")
Back to Journals » International Journal of Nanomedicine » Volume 14
Superiority of TPGS-loaded micelles in the brain delivery of vinpocetine via administration of thermosensitive intranasal gel
Authors Ahmed TA , El-Say KM , Ahmed OAA , Aljaeid BM
Received 22 April 2019
Accepted for publication 27 June 2019
Published 23 July 2019 Volume 2019:14 Pages 5555—5567
DOI https://doi.org/10.2147/IJN.S213086
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Tarek A Ahmed,1,2 Khalid M El-Say,1,2 Osama AA Ahmed,1,3 Bader M Aljaeid1
1Department of Pharmaceutics, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Al-Azhar University, Cairo 11651, Egypt; 3Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Minia University, Minia, Egypt
Background: Vinpocetine (VPN) is a synthetic derivative of the Vinca minor alkaloids. The drug is characterized by a short half-life, limited water solubility and high hepatic first-pass effect. The objective was to develop different lipid-based nanocarriers (NCs) loaded into a thermosensitive in situ gelling (ISG) system to improve VPN bioavailability and brain targeting via intranasal (IN) delivery.
Methods: Different lipid-based NCs were developed and characterized for vesicle size, zeta potential, VPN entrapment efficiency (EE) and morphological characterization using transmission electron microscope (TEM). The prepared NCs were loaded into ISG formulations and characterized for their mucoadhesive properties. Ex-vivo permeation and histological study of the nasal mucosa were conducted. Pharmacokinetic and brain tissue distribution were investigated and compared to a marketed VPN product following administration of a single dose to rats.
Results: VPN-D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) micelles nano-formulation showed the smallest particle size, highest EE among the studied NCs. TEM images revealed an almost spherical shape for all the prepared NCs. Among the NCs studied, VPN-loaded TPGS micelles demonstrated the highest percent cumulative VPN ex vivo permeation. All the prepared ISG formulations revealed the presence of mucoadhesive properties and showed no signs of inflammation or necrosis upon histological examination. Rats administered IN VPN-loaded TPGS-micelles ISG showed superior VPN concentration in the brain tissue and significant high relative bioavailability when compared to that received raw VPN-loaded ISG and marketed drug oral tablets. VPN-D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) micelles nano-formulation showed the smallest particle size, highest EE among the studied NCs. TEM images revealed an almost spherical shape for all the prepared NCs. Among the NCs studied, VPN-loaded TPGS micelles demonstrated the highest percent cumulative VPN ex vivo permeation. All the prepared ISG formulations revealed the presence of mucoadhesive properties and showed no signs of inflammation or necrosis upon histological examination. Rats administered IN VPN-loaded TPGS-micelles ISG showed superior VPN concentration in the brain tissue and significant high relative bioavailability when compared to that received raw VPN-loaded ISG and marketed drug oral tablets.
Conclusion: VPN-loaded TPGS-micelles ISG formulation is a successful brain drug delivery system with enhanced bioavailability for drugs with poor bioavailability and those that are frequently administered.
Keywords: brain distribution, lipid based nanocarriers, in situ gel, intranasal, vinpocetine
Introduction
Intranasal (IN) drug administration is characterized by enhanced bioavailability, especially for drugs that easily cross the mucous membranes, due to the enriched vascular supply in the nasal cavity. This route avoids the drug destruction either by gastrointestinal enzymes or by hepatic first-pass effect. Moreover, the rate of absorption and plasma concentration of IN administered drugs are comparable to intravenous administration and are usually better than subcutaneous or intramuscular routes.1,2 It is a noninvasive, simple, convenient and safe route for all patients.3 In addition, IN administration may achieve an effective therapeutic brain drug concentration, through nose–brain pathway, that allows direct delivery to the cerebrospinal fluid.4,5 Therefore, IN route can diminish drug distribution to nontargeted sites and decreases systemic adverse effects.2,6,7 Despite these advantages, the IN delivery suffers from some restrictions such as poor drug permeability from nasal mucosa, mucociliary clearance, low drug retention time and nasomucosal toxicity.8,9 To overcome these restrictions, various colloidal drug nanocarriers (NCs) especially the lipid-based ones have been utilized to improve the drug permeability and absorption.7,10,11 The rapid nasal mucociliary clearance, that results in low absorption and poor bioavailability, could be overcome by the development of mucoadhesive in situ nasal gelling drug delivery systems that prolong the retention time and control the drug delivery via this route.12 Such formulations are supposed to help the IN drug delivery but, the clinical success of IN therapy is limited owing to the irritation of the nasal mucosa or owing to the frequent and high dose of the formulation. Hence, our research focused on the development of a suitable lipid-based NCs to overcome these barriers.
Biomolecules as lipids, proteins and carbohydrates have been utilized to fabricate different types of nanoparticulate drug delivery systems that are suitable for delivery of many active pharmaceutical ingredients and appropriate for clinical applications.13 Among these systems, lipid-based NCs are considered effective drug delivery systems due to their unique formulation components. The classic example of the lipid-based NCs is liposomes that mainly consist of phospholipid bilayers which are the main components of the biological cell membranes.14 Currently, many liposomal formulations are in clinical use or awaiting clinical trial results.15 In addition to liposomes, transfersomes (TFs) and ethosomes are also phospholipid-based NCs that have been reported to be successful alternatives for liposomes.16–18 Recently, solid lipid nanoparticles (SLNs) and nanostructure lipid carriers (NLCs) have been investigated. They are ease to scale up, but both formulations depend on the presence of other lipid components in addition to the phospholipids. Many reports have described the successful development of drug-loaded SLNs and NLCs and their potential application in drug-delivery process.19–21
Polymeric micelles are nanostructured assemblies that consist of block copolymers of amphiphilic macromolecules. They represent an effective delivery system for poorly soluble drugs.22 The amphiphilic macromolecules are spontaneously assembled into submicroscopic nanostructures that enclose water-insoluble drugs. D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) is formed by conjugation of the hydrophobic vitamin E succinate with the hydrophilic polyethylene glycol.23 TPGS is a pharmaceutical additive, approved by the United State Food and Drug Administration and is used in different drug formulations to solubilize poorly soluble drugs, enhances cellular drug uptake and prolongs the drug blood circulation time.24 TPGS is a surfactant (nonionic type) that can be used in the preparation of other nanostructured formulations that utilize surfactants.25 Moreover, TPGS has been reported to enhance the transport of drug across biological membrane barriers such as brain endothelium and to modify biological response by inhibiting P-glycoprotein (P-gp) that is responsible for decreasing efflux of the drugs from the cells.26
Vinpocetine (VPN) is a synthetic alkaloid obtained from the Periwinkle plant. It is used effectively to improve brain blood flow.27 VPN inhibits phosphodiesterase type-1 enzyme that selectively improves brain blood supply. VPN enhances cerebral circulation and brain oxygen utilization and facilitates blood flow redistribution to ischemic areas. VPN has a limited aqueous solubility (2.4 μg/mL) with pKa =7.31 (weak base), short half-life (1–2 hrs) and poor bioavailability (75% metabolized in liver).28,29 So, it would be a benefit for patients utilizing VPN to develop a formulation that improves the drug bioavailability. To the best of our knowledge, no previous studies have investigated the pharmacokinetics and brain concentration of VPN from Pluronic-based in situ gel (ISG) preparation loaded with drug lipid-based NCs following IN administration.
In this study, different VPN-loaded lipid-based NCs were formulated and loaded into the ISG system suitable for IN delivery to enhance the drug bioavailability and brain tissue distribution.
