")
Back to Journals » OncoTargets and Therapy » Volume 11
Suppression of c-MET overcomes erlotinib resistance in tongue cancer cells
Received 12 March 2018
Accepted for publication 18 May 2018
Published 5 September 2018 Volume 2018:11 Pages 5499—5508
DOI https://doi.org/10.2147/OTT.S167936
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sanjeev K. Srivastava
Keqiang Huang, Dongxu Liu
Department of Orthodontics, School of Stomatology, Shandong University, Shandong Provincial Key Laboratory of Oral Tissue Regeneration, Jinan 250012, China
Background: Erlotinib is a commonly used molecular-targeted drug for the treatment of tongue cancer. However, the development of acquired resistance to erlotinib hampers its therapeutic use.
Materials and methods: To analyze the erlotinib resistance, long-term and short term survival assay were used to compare the resistance between parental and resistant tongue cancer cells. Flow cytometry, Hochest staining and western blot were used to analyze the apoptosis among the cells. Moreover, Transwell and wound healing assay were used to compare the invasion ability of the cells. To deeply explore the drug resistance in vivo, orthotopic tumor studies were applied. Finally, to explain the mechanism of c-met in erlotinib resistance, shRNA against c-met was used to down-regulate the expression of c-met. And SU11274 also used in orthotopic model.
Results: We established erlotinib-resistant human tongue cancer cell line by chronic exposure of TCA-8113 cells to increasing concentrations of erlotinib and determined the role of c-MET and EGFR in the development of acquired resistance. We found a significant increase in the phosphorylation of c-MET and an obvious decrease of the phosphorylation of EGFR in erlotinib-resistant cells. Our results also revealed that inhibition of c-MET alone with SU11274 exerted an inhibitory effect on the proliferation of erlotinib-resistant cells in the short term; however, it failed to sustain the inhibitory effect in the long term. Simultaneous inhibition of c-MET and EGFR significantly inhibited the proliferation of erlotinib-resistant cells in both a short and long period. Furthermore, we explored the underlying mechanism and found that treatment of erlotinib-resistant cells with SU11274 or shRNA against c-MET induced the phosphorylation of EGFR. Moreover, our results demonstrated that simultaneous inhibition of c-MET and EGFR significantly inhibited the migration and invasion of erlotinib-resistant cells.
Conclusion: Taken together, our results suggested that c-MET is involved in acquired drug resistance to erlotinib and that cotargeting of EGFR and c-MET could overcome acquired resistance to erlotinib and inhibit the invasion and metastasis of erlotinib-resistant cells.
Keywords: tongue cancer, erlotinib, acquired resistance, EGFR, c-MET
Introduction
The incidence of tongue cancer is about 40% in oral cancer, and the most common type of tongue cancer is squamous cell carcinoma. So far, oral tongue cancer has a poor prognosis, and increasing incidence is seen among young adults.1 Presently, due to chemotherapeutic resistance, the survival rates of tongue cancer patients is below the median.2,3 Not only that, limited information regarding the regulatory mechanisms of acquired chemoresistance exists.
EGFR, a member of the ERBB family of cell-surface tyrosine kinases is overexpressed in tongue cancer, and the overexpression of EGFR has been associated with poor clinical outcomes.4–7 After ligand binding, EGFR dimerized homo- and/or heterologously and triggered downstream intracellular signaling cascades such as the PI3K/Akt, Raf/MEK/ERK, and STAT signaling pathways, leading to cellular proliferation,8 angiogenesis,9 metastasis,10 and inhibition of apoptosis.11 Therefore, targeting EGFR for chemoprevention of tongue cancer has received considerable attention. Small molecule EGFR-targeted tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib have been used for the clinical treatment of many human cancers,12–14 and they are currently undergoing clinical evaluation for treatment. Currently, the therapeutic effects are being studied in patients with advanced tongue squamous cell carcinoma.15–17 However, for many reasons, the cancer cells often acquire drug resistance to EGFR-TKI drugs.7 This acquired resistance severely disrupted the application of EGFR-TKI drugs. So, it is imperative to develop new strategies or alternative therapies to reverse this process. For example, using combination treatment to lower the doses of toxic drugs and overcome drug resistance.
c-MET is a membrane spanning receptor tyrosine kinase for hepatocyte growth factor (HGF) also termed scatter factor. Overactivation of HGF/c-MET signaling is reported to be involved in tumor progression and metastasis, and it has been identified as a poor prognosis factor in non-small-cell lung cancer.18–21 Engelman et al22 report that acquired EGFR-TKI resistance could be reversed by the combination of c-MET inhibitor with gefitinib in EGFR-TKI-resistant lung cancer cells. However, the effect of c-MET in erlotinib drug resistance of tongue cancer has not been reported.
In this study, we investigated the role of c-MET in the acquired resistance to erlotinib and explored the possibility of c-MET as a therapeutic target to reverse the acquired resistance to erlotinib in human tongue cancer cells.
Materials and methods
Cell culture
Human tongue cancer cell line TCA-8113 was purchased from the Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; 1995065, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; FB15015, Clark Bioscience, Richmond, VA, USA), 1 mM L-glutamine, and 1% penicillin/streptomycin antibiotics (15140-122, Thermo Fisher Scientific).
