Back to Journals » OncoTargets and Therapy » Volume 13
Suppression of CXCL-1 Could Restore Necroptotic Pathway in Chronic Lymphocytic Leukemia
Authors Xu Z, Sun Y, Wei Z ![]() , Jiang J, Xu J
, Jiang J, Xu J ![]() , Liu P
, Liu P
Received 18 April 2020
Accepted for publication 17 June 2020
Published 16 July 2020 Volume 2020:13 Pages 6917—6925
DOI https://doi.org/10.2147/OTT.S256993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Zhao Xu,* Yifeng Sun,* Zheng Wei,* Jifeng Jiang, Jiadai Xu, Peng Liu
Department of Hematology, Zhongshan Hospital, Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Peng Liu
Department of Hematology, Zhongshan Hospital, Fudan University, 180 Fenglin Road, Shanghai 200032, People’s Republic of China
Email [email protected]
Purpose: To clarify the role of different cytokines and selenite in the defective necroptotic pathway of chronic lymphocytic leukemia (CLL).
Patients and Methods: We randomly collected the peripheral blood samples of 11 untreated CLL patients and 10 healthy volunteers, and then separated B lymphocytes from peripheral blood. Then, real-time polymerase chain reaction (PCR), enzyme-linked immunosorbent assay (ELISA) and Western Blot were performed to detect the expression of different cytokines, including CXC-motif chemokine ligand 1 (CXCL-1). Finally, we used flow cytometry to analyze the percentage of surviving cells to figure out whether CLL cells or normal B lymphocytes underwent necroptosis.
Results: 1) The high expression of CXCL-1 was seen in CLL cells compared with normal B lymphocytes (p = 0.0001, adjusted p =0.0012); 2) The downregulation of CXCL-1 was shown in normal B lymphocytes after induction by TNF-α and z-VAD; 3) CLL cells could restore necroptosis induced by TNF-α and z-VAD after knockdown of CXCL-1; 4) The transcriptional and translational expression of LEF-1 were downregulated after the knockdown of CXCL-1 in CLL cells; 5. 3.2μM selenite could help CLL cells restore necroptosis (p = 0.0102) and inhibit the transcriptional and translational expression of CXCL-1.
Conclusion: CXCL-1 played an important role in the defective necroptosis of CLL cells and regulated the expression of LEF-1. Selenite could inhibit the expression of CXCL-1 and help CLL cells restore necroptosis together with TNF-α and z-VAD. Selenite might be the potential medication of CLL in the future.
Keywords: chronic lymphocytic leukemia (CLL), CXC-motif chemokine ligand 1 (CXCL-1), selenite, necroptosis
Introduction
Chronic lymphocytic leukemia (CLL) is one of the most common hematological malignancies worldwide. CLL is characterized by the progressive accumulation of a monoclonal CD5-positive subgroup of B lymphocytes. The aggregation of these B cells leads to various clinical manifestations, such as lymphadenopathy, hepatosplenomegaly, and bone marrow failure.1 Although the overall survival and progression-free survival has seen huge improvement among CLL patients with the emergence of rituximab and ibrutinib,2 CLL is still incurable. A deeper understanding of the pathogenesis might be helpful to explore novel strategies for CLL patients.
When normal B cells fail to undergo apoptosis with the induction of tumor necrosis factor-α (TNF-α) and caspase inhibitor such as benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl-ketone (z-VAD),3,4 necroptosis often occurs as the alternative programmed cell death pathway. However, both apoptosis and necroptosis are impaired in CLL cells, which explains why malignant B lymphocytes accumulate in CLL patients.5 As the key regulator of canonical wingless-type (Wnt) pathway, the lymphoid enhancer-binding factor 1 (LEF-1) is overexpressed in various hematological malignancies.6–9 The high expression of LEF-1 in CLL cells downregulates deubiquitinase cylindromatosis (CYLD), a deubiquitinating enzyme important in the necroptotic pathway.10 CYLD dismantles the ubiquitination from RIPK1, leading to necroptosis. The suppression of CYLD by overexpression of LEF-1 stimulates sustained ubiquitination of RIPK1, causing the defection of necroptosis and survival of CLL cells. Therefore, the restoration of necroptosis will be another aim for CLL treatment strategies.
Selenite is associated with both necroptosis and prevention of tumor development. Selenite induced reactive oxygen species (ROS) generation in the necroptotic pathway of the HeLa cells.11 Besides, the biogenic selenium nanoparticles stimulated cell death in the prostate adenocarcinoma cells by the ROS-mediated activation of necroptosis.12 Furthermore, selenite is selectively toxic to tumor cells at a concentration that does not affect normal cells.13 Thus, selenite might become an ideal chemotherapeutic medicine in the future.
On the other hand, different cytokines also play an important role in the pathogenesis of CLL. CLL cells receive signals from cytokines, which were secreted by accessory cells in the microenvironment.14 The interaction between cytokines and its receptors is critical for the homing and retention of CLL cells.15 However, the relationship between cytokines and defective necroptosis in CLL cells remains unclear. In addition, the impact of selenite on either cytokines or necroptosis has received little attention.
Our research was designed to illustrate the association between different cytokines and the defective necroptotic pathway in CLL cells. Moreover, we managed to discover the influence of selenite on the cytokines and defective necroptosis in the CLL cells.