Materials and methods
Materials
VPN was procured from Wuhan Trustchem Fine Chemical Co., Ltd. (Wuhan, China). Glyceryl monostearate (GMS) was gifted from Nikkol Chemicals Co., Ltd (Tokyo, Japan). Hydrogenated phosphatidylcholine (PC) (95% hydrogenated phosphatidylcholine, 0.5% hydrogenated lyso-phophatidylcholine) was obtained as a kind gift from American Lecithin company (Oxford, CT, USA). Stearic acid was procured from Fischer Scientific (Loughborough, UK). Dicetyl phosphate (DCP), cholesterol, ethanol and methanol were purchased from Fisher Scientific (Pittsburgh, PA, USA). Poloxamer 407 was obtained from Xi’an Lyphar Biotech Co., Ltd (Xi’an, China). Tween 80, Span 80, polyethylene glycol (PEG) 4000, chloroform and D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) were procured from Sigma-Aldrich (St. Louis, MI, USA). Carbopol 940 was obtained from Acros Organics (Morris Plains, NJ, USA).
Preparation of different VPN-loaded lipid-based NCs
Different optimized VPN-loaded lipid-based NCs, namely, SLNs, TFs, PEGylated liposomes (Peg-Lips) and TPGS-micelles were prepared as described in our previously published work.17,18,30–33 Table 1 illustrates the composition of the prepared lipid-based NCs.
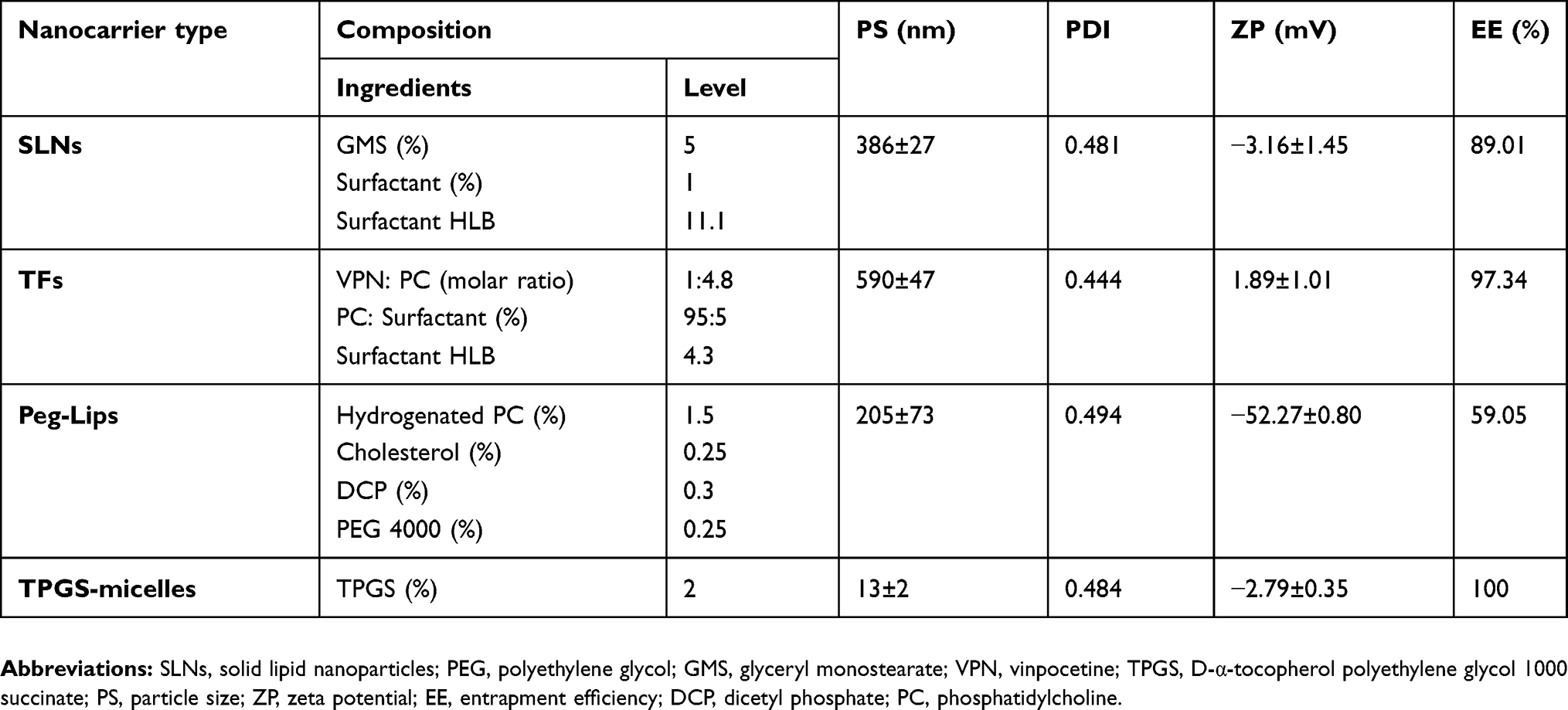 |
Table 1 Composition and characterization of VPN-loaded lipid-based NCs |
Preparation of VPN-loaded SLNs
Melt-emulsion ultrasonication and low-temperature solidification were the techniques utilized to develop VPN-loaded SLNs.34,35 Briefly, the specified amount of GMS was heated at 80°C and 0.25% w/v of VPN (based on the total volume of the formulation) was solubilized in the lipid phase. An aqueous surfactant (Tween 80, span 80 or both) solution (80°C) was prepared, added to the melted lipid phase and the mixture was stirred at 1200 rpm for 15 mins at 80°C until a milky color emulsion was formed. The prepared primary emulsion was then ultrasonicated for 10 mins using a Sonics VCX 750, Sonics & Materials INC. (Newtown, CT, USA) until a uniform nano-dispersion was formed. The colloidal dispersion was promptly cooled by immersing the container into ice-cold water while stirring on a magnetic stirrer for 15 mins until a homogenous dispersion was obtained.31
Preparation of VPN-loaded TFs
Preparation of VPN-loaded TFs was achieved using lipid film hydration technique,36 with some modifications. The specified amount of VPN (250 mg), PC and nonionic surfactant, described in Table 1, were dissolved in methanol using an ultrasonic bath. The obtained dispersion was subjected to rotary evaporation under reduced pressure at 45°C until the complete formation of a thin film on the flask wall. This film was maintained overnight in a vacuum oven to confirm complete removal of organic solvent. Finally, the film was then hydrated with 100 mL PBS (pH =7.5) for about 2 hrs at 19.8°C.18
Preparation of VPN-loaded Peg-Lips
Thin-film hydration technique was utilized to develop VPN-loaded Peg-Lips as previously described30 with slight modifications. The calculated amounts of VPN (250 mg), hydrogenated PC, cholesterol, DCP and PEG 4000 were dissolved in 40 mL of methanol in chloroform mixture (40% v/v). Organic solvents were removed under vacuum using Buchi Rotavapor R-200; BÜCHI Labortechnik AG (Flawil, Switzerland) at 60°C until a thin film was formed. Traces of the organic solvents were removed after the film was kept overnight in a vacuum oven at 30°C. Multilamellar Peg-Lips were assembled by hydrating the dried lipid film at 60°C with 100 mL of isotonic buffer for about 10 mins. Finally, VPN-loaded Peg-Lips vesicles were subjected to vesicle size reduction using a probe sonicator (Qsonica, LLC., Newtown, CT, USA) for 30 s.30
Preparation of VPN-loaded TPGS-micelles
VPN-loaded TPGS-micelles were prepared by dissolving VPN (0.25% w/v) and TPGS (2% w/v) in a 50 mL of ethanol over a magnetic stirrer for about 5 mins. Distilled water (100 mL) was added to the prepared dispersion. Ethanol was completely removed using Buchi Rotavapor R-200; BÜCHI Labortechnik AG (Flawil, Switzerland). The obtained micellar dispersion was kept in a refrigerator at 4°C until further characterization.33
NCs characterization
Particle size and zeta potential determination
Malvern Zetasizer Nano ZSP, Malvern Panalytical Ltd (Malvern, United Kingdom), that utilizes the dynamic light scattering with non-invasive backscatter (NIBS) optics technique, was used to determine the particle size and zeta potential of the prepared lipid-based NCs. Measurement for each sample was done in triplicate.