Establishment of erlotinib-resistant cells
The erlotinib-resistant cells were established by chronic exposing of TCA8113 cells to increasing concentrations of erlotinib (S1023, Selleck, Houston, TX, USA). The initial concentration was 5 μM; when the cells became resistant to this concentration, the concentration was increased. Thereafter, the concentration was gradually increased (each time 0.5 μM) every 3–5 days, reaching a final concentration to 10 μM. The concentration was maintained at 10 μM for at least 30 days. The remaining clones were harvested, expanded, and identified using the short- and long-term survival assay. The erlotinib-resistant cells were cultured in complete DMEM containing 5 μM of erlotinib.
Short-term survival assay
Cells were cultured in 96-well tissue culture plates (5,000 cells each well). After 24 h of plating, cells were washed with PBS 3 times and then treated with erlotinib or SU11274 (S1080, Selleck) for 48 h in DMEM containing 0.5% FBS. At the end of the culture period, cells were washed with PBS, and 20 μL of MTT solution (5 mg/mL, Sigma, St Louis, MO, USA) was added to each well, Cells were incubated for another 4 h. The absorbance was measured at 570 nm using a microplate reader. Mean values were calculated from three independent experiments and represented as means ± standard error.
Long-term survival assay
Cells were seeded into 6-well plates (104 cells per well) and allowed to adhere overnight in regular growth media. Cells were then cultured in the absence or presence of erlotinib/SU11274 at different concentrations in complete media for 2 weeks. Growth media with or without erlotinib/SU11274 was replaced every 3 days. After 2 weeks, tumor cells were fixed with ethanol (70%), stained with 0.5% crystal violet, and photographed using a digital camera.
Flow cytometry
Cells were seeded in complete medium in 6-well culture plates at a density of 106 cells per well. After 24 h of plating, cells were washed 3 times with PBS and then treated with the indicated drugs for 48 h in DMEM containing 0.5% FBS. After 72 h, cells were trypsinized, stained with Annexin V-FITC and propidium iodide for 15 min, and fixed with 70% ethanol. The cells undergoing apoptosis were determined by flow cytometry (FACS Calibur™, Becton Dickinson, Franklin Lakes, NJ, USA). Experiments were repeated 3 times.
Western blot analysis
For extraction of total cellular protein, cells were lysed in RIPA buffer with PMSF. Protein concentration was quantified using the BCA kit (Pierce Biotechnology, Inc., Rockford, IL, USA). Proteins were separated and transferred to PVDF membranes. The membranes were incubated overnight at 4°C with cleaved caspase-3 (9664#), caspase 3 (9662#), EGFR (2232#), p-EGFR (3777#), c-MET (3127#), p-c-MET (3129#), ERK (4696#), p-ERK (4370#), Akt (4685#), p-Akt (9271#), and β-actin (1:1,000) (Cell Signaling Technology, Danvers, MA, USA). Thereafter, the membranes were incubated with HRP-labeled anti-rabbit secondary antibodies (1:1,000) for 1 h at room temperature. Finally, the membrane was visualized using an enhanced chemiluminescence kit (Thermo Fisher Scientific Inc., Rockford, IL, USA).
Hochest 33342 staining
After treatment, cells were stained with Hochest 33342 (Beyotime, China) and photographed using a fluorescence microscope. The incidence of DNA condensation in each preparation was analyzed by counting 300 cells and determining the percentage of DNA condensed cells.
Orthotopic tumor studies
Six to eight weeks old male BALB/C nude mice (Beijing Weitong Lihua Animal Co. Ltd., Beijing, China) were used in the experiment. All animal experiments were approved and supervised by the Animal Care and Use Committees of Shandong Medical University and were performed according to the institutional ethical guidelines for animal experiments. The erlotinib-resistant cells 107 in 200 μL PBS was injected subcutaneously to the dorsal midline in nude mice. Once the tumors reached ~100 mm3, mice were randomly divided into 4 groups and treated with SU11274 (30 mg/kg), erlotinib (30 mg/kg), or SU11274 in combination with erlotinib (SU11274: 30 mg/kg, erlotinib: 30 mg/kg) by intragastric administration twice a week for 14 days. After 2 weeks, the xenografts were removed and tumor weights were evaluated.
shRNA transfection
The erlotinib-resistant cells derived from TCA-8113 were cultured in a 6-well culture plate and allowed to grow until 90% confluency was achieved. Transfection was performed with Lipofectamine 2000 following the manufacturer’s instructions (12566014, Thermo Fisher Scientific). The sequence of shRNAs against c-MET used were as follows:
shMET#1:
5′-CGGAGACTCATAATCCAACTGTAACTCGAGTTACAGTTGGATTATGAGTCTTTTTTG-3′
shMET#2:
5′-CCGGGCACTATTATAGGACTTGTATCTCGAGATACAAGTCTTATAATAGTGCTTTTG-3′
nontargeting (NT) control:
5′-CTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′.
Cells were transfected with 4 μg of shRNA using 10 μL Lipofectamine 2000 in 100 μL DMEM containing 10% FBS. After 48 h, the expression of c-MET was determined using Western blot.
Wound healing assay
The erlotinib-resistant cells were harvested and seeded in a 6-well plate (1×105 each well) and allowed to grow until confluent. The monolayer was carefully wounded using a sterile pipette and washed with PBS three times to remove the debris. The wounded monolayer was cultured in DMEM containing 0.5% FBS for 24 h, and photographed with an inverted microscope (100×). The status of wound closure was evaluated.