Patients and Methods
Patients
We enrolled 10 healthy volunteers and 11 untreated CLL patients diagnosed in our hospital between 2017 and 2019. The protocol was approved by the Review Board of Zhongshan Hospital of Fudan University. All patients and volunteers provided written informed consent in accordance with the Declaration of Helsinki.
Cells and Reagents
Peripheral blood samples were obtained from the patients and volunteers above. Peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood samples by Ficoll-isopaque centrifugation. Magnetic cell sorting (MACS, Miltenyi Biotec, Germany) were performed to isolate CLL cells and normal B cells. Cells were cultured in RPMI-1640 medium with 10% heat-inactivated fetal bovine serum (FBS) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. TNF-α was from Sigma (St. Louis, MO, USA) and z-VAD was from Alexis Biochemicals (San Diego, CA, USA). Antibodies against LEF-1 were from Abcam (Cambridge, MA, USA) and β-actin was from Cell Signaling technology (Beverly, MA, USA). Sodium selenite was dissolved in water treated by diethyl pyrocarbonate (DEPC) with the concentration of 32μM, 3.2μM, 0.32μM and 0.032μM respectively.
Gene Expression Detection
Total RNA was extracted by Trizol agent (Invitrogen, Carlsbad, CA, USA) and cDNA was reverse transcribed by the reverse transcription kit (Thermo Scientific, Lithuania). The transcriptional expression of CXC-motif chemokine ligand 1 (CXCL-1), monocyte chemotactic protein 1 (MCP-1), interleukin 6 (IL-6), granulocyte-macrophage colony-stimulating factor (GM-CSF), C-X-C motif chemokine ligand 2 (CXCL-2), C-C motif chemokine ligand 8 (CCL-8), colony stimulating factor 1(CSF-1), interleukin 9 (IL-9), C-X-C motif chemokine ligand 9 (CXCL-9), interleukin 1 receptor, type I (IL-1R1), interleukin 23 receptor (IL-23R), interleukin 15 (IL-15), C-C motif chemokine ligand 3 (CCL-3), interleukin 6 receptor (IL-6R), C-C motif chemokine ligand 2 (CCL-2), C-C motif chemokine receptor 4 (CCR-4), C-X-C motif chemokine ligand 5 (CXCL-5), interleukin 1 beta (IL-1β), C-X-C motif chemokine receptor 1 (CXCR-1), C-X-C motif chemokine ligand 8 (CXCL-8), C-X-C motif chemokine ligand 10 (CXCL-10), C-X-C motif chemokine receptor 2 (CXCR-2), and C-C motif chemokine receptor 1 (CCR-1) were analyzed by real-time quantitative polymerase chain reaction (PCR) using a 7500HT fast real-time PCR system (Applied Biosystem, Foster City, CA, USA). The primers of different cytokines are shown in Table 1. Relative transcriptional expressions were calculated by the method of ΔΔCT.
 |
Table 1 Primers of Cytokines Associated with Malignancies |
A Western Blot was performed based on the procedure described previously to evaluate the translational expression of LEF-1.16 β-actin was used to ensure equivalent protein loading. The concentrations of CXCL-1 of supernatants after cell transfection were determined by enzyme-linked immunosorbent assay (ELISA) test by using ELISA kit (Solarbio, Beijing, China).
Flow Cytometry
Cell apoptosis and necroptosis were assessed by the PI-FITC apoptosis detection kit I (BD Pharmingen, Franklin Lakes, NJ, USA) as described previously.4
Cell Transfection
Small interfering RNA (siRNA) of CXCL-1, MCP-1, or LEF-1 and negative control siRNA (nc siRNA) for cell transfection were synthesized by Biotend (Shanghai, China). Besides siRNA, 30ng/mL TNF-α and 20μM z-VAD might also be added to induce necroptosis if necessary. PBMCs were transfected by siRNA with an ultimate concentration of 100nM according to the manufacturer’s protocol. The transfected clones were detected after 24-hour transfection.
Statistical Analysis
Differences of cytokines and LEF-1 expression between groups were assessed via Student’s t-test. The differential expression of cytokines between CLL cells and normal B lymphocytes was adjusted for multiple testing by using False Discovery Rate. All statistical tests were two-sided, and the analysis was made by R software, version 3.6.0 (R Core Team, R Foundation for Statistical Computing). P < 0.05 was considered to be statistically significant.
Results
CXCL-1 and MCP-1 Might Have a Correlation with Defective Necroptosis of CLL Cells
First, we isolated CLL cells and normal B lymphocytes from peripheral blood samples of 3 untreated CLL patients and 3 healthy volunteers, respectively. Then, real-time RT-PCR was performed to detect the expression of 23 different cytokines or their receptors associated with various malignancies.17–37 Only the relative expression of CXCL-1 (P = 0.0001, adjusted P =0.0012), MCP-1 (P = 0.0003, adjusted P = 0.0023), IL-6 (P = 0.0001, adjusted P = 0.0477) and GM-CSF (P = 0.0083, adjusted P = 0.0012) was significantly upregulated in CLL cells compared with normal B lymphocytes (Figure 1A and B). On the other hand, necroptotic pathway is defective in CLL cells.4 To figure out whether these cytokines were associated with necroptosis, we managed to induce necroptosis of normal B lymphocytes from other 6 healthy volunteers with TNF-α and z-VAD. Besides necroptosis, the expression of CXCL-1 and MCP-1 were downregulated after induction of TNF-α and z-VAD (Figure 1C). Therefore, CXCL-1 and MCP-1 might associate with necroptotic pathway.