Entrapment efficiency (EE) determination
For VPN-loaded SLNs, TFs and Peg-Lips, the percent EE was determined using an indirect centrifugation method. To isolate the free unentrapped VPN from the prepared NCs, aliquots from each formulation were centrifuged at 20,000 rpm for 1 hr at 4°C. The supernatant was collected and passed through a 0.2-µm filter. VPN concentration was determined using a reported HPLC method,37 except for slight modifications. Briefly, the mobile phase consisted of methanol and 0.05 ammonium acetate buffer mixture of pH 5.5 (80: 20 v/v). The mobile phase was flowing at a rate of 1 mL/min through a Ponapak C18 analytical column, 4.6×250 I.D. mm, particle size 125A (Waters Associates, Dublin, Ireland). The injection volume was adjusted at 20 µL and the detection wavelength was set at 273 nm. The % VPN entrapped in each NCs formulation was calculated using Equation 1.
(1)
For VPN-loaded TPGS-micelles, the prepared micellar dispersion was centrifuged at 20,000 rpm for 5 mins at 4°C to separate unloaded VPN.38 The supernatant, which contained VPN-loaded TPGS-micelles, was collected, diluted with anhydrous ethanol and the drug concentration was determined using the HPLC method described above. EE% was calculated using Equation 2.
(2)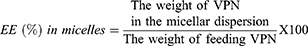
Examination using transmission electron microscope (TEM)
Few drops of each NCs formulation were mounted on a carbon-coated grid and left for approximately 2 mins before examination using TEM model JEM-1230 (JOEL, Tokyo, Japan).
Incorporation of VPN-loaded NCs into thermosensitive ISG
ISG formulations loaded with NCs containing VPN equivalent to 0.25% w/v were developed using a combination of poloxamer 407 (22% w/v) and carbopol 940 (0.5 % w/v) by cold method. Selection of poloxamer and carbopol levels was based on the polymeric concentrations that achieve good ISG formulation characterization, which are in good agreement with our previously published work.31 Briefly, the calculated amount of poloxamer and carbopol was successively added to aqueous cold dispersion of VPN-loaded NCs at 4°C on a magnetic stirrer. The obtained dispersions were kept in the refrigerator (6°C) overnight. ISG formulation loaded with raw VPN (0.25% w/v) was also prepared, using the same method described above.
Characterization of VPN-loaded ISG formulations
Gelation temperature determination
The sol–gel temperature (Tsol-gel) for the prepared ISG formulations was evaluated by the tube inversion method previously published.39,40 Briefly, 1 mL of each formulation was placed in a glass vial and stored overnight in the fridge. Each ISG formulation was heated, with 1°C increments per step, in a thermostatic control heating water bath. The Tsol-gel was recorded in the temperature range 20–30°C. The Tsol-gel was considered as the temperature at which the ISG solution stopped flowing after complete inversion of the tube.
Determination of the pH
The pH of the prepared ISG formulations was determined using a calibrated pH meter using pH/mV/°C meter pH 11 series Oakton®, manufactured by Euteoh Instruments (Thermo Fisher Scientific, Singapore). Determinations were carried out in triplicate.
Evaluation of the mucoadhesive strength
The mucin particle method was utilized to study the mucoadhesiveness of the ISG formulations.31,41 Briefly, bovine mucin suspension was prepared by dispersing a known weight (10 mg) of the mucin powder in 100 mL phosphate buffer solution of pH 7.4. Then, a specified weight (150 mg) of the ISG formulation was added to 10 mL of the prepared mucin suspension with continuous shaking. After incubation for 48 hrs, zeta potential of each mixture was estimated using a Malvern Zetasizer Nano ZSP (Malvern Panalytical Ltd), and the obtained value was compared to that of the raw bovine mucin suspension. The mucoadhesiveness, interaction of the studied formulation components with mucin, was determined by identifying the change in zeta potential value.31,41
Ex vivo permeation studies
The permeation of VPN, from the prepared ISG formulations loaded with either VPN-NCs or raw VPN, through excised bovine nasal mucosa was carried out using Franz diffusion cells, Microette Plus (Hanson Research, CA, USA). Fresh nasal mucosa was carefully excised from the nasal cavity of a bovine snout that was obtained from a local slaughterhouse. The prepared nasal mucosal samples were placed in the diffusion apparatus. The donor compartment was filled with 250 μL of the ISG formulation. Cells of the receptor compartments were filled with 7 mL phosphate buffer of pH 6.8. The temperature was kept at 34ºC and stirring speed was adjusted at 400 rpm. Aliquots were automatically collected from the receptor media for 12 hrs at specified time intervals and replaced with fresh media. The concentration of VPN was determined using HPLC method described above. The experiment was performed in triplicate.
The permeation profiles of VPN were constructed by plotting the cumulative VPN amount permeated (Q) per unit area as a function of time. The steady-state flux (JSS) was calculated for both the initial and the delayed permeation phases from the corresponding slopes. The permeability coefficients (Pc) were calculated by dividing the delayed flux by the initial drug load (Co). The diffusivity (D) was also obtained by plotting the cumulative amount of VPN permeated versus the square root of time and applying Equation 3.
(3)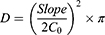
Histological examination of nasal mucosa
To explore any change or modification in the intra-nasal tissues that arises due to IN application of the VPN-loaded TPGS-micelle ISG formulation, microscopic examination of the treated nasal epithelium was accomplished. The studied ISG formulation was applied on freshly separated excised bovine nasal mucosa for 12 hrs in the Franz diffusion cells as previously stated in the ex vivo permeation study. Following treatment, nasal mucosal samples were removed and stored in formalin (10%), dehydrated and finally embedded in paraffin wax. Samples were cut into 4-micron sections, stained with hematoxylin and eosin and Gomori’s Trichrome and examined using Nikon Eclipse 80i digital imaging light microscopy (Kanagawa, Japan). For comparative study, the control sample was also investigated. Each tissue sample was assessed for any sign of irritation, inflammation, and the appearance of epithelial and goblet cells.42
Pharmacokinetic and brain tissue distribution after IN administration
The pharmacokinetics and brain tissue distribution of VPN following IN administration of ISG formulations containing either VPN-loaded TPGS-micelles or raw VPN was evaluated. The study was conducted in comparison with a marketed oral VPN tablet, Vinporal® 5 mg (Amriya Pharmaceutical Industries Company, Alexandria, Egypt), to determine the relative bioavailability. The concentration of VPN in the brain was calculated using Equation 4.