Transwell assay
Transwell assay was performed using Costar’s 24 well Transwell (Costar #3422). Cells were plated on the upper chamber of a 96-well-plate at a concentration of 1×104/well and allowed to invade for 24 h. The inserts were inverted and stained with 0.5% crystal violet. The number of invaded cells were observed and counted using inverted microscope. Five fields were randomly chosen and the numbers of penetrated cells were counted. The invasion potentiality of tumor cells was represented by the average value of penetrated cells in these fields.
Statistical analysis
Comparison of the data among multiple groups was performed using one-way analysis of variance. Comparison of the data between two groups was performed by Student’s t-test. A P-value <0.05 was considered to be statistically significant.
Results
c-MET is overactivated in erlotinib-resistant cells
To establish erlotinib-resistant cancer cells, we exposed tongue cancer cells TCA8113 to increasing concentrations of erlotinib (Figure 1A). Two resistant clones were chosen for further experiments. Compared with parent cells, erlotinib-resistant TCA8113 (TCA8113-R) cells exhibited an asymmetrically elongated morphology, whereas wild-type TCA8113 cells presented a symmetrically polygonal appearance (Figure 1B). Short-term and long-term survival analyses showed that although wild-type cells were highly sensitive to erlotinib, erlotinib-resistant cells required higher doses of the drug for partial growth inhibition (Figure 1C and D). From the short-term survival assay, we found the IC50 of erlotinib in TCA-8113 to be about 5 μM; however, the IC50 in erlotinib-resistant cells was >20 μM. We also determined the invasion potentials of 2 clones of resistant cells and found that the invasion potential of the resistant cells was significantly greater than that of parent cells (Figure 1E). Then, we compared the phosphorylation levels of EGFR and c-MET in wild-type and the resistant cells. We found that the phosphorylation level of c-MET was significantly increased, whereas the phosphorylation level of EGFR was obviously decreased in erlotinib-resistant cells as compared with that in wild-type cells (Figure 1F).
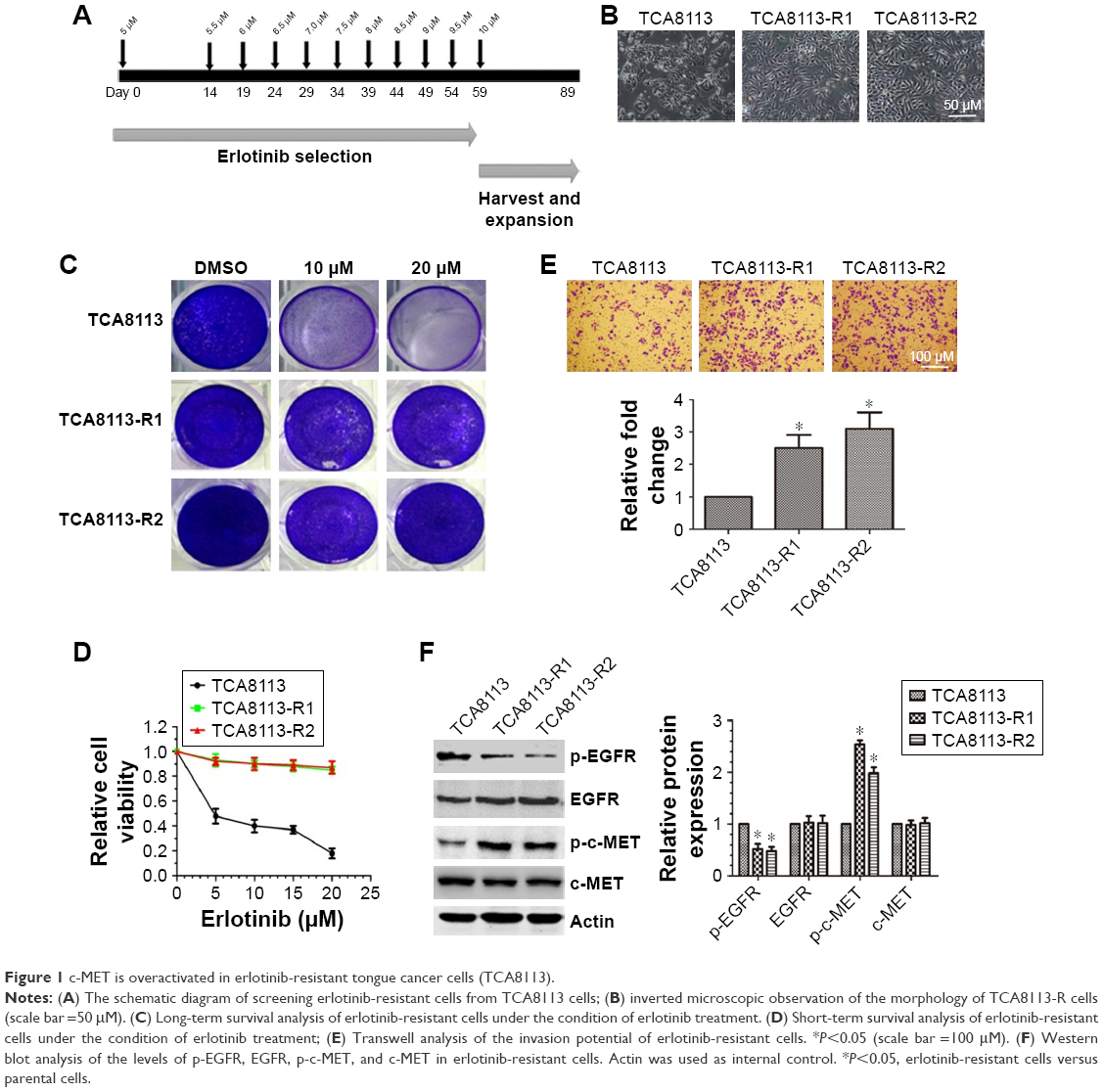 | Figure 1 c-MET is overactivated in erlotinib-resistant tongue cancer cells (TCA8113). |
Cotargeting EGFR and c-MET inhibited the growth of erlotinib-resistant cells
Since c-MET is overactivated in erlotinib-resistant cells, we then treated erlotinib-resistant cells with c-MET kinase inhibitor SU11274 and examined whether it could inhibit the growth of TCA8113-R cells. Short-term survival analysis showed that treatment of the resistant cells with SU11274 caused a significant growth inhibition (Figure 2A). From the assay, we determined the IC50 of SU11274 as about 5 μM, and we used this concentration for further experiments. However, long-term survival analysis revealed that SU11274 treatment could not lead to an obvious growth inhibition (Figure 2B). These data indicated that although inhibition of c-MET alone could inhibit the growth of erlotinib-resistant cells in a short time, it could not efficiently inhibit the growth of the resistant cells for a long time.