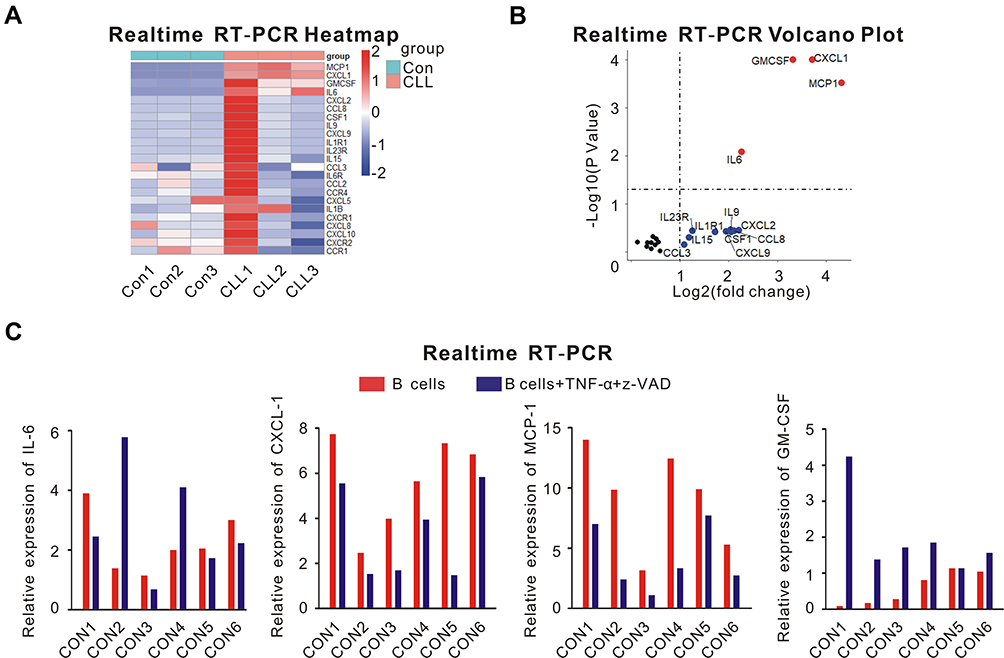 |
Figure 1 The screen of different cytokines that might associate with necroptotic pathway. (A) real-time RT-PCR analysis of 23 cytokines or their receptors was performed using peripheral blood samples of 3 healthy volunteers (Con1, Con2, Con3) and 3 untreated CLL patients (CLL1, CLL2, CLL3). PBMCs were isolated by Ficoll centrifugation, then CLL cells and normal B lymphocytes were segregated from PBMCs by MACS. (B) Volcano plot of the different cytokine expression was shown. The differentially expressed cytokines with log fold change >1 were plotted as red; Cytokines with log fold change >1 but without significantly differential expression (P > 0.05) were plotted as blue; Others were plotted as black. The relative expression of CXCL-1 (P = 0.0001, adjusted P =0.0012), MCP-1 (P = 0.0003, adjusted P = 0.0023), IL-6 (P = 0.0001, adjusted P = 0.0477) and GM-CSF (P = 0.0083, adjusted P = 0.0012) was significantly upregulated in CLL cells compared with normal B lymphocytes. (C) The expression of IL-6, CXCL-1, MCP-1 and GM-CSF from normal B lymphocytes of other 6 healthy volunteers was shown before (red) and after (blue) the induction of TNF-α and z-VAD. Besides necroptosis, TNF-α and z-VAD also induced the downregulation of CXCL-1 and MCP-1. Therefore, CXCL-1 and MCP-1 might have correlation with necroptosis. |
CLL Cells Restored Necroptosis After Knockdown of CXCL-1 Rather Than MCP-1
Through flow cytometry, we were able to estimate whether CLL cells underwent necroptosis. Nc siRNA with TNF-α and z-VAD or only CXCL-1 siRNA could not induce necroptosis of CLL cells. However, siRNA of CXCL-1 with TNF-α and z-VAD restored necroptotic pathway of CLL cells (Figure 2A, P = 0.0004). Similarly, neither nc siRNA with TNF-α and z-VAD nor only MCP-1 siRNA could induce necroptosis of CLL cells. Even after the induction of MCP-1 siRNA together with TNF-α and z-VAD, the percentage of surviving CLL cells did not change significantly (Figure 2B). Therefore, knockdown of CXCL-1 rather than MCP-1 could help induce the necroptotic pathway of CLL cells.