(4)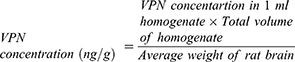
Animal population
Male Sprague-Dawley rats, with an average weight of 260 g, were kept in a pathogen-free space in the Department of Pharmacology and Toxicology, Faculty of Pharmacy, King Abdulaziz University (KAU), Jeddah, KSA. The animal experimental protocol was revised and approved by the Animal Ethics and Animal Care Committee, Faculty of Pharmacy, KAU (Approval No. 1031439). The study fulfilled with the Declaration of Helsinki, the Guiding Principle in Care and Use of Animals (DHEW production NIH 80±23) and the “Standards of Laboratory Animal Care” (NIH distribution #85±23, reconsidered in 1985). Rats were divided into three groups (15 per group). Group I administered IN ISG formulation loaded with VPN-TPGS micelles; group II given IN ISG formulation containing raw VPN; group III administered oral marketed VPN tablet that was crushed and suspended in 0.25% sodium carboxymethyl cellulose solution. Each rat was administered a VPN dose of 10 mg/kg.
Sample collection
Animals were anesthetized and blood samples of 0.5 mL (n=6) were taken at different time points of 0.5, 1, 1.5, 2, 4, 6, 8, 12 and 24 hrs. For brain tissue samples, animals were euthanized by cervical dislocation, brain tissues (n=3) were harvested after 6, 12 and 24 hrs and stored at −80ºC. Brain tissues were homogenized in PBS. Plasma and brain tissue homogenate samples were treated and analyzed as described in the following section.
Chromatographic quantification of VPN
The concentrations of VPN in the plasma and brain tissue samples were analyzed by liquid chromatography-tandem mass spectrometry method. HPLC Agilent 1200 system equipped with Agilent 6420, triple quad mass spectrometer and controlled by Mass Hunter software was used. The separation was performed on a Nacherey Nagel, Nucleodur C18 column, 5 µm, 4.6×250 mm (Duren, Germany). The mobile phase comprised of 0.1% formic acid and acetonitrile (29:71, v/v) and the flow rate was adjusted at 0.5 mL/min. VPN and the internal standard (IS) Valsartan were detected in a single-ion monitoring (SIM) scan mode with positive ion detection. The ions used for the SIM detection were m/z 351.1 for VPN and m/z 436.1 for Valsartan.
Linearity and recoveries
Linearity of the assay method within a VPN concentration range of 200–800 pg/µL was verified with a regression coefficient (R2=0.9991). All the obtained results were within the acceptable criteria as previously stated in the recommended guidelines. The mean recovery of VPN was 102.2% at 200 pg/µL (LLOQ) and 97.5% at 800 pg/µL (ULOQ).
Sample extraction procedure
To a 200 μL of the plasma samples or brain tissue homogenate, 50 μL of Valsartan (IS) and 1 mL of acetonitrile were added. The resulting solution was thoroughly vortex-mixed for 10 s. After centrifugation at 5000 rpm for 5 mins, 5 μL of the supernatant was injected into the HPLC system for analysis. The concentration of VPN in the unknown samples was calculated from the regression equation obtained from the constructed calibration curve.
Pharmacokinetic treatment
The pharmacokinetic parameters of VPN in the collected plasma data were assessed using non-compartmental pharmacokinetic treatment utilizing KineticaTM software (version 4, Thermo Scientific, MA, USA). The maximum plasma VPN concentration (Cmax), time to reach maximum VPN plasma concentration (tmax), area under the plasma VPN concentration time curve from zero to the last measurable VPN concentration (AUC0–t), area under the plasma VPN concentration time curve from zero to infinity (AUC0–α), mean residence time (MRT), the elimination rate constant, elimination half-life and total body clearance were measured. The data were expressed as the mean ± SD.
Statistical analysis of the data
The data obtained was analyzed using GraphPad Prism 6 (GraphPad Software, San Diego, CA) software. Two-way ANOVA followed by Tukey’s multiple comparisons test was used to assess the significance of the difference between the investigated groups.
Results and discussion
Evaluation of the prepared VPN-loaded lipid-based NCs
Table 1 shows the results of the particle size for the prepared NCs formulations. VPN-loaded TFs revealed the largest size (590±47 nm), while VPN-loaded TPGS-micelles displayed the smallest size (13±2 nm). Furthermore, the polydispersity index of the prepared VPN-loaded lipid-based NCs was between 0.444 and 0.494, which indicates acceptable size distribution. TEM images (Figure 1) showed a spherical particle morphology for all the prepared colloidal dispersion. The obtained images confirmed the uniformity in size distribution that were comparable with the data obtained by the Zetasizer Nano ZSP. Due to its content of the charge-inducing agent (DCP), the colloidal dispersion of VPN-loaded Peg-Lips showed the highest stability with a zeta potential value of −52.27±0.80 mV. This Peg-Lips displayed the lowest percentage of VPN entrapped (59.05%), whereas the remaining NCs formulations exhibited high VPN EE that ranged from 89.01% to 100%. TPGS-micelles demonstrated the highest EE of about 100% which could be attributed to the nature of the drug loaded. Yang et al reported high drug encapsulation efficiency for TPGS-based fabricated NCs.43 Zhu et al also stated 85–95% EE for docetaxel vitamin E TPGS NPs.44 Similarly, Muthu et al mentioned an encapsulation efficiency up to 84.30±0.80% for docetaxel-loaded vitamin E TPGS micelles.45 The prepared TFs and SLNs formulations showed high drug entrapment of 97.01% and 89.34%, respectively. The Peg-Lips displayed the lowest drug EE of 59.05% which is in a good accordance with the previously reported EE results of the same formulation.30
 |
Figure 1 Transmission electron microscope images of the prepared lipid-based nanocarriers. |
Evaluation of the prepared VPN-loaded ISG formulations
Poloxamer 407 (22% w/v) and carbopol 940 (0.5% w/v) combination was found to be the optimum concentration for the polymeric solution to form ISG formulations loaded with different NCs. Addition of hydrophilic polymer such as carbopol 940 to poloxamer 407 enforces the mechanical strength and overcomes the possibility of the gel erosion.46
The sol–gel transition temperature of the colloidal dispersions ranged from 25.33±1.53°C for TFs-ISG to 30.00±1.00°C for TPGS-micelles-ISG. It has been previously reported that an ISG system with a sol–gel transition temperature higher than but close to 25°C is anticipated to be highly viscous at room temperature, the favorable sol–gel transition temperature should be close to but not exceed 30°C.47 In addition, the reported temperature in the human nose ranged from 30.2±1.7°C to 34.4±1.1°C,48–50 and the sol–gel transition temperature of the prepared formulations ensures the suitability of their application on the nasal mucosa. Our results revealed that the pH of the formulations ranged from 5.54±0.02 to 7.06±0.02. This pH range indicated that no irritancy is expected from the formulations following application on the nasal mucosa. All the prepared ISG formulations revealed the presence of mucoadhesive properties as indicated by the change in zeta potential value for all the studied ISG formulation when compared with raw bovine mucin suspension (data not shown). The prepared mucin suspension showed a negative zeta potential value of −9.88 mv. This value is attributed to ionization of the mucin carboxyl groups as previously stated.31 A marked decrease in the mucin zeta potential value was noticed upon mixing with the prepared ISG formulations. This finding is an indication of the formulation mucoadhesive properties brought about by the interaction between mucin and the polymeric ISG formulation.
