Back to Journals » OncoTargets and Therapy » Volume 9
Sunitinib: the antiangiogenic effects and beyond
Received 7 May 2016
Accepted for publication 2 August 2016
Published 8 September 2016 Volume 2016:9 Pages 5495—5505
DOI https://doi.org/10.2147/OTT.S112242
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Zhonglin Hao, Ibrahim Sadek
Department of Medicine, Section of Hematology and Oncology, Georgia Cancer Center, Medical College of Georgia, Augusta University, Augusta, GA, USA
Abstract: As a multitargeted kinase inhibitor, sunitinib has carved its way into demonstrating itself as a most effective tyrosine kinase inhibitor in the treatment of metastatic renal cell carcinoma. Mechanistically, sunitinib inhibits multiple receptor tyrosine kinases, especially those involved in angiogenesis, that is, vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and proto-oncogene cKIT. Sunitinib has also been implicated in enhancing cancer invasiveness and metastasis. Mechanisms of resistance are poorly understood, but both intrinsic and acquired mechanisms are thought to be involved. While the side effects are manageable, sunitinib, like many other tyrosine kinase inhibitors, can be associated with serious toxicities that require careful management including frequent dose reductions. Although still in the early stage, emerging evidence points to an immunomodulatory role for sunitinib. It is also likely to contribute to the overall outcomes, especially those seen in metastatic renal cell carcinoma, and such effects are thought to be mediated by the proto-oncogene cKIT receptor. Combination with other modalities such as stereotactic body radiation therapy, therapeutic vaccines, and checkpoint inhibitors is being pursued for improved efficacy.
Keywords: sunitinib, angiogenesis, MDSC, cancer
Introduction
Sunitinib is an oral, small-molecule, multitargeted receptor tyrosine kinase (RTK) inhibitor. Several clinical trials have demonstrated that sunitinib has remarkable objective response rates (ORRs) and clinical benefit in the treatment of metastatic renal cell carcinoma (mRCC) in the first-line setting and for treating unresectable/metastatic gastrointestinal stromal tumor (GIST) in the second-line setting after imatinib.1 Sunitinib has also demonstrated efficacy in extending progression-free survival among patients with unresectable, locally advanced, or metastatic well-differentiated pancreatic neuroendocrine tumors (pNETs).2 Furthermore, sunitinib has shown potential antitumor activity in various other malignancies, such as the thyroid, lung, pancreatic, esophageal and bladder carcinomas, gliomas, and sarcomas. Here we review what has been achieved to date from discovering the drug and defining its pharmacology, mechanisms of action, and resistance, to conducting the pivotal trials that led to its US Food and Drug Administration (FDA) approval. We also summarize the latest preclinical and clinical efforts attempting to boost sunitinib’s efficacy by combining it with chemotherapy, radiation, and immunotherapeutic strategies.
Drug discovery
Sunitinib was discovered from a screen of indolin-2-one analogs, based on its ability to potently and selectively inhibit four RTKs including vascular endothelial growth factor receptor (VEGFR)-2, platelet-derived growth factor receptor (PDGFR)-β, fibroblast growth factor receptor 1, and epidermal growth factor receptor.3 Earlier prototype compounds, SU-5416 and SU-6668, had low water solubility and high protein-binding properties, which made them less desirable in clinical trials. Hence, attempts were made to modify the compounds.4,5 Modifications made on the C4′ position of SU-5416 introduced side chains to this position. This created the most potent inhibitor against VEGFR and PDGFR with high water solubility. The final compound, SU-11248, was given the name sunitinib. This compound is supplied as the malate salt (sunitinib malate). Chemically, it is a butanedioic acid, hydroxy-, (2S)-, compound with N-[2-(diethylamino) ethyl]-5-[(Z)-(5-fluoro-2-oxo-1H-indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide. The molecular formula is C22H27FN4O2·C4H6O5 and the molecular weight is 532.6 Da. Chemical structure of sunitinib is shown in Figure 1.
 | Figure 1 Chemical structure of sunitinib malate. |
Sunitinib capsules on the market are supplied as printed hard shell capsules containing sunitinib malate equivalent to 12.5, 25, 37.5, or 50 mg of sunitinib together with mannitol, croscarmellose sodium, povidone (K-25), and magnesium stearate provided as inactive ingredients.
Pharmacokinetics and drug metabolism
Sunitinib bioavailability is not affected by the diet. Absorption is the same whether administered in the fasting state or after a meal, regardless of its fat or calorie content (from the package insert).6 Following its oral adminstration, sunitinib’s maximal concentration (Cmax) is usually achieved 6–12 hours later. Ninty-five percent of sunitinib and 90% of its primary metabolite, N-desethyl, bind to plasma proteins in the blood stream. The volume distribution is 2,230 L. The Cmax and the area under the curve are proportional to dosing when the drug is administered at doses between 25 and 100 mg daily. The drug and its metablite are metabolized by cytochrome P450 3A4 (CYP3A4). Therefore, CYP3A4 inducers decrease and inhibitors increase its blood concentration. Coadministration with a drug affecting CYP3A4 should be avoided or appropriate dose adjustments made. In healthy voluteers,7 the half-life for sunitinib is 40–60 hours and for its primary metabolite is 80–110 hours. On repeated administration, the drug concentration increases 3–4 fold and the concentration of its primary metabolite increases 7–10 fold. Sixty-one percent of the drug is eliminated through feces and 16% excreted in the urine. No adjustment is needed for renal impairment or for mild hepatic impairment (Child–Pugh class A).
