetty S, Bhave VS, Mars WM, et al. Role of PINCH and its partner tumor suppressor Rsu-1 in regulating liver size and tumorigenesis. PLoS One. 2013;8(9):e74625. | ||
Giotopoulou N, Valiakou V, Papanikolaou V, et al. Ras suppressor-1 promotes apoptosis in breast cancer cells by inhibiting PINCH-1 and activating p53-upregulated-modulator of apoptosis (PUMA); verification from metastatic breast cancer human samples. Clin Exp Metastasis. 2015;32(3):255–265. |
Supplementary materials
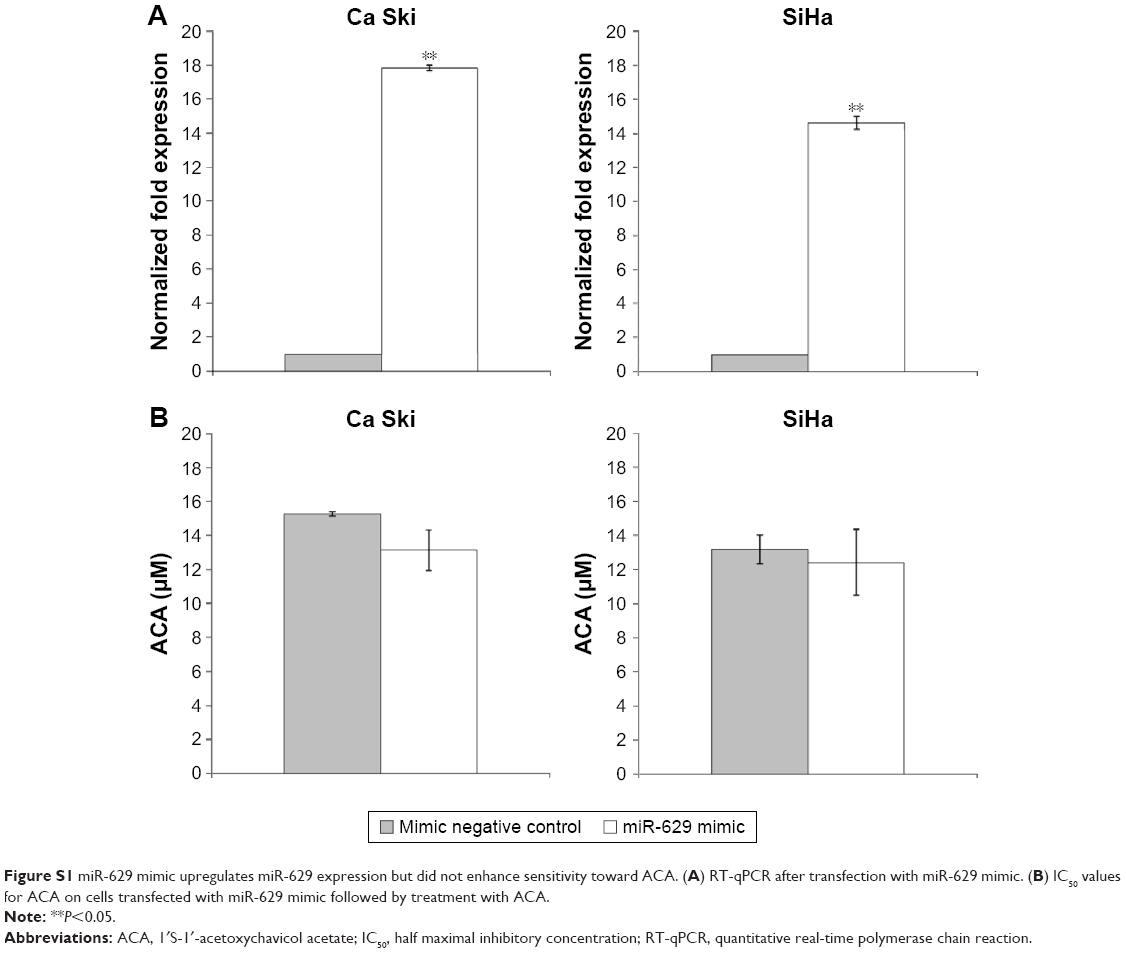 | Figure S1 miR-629 mimic upregulates miR-629 expression but did not enhance sensitivity toward ACA. (A) RT-qPCR after transfection with miR-629 mimic. (B) IC50 values for ACA on cells transfected with miR-629 mimic followed by treatment with ACA. |
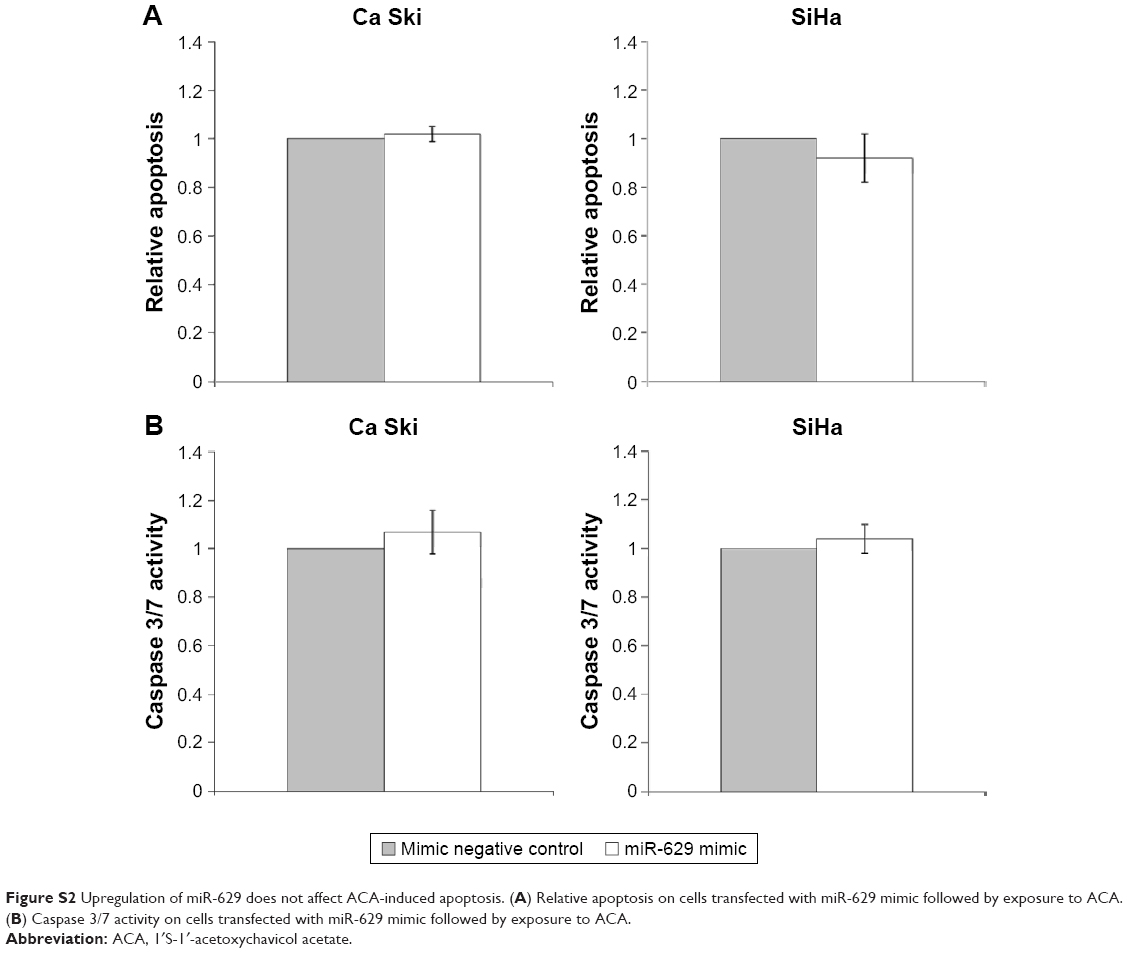 | Figure S2 Upregulation of miR-629 does not affect ACA-induced apoptosis. (A) Relative apoptosis on cells transfected with miR-629 mimic followed by exposure to ACA. (B) Caspase 3/7 activity on cells transfected with miR-629 mimic followed by exposure to ACA. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.