Ex vivo permeation studies
Table 2 shows the permeation parameters of VPN from different NCs-loaded ISG formulations compared with raw VPN-loaded ISG across the bovine nasal mucosa. The permeation studies showed that higher permeability coefficient, diffusion coefficient and maximum amount of drug permeated (Dmax) were noticed from VPN-loaded Peg-Lips-ISG followed by VPN-loaded TPGS-micelles-ISG which is an indication of the ability of these NCs to penetrate the nasal mucosa to a greater extent when compared with the other NCs-loaded ISG formulations and the raw VPN-loaded ISG. Also, the Peg-Lips-ISG and TPGS-micelles-ISG formulation recorded the highest steady-state permeation fluxes (Jss) of 0.7913 and 0.7258 µg/cm2.h, respectively. While their permeability coefficients (Kc) reaching 0.0012 and 0.0011 cm/h, respectively, indicating a general better permeation results than other NC formulations (Figure 2). The highest amount of drug diffused (639.029 µg) with the lowest percentage of VPN permeated over 12 hrs (58.38%) reflect the superiority of TPGS-micelles-ISG in the diffusivity and in sustaining the permeation over the other NCs formulations. The lipophilic nature and the small particle size of micelles could explain this nasal permeation enhancement. Another reason for this finding encompasses the fact that the formation of occlusive monolayer film on a large surface area of the mucous membrane diminishes the loss of moisture due to evaporation, which can enhance the drug permeation.51 Additionally, the surfactant activity of TPGS can fluidize or loosen the intercellular lipid layer of the nasal mucosa and so enhances the drug permeation.52,53 Also, the enhanced permeability of TPGS-micelles might be attributed to the P-gp inhibitory effects which has been approved by the FDA.43,54,55
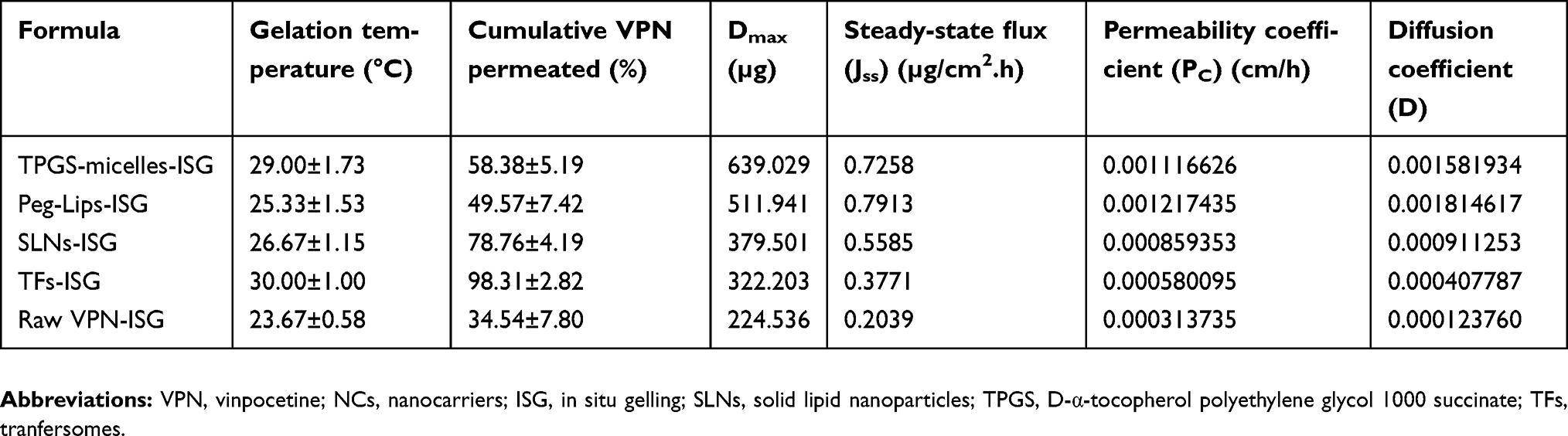 |
Table 2 Gelation temperature and ex vivo permeation parameters of VPN from different NCs-loaded ISG compared with raw VPN-loaded ISG across bovine nasal mucosa |
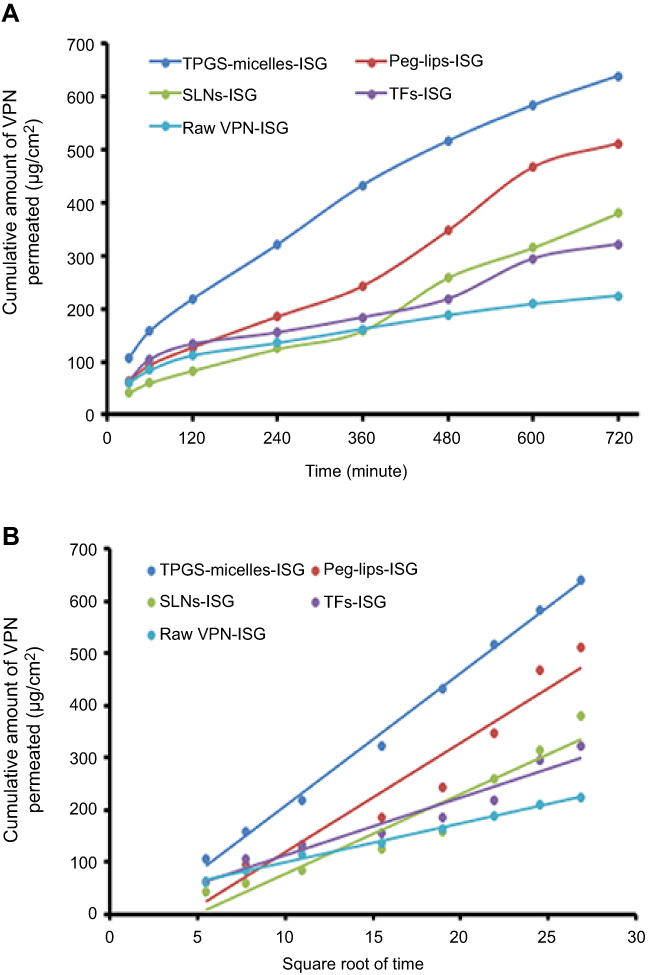 |
Figure 2 Cumulative amount of VPN permeated per unit area against time (A) and against square root of time (B) from different NCs-loaded ISG formulations compared with the raw VPN-ISG. Abbreviations: VPN, vinpocetine; NCs, nanocarriers; ISG, in situ gelling; SLNs, solid lipid nanoparticles; TPGS, D-α-tocopherol polyethylene glycol 1000 succinate; TFs, tranfersomes. |
Assessment of nasal mucosa irritancy upon application of TPGS-micelles-ISG formulation
Figure 3 demonstrates the histological photomicrographs of the nasal mucosa after 12 hrs of treatment with VPN-loaded TPGS-micelle ISG (Figure 3A) and the normal (untreated) nasal mucosa (Figure 3B). No signs of irritancy or inflammation have been observed on the treated nasal tissue. Also, the normal appearance of both ciliated respiratory epithelium and goblet cells have been noticed. Therefore, the poloxamer-based ISG formulation is considered safe to use with respect to nasal administration which is in good accordance with a previous study.31
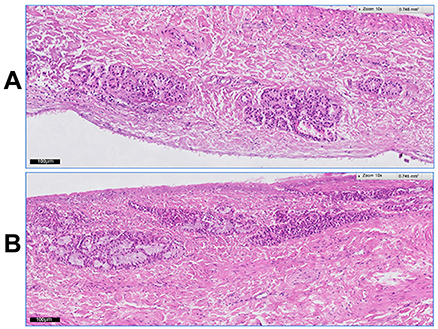 |
Figure 3 Histological images of the bovine nasal mucosa: (A) after 12-hr treatment with VPN-loaded TPGS-micelle ISG; (B) the normal (untreated) nasal mucosa.Abbreviations: VPN, vinpocetine; ISG, in situ gelling; TPGS, D-α-tocopherol polyethylene glycol 1000 succinate. |
Brain tissue distribution and pharmacokinetics
To evaluate the in vivo effectiveness of the developed VPN-loaded TPGS-micelles ISG formulation after IN administration to male Sprague Dawley rats, the brain distribution and the pharmacokinetics were studied and compared to raw VPN-loaded ISG formulation and a marketed oral VPN tablet. The concentration of VPN in the rat’s brain tissue at different time points is shown in Figure 4. The nasal administration of VPN-loaded TPGS-micelles ISG formulation exhibited approximately 10-fold higher in brain tissue than the corresponding raw VPN-loaded ISG formulation and the marketed oral VPN tablets after 6 hrs. The plasma concentration versus time profiles after IN administration of VPN-loaded TPGS-micelles ISG and the raw VPN-loaded ISG as well as the oral administration of VPN tablet is shown in Figure 5. The pharmacokinetic parameters for each group are summarized in Table 3. In the brain tissue, rats administered VPN-loaded TPGS-micelles-ISG showed a much higher VPN concentration profile in comparison to that received oral tablet which displayed irrelevant difference with rats administered raw VPN-loaded ISG (Figure 4). It was reported that the human nasal respiratory and olfactory mucosa contain an efflux transporter known as P-gp which plays an important role in preventing actively the drugs influx from the nasal membrane.56,57 Therefore, the incorporation of P-gp inhibitor as TPGS in the micelles could influence the penetration of the blood–brain barrier and enhance the brain uptake with higher concentrations after IN administration.58 This finding indicates that development of VPN in the form of TPGS-micelles ISG played a major role in the enhancement of the drug nasal absorption and results in enhanced drug brain circulation using the TPGS micelles.