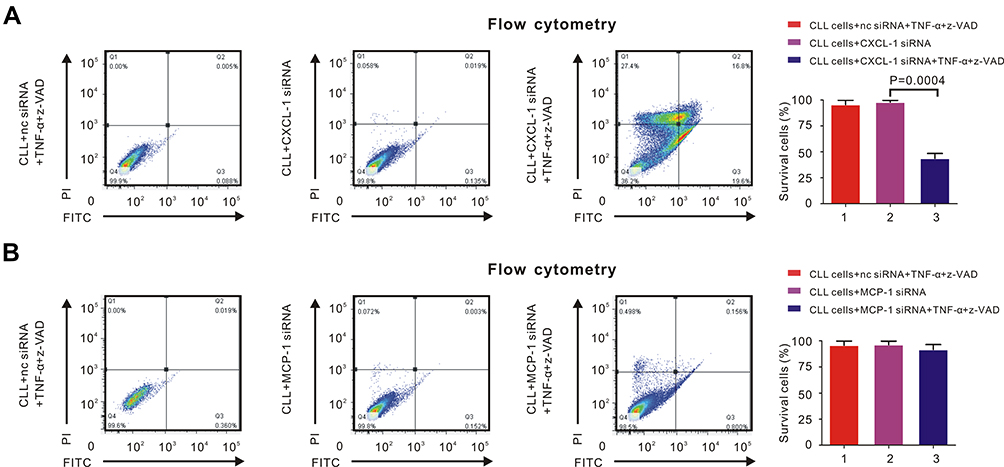 |
Figure 2 CXCL-1 rather than MCP-1 had correlation with necroptotic pathway of CLL cells. PBMCs were isolated by Ficoll and CLL cells were isolated by MACS. Percentage of necroptosis cells was detected by flow cytometry. (A) The percentage of survival CLL cells was over 99% after adding 30ng/mL TNF-α and 20μM z-VAD (left panel) or CXCL-1 siRNA (middle panel). However, the percentage of survival CLL cells decreased significantly after adding TNF-α, z-VAD and siRNA of CXCL-1 (right panel), indicating that high expression of CXCL-1 had correlation with defective necroptosis in CLL cells. (B) The percentage of survivaing CLL cells did not change significantly after adding TNF-α and z-VAD (left panel), siRNA of MCP-1 (middle panel) or both of them (right panel), demonstrating that MCP-1 was not associated with the necroptotic pathway of CLL cells. |
Knockdown of CXCL-1 Downregulated LEF-1 in CLL Cells
Measured by flow cytometry, the percentage of surviving CLL cells increased after knockdown of LEF-1 by siRNA with TNF-α and z-VAD (Figure 3A, P = 0.0001). This phenomenon had already been observed in other research, in which LEF-1 was the core element in the defective necroptosis of CLL cells.4 Furthermore, TNF-α/z-VAD induced necroptosis could also be restored in CLL cells after CXCL-1 was knocked down by siRNA (Figure 3A, P = 0.0008). Therefore, both LEF-1 and CXCL-1 were important in the necroptotic pathway of CLL cells.
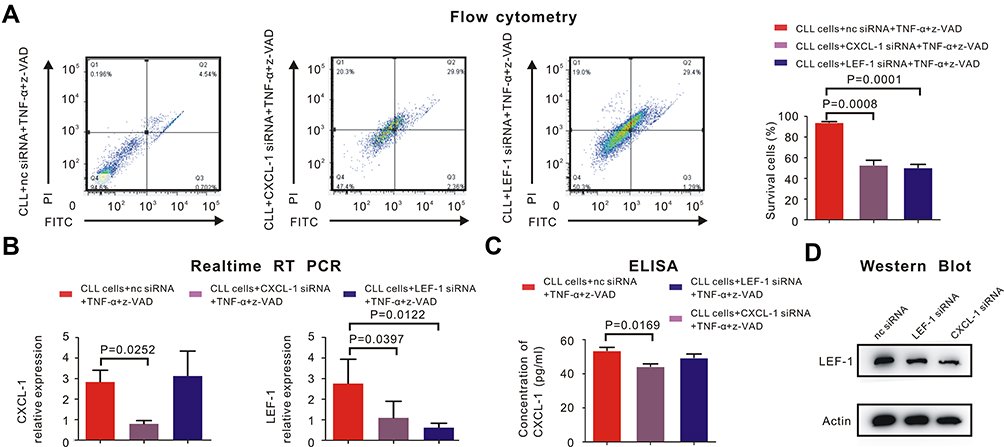 |
Figure 3 CXCL-1 activated LEF-1 in CLL cells to inhibit necroptosis. PBMCs were isolated by Ficoll and CLL cells were isolated by MACS. (A) Percentage of survival CLL cells was detected by flow cytometry. CLL cells did not undergo necroptosis with the induction of negative control siRNA (nc siRNA), 30ng/mL TNF-α and 20μM z-VAD. The percentage of survival CLL cells was 94.6%. (left panel) However, percentage of survival CLL cells significantly decreased after adding siRNA of CXCL-1 (P = 0.0008, middle panel) or siRNA of LEF-1 (P = 0.0001, right panel) with TNF-α and z-VAD, which indicated both CXCL-1 and LEF-1 played an important role in the defective necroptotic pathway of CLL cells. (B) The expression of LEF-1 and CXCL-1 were verified by real-time RT-PCR. The red bar indicated the expression of LEF-1 or CXCL-1 after adding nc siRNA, TNF-α and z-VAD; The gray bar referred to the expression of LEF-1 or CXCL-1 after adding siRNA of CXCL-1, TNF-α and z-VAD; The blue bar showed the expression of LEF-1 or CXCL-1 after adding siRNA of LEF-1, TNF-α and z-VAD. P-values were shown in the histogram when P < 0.05. The expression of LEF-1 was downregulated (P = 0.0397) when CXCL-1 was knocked down, but the expression of CXCL-1 did not change significantly after knockdown of LEF-1. (C) The translational expression of CXCL-1 was measured by ELISA. The red bar referred to the expression of CXCL-1 after CLL cells were treated by nc siRNA, TNF-α and z-VAD; The gray bar demonstrated CXCL-1 expression after CXCL-1 was knocked down by siRNA; The blue bar showed the CXCL-1 expression when LEF-1 was downregulated by siRNA with the induction of TNF-α and z-VAD. There existed no significantly statistical difference between the concentration of CXCL-1 before and after adding LEF-1 siRNA. (D) Measured by Western Blot, the translational expression of LEF-1 was downregulated after inhibiting the expression of CXCL-1 or LEF-1 by siRNA. |
We were able to discover the relationship between LEF-1 and CXCL-1. After knocking down CXCL-1 by siRNA, the expression of LEF-1 was downregulated (Figure 3B, P = 0.0397) and Western Blot showed the similar results (Figure 3D). However, both transcriptional and translational expression of CXCL-1 did not change significantly after knockdown of LEF-1 (Figure 3B and C). This phenomenon demonstrated that CXCL-1 was in the upstream of LEF-1, and the expression of LEF-1 was decreased after knockdown of CXCL-1.