Mechanism of action
Sunitinib inhibits multiple RTKs in biochemical and cell-based assays and exerts potent antiangiogenesis and antitumor effects in animal experiments. In vivo, sunitinib downregulates myeloid-derived suppressor cells (MDSC), which are thought to contribute to the antitumor effects also.
Of the ~80 kinases tested in the initial characterization, activities against VEGFR-1, -2 and -3, (the inhibitor constant [Ki], a constant that indicates how potent is an inhibitor, was 0.009 μM), PDGFR-α, PDGFR-β (Ki=0.008 μM), and fibroblast growth factor receptor 1 (Ki=0.83 μM) were the most impressive. In addition, proto-oncogene cKIT (KIT), fms-related tyrosine kinase 3 (FLT3), ret proto-oncogene, rearranged during transfection (RET), and colony stimulating factor 1 receptor were also inhibited.8 In comparison, there were no demonstrable activities against epidermal growth factor receptor, MET, or insulin growth factor receptor. In engineered cell lines, sunitinib also inhibited ligand-dependent RTK phosphorylation and proliferation of human endothelial cells and mouse fibroblasts in response to vascular endothelial growth factor (VEGF), fibroblast growth factor, and PDGF stimulation.9
Antiangiogenic effects mediated by VEGFR-2 and PDGFR
The antiangiogenic effects of sunitinib are thought to be mediated by VEGFR-2 and PDGFR blockade, whereas KIT inhibition is thought to mediate the antitumor effects in GIST, which often harbors the gain-of-function KIT and PDGFR-α mutation.
In vivo, experiments were conducted after tumors were established in animals. Sunitinib, given orally at 20–80 mg/kg, inhibited growth of many tumors grown as xenografts and induced regression among animal models employing human cancer cell lines (HT-29 colon, MDA-MB-435 breast, A375 melanoma, H460 lung, A431 skin epidermoid, Colo205 colon, and SF763T glioblastoma). In tumors expressing VEGFR-2 and PDGFR-β, sunitinib significantly inhibited the phosphorylation of these receptors and downstream events. Further studies of tumor angiogenesis using immunohistochemical staining of CD31 revealed a reduction in microvessel density after the tumor resection in tumors treated with sunitinib compared with untreated group. These findings are consistent with an antiangiogenic role for sunitinib.9 Examination of the treated tumors also demonstrated extensive tumor necrosis, which resembled those found later in the imaging studies of patients treated with sunitinib. In studies conducted with the human umbilical vein endothelial cell model, sunitinib inhibited cell migration, tubule formation, and caused endothelial cell death.10 Sunitinib administration also significantly decreased the tumor burden of animals compared to control animals in a Lewis lung carcinoma lung metastasis model. The decrease in vascular density in sunitinib-treated animals occurred before the establishment of blood vessels.10 Interestingly, sunitinib significantly decreased the vascular permeability and blood flow in a study using dynamic contrast-enhanced magnetic resonance imaging conducted in a subcutaneous HT-29 colon cancer murine model.11 Detailed studies also revealed that the biological effects of sunitinib could be achieved when the plasma concentration was maintained at 50–100 ng/mL or above for 12 hours on a daily dosing regimen.
In studies conducted using a human GIST cell line,12 sunitinib inhibited phosphorylation of KIT in these cells, resulting in growth arrest and apoptosis. Furthermore, concomitant blockade of phosphatidylinositol-4,5-bisphosphate 3-kinase/proto-oncogene cAKT (AKT)/mammalian target of rapamycin, which lies downstream from KIT, strengthened the growth suppression effects of sunitinib. Sunitinib can inhibit other RTKs relevant to its antitumor effects, including FLT3 and RET. FLT3 is frequently activated in acute myeloid leukemia,13 and RET14 contributes to the development of medullary thyroid carcinomas.
Anti-MDSC effect mediated by KIT
In the tumor microenvironment, MDSC and T-regulatory cells (Tregs) are among the suppressor cells that are significantly increased in hosts with advanced malignancies, and they are known to mediate tumor-induced immunosuppression.15–17 MDSC mediate the suppression of the tumor antigen-specific T-cell response through induction of T-cell anergy.18–21 By inducing other immunosuppressive cell populations such as Tregs and regulating inflammation in the tumor microenvironment, MDSC promote changes of the immune system to increase tolerance of tumor cells and enhance their growth. Therefore, they may serve as major obstacles for natural antitumor immunity.