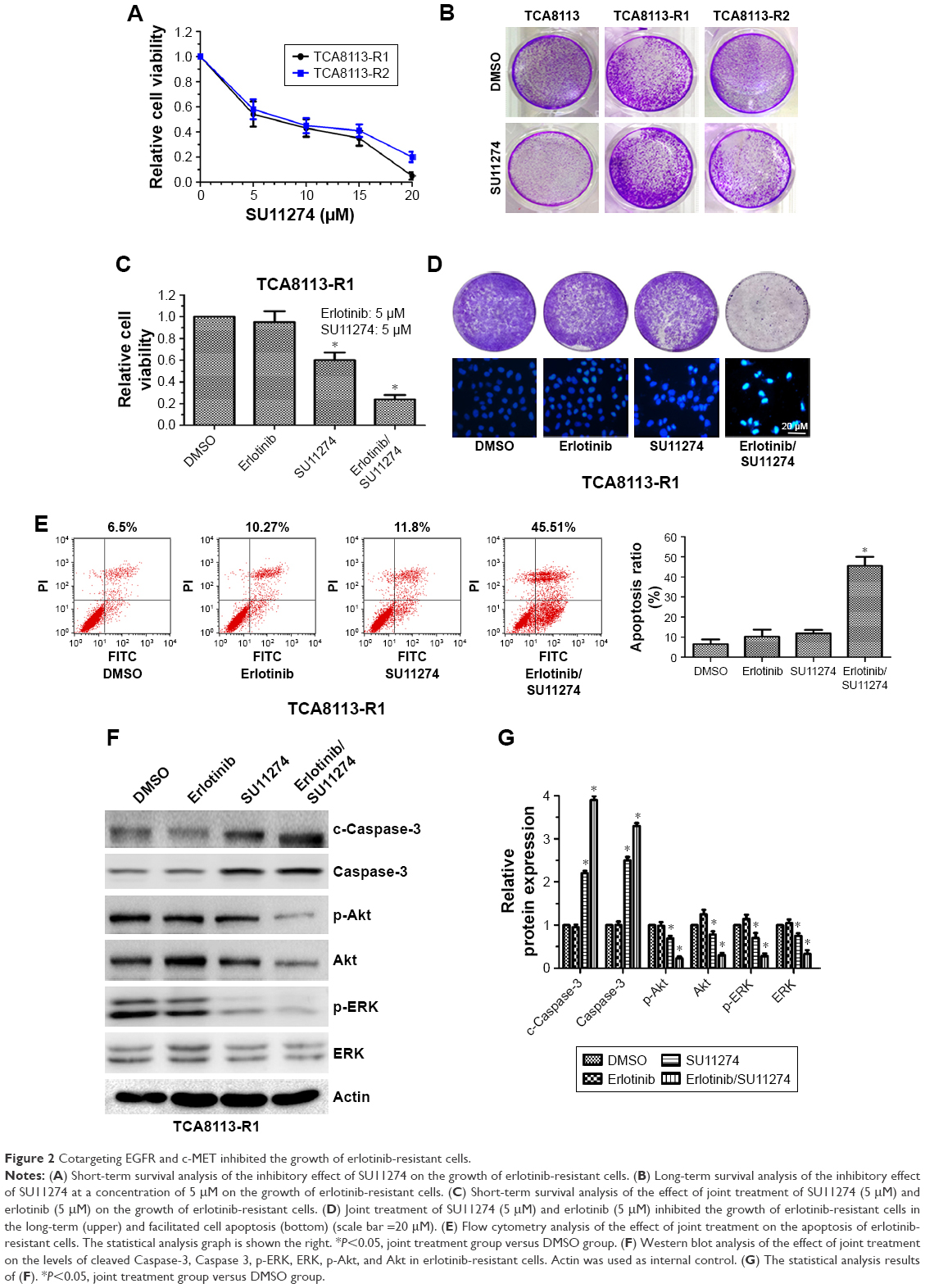 | Figure 2 Cotargeting EGFR and c-MET inhibited the growth of erlotinib-resistant cells. |
We next tried to explore whether cotargeting of EGFR and c-MET could overcome the acquired resistance to erlotinib in human tongue cancer. For this purpose, the resistant cells were treated by SU11274 (5 μM) in combination with erlotinib (5 μM) and the inhibitory effects were investigated using short and long time survival experiments. We found that joint treatment of erlotinib-resistant cells with SU11274 and erlotinib significantly inhibited the growth of tumor cells in the short- (Figure 2C) and long-term as compared with treatment with SU11274 alone (Figure 2D upper). Furthermore, to investigate whether joint treatment with SU11274 and erlotinib could induce cell apoptosis in the resistant cells, we used Hochest 33258 staining and found that this joint treatment caused a significant increase in the percentage of cell apoptosis as compared with treatment with SU11274 or erlotinib alone (Figure 2D bottom). Furthermore, these results were further demonstrated by flow cytometry analysis (Figure 2E). Western blot analysis showed that treatment of erlotinib-resistant cells with SU11274 in combination with erlotinib significantly promoted the cleavage of Caspase-3 and suppressed the phosphorylation of ERK and Akt (Figure 2F and G).