Sodium Selenite Restored Necroptosis of CLL Cells and Downregulated the Expression of CXCL-1
Sodium selenite with different concentrations (0.032μM, 0.32μM and 3.2μM) was added to the CLL cells together with TNF-α and z-VAD. Then, RT-PCR was performed to evaluate the expression of CXCL-1 and LEF-1. We found that sodium selenite downregulated the expression of CXCL-1 but had little influence on LEF-1 (Figure 4A).
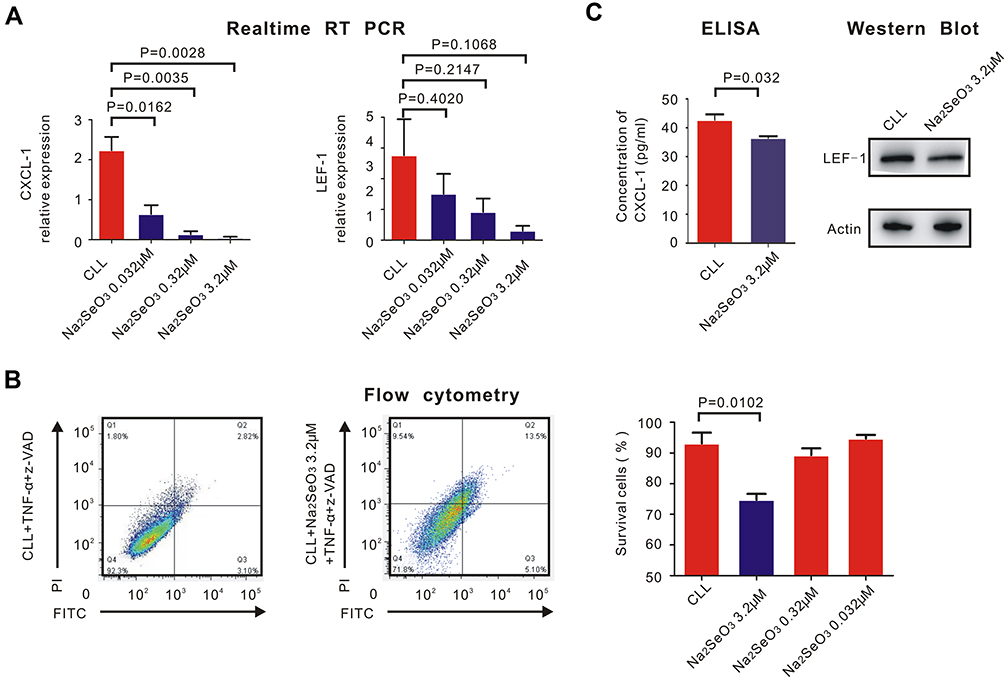 |
Figure 4 Sodium selenite helped restore necroptosis of CLL cells and inhibit the expression of CXCL-1. PBMCs were isolated by Ficoll and CLL cells were isolated by MACS. (A) real-time RT-PCR was performed to detect the expression of CXCL-1 (left) and LEF-1 (right). P-values were also shown in the histogram. Sodium selenite inhibited the expression of CXCL-1 but had little impact on the expression of LEF-1 (B) Flow cytometry was performed to detect the percentage of CLL survival cells before (left) and after (right) adding sodium selenite with different concentrations with TNF-α and z-VAD. Only 3.2μM sodium selenite helped restore necroptosis of CLL cells (P = 0.0102). (C) The translational expression of CXCL-1 measured by ELISA was downregulated with the contribution of 3.2μM sodium selenite, TNF-α and z-VAD (P = 0.032). However, the translational expression of LEF-1 did not change significantly before and after adding 3.2μM sodium selenite measured by Western Blot. |
Measured by flow cytometry, the percentage of surviving CLL cells was calculated after adding sodium selenite with TNF-α and z-VAD. Only 3.2μM sodium selenite significantly induced necroptosis of CLL cells (Figure 4B, P = 0.0102), but 3.2μM sodium selenite had little impact on normal B lymphocytes (Figure S1). Western Blot and ELISA confirmed the fact that 3.2μM sodium selenite downregulated the translational expression of CXCL-1 (P = 0.032) but had little impact on LEF-1 (Figure 4C).