Emerging evidence suggests that sunitinib downregulates the immunosuppressive effects mediated by MDSC in human and various animal models of cancer. A comparison of the number of peripheral blood MDSC in renal cell cancer patients has shown that sunitinib treatment reduces the MDSC and reverses type 1 T-cell suppression. The scenario has been reproduced in vitro by depletion of MDSC. The reduction of MDSC in response to sunitinib administration also correlated with a reversal of Treg elevation.22 In addition, the expression of immune checkpoint molecules, cytotoxic T-cell lymphocyte-associated antigen-4 and programmed death-1, in both CD4 and CD8 T cells and the expression of programmed death ligand-1 on MDSC and plasmacytoid dendritic cells are also significantly reduced by sunitinib treatment.23 In the human glioma model, treatment with sunitinib decreased the infiltration of MDSC in the tumor and prolonged the survival of tumor-carrying animals.24 In a renal cell tumor model, sunitinib pretreatment improved tumor infiltrating lymphocyte expansion by reducing intratumoral MDSC.25 Sunitinib also enhanced the effects of therapeutic cancer vaccine and stereotactic radiation therapy through modulation of MDSC. Hence, sunitinib can be a valuable tool for boosting the antitumor effects where MDSC-mediated immunosuppression is crucial. Its activity appears to be mediated by KIT blockade, since KIT ligand is required for MDSC accumulation and Treg development.26
It is also worth noting that beside the role of MDSC on adaptive immune response, MDSC can also promote tumor growth independent of the adaptive immune response mechanism.27–29 In addition, MDSC were found to facilitate angiogenesis30 and tumor metastasis.31,32 All of these actions point to a strong rationale for downregulation of MDSC in cancer.
Mechanism of sunitinib resistance
Factors associated with sunitinib primary resistance have been investigated in renal cell carcinoma (RCC). Of the 25% (33/134) of patients resistant to sunitinib, the number or size of metastases, neutrophilia (odds ratio 7.4), deteriorating performance status, and elevated lactate dehydrogenase levels stood out. The neutrophilia is thought to be associated with increased MDSC and is linked to primary resistance.33 Studies on how cells adapt to VEGF-targeted antiangiogenesis in vitro revealed that the resistance is the result of activation of VEGF-independent angiogenesis pathways through interactions with VEGF-targeted drugs, but not hypoxia.34 According to another report, intratumoral sequestration of sunitinib in lysozymes also contributed to sunitinib resistance.35
At the molecular level, MET and AXL expression levels increase during chronic sunitinib treatment. Those patients with more elevated levels had inferior outcome when compared to those without. Targeting MET and AXL eradicated this resistance.36 Cell lines with acquired sunitinib resistance have uninhibited Akt and p44/42. Axitinib use eliminated the resistance and brought Akt and p44/42 levels down.37 Overexpression of IL13RA2 has also been linked to sunitinib resistance.38 MDM2 oncogene (MDM2) is a ubiquitin protein ligase that antagonizes p53. The role of MDM2 antagonism in sunitinib resistance and p53 activation has been investigated. It was found that treatment with sunitinib increased p53 levels initially. With continued use, resistance developed with induction of MDM2 that antagonizes p53. Concurrent administration of the MDM2 inhibitor MI-319 markedly increased the antitumor and antiangiogenic activities of sunitinib and restored p53-dependent gene expression. In addition, MI-319 also suppressed the expression of stromal cell derived factor-1 and the influx of MDSC induced by sunitinib. All these suggest that evasion of p53 function might be contributing to sunitinib resistance.39
Resistance to sunitinib in GIST shares similar pathogenetic mechanisms to those identified in imatinib failure, with acquisition of secondary mutations after an extended initial response to the drug. In GIST patients who are resistant or intolerant to imatinib, sunitinib response correlated with primary tumor mutational status (before imatinib). Progression-free survival (PFS) and overall survival (OS) were significantly longer in patients who had wild type or mutation in KIT exon 9, compared to those who were mutated in exon 11. Exon 9 is in the ligand-binding domain, whereas exon 11 sits in the juxtamembrane domain. When the mutational status is checked following initiation of imatinib therapy, those patients expressing secondary mutations in exon 13 and 14 did better than those who acquired mutations in exon 17 or 18 in addition to the primary mutation in KIT. These findings are consistent with the in vitro data demonstrating that cells are more resistant to sunitinib if the secondary mutation occurs in the activation loop (exon 17, 18) of KIT RTK than the mutations expressed in the adenosine triphosphate-binding pocket (exon 13, 14).40
Potential harm of antiangiogenic therapy
As stated earlier, sunitinib is developed as a potent antiangiogenic drug that targets a rate-limiting step in tumor growth. Despite the regulatory approval of multiple antiangiogenic drugs in cancer therapy against different types of cancer including lung, renal, glioblastoma multiforme, and others, clinical benefits are limited by rapid emergence of resistance. Recently, antiangiogenic interventions including sunitinib treatment have been linked to increased tumor invasiveness and metastasis. In one report,41 sunitinib administration before or after intravenous injection of tumor cells into the circulation or following the removal of orthotopically grown tumors accelerated multiorgan metastasis, although the antitumor effects were more effective when sunitinib was used to treat animals carrying breast cancer or melanoma grown orthotopically. An independent report from another laboratory42 concluded the same. Here, VEGFA ablation and sunitinib use both increased the tumor invasiveness and incidence of hematogenous metastases in glioblastoma multiforme and pNET models. If these observations are confirmed, clinical use of sunitinib or other antiangiogenic drugs as a single agent in the adjuvant settings should be discouraged. Single-agent use as maintenance after combination use with chemotherapy should be carefully studied, given the risk of increased systemic metastasis and invasiveness associated with such antiangiogenic drugs.