For further validating these in vitro results, we established in vivo xenograft model by injecting erlotinib-resistant cells (107 each animal) subcutaneously. We found that joint treatment of the resistant cells with SU11274 in combination with erlotinib caused a significant decrease in the average weight of xenografts as compared with treatment with SU11274 or erlotinib alone (Figure 3A and B). Western blot analysis showed that joint treatment with SU11274 and erlotinib significantly inhibited the phosphorylation of ERK and Akt in the xenograft tumors (Figure 3C and D). Taken together, cotargeting of c-MET and EGFR could inhibit the growth of erlotinib-resistant cells in the long term, whereas inhibition of c-MET alone only caused a transient growth inhibition in the short term.
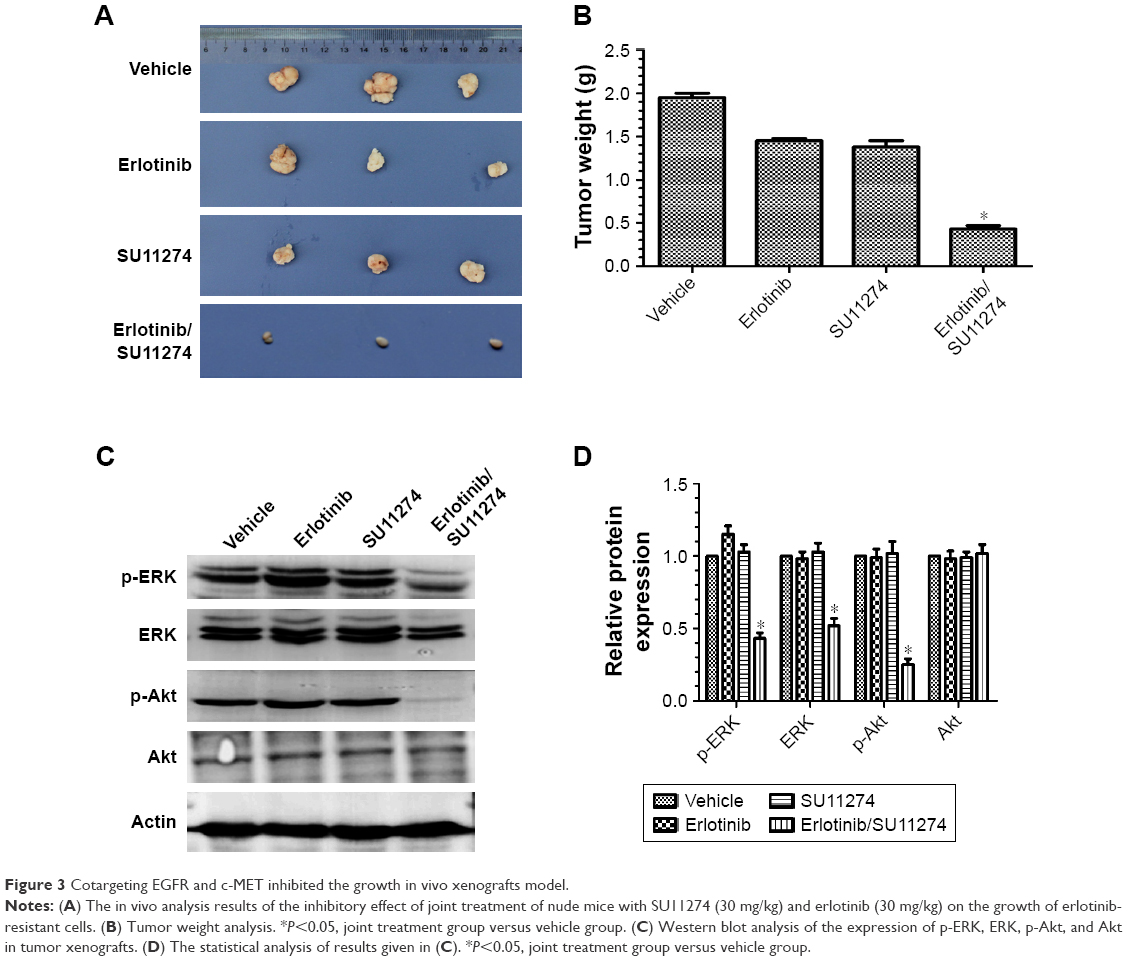 | Figure 3 Cotargeting EGFR and c-MET inhibited the growth in vivo xenografts model. |
Knockdown of c-MET sensitized erlotinib-resistant cells to erlotinib
To explore the underlying molecular mechanism, we treated the resistant cells with SU11274 at a concentration of 5 μM and then the phosphorylation levels of c-MET and EGFR were examined dynamically by Western blot. We found that treatment of the resistant cells with SU11274 caused a significant inhibition of c-MET phosphorylation, whereas coincidently this caused a significant increase in the phosphorylation levels of EGFR (Figure 4A).