Discussion
Necroptosis always occurs when cell apoptosis is defective and the mechanism induced by TNF-α is fully understood. The combination of TNF-α and TNF receptor recruits various proteins to form complex I, including cellular inhibitor of apoptosis (cIAP) and receptor-interacting protein 1 (RIP1). RIP1 was added the lysine 63-linked ubiquitin by cIAP.36 When the ubiquitin on RIP1 is removed by CYLD, the apoptotic pathway is activated.3 Furthermore, normal B lymphocytes undergo necroptosis when apoptosis is inhibited by caspase inhibitor, such as z-VAD.37 However, neither necroptosis nor apoptosis could be induced in CLL cells even with the cooperation of TNF-α and z-VAD. This phenomenon is due to the downregulation of CYLD expression caused by the upregulation of LEF-1, which might be one reason of CLL pathogenesis and drug resistance.4
On the other hand, some certain cytokines in the CLL microenvironment also contribute to defective apoptosis of CLL cells.38–40 However, very few articles focused on the impact of cytokines on the defective necroptotic pathway of CLL cells. We considered that some cytokines regulated the expression of LEF-1 to participate in the pathogenesis of defective necroptosis in CLL cells.
As a member of CXC family, CXCL-1 plays an important role in the angiogenesis, survival and metastasis of various solid malignancies. The high expression of CXCL-1 is observed in different malignant tumors, such as melanoma, prostate cancer, breast cancer and pancreatic carcinoma.41–44 CXCL-1 inhibitor might become a member of CLL treatment strategies in the future but it still needs further research.
Besides, MCP-1, IL-6 and GM-CSF also regulate the pathogenesis and metastasis of many malignant tumors. MCP-1 interacts with CCR2 receptor on the circulating fibroblast precursors of hematopoietic stem cells. The inhibition of MCP-1/CCR2 pathway was confirmed to reduce the tumor burden.17,45,46 IL-6 secreted by tumor cells promotes the development of tumor cells and inhibits the anti-tumor effect of CD4+ T lymphocytes.18,19,47 Furthermore, the monoclonal antibody of IL-6 enhances the efficacy and extends the indication of cancer immunotherapies including anti-PD-L1 antibody.48 GM-CSF combined with FLt3 ligand promotes the proliferation and activation of tumor-infiltrating dendrite cells with anti-tumor effect.49 However, GM-CSF also enhances the proliferation and metastasis of various malignant tumors, including skin carcinoma, glioma, head and neck squamous cell carcinoma, and lung cancer.50–53
In our research, the upregulation of CXCL-1, MCP-1, IL-6 and GM-CSF was observed in CLL cells compared with normal B lymphocytes. However, only CXCL-1 and MCP-1 of normal B lymphocytes were downregulated after induced by TNF-α and z-VAD, which also led to necroptotic pathway. This phenomenon demonstrated that the downregulation of CXCL-1 and MCP-1 might have correlation with necroptotic pathway. Then, we knocked down CXCL-1 and MCP-1 by siRNA respectively, and only the inhibition of CXCL-1 rather than MCP-1 helped restore necroptosis of CLL cells. Next, we clarified the fact that CXCL-1 located the upstream of LEF-1, which had been confirmed as the key protein in the defective necroptotic pathway of CLL.4 Therefore, the high expression of CXCL-1 in CLL cells upregulated the expression of LEF-1 to cause defective necroptosis.
Sodium selenite induced necroptosis by promoting the generation of ROS and was very selective to tumor cells.11,54 Our research first discovered that sodium selenite both inhibited the expression of CXCL-1 and restored the defective necroptotic pathway of CLL cells together with TNF-α and z-VAD. However, selenite had little impact on expression of LEF-1, which might be explained as the indirect influence or the existence of another necroptotic pathway controlled by CXCL-1. Therefore, selenite will be promising in the treatment of CLL patients, and the combination of selenite and other new drugs, such as ibrutinib, still needs further research.
Conclusion
CXCL-1 played an important role in the defective necroptosis of CLL cells and regulated the expression of LEF-1. Selenite inhibited the expression of CXCL-1 and helped CLL cells restore necroptosis together with TNF-α and z-VAD. Selenite might be the potential medication of CLL in the future.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81570123), the Natural Science Foundation of Shanghai (16ZR1405800), the National Key New Drug Creation Special Programs (2017ZX09304-021) and Academic Pacesetters Program of Shanghai Healthcare System (2017BR033).
Use of patient and volunteer peripheral blood samples were approved by the Review Board of Zhongshan Hospital, Fudan University (Shanghai, China).
Disclosure
The abstract of this paper was submitted to the American Society of Hematology (ASH) 2019 Conference, and the abstract was published in Blood; 2019;134:(Supplement_1): 5466 (https://doi.org/10.1182/blood-2019-123748).