First- and second-line therapy for mRCC
Sunitinib efficacy was first shown in a multicenter Phase 2 study in the first-line settings for RCC in 2006. With a 6 weeks schedule administered at 50 mg daily for 4 weeks on and 2 weeks off, the ORR, which served as the primary endpoint, was 40% (25/63). An additional 17 patients had stable disease (SD) for more than 3 months. Time to tumor progression was 8.7 months.43 In another study, patients with metastatic clear cell RCC that had progressed on cytokine treatment were given sunitinib in an open-label, single-arm, and multicenter fashion, until progression or unacceptable toxicity. Of the 105 evaluable patients, 36 had partial response (PR; 34%) and the median PFS (mPFS) was 8.3 months. Toxicities, all manageable, included fatigue (28%), diarrhea (20%), neutropenia (42%), increased lipase (28%), and anemia (26%).44 Based on these results, sunitinib received accelerated FDA approval in January 2006. A retrospective analysis of an Asian population reported similar findings.45
The pivotal Phase 3 study, comparing sunitinib and interferon (IFN)-α, was conducted on 750 patients with largely good- or intermediate-prognosis metastatic clear cell RCC who had not received prior systemic therapy. Sunitinib was administered once daily for 4 weeks on and 2 weeks off and 9 million units of IFN was given three times a week. The mPFS was 11 months in the sunitinib arm and 5 months in the IFN arm (hazard ratio [HR] 0.42, 95% confidence interval [CI] 0.32–0.54, P<0.001). ORR was significantly higher in the sunitinib group (31% vs 6%, P<0.01).46 In the final updated analysis, there was a significantly higher ORR (47% vs 12%, respectively, P<0.001), longer PFS (median, 11 vs 5 months, HR 0.42, P<0.001), and longer OS (median, 26.4 vs 21.8 months, HR 0.82, 95% CI 0.67–1.00, P<0.051) in the sunitinib group. Multivariate analysis of survival based upon the pretreatment stratification parameters and treatment assignment found that initial treatment with sunitinib was a statistically significant predictor for prolonged survival.46–48
This Phase 3 study (N=750) also measured the health-related quality of life and compared sunitinib (n=375) with IFN-α (n=375) in patients with mRCC, and showed that the health-related quality of life in patients treated with sunitinib was better than in patients treated with IFN-α.49 This means that sunitinib is better than IFN-α in terms of both tolerability and efficacy. However, in patients with untreated brain metastases from mRCC, sunitinib cannot be recommended as a valid option, due to its limited efficacy in brain metastases. Nevertheless, no neurologic complication has been observed with sunitinib therapy.48
The majority of RCCs have clear cell histology. The nonclear cell histology patients are generally excluded from clinical trials. A systemic analysis was recently performed using data available till June 1, 2014. The survival and response data from six Phase 2, one expanded access, and five retrospective studies involving 980 patients were reported. The mPFS was 1.6–8.9 months (eleven studies), median OS (mOS) 12–22 months (nine studies), disease control rate 35%–91% (ten studies), and the ORR was 0%–36% (ten studies). Grade 3/4 toxicities included those gastrointestinal, dermatological, and hematological in nature. It was concluded that there is only level C evidence to recommend sunitinib (insufficient as compared to level A evidence for clear cell histology) in this patient population.50
A retrospective study was later completed to determine whether later sunitinib rechallenge is effective.51 These patients (23 of them) received sunitinib after progression of disease on sunitinib. The PR was 22%. Compared to a PFS of 13.7 months for sunitinib-naïve patients, rechallenge yielded a PFS of 7.2 months. Patients who had more than 6 months of response had significantly longer PFS than those who had less than 6 months of response (mPFS 16.5 vs 6.0 months, P<0.03).
In the initial trial done to establish the efficacy of sunitinib, sunitinib was given as 50 mg for 4 weeks followed by 2 weeks off.46–48 In subsequent Phase 2 studies, however,52–55 a dosage schedule of 50 mg for 2 weeks followed by 1 week off was found to be better tolerated, with reduced side effects, fewer dose reductions, and similar efficacy. A retrospective analysis of patients switched from the 4-week-on/2-week-off schedule to a 2-week-on/1-week-off schedule also supported this alternative approach.56
In a separate study, the continuous daily dosing (CDD; 37.5 mg/day) was compared with the 4-week-on/2-week-off schedule in a randomized trial that included 292 patients with advanced RCC.57 The 4-week-on/2-week-off schedule demonstrated a significantly longer time to disease progression and no higher, commonly reported adverse events. The CDD was not recommended. Thus, based upon the results of the above-cited trials, the preferred regimen is to give 3-week cycles of 2 weeks on drug (50 mg/day), followed by 1 week without therapy.
Sunitinib appears to be efficacious regardless of age. In a retrospective study that included over 1,000 previously untreated patients, there were no significant differences in PFS (median, 11 vs 10 months) and OS (26 vs 24 months) among patients <70 and ≥70 years, respectively.58 Similar results were reported among patients receiving sunitinib following progression on prior cytokine treatment; however, patients ≥70 years experienced significantly more treatment-related toxicity. Decisions on the administration of sunitinib among older patients should be based on considerations of the treatment-related risks and the goals and preferences of the individual patient.