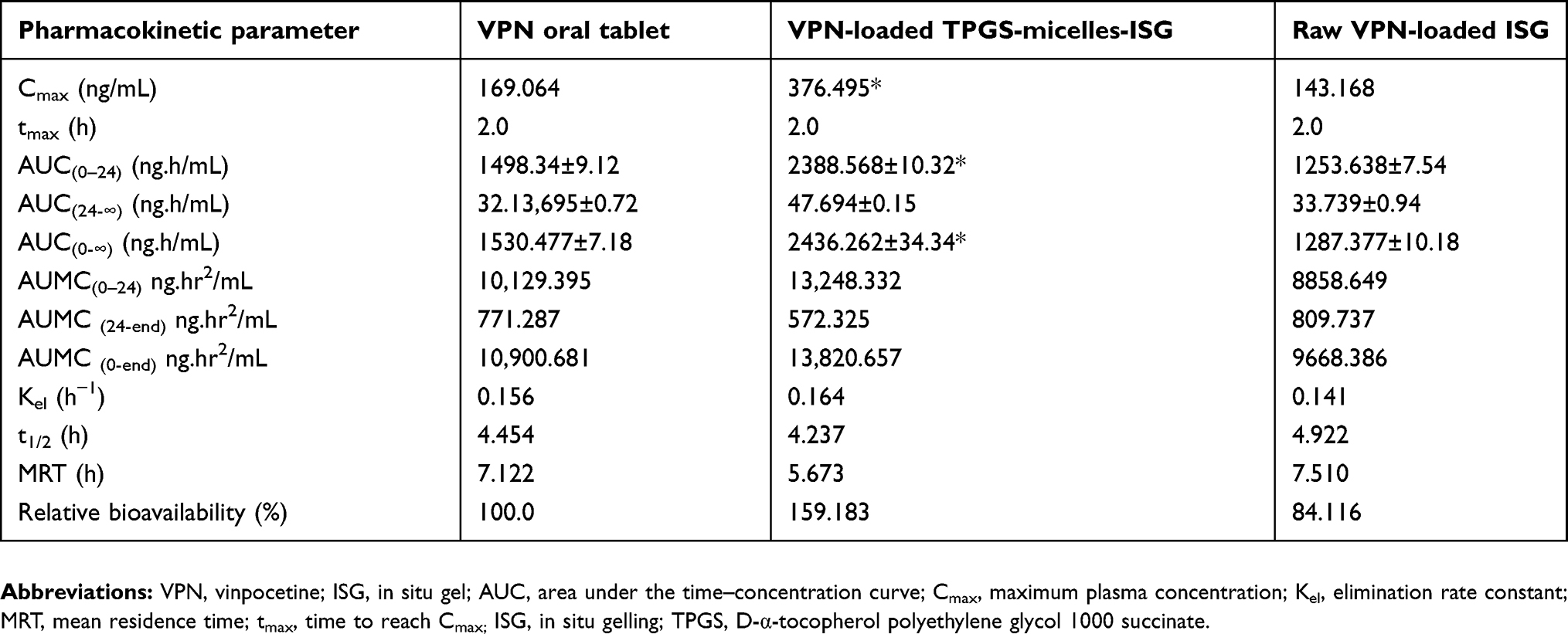 |
Table 3 Pharmacokinetic parameters ± SD of VPN following the intranasal administration of a single dose (10 mg/kg) of VPN-loaded TPGS-micelles-ISG in comparison with the intranasal administration of raw VPN-loaded ISG and the oral administration of VPN tablet by rats |
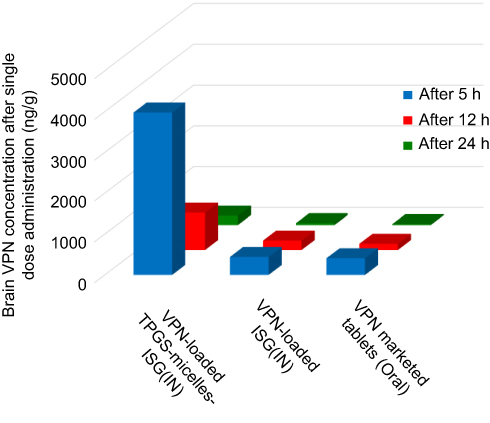 |
Figure 4 Brain concentration of VPN in rats after intranasal administration of VPN-loaded TPGS-micelle ISG in comparison with raw VPN-loaded ISG and oral administration of the marketed VPN tablet (10 mg/kg) at different time points.Abbreviations: VPN, vinpocetine; ISG, in situ gelling; TPGS, D-α-tocopherol polyethylene glycol 1000 succinate. |
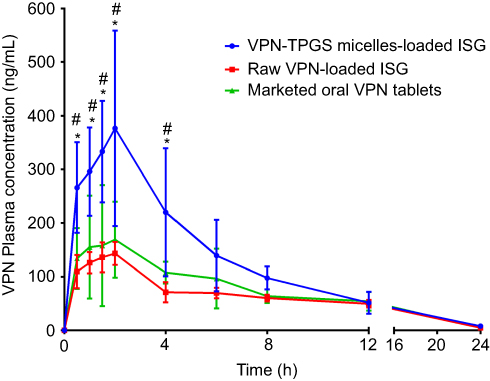 |
Figure 5 Plasma concentration versus time profiles of VPN in rats after intranasal administration of VPN-loaded TPGS-micelle ISG in comparison with raw VPN-loaded ISG and oral administration of the marketed VPN tablet (10 mg/kg). Each point represents mean ± S.D. (n=6). * and #: p<0.05 vs raw VPN-loaded ISG and marketed VPN tablet, respectively.Abbreviations: VPN, vinpocetine; ISG, in situ gelling; TPGS, D-α-tocopherol polyethylene glycol 1000 succinate. |
Finally, the developed thermosensitive ISG loaded with TPGS micelles exhibited enhancement in the pharmacokinetic parameters with significant high relative bioavailability. VPN-loaded TPGS-micelles ISG showed a significant increase in VPN Cmax by 2.2- and 2.6-fold when compared with the oral VPN tablet and the raw VPN-loaded ISG, respectively. All formulations reached the maximum plasma concentration after 2 hrs with no significant difference in the elimination half-life (P-value >0.05). The relative bioavailability of VPN from TPGS-micelles ISG was 159.183% compared with the oral marketed tablet. Whereas, the bioavailability of VPN was reduced to 84.116% after IN administration of the raw VPN-loaded ISG when compared with the marketed tablet. This finding confirms the superiority of TPGS-micelles in the brain delivery of VPN via administration of thermosensitive IN ISG although a long-term study is required.