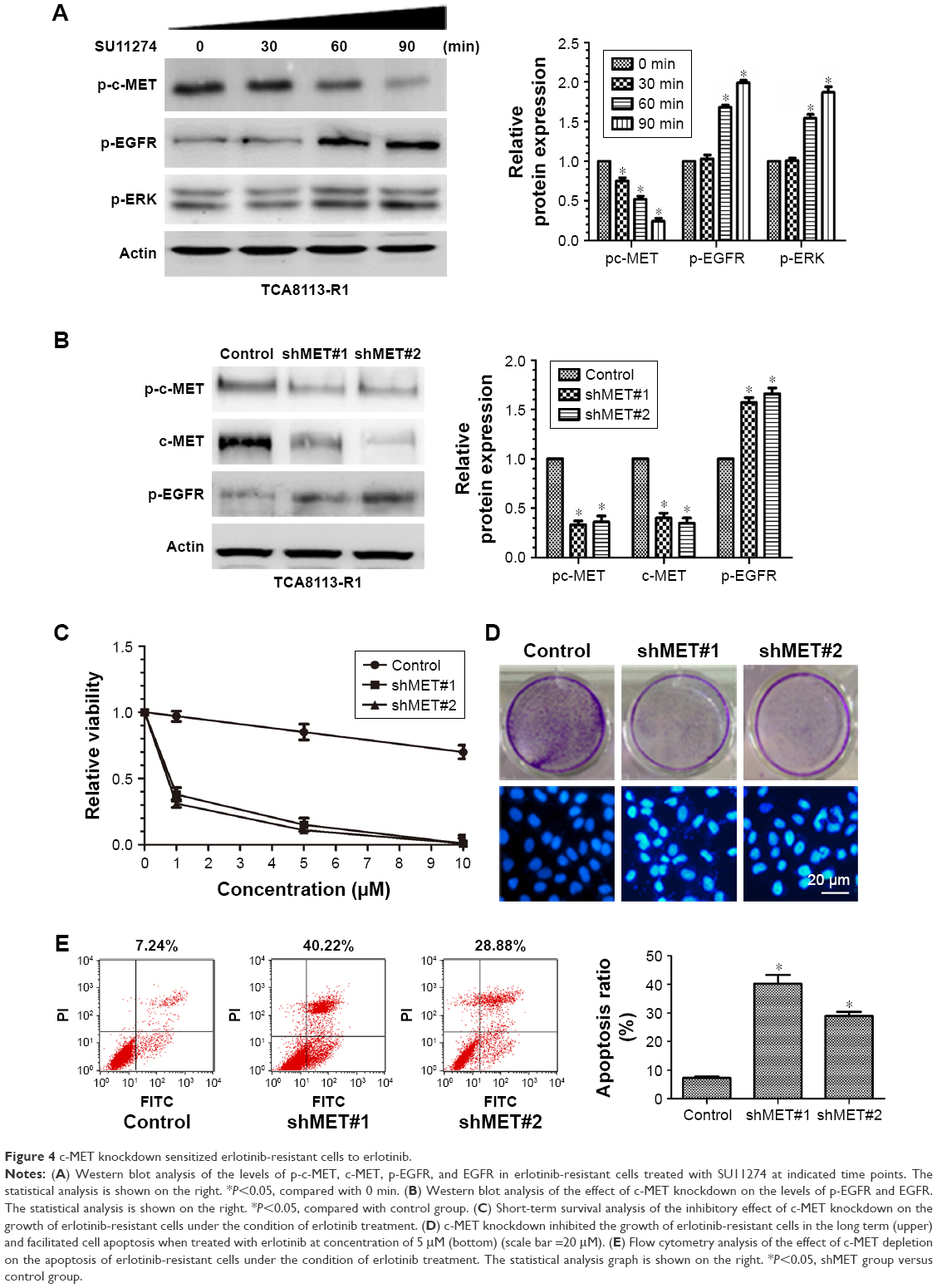 | Figure 4 c-MET knockdown sensitized erlotinib-resistant cells to erlotinib. |
To further elucidate the role of c-MET in the development of acquired resistance, we then knocked down c-MET in the resistant cells using shRNA against c-MET and examined the sensitivity of c-MET knockdown cells to erlotinib (Figure 4B). Knockdown of c-MET significantly inhibited the growth of the resistant cells in the short and long term when treated with erlotinib (Figure 4C and D upper). We also observed that erlotinib could induce the apoptosis of the resistant cells under the condition of c-MET knockdown, as assessed by Hochest 33258 staining and flow cytometry (Figure 4D bottom and E).
We also explored whether c-MET affected the phosphorylation of EGFR. As shown in Figure 4A, although treatment of the resistant cells with SU11274 inhibited the phosphorylation of c-MET, it promoted the phosphorylation of EGFR coincidently. This conclusion was further demonstrated by the fact that c-MET knockdown facilitated the phosphorylation of EGFR in the resistant cells (Figure 4B).