GIST refractory or intolerant to imatinib
Sunitinib is approved for treating GIST that is imatinib-refractory or -intolerant. The first sign of sunitinib efficacy was shown in a randomized, double-blinded, placebo-controlled, international Phase 3 study in 312 patients with refractory disease in a 2:1 ratio. After 42 months of follow-up, median time to tumor progression, the primary endpoint, was fourfold higher as compared with the placebo group (27.3 vs 6 weeks, HR 0.33, P<0.0001). Sunitinib showed significant clinical benefit in terms of disease control rate and survival.59 In the final analyses of a Phase 3 trial involving 243 patients in the sunitinib group and 118 in placebo (103 crossed over to sunitinib and converted to open labeled), the mOS was 72.7 weeks for sunitinib and 64.9 weeks for placebo receiving also sunitinib after crossover (HR 0.876, P=0.306). A software program which estimates the impact of crossover determined that the mOS was 39 weeks, well below the sunitinib group.60 Sunitinib benefit was associated with suppression of KIT activity.61
A German study of 1,124 patients recently pointed out the importance of prompt dose adjustment among the 15% whose performance is less than optimal (Eastern Cooperative Oncology Group [performance scale] 2 or above). Patients in whom the initial dosing schedule was modified exhibited a longer mOS (23.5 months) than those treated strictly as per the initial dosing schedule (11.1 months).62 Grade 3/4 toxicities included hand and foot syndrome in 11%, fatigue in 9%, hypertension in 7%, neutropenia in 8%, and thrombocytopenia in 6%.
The clinical activity of sunitinib against specific mutations in the KIT gene was shown in a Phase 1/2 trial performed in 97 patients with metastatic, imatinib-refractory or -intolerant GIST. Clinical benefit rate (PR or SD for longer than 6 months) was significantly higher for those with a primary KIT exon 9 (58%) or wild type (56%) than for those with a KIT exon 11 mutation (34%).63 Patients with KIT exon 9 and a PDGFR-α mutation had a PFS and OS of 19.4 and 26.9 months, respectively, while those with exon 11 mutations had only 5.1 and 12.3 months, respectively. Following progression on imatinib, both PFS and OS were significantly longer for patients with secondary KIT exon 13 or 14 mutations than for those with exon 17 or 18 mutations (7.8 vs 2.3 months and 13 vs 4.0 months, respectively) on sunitinib. Mutations in different positions affect different KIT domains (refer to section of mechanism of sunitinib resistance above).
Positron emission tomography scan seems to predict sunitinib response after imatinib failure. In this study, PFS was correlated with the positron emission tomography response demonstrated 4 weeks after initiation of sunitinib (N=23). The PFS was 29 weeks for those with a standard uptake value (SUV) of less than eight, whereas it was only 4 weeks when the SUV was eight or higher. None of the patients who had metabolically progressive disease subsequently responded to sunitinib. Those who had higher residual standard uptake value (on positron emission tomography) after 4 weeks of treatment, primary refractory to imatinib and nongastric GIST had shorter PFS, regardless of the KIT or PDGFR mutational status.64 Another study concluded that in addition to genotype, sunitinib-induced hypertension is predictive of good response and better PFS and OS.65
Sunitinib is approved in the US for the treatment of imatinib-refractory or -intolerant advanced GISTs. The approved dose is 50 mg daily for four of every 6 weeks; however, CDD (37.5 mg daily) appears similarly safe and effective in a separate study.66
Advanced pNETs
Sunitinib was approved in the US for the treatment of progressive, well-differentiated pNETs in patients with unresectable, locally advanced, or metastatic disease. This approval was based on two trials. In the first (Phase 2) trial in patients with advanced neuroendocrine tumors (NETs), sunitinib at 50 mg daily for four of every 6 weeks was administered to 109 patients.67 Eighteen percent had a PR and 68% had prolonged periods of SD. Of the 61 patients with pancreatic NETs, the median time to tumor progression was 7.7 months. Rates of symptom control for patients with functioning tumors and refractory symptoms were not reported.
A continuous daily dose schedule of sunitinib 37.5 mg was compared to placebo in a Phase 3 trial of 171 patients with progressing pNETs.68 The mPFS was significantly longer with sunitinib (11.4 vs 5.5 months). There were eight objective responses with sunitinib (vs none in the placebo group), two of which were complete. Twenty-three percent of patients developed hand–foot skin reaction and 26% developed hypertension of any grade among the group receiving sunitinib. The most common grade 3 or 4 adverse events in this group were neutropenia (12%) and hypertension (10%). Despite these side effects, there were no differences in the quality-of-life index with sunitinib. Rates of symptom control for patients with functioning tumors and refractory symptoms were not reported.
In two patients with VIPomas, sunitinib provided control for refractory symptoms due to tumor hormone production.69 In contrast, blood glucose concentrations were not increased in a single patient treated with sunitinib for a functioning metastatic insulinoma.70 Please refer to Table 1 for a summary of clinical trials that have established the efficacy of sunitinib.