Conclusion
Successful development of VPN-loaded TPGS-micelles ISG formulation could be considered a key solution for VPN short half-life and poor bioavailability. Moreover, the prepared formulation enhanced the drug brain delivery and was superior to the marketed drug oral product. So, this formulation could represent a good alternative for the currently available marketed drug products.
Acknowledgments
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. G 57-166-38. The authors, therefore, acknowledge with thanks DSR technical and financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Henry R, Ruano N, Casto D, Wolf R. A pharmacokinetic study of midazolam in dogs: nasal drop vs. atomizer administration. Pediatr Dent. 1998;20(5):321–326.
2. Pires PC, Santos AO. Nanosystems in nose-to-brain drug delivery: a review of non-clinical brain targeting studies. J Control Release. 2018;270:89–100. doi:10.1016/J.JCONREL.2017.11.047
3. Dhuria SV, Hanson LR, Frey WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–1673. doi:10.1002/jps.21924
4. Agrawal M, Saraf S, Saraf S, et al. Nose-to-brain drug delivery: an update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J Control Release. 2018;281:139–177. doi:10.1016/j.jconrel.2018.05.011
5. Liu Z, Jiang M, Kang T, et al. Lactoferrin-modified PEG-co-PCL nanoparticles for enhanced brain delivery of NAP peptide following intranasal administration. Biomaterials. 2013;34(15):3870–3881. doi:10.1016/j.biomaterials.2013.02.003
6. El-Zaafarany GM, Soliman ME, Mansour S, Awad GAS. Identifying lipidic emulsomes for improved oxcarbazepine brain targeting: in vitro and rat in vivo studies. Int J Pharm. 2016;503(1–2):127–140. doi:10.1016/j.ijpharm.2016.02.038
7. Salama HA, Mahmoud AA, Kamel AO, Abdel HM, Awad GAS. Phospholipid based colloidal poloxamer-nanocubic vesicles for brain targeting via the nasal route. Colloids Surf B. 2012;100:146–154. doi:10.1016/j.colsurfb.2012.05.010
8. Illum L. Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci. 2000;11(1):1–18. doi:10.1016/S0928-0987(00)00087-7
9. Minn A, Leclerc S, Heydel JM, et al. Drug transport into the mammalian brain: the nasal pathway and its specific metabolic barrier. J Drug Target. 2002;10(4):285–296. doi:10.1080/713714452
10. Lockman PR, Mumper RJ, Khan MA, Allen DD. Nanoparticle technology for drug delivery across the blood-brain barrier. Drug Dev Ind Pharm. 2002;28(1):1–13. doi:10.1081/DDC-120001481
11. Feng Y, He H, Li F, Lu Y, Qi J, Wu W. An update on the role of nanovehicles in nose-to-brain drug delivery. Drug Discov Today. 2018;23(5):1079–1088. doi:10.1016/J.DRUDIS.2018.01.005
12. Ugwoke MI, Verbeke N, Kinget R. The biopharmaceutical aspects of nasal mucoadhesive drug delivery. J Pharm Pharmacol. 2001;53(1):3–22. doi:10.1211/0022357011775145
13. Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. doi:10.1038/nrd2614
14. Fenske DB, Cullis PR. Liposomal nanomedicines. Expert Opin Drug Deliv. 2008;5(1):25–44. doi:10.1517/17425247.5.1.25
15. Puri A, Loomis K, Smith B, et al. Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit Rev Ther Drug Carrier Syst. 2009;26(6):523–580. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20402623. Accessed October 10, 2018. doi:10.1615/CritRevTherDrugCarrierSyst.v26.i6.10
16. Ahmed TA, El-Say KM, Aljaeid BM, Fahmy UA, Abd-Allah FI. Transdermal glimepiride delivery system based on optimized ethosomal nano-vesicles: preparation, characterization, in vitro, ex vivo and clinical evaluation. Int J Pharm. 2016;500(1–2):245–254. doi:10.1016/j.ijpharm.2016.01.017
17. Badr-Eldin SM, Ahmed OAA. Optimized nano-transfersomal films for enhanced sildenafil citrate transdermal delivery: ex vivo and in vivo evaluation. Drug Des Devel Ther. 2016;10:1323–1333. doi:10.2147/DDDT.S103122
18. Ahmed TA. Preparation of transfersomes encapsulating sildenafil aimed for transdermal drug delivery: plackett–burman design and characterization. J Liposome Res. 2015;25(1):1–10. doi:10.3109/08982104.2014.950276
19. Zhang X-G, Miao J, Dai Y-Q, Du Y-Z, Yuan H, Hu F-Q. Reversal activity of nanostructured lipid carriers loading cytotoxic drug in multi-drug resistant cancer cells. Int J Pharm. 2008;361(1–2):239–244. doi:10.1016/j.ijpharm.2008.06.002
20. Martins S, Sarmento B, Ferreira DC, Souto EB. Lipid-based colloidal carriers for peptide and protein delivery–liposomes versus lipid nanoparticles. Int J Nanomedicine. 2007;2(4):595–607. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18203427. Accessed October 10, 2018.