Cotargeting of EGFR and c-MET inhibited the invasion of erlotinib-resistant cells
Having demonstrated that cotargeting of EGFR and c-MET sensitizes erlotinib-resistant cells to erlotinib, we sought to determine whether cotargeting of EGFR and c-MET inhibited the invasion of the resistant cells. To study this goal, the resistant cells were treated with erlotinib and/or SU11274, and the migratory and invasive potentials were determined by wound healing assay and Transwell assay, respectively. Wound healing assay showed that joint treatment with erlotinib and SU11274 inhibited the migration of the resistant cells to a greater extent as compared with treatment with SU11274 or erlotinib alone (Figure 5A). Consistently, Transwell assay showed that simultaneous treatment with erlotinib and SU11274 inhibited the invasion potential of TCA8113-R cells to a greater extent (Figure 5B and C).
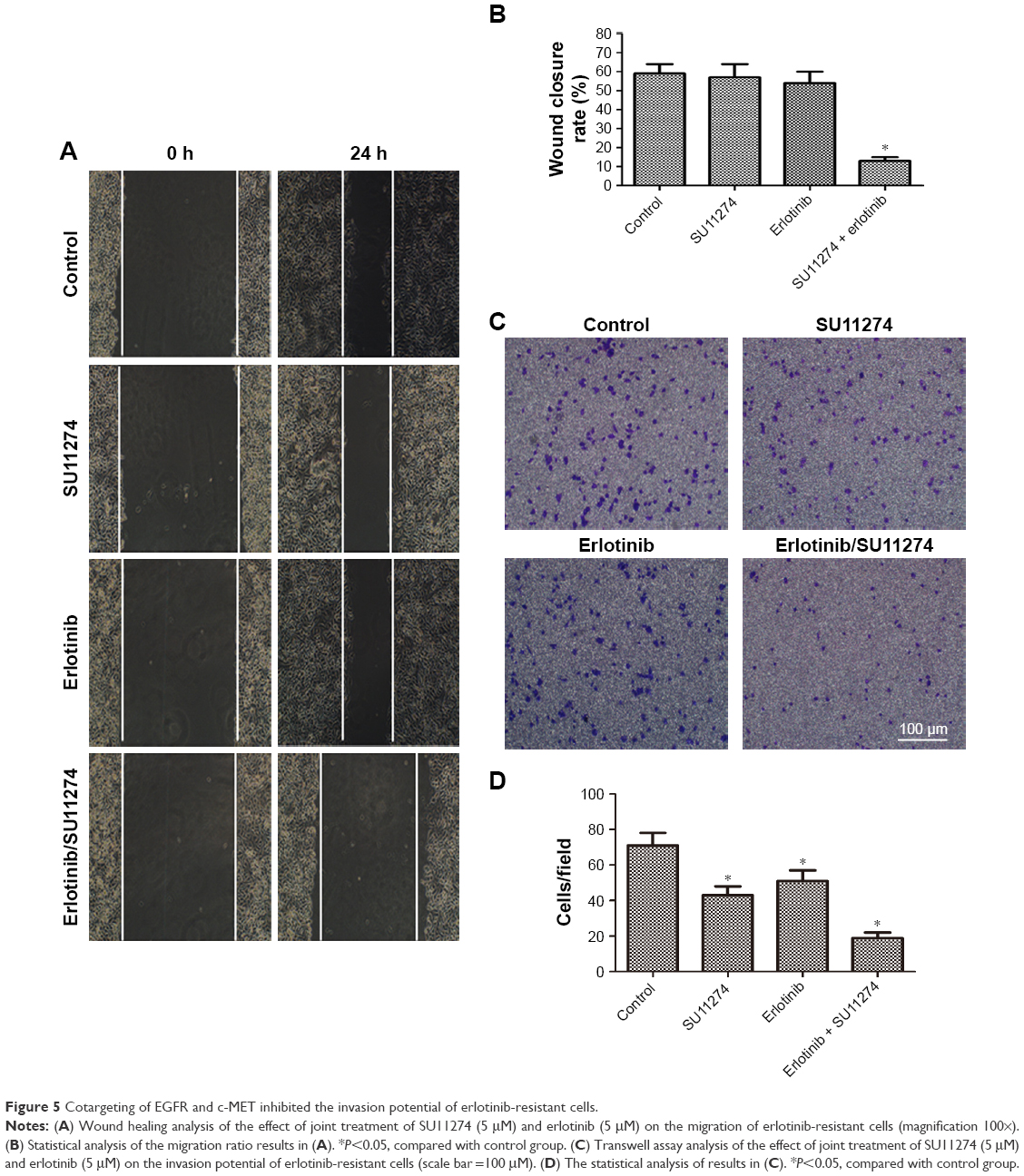 | Figure 5 Cotargeting of EGFR and c-MET inhibited the invasion potential of erlotinib-resistant cells. |
Discussion
Erlotinib is an important chemotherapeutic drug in tongue cancer patients.17,23 However, the long-term effect is unsatisfactory due to the development of acquired resistance. So, researchers have been trying to explore the underlying mechanism and effective treatment options to overcome the acquired resistance. Herein, we found that c-MET is involved in the development of acquired resistance and could be used as a potential therapeutic target for overcoming the acquired resistance.