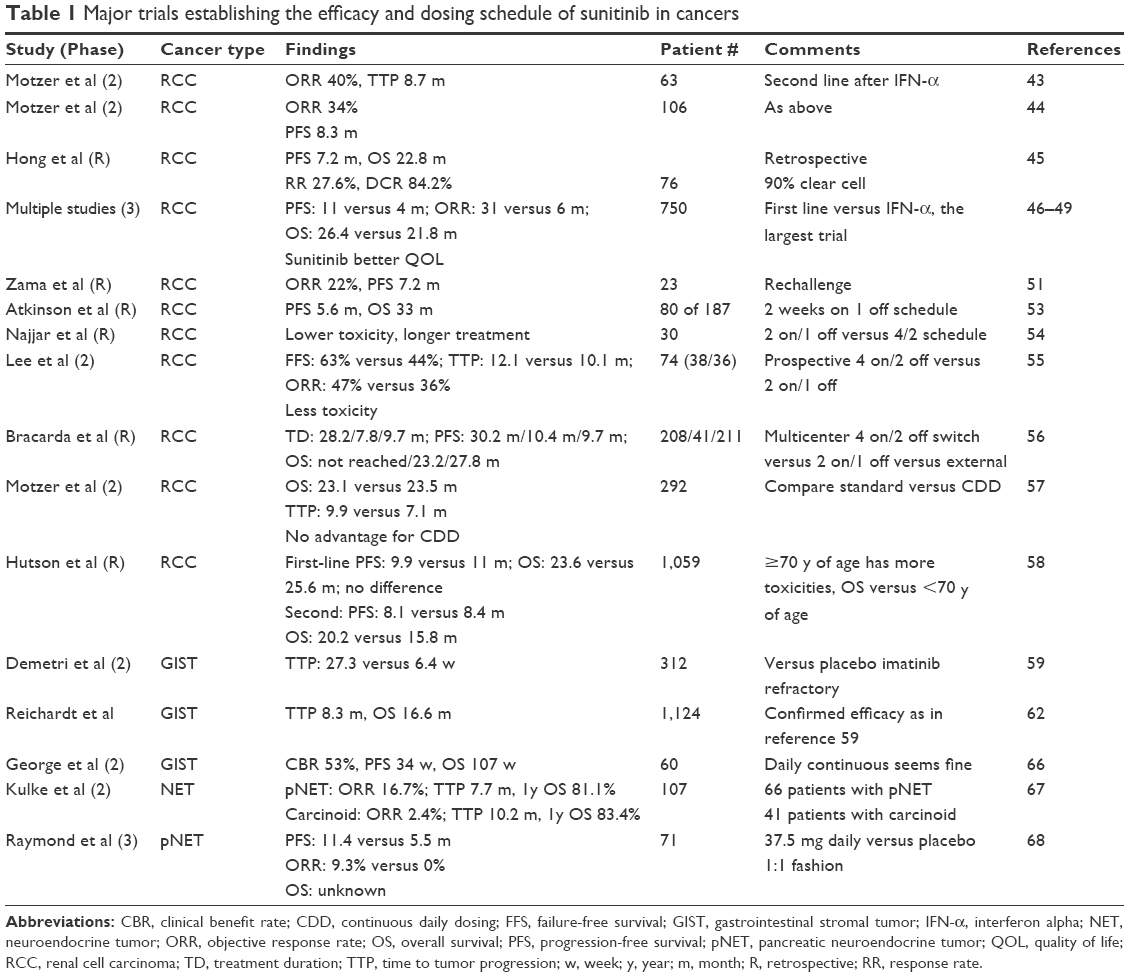 | Table 1 Major trials establishing the efficacy and dosing schedule of sunitinib in cancers |
Management of thyroid cancer
Many small-molecule tyrosine kinase inhibitors including sunitinib have been tested in thyroid cancer. In one Phase 2 study,71 sunitinib was given to 28 differentiated and seven medullary thyroid cancer patients. One complete response (3%), ten PR (28%), and 16 SD (46%) with 77% clinical benefit was recorded (complete response/PR/SD). The most common toxicities included fatigue (11%), neutropenia (34%), hand–foot syndrome (17%), diarrhea (17%), and leukopenia (31%). One patient on anticoagulation died of gastrointestinal bleeding. Sixty percent of patients required dose reductions from 37.5 to 25 mg once daily. In another Phase 2 study of differentiated thyroid cancer patients who had received at least one course of radioactive ablation, 23 patients were given 37.5 mg daily. The mean best response achieved was a 17.2% reduction in the tumor size from the baseline level. PR was achieved in 6 (26%) and SD in 13 (57%). The overall clinical benefit rate was 83% with a PFS of 241 days (range 114–518 days).72 Currently, vandetanib73 and cabozantinib74 are approved for progressive, metastatic medullary thyroid cancer. Sorafenib75 and lenvatinib76 are approved for treating unresectable, locally advanced, or metastatic well-differentiated thyroid cancer refractory to radioactive iodine therapy. Sunitinib is not currently approved by the FDA for the treatment of thyroid cancer.
Concurrent or sequential use with chemotherapy in solid tumors
Although sunitinib given concurrently with chemotherapy has shown toxicities that were manageable in early-phase clinical trials,77,78 so far no Phase 3 trial has shown it to improve the efficacy of chemotherapy. A Phase 2 sunitinib maintenance trial79 conducted in extensive stage small cell lung cancer following platinum-based chemotherapy reported an mPFS benefit from 2.1 to 3.7 in 85 patients randomized to receiving either placebo or sunitinib, respectively (HR 1.62, 95% CI 1.02–2.60, P=0.02). No mOS benefit was seen (6.9 months in control arm and 9.0 months in treatment arm, HR 1.28 and 95% CI 0.79–2.10, P=0.16). Further study will hopefully clarify the benefit of sunitinib in maintenance for small cell lung cancer. Adding sunitinib to fluorouracil, leucovorin, irinotecan in the treatment of esophageal/gastric cancer failed to improve ORR or PFS, compared to fluorouracil, leucovorin, irinotecan alone; however, hepatotoxicity was worse. Therefore, the role of sunitinib in combination with or following chemotherapy remains unclear at this point.