References
1. Wierda WG, Zelenetz AD, Gordon LI, et al. NCCN guidelines insights: chronic lymphocytic leukemia/small lymphocytic lymphoma, version 1.2017. J Natl Compr Canc Netw. 2017;15(3):293–311. doi:10.6004/jnccn.2017.0030
2. Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432–443. doi:10.1056/NEJMoa1817073
3. Hitomi J, Christofferson DE, Ng A, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135(7):1311–1323. doi:10.1016/j.cell.2008.10.044
4. Liu P, Xu B, Shen W, et al. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2012;26(6):1293–1300. doi:10.1038/leu.2011.357
5. Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016;23(5):748–756. doi:10.1038/cdd.2016.8
6. Gruszka AM, Valli D, Alcalay M. Wnt signalling in acute myeloid leukaemia. Celss-BASEL. 2019;8(11).
7. Gutierrez AJ, Tschumper RC, Wu X, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116(16):2975–2983. doi:10.1182/blood-2010-02-269878
8. Kuhnl A, Gokbuget N, Kaiser M, et al. Overexpression of LEF1 predicts unfavorable outcome in adult patients with B-precursor acute lymphoblastic leukemia. Blood. 2011;118(24):6362–6367. doi:10.1182/blood-2011-04-350850
9. Spaulding C, Reschly EJ, Zagort DE, et al. Notch1 co-opts lymphoid enhancer factor 1 for survival of murine T-cell lymphomas. Blood. 2007;110(7):2650–2658. doi:10.1182/blood-2007-04-084202
10. Vanlangenakker N, Vanden BT, Bogaert P, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18(4):656–665. doi:10.1038/cdd.2010.138
11. Wallenberg M, Misra S, Wasik AM, et al. Selenium induces a multi-targeted cell death process in addition to ROS formation. J Cell Mol Med. 2014;18(4):671–684. doi:10.1111/jcmm.12214
12. Sonkusre P, Cameotra SS. Biogenic selenium nanoparticles induce ROS-mediated necroptosis in PC-3 cancer cells through TNF activation. J Nanobiotechnology. 2017;15(1):43. doi:10.1186/s12951-017-0276-3
13. Bjorkhem-Bergman L, Jonsson K, Eriksson LC, et al. Drug-resistant human lung cancer cells are more sensitive to selenium cytotoxicity. Effects on thioredoxin reductase and glutathione reductase. Biochem Pharmacol. 2002;63(10):1875–1884. doi:10.1016/S0006-2952(02)00981-4
14. Zhang S, Kipps TJ. The pathogenesis of chronic lymphocytic leukemia. Annu Rev Pathol. 2014;9:103–118. doi:10.1146/annurev-pathol-020712-163955
15. Till KJ, Lin K, Zuzel M, Cawley JC. The chemokine receptor CCR7 and alpha4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood. 2002;99(8):2977–2984. doi:10.1182/blood.V99.8.2977
16. Xu PP, Sun YF, Fang Y, et al. JAM-A overexpression is related to disease progression in diffuse large B-cell lymphoma and downregulated by lenalidomide. Sci Rep. 2017;7(1):7433. doi:10.1038/s41598-017-07964-5
17. Abangan RJ, Williams CR, Mehrotra M, Duncan JD, Larue AC. MCP1 directs trafficking of hematopoietic stem cell-derived fibroblast precursors in solid tumor. Am J Pathol. 2010;176(4):1914–1926. doi:10.2353/ajpath.2010.080839
18. Tsukamoto H, Fujieda K, Hirayama M, et al. Soluble IL6R expressed by myeloid cells reduces tumor-specific Th1 differentiation and drives tumor progression. Cancer Res. 2017;77(9):2279–2291. doi:10.1158/0008-5472.CAN-16-2446
19. Tsukamoto H, Nishikata R, Senju S, Nishimura Y. Myeloid-derived suppressor cells attenuate TH1 development through IL-6 production to promote tumor progression. Cancer Immunol Res. 2013;1(1):64–76. doi:10.1158/2326-6066.CIR-13-0030
20. Mueller MM, Fusenig NE. Constitutive expression of G-CSF and GM-CSF in human skin carcinoma cells with functional consequence for tumor progression. Int J Cancer. 1999;83(6):780–789. doi:10.1002/(SICI)1097-0215(19991210)83:6<780::AID-IJC14>3.0.CO;2-C
21. Zhang H, Ye YL, Li MX, et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. 2017;36(15):2095–2104. doi:10.1038/onc.2016.367
22. Tang J, Yang Q, Cui Q, et al. Weighted gene correlation network analysis identifies RSAD2, HERC5, and CCL8 as prognostic candidates for breast cancer. J Cell Physiol. 2019;235:394–407.