21. El-Say KM, Hosny KM. Optimization of carvedilol solid lipid nanoparticles: an approach to control the release and enhance the oral bioavailability on rabbits. Ahmad A, ed. PLoS One. 2018;13(8):e0203405. doi:10.1371/JOURNAL.PONE.0203405
22. Zhang Y, Huang Y, Li S. Polymeric micelles: nanocarriers for cancer-targeted drug delivery. AAPS PharmSciTech. 2014;15(4):862–871. doi:10.1208/s12249-014-0113-z
23. Varma MVS, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci. 2005;25(4–5):445–453. doi:10.1016/j.ejps.2005.04.003
24. Zhang Z, Tan S, Feng -S-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. doi:10.1016/j.biomaterials.2012.03.046
25. Tan S, Zou C, Zhang W, Yin M, Gao X, Tang Q. Recent developments in d -α-tocopheryl polyethylene glycol-succinate-based nanomedicine for cancer therapy. Drug Deliv. 2017;24(1):1831–1842. doi:10.1080/10717544.2017.1406561
26. Dintaman JM, Silverman JA, Inhibition of P-glycoprotein by D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS). Pharm Res. 1999;16(10):1550–1556. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10554096. Accessed October 12, 2018. doi:10.1023/A:1015000503629
27. Ogunrin A. Effect of vinpocetine (cognitolTM) on cognitive performances of a nigerian population. Ann Med Health Sci Res. 2014;4(4):654–661. doi:10.4103/2141-9248.139368
28. Luo Y, Chen D, Ren L, Zhao X, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Release. 2006;114:53–59. doi:10.1016/j.jconrel.2006.05.010
29. Liu M, Zhang S, Cui S, et al. Preparation and evaluation of Vinpocetine self-emulsifying pH gradient release pellets. Drug Deliv. 2017;24(1):1598–1604. doi:10.1080/10717544.2017.1388453
30. Harbi I, Aljaeid B, El-Say KM, Zidan AS. Glycosylated sertraline-loaded liposomes for brain targeting: qbD study of formulation variabilities and brain transport. AAPS PharmSciTech. 2016;17(6):1404–1420. doi:10.1208/s12249-016-0481-7
31. Ahmed TA, Badr-Eldin SM, Ahmed OAA, Aldawsari H. Intranasal optimized solid lipid nanoparticles loaded in situ gel for enhancing trans-mucosal delivery of simvastatin. J Drug Deliv Sci Technol. 2018;48(July):499–508. doi:10.1016/J.JDDST.2018.10.027
32. Ahmed OAA, Badr-Eldin SM. In situ misemgel as a multifunctional dual-absorption platform for nasal delivery of raloxifene hydrochloride: formulation, characterization, and in vivo performance. Int J Nanomedicine. 2018;13:6325–6335. doi:10.2147/IJN.S181587
33. Ahmed OAA, El-Say KM, Aljaeid BM, Badr-Eldin SM, Ahmed TA. Optimized vinpocetine-loaded vitamin E D- α - tocopherol polyethylene glycol 1000 succinate- alpha lipoic acid micelles as a potential transdermal drug delivery system : in vitro and ex vivo studies. Int J Nanomedicine. 2019;14:33–43. doi:10.2147/IJN.S187470
34. Hao J, Wang X, Bi Y, et al. Fabrication of a composite system combining solid lipid nanoparticles and thermosensitive hydrogel for challenging ophthalmic drug delivery. Colloids Surf B. 2014;114:111–120. doi:10.1016/j.colsurfb.2013.09.059
35. Silva AC, González-Mira E, García ML, et al. Preparation, characterization and biocompatibility studies on risperidone-loaded solid lipid nanoparticles (SLN): high pressure homogenization versus ultrasound. Colloids Surf B. 2011;86(1):158–165. doi:10.1016/j.colsurfb.2011.03.035
36. Cevc G, Blume G, Schätzlein A. Transfersomes-mediated transepidermal delivery improves the regio-specificity and biological activity of corticosteroids in vivo1Dedicated to the late Dr. Henri Ernest Bodde.1. J Control Release. 1997;45(3):211–226. doi:10.1016/S0168-3659(96)01566-0
37. Ding J, Li J, Mao S. Development and evaluation of vinpocetine inclusion complex for brain targeting. Asian J Pharm Sci. 2015;10:114–120. doi:10.1016/j.ajps.2014.08.008
38. Duan Y, Wang J, Yang X, Du H, Xi Y, Zhai G. Curcumin-loaded mixed micelles: preparation, optimization, physicochemical properties and cytotoxicity in vitro. Drug Deliv. 2015;22(1):50–57. doi:10.3109/10717544.2013.873501
39. Choi SG, Lee S-E, Kang B-S, Ng CL, Davaa E, Park J-S. Thermosensitive and mucoadhesive sol-gel composites of paclitaxel/dimethyl-β-cyclodextrin for buccal delivery. Xu B, ed. PLoS One. 2014;9(10):e109090. doi:10.1371/journal.pone.0109090
40. Gilbert JC, Richardson JL, Davies MC, Palin KJ, Hadgraft J. The effect of solutes and polymers on the gelation properties of pluronic F-127 solutions for controlled drug delivery. J Control Release. 1987;5(2):113–118. doi:10.1016/0168-3659(87)90002-2
41. García MC, Aldana AA, Tártara LI, et al. Bioadhesive and biocompatible films as wound dressing materials based on a novel dendronized chitosan loaded with ciprofloxacin. Carbohydr Polym. 2017;175:75–86. doi:10.1016/j.carbpol.2017.07.053
42. Karasulu E, Yavaşoǧlu A, Evrenşanal Z, Uyanikgil Y, Karasulu HY. Permeation studies and histological examination of sheep nasal mucosa following administration of different nasal formulations with or without absorption enhancers. Drug Deliv. 2008;15(4):219–225. doi:10.1080/10717540802006377
43. Yang C, Wu T, Qi Y, Zhang Z. Recent advances in the application of vitamin E TPGS for drug delivery. Theranostics. 2018;8(2):464–485. doi:10.7150/thno.22711
44. Zhu H, Chen H, Zeng X, et al. Co-delivery of chemotherapeutic drugs with vitamin E TPGS by porous PLGA nanoparticles for enhanced chemotherapy against multi-drug resistance. Biomaterials. 2014;35(7):2391–2400. doi:10.1016/j.biomaterials.2013.11.086
45. Muthu MS, Avinash Kulkarni S, Liu Y, Feng -S-S. Development of docetaxel-loaded vitamin E TPGS micelles: formulation optimization, effects on brain cancer cells and biodistribution in rats. Nanomedicine. 2012;7(3):353–364. doi:10.2217/nnm.11.111
46. Chaudhary B, Verma S. Preparation and evaluation of novel in situ gels containing acyclovir for the treatment of oral herpes simplex virus infections. Sci World J. 2014;2014:1–7. doi:10.1155/2014/280928
47. Qian S, Wong YC, Zuo Z. Development, characterization and application of in situ gel systems for intranasal delivery of tacrine. Int J Pharm. 2014;468(1–2):272–282. doi:10.1016/j.ijpharm.2014.04.015
48. Lindemann J, Leiacker R, Rettinger G, Keck T. Nasal mucosal temperature during respiration. Clin Otolaryngol Allied Sci. 2002;27(3):135–139. doi:10.1046/j.1365-2273.2002.00544.x
49. Proctor DF, Andersen I, Lundqvist GR. Human nasal mucosal function at controlled temperatures. Respir Physiol. 1977;30(1–2):109–124. doi:10.1016/0034-5687(77)90025-1
50. Jacky JP. Barometric measurement of tidal volume: effects of pattern and nasal temperature. J Appl Physiol. 1980;49(2):319–325. doi:10.1152/jappl.1980.49.2.319
51. Wissing SA, Müller RH. The influence of solid lipid nanoparticles on skin hydration and viscoelasticity - In vivo study. Eur J Pharm Biopharm. 2003;56(1):67–72. doi:10.1016/S0939-6411(03)00040-7
52. Fang JY, Fang CL, Liu CH, Su YH. Lipid nanoparticles as vehicles for topical psoralen delivery: solid lipid nanoparticles (SLN) versus nanostructured lipid carriers (NLC). Eur J Pharm Biopharm. 2008;70(2):633–640. doi:10.1016/j.ejpb.2008.05.008
53. Aboud HM, El Komy MH, Ali AA, El Menshawe SF, Abd Elbary A. Development, optimization, and evaluation of carvedilol-loaded solid lipid nanoparticles for intranasal drug delivery. AAPS PharmSciTech. 2016;17(6):1–13. doi:10.1208/s12249-015-0440-8
54. Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin e TPGS in drug delivery. Eur J Pharm Sci. 2013;49(2):175–186. doi:10.1016/j.ejps.2013.02.006
55. Beig A, Fine-Shamir N, Porat D, Lindley D, Miller JM, Dahan A. Concomitant solubility-permeability increase: vitamin E TPGS vs. amorphous solid dispersion as oral delivery systems for etoposide. Eur J Pharm Biopharm. 2017;121:97–103. doi:10.1016/j.ejpb.2017.09.012
56. Illum L. Nasal drug delivery - Possibilities, problems and solutions. J Controlled Release. 2003;87:187–198. doi:10.1016/S0168-3659(02)00363-2
57. Graff CL, Pollack GM. Functional evidence for P-glycoprotein at the nose-brain barrier. Pharm Res. 2005;22(1):86–93. doi:10.1007/s11095-004-9013-3
58. Graff CL, Pollack GM. P-glycoprotein attenuates brain uptake of substrates after nasal instillation. Pharm Res. 2003;20(8):1225–1230. doi:10.1023/A:1025053115583
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.