Improved outcome in patients treated in combination with stereotactic body radiation therapy (SBRT)
In a Phase 1/2 trial conducted in patients with solid tumors with oligometastases (<5), it was found that sunitinib may be safely administered to patients treated concomitantly with SBRT. Of the 46 patients treated with concurrent sunitinib, no grade 5 toxicity was observed. Three grade 3 toxicities were reported, including anemia and thrombocytopenia which occurred when liver and large areas of bone were irradiated. Sunitnib appeared to improve the efficacy of SBRT, especially in renal and prostate cancer.80 Sunitinib mediated decrease of MDSC accumulation and functional suppression, which correlated with Treg reduction in responders. SBRT synergized with sunitinib, leading to decrease in monocytic MDSC, Treg, B cells, and augmented T-bet expression in primary CD4 and CD8 T cells. PFS and cause-specific survival were improved in responders (decreased MDSC) in the combination group. but not in the SBRT alone group.81
Improved efficacy of therapeutic cancer vaccine
A number of hurdles need to be overcome before a therapeutic vaccine can work. Not only will the vaccine need to induce antitumor immune responses, but also it should bypass the tumor-mediated immune escape mechanisms. As stated earlier, MDSC and Tregs are among the suppressor cells that mediate immune escape. In a study of a therapeutic vaccine made from human papilloma virus antigens E6 and 7 expressed in a Semliki Forest virus vector, SBRT or sunitinib each alone boosted the efficacy of the vaccine and increased the antitumor effector cell to MDSC ratio. Interestingly, when the three modalities were combined, the E7-specific CD8+ T cell for every one of MDSC cell increased from 0.02 to 200, a 10,000 fold change. This phenomenon eventually led to 100% tumor-free survival.82 In two murine tumor models of colon (MC38CEA) and breast cancer (4T1), sunitinib decreased tumor vasculature and compactness. Combination with a therapeutic vaccine led to increased tumor-infiltrating lymphocyte and tumor antigen-specific CD8 T cells, decreased tumor size, and improved survival, supporting concurrent use of sunitinib in combination with a therapeutic vaccine.83
Tolerance
Sunitinib is generally well tolerated. The most common adverse reactions, occurring in more than 20% of patients, are fatigue, asthenia, diarrhea, nausea, mucositis/stomatitis, vomiting, dyspepsia, abdominal pain, constipation, hypertension, rash, hand–foot syndrome, skin discoloration, altered taste, anorexia, and mild bleeding.
Among patients treated with sunitinib, the incidence of hypertension was 28% (6% had grade 3) in Phase 2 trials in mRCC44 and 15% (4% had grade 3) in a Phase 3 GIST trial.59
An important part of the mechanism of hypertension associated with all VEGF inhibitors including sunitinib is thought to involve decreased production of nitric oxide (NO) in the wall of arterioles and other resistance vessels. VEGF increases NO synthesis through upregulation of endothelial NO synthase, while VEGF inhibition diminishes NO synthesis84 leading to increased peripheral resistance and blood pressure.
The most common cutaneous toxicity is hand-and-foot skin reaction (HFSR). Other cutaneous toxicities include alopecia, stomatitis, and skin discoloration. HFSR and severe stomatitis manifest early compared with other cutaneous toxicities and require therapy modifications to relieve symptoms; however, other cutaneous toxicities do not affect the treatment course. HFSR was observed in 36% of those treated with sunitinib and median time to onset was 32.4 days. A significant correlation between the severity of HFSR and development of alopecia and stomatitis was found.85
Conclusion
As a multitargeted kinase inhibitor, sunitinib was formally approved in 2006 for the treatment of GIST patients who are intolerant or refractory to imatinib and in 2007 for advanced RCC. Its role in nonclear cell RCC appears to be limited. Over the years, sunitinib changed the way we manage these tumors. More recently, sunitinib has received approval for pNET. Despite the common perception that downregulation of angiogenesis may improve outcome, no role for sunitinib has been found in the management of solid tumors in patients receiving chemotherapy. In recent years, increasing evidence has indicated a role for sunitinib in downregulating the MDSC. Given the importance of MDSC in promoting tumor growth, metastasis, and suppression of immune response, synergy between sunitinib and immune checkpoint inhibitors, SBRT, or therapeutic vaccine is being hotly pursued. Preclinical studies appear very promising. Sunitinib may exert toxic effects; however, the general benign nature of SBRT and several immunotherapies provides promise that additive side effects will be limited.