23. You FP, Zhang J, Cui T, et al. Th9 cells promote antitumor immunity via IL-9 and IL-21 and demonstrate atypical cytokine expression in breast cancer. Int Immunopharmacol. 2017;52:163–167. doi:10.1016/j.intimp.2017.08.031
24. Bronger H, Singer J, Windmuller C, et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br J Cancer. 2016;115(5):553–563. doi:10.1038/bjc.2016.172
25. De Marco P, Lappano R, De Francesco EM, et al. GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1beta/IL1R1 response. Sci Rep. 2016;6:24354. doi:10.1038/srep24354
26. Chen J, Lu Y, Zhang H, et al. A nonsynonymous polymorphism in IL23R gene is associated with risk of gastric cancer in a Chinese population. Mol Carcinog. 2010;49(10):862–868. doi:10.1002/mc.20661
27. IL15 is linked to antitumor immune responses in colorectal cancer. Cancer Discov. 2014;4(5):F9. doi:10.1158/2159-8290.CD-RW2014-077
28. Farmaki E, Kaza V, Papavassiliou AG. Chatzistamou I, Kiaris H. Induction of the MCP chemokine cluster cascade in the periphery by cancer cell-derived Ccl3. Cancer Lett. 2017;389:49–58. doi:10.1016/j.canlet.2016.12.028
29. Oner MG, Rokavec M, Kaller M, et al. Combined inactivation of TP53 and MIR34A promotes colorectal cancer development and progression in mice via increasing levels of IL6R and PAI1. Gastroenterology. 2018;155(6):1868–1882. doi:10.1053/j.gastro.2018.08.011
30. Zhou L, Jiang Y, Liu X, et al. Promotion of tumor-associated macrophages infiltration by elevated neddylation pathway via NF-kappaB-CCL2 signaling in lung cancer. Oncogene. 2019;38(29):5792–5804. doi:10.1038/s41388-019-0840-4
31. Maeda S, Murakami K, Inoue A, Yonezawa T, Matsuki N. CCR4 blockade depletes regulatory T cells and prolongs survival in a canine model of bladder cancer. Cancer Immunol Res. 2019;7(7):1175–1187. doi:10.1158/2326-6066.CIR-18-0751
32. Chen C, Xu ZQ, Zong YP, et al. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-kappaB pathway in colorectal cancer. Cell Death Dis. 2019;10(3):178. doi:10.1038/s41419-019-1431-6
33. Tulotta C, Lefley DV, Freeman K, et al. Endogenous production of IL1B by breast cancer cells drives metastasis and colonization of the bone microenvironment. Clin Cancer Res. 2019;25(9):2769–2782. doi:10.1158/1078-0432.CCR-18-2202
34. Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics. 2017;7(6):1543–1588. doi:10.7150/thno.15625
35. Akram IG, Georges R, Hielscher T, Adwan H, Berger MR. The chemokines CCR1 and CCRL2 have a role in colorectal cancer liver metastasis. Tumour Biol. 2016;37(2):2461–2471. doi:10.1007/s13277-015-4089-4
36. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–190. doi:10.1016/S0092-8674(03)00521-X
37. Vandenabeele P, Vanden BT, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;2006(358):e44. doi:10.1126/stke.3582006pe44
38. Kurtova AV, Balakrishnan K, Chen R, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114(20):4441–4450. doi:10.1182/blood-2009-07-233718
39. Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. doi:10.1182/blood.V96.8.2655
40. Burger JA. Targeting the microenvironment in chronic lymphocytic leukemia is changing the therapeutic landscape. Curr Opin Oncol. 2012;24(6):643–649. doi:10.1097/CCO.0b013e3283589950
41. Dhawan P, Richmond A. Role of CXCL1 in tumorigenesis of melanoma. J Leukoc Biol. 2002;72(1):9–18.
42. Kogan-Sakin I, Cohen M, Paland N, et al. Prostate stromal cells produce CXCL-1, CXCL-2, CXCL-3 and IL-8 in response to epithelia-secreted IL-1. Carcinogenesis. 2009;30(4):698–705. doi:10.1093/carcin/bgp043
43. Wang N, Liu W, Zheng Y, et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis. 2018;9(9):880. doi:10.1038/s41419-018-0876-3
44. Seifert L, Werba G, Tiwari S, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532(7598):245–249. doi:10.1038/nature17403
45. Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5(15):1597–1601. doi:10.4161/cc.5.15.3112
46. Ogawa M, LaRue AC, Drake CJ. Hematopoietic origin of fibroblasts/myofibroblasts: its pathophysiologic implications. Blood. 2006;108(9):2893–2896. doi:10.1182/blood-2006-04-016600
47. Tsukamoto H, Senju S, Matsumura K, Swain SL, Nishimura Y. IL-6-mediated environmental conditioning of defective Th1 differentiation dampens antitumour immune responses in old age. Nat Commun. 2015;6:6702. doi:10.1038/ncomms7702
48. Ohno Y, Toyoshima Y, Yurino H, et al. Lack of interleukin-6 in the tumor microenvironment augments type-1 immunity and increases the efficacy of cancer immunotherapy. Cancer Sci. 2017;108(10):1959–1966. doi:10.1111/cas.13330
49. Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, Dranoff G. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 2000;60(12):3239–3246.
50. Obermueller E, Vosseler S, Fusenig NE, Mueller MM. Cooperative autocrine and paracrine functions of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the progression of skin carcinoma cells. Cancer Res. 2004;64(21):7801–7812. doi:10.1158/0008-5472.CAN-03-3301
51. Mueller MM, Herold-Mende CC, Riede D, Lange M, Steiner HH, Fusenig NE. Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. Am J Pathol. 1999;155(5):1557–1567. doi:10.1016/S0002-9440(10)65472-7
52. Gutschalk CM, Herold-Mende CC, Fusenig NE, Mueller MM. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor promote malignant growth of cells from head and neck squamous cell carcinomas in vivo. Cancer Res. 2006;66(16):8026–8036. doi:10.1158/0008-5472.CAN-06-0158
53. Pei XH, Nakanishi Y, Takayama K, Bai F, Hara N. Granulocyte, granulocyte-macrophage, and macrophage colony-stimulating factors can stimulate the invasive capacity of human lung cancer cells. Br J Cancer. 1999;79(1):40–46. doi:10.1038/sj.bjc.6690009
54. Nilsonne G, Olm E, Szulkin A, et al. Phenotype-dependent apoptosis signalling in mesothelioma cells after selenite exposure. J Exp Clin Cancer Res. 2009;28:92. doi:10.1186/1756-9966-28-92
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.