To study what happened in erlotinib-resistant tongue cancer cells, we screened TCA-8113R and compared the phenotype with parental cells. After examination of the resistance extent, we found the TCA8113-R cells were more resistant to erlotinib than parental cells. Interestingly, in the TCA8113-R cells, c-MET activity could not be inhibited in erlotinib treatment, though EGFR activity already has been inhibited. It is suggested that c-MET is associated with erlotinib resistance. After c-MET was inhibited by SU11274, we found that TCA-8113R could be inhibited by erlotinib. In vivo xenografts model in BALB/C nude mice also helped us determine this conclusion. To explore the mechanism of c-MET in TCA-8113R, we examine the phosphorylation levels of c-MET and EGFR by Western blot dynamically in erlotinib- or SU11274-treated TCA-8113 cells. We found that erlotinib significantly inhibited EGFR phosphorylation, but promoted the phosphorylation of c-MET. Therefore, the mutual compensation of c-MET and EGFR in the development and reverse of acquired resistance to erlotinib. Knocking down the expression of c-MET, the acquired resistance to erlotinib disappeared in TCA-8113 cells. In addition, cotargeting of EGFR and c-MET could obviously inhibit the invasion potential of TCA8113-R cells. It is suggested only cotargeting c-MET and EGFR could inhibit the erlotinib drug resistance of tongue cancer cells and invasion.
Taken together, c-MET is involved in erlotinib-resistant tongue cancer, and cotargeting of c-MET and EGFR could reverse the erlotinib resistance in TCA-8113 cells.
Conclusion
c-MET is involved in acquired drug resistance to erlotinib and cotargeting, of EGFR and c-MET could overcome this process and also inhibit the invasion potential of erlotinib-resistant cells.
Disclosure
The authors report no conflicts of interest in this work.
References
Shiboski CH, Schmidt BL, Jordan RC. Tongue and tonsil carcinoma: increasing trends in the U.S. population ages 20–44 years. Cancer. 2005;103(9):1843–1849. | ||
Zhang Q, Shi S, Yen Y, Brown J, Ta JQ, Le AD. A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett. 2010;289(2):151–160. | ||
Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Diz PG, Rey JM, García-García A. Multidrug resistance in oral squamous cell carcinoma: the role of vacuolar ATPases. Cancer Lett. 2010;295(2):135–143. | ||
Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–1400. | ||
Rabinowits G, Haddad RI. Overcoming resistance to EGFR inhibitor in head and neck cancer: a review of the literature. Oral Oncol. 2012;48(11):1085–1089. | ||
Ford AC, Grandis JR. Targeting epidermal growth factor receptor in head and neck cancer. Head Neck. 2003;25(1):67–73. | ||
Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45(4–5):324–334. | ||
Mueller KL, Powell K, Madden JM, Eblen ST, Boerner J. EGFR tyrosine 845 phosphorylation-dependent proliferation and transformation of breast cancer cells require activation of p38 MAPK. Transl Oncol. 2012;5(5):327–334. | ||
Larsen AK, Ouaret D, El Ouadrani K, Petitprez A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol Ther. 2011;131(1):80–90. | ||
Lindzen M, Carvalho S, Starr A, et al. A recombinant decoy comprising EGFR and ErbB-4 inhibits tumor growth and metastasis. Oncogene. 2012;31(30):3505–3515. | ||
Donev IS, Wang W, Yamada T, et al. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clin Cancer Res. 2011;17(8):2260–2269. | ||
Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26(15):2442–2449. | ||
Ciardiello F, Caputo R, Bianco R, et al. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res. 2001;7(5):1459–1465. | ||
Cappuzzo F, Varella-Garcia M, Shigematsu H, et al. Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non-small-cell lung cancer patients. J Clin Oncol. 2005;23(22):5007–5018. | ||
Tan DS, Wang W, Leong HS, et al. Tongue carcinoma infrequently harbor common actionable genetic alterations. BMC Cancer. 2014;14:679. | ||
Ishiguro Y, Ishiguro H, Miyamoto H. Epidermal growth factor receptor tyrosine kinase inhibition up-regulates interleukin-6 in cancer cells and induces subsequent development of interstitial pneumonia. Oncotarget. 2013;4(4):550–559. | ||
Gusenbauer S, Vlaicu P, Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene. 2013;32(33):3846–3856. | ||
Tanaka A, Sueoka-Aragane N, Nakamura T, et al. Co-existence of positive MET FISH status with EGFR mutations signifies poor prognosis in lung adenocarcinoma patients. Lung Cancer. 2012;75(1):89–94. | ||
Finocchiaro G, Toschi L, Gianoncelli L, et al. Prognostic and predictive value of MET deregulation in non-small cell lung cancer. Ann Transl Med. 2015;3(6):83. | ||
Sun W, Song L, Ai T, et al. Prognostic value of MET, cyclin D1 and MET gene copy number in non-small cell lung cancer. J Biomed Res. 2013;27(3):220–230. | ||
Noro R, Seike M, Zou F, et al. MET FISH-positive status predicts short progression-free survival and overall survival after gefitinib treatment in lung adenocarcinoma with EGFR mutation. BMC Cancer. 2015;15:31. | ||
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | ||
Huang K, Liu D. Targeting non-canonical autophagy overcomes erlotinib resistance in tongue cancer. Tumour Biol. 2016;37(7):9625–9633. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.