Acknowledgment
The authors wish to thank Paul M Dainer, MD, for his critical reading and editing of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Goodman VL, Rock EP, Dagher R, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13(5):1367–1373. | ||
Blumenthal GM, Cortazar P, Zhang JJ, et al. FDA approval summary: sunitinib for the treatment of progressive well-differentiated locally advanced or metastatic pancreatic neuroendocrine tumors. Oncologist. 2012;17(8):1108–1113. | ||
Sun L, Liang C, Shirazian S, et al. Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46(7):1116–1119. | ||
Sun H, Zhi C, Wright GE, et al. Molecular modeling and synthesis of inhibitors of herpes simplex virus type 1 uracil-DNA glycosylase. J Med Chem. 1999;42(13):2344–2350. | ||
Laird AD, Vajkoczy P, Shawver LK, et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000;60(15):4152–4160. | ||
Sutent® (Sunitinib malate capsule) [package insert]. Pfizer Inc; 2016. | ||
O’Farrell AM, Foran JM, Fiedler W, et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin Cancer Res. 2003;9(15):5465–5476. | ||
Rini BI. Sunitinib. Expert Opin Pharmacother. 2007;8(14):2359–2369. | ||
Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337. | ||
Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor SU11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis. 2004;7(3):225–233. | ||
Marzola P, Degrassi A, Calderan L, et al. Early antiangiogenic activity of SU11248 evaluated in vivo by dynamic contrast-enhanced magnetic resonance imaging in an experimental model of colon carcinoma. Clin Cancer Res. 2005;11(16):5827–5832. | ||
Ikezoe T, Yang Y, Nishioka C, et al. Effect of SU11248 on gastrointestinal stromal tumor-T1 cells: enhancement of growth inhibition via inhibition of 3-kinase/Akt/mammalian target of rapamycin signaling. Cancer Sci. 2006;97(9):945–951. | ||
Coombs CC, Tallman MS, Levine RL. Molecular therapy for acute myeloid leukaemia. Nat Rev Clin Oncol. 2016;13(5):305–318. | ||
Prescott JD, Zeiger MA. The RET oncogene in papillary thyroid carcinoma. Cancer. 2015;121(13):2137–2146. | ||
Arina A, Corrales L, Bronte V. Enhancing T cell therapy by overcoming the immunosuppressive tumor microenvironment. Semin Immunol. 2016;28(1):54–63. | ||
Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128:95–139. | ||
Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–3364. | ||
Bronte V, Chappell DB, Apolloni E, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162(10):5728–5737. | ||
Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol. 2005;175(7):4583–4592. | ||
Watanabe S, Deguchi K, Zheng R, et al. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J Immunol. 2008;181(5):3291–3300. | ||
Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184(6):3106–3116. | ||
Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. | ||
Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69(6):2514–2522. | ||
Raychaudhuri B, Rayman P, Huang P, et al. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol. 2015;122(2):293–301. | ||
Guislain A, Gadiot J, Kaiser A, et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol Immunother. 2015;64(10):1241–1250. | ||
Kao J, Ko EC, Eisenstein S, Sikora AG, Fu S, Chen SH. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit Rev Oncol Hematol. 2011;77(1):12–19. | ||
Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. J Exp Med. 1995;181(1):435–440. | ||
Seung LP, Rowley DA, Dubey P, Schreiber H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Nati Acad Sci U S A. 1995;92(14):6254–6258. | ||
Seung LP, Seung SK, Schreiber H. Antigenic cancer cells that escape immune destruction are stimulated by host cells. Cancer Res. 1995;55(21):5094–5100. | ||
Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–421. | ||
Mauti LA, Le Bitoux MA, Baumer K, et al. Myeloid-derived suppressor cells are implicated in regulating permissiveness for tumor metastasis during mouse gestation. J Clin Invest. 2011;121(7):2794–2807. | ||
Yang L, Huang J, Ren X, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23–35. | ||
Lim SH, Hwang IG, Ji JH, et al. Intrinsic resistance to sunitinib in patients with metastatic renal cell carcinoma. Asia Pac J Clin Oncol. Epub 2016 Mar 31. | ||
Han KS, Raven PA, Frees S, et al. Cellular adaptation to VEGF-targeted antiangiogenic therapy induces evasive resistance by overproduction of alternative endothelial cell growth factors in renal cell carcinoma. Neoplasia. 2015;17(11):805–816. | ||
Gotink KJ, Broxterman HJ, Labots M, et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res. 2011;17(23):7337–7346. | ||
Zhou L, Liu XD, Sun M, et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene. 2016;35(21):2687–2697. | ||
Matsuyama H, Ikemoto K, Eguchi S, et al. Copy number aberrations using multicolour fluorescence in situ hybridization (FISH) for prognostication in non-muscle-invasive bladder cancer (NIMBC). BJU Int. 2014;113(4):662–667. | ||
Shibasaki N, Yamasaki T, Kanno T, et al. Role of IL13RA2 in sunitinib resistance in clear cell renal cell carcinoma. PLoS One. 2015;10(6):e0130980. | ||
Panka DJ, Liu Q, Geissler AK, Mier JW. Effects of HDM2 antagonism on sunitinib resistance, p53 activation, SDF-1 induction, and tumor infiltration by CD11b+/Gr-1+ myeloid derived suppressor cells. Mol Cancer. 2013;12:17. | ||
Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15(24):7510–7518. | ||
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–239. | ||
Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. | ||
Motzer RJ, Michaelson MD, Redman BG, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24(1):16–24. | ||
Motzer RJ, Rini BI, Bukowski RM, et al. Sunitinib in patients with metastatic renal cell carcinoma. JAMA. 2006;295(21):2516–2524. | ||
Hong MH, Kim HS, Kim C, et al. Treatment outcomes of sunitinib treatment in advanced renal cell carcinoma patients: a single cancer center experience in Korea. Cancer Res Treat. 2009;41(2):67–72. | ||
Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–124. | ||
Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27(22):3584–3590. | ||
Patil S, Figlin RA, Hutson TE, et al. Prognostic factors for progression-free and overall survival with sunitinib targeted therapy and with cytokine as first-line therapy in patients with metastatic renal cell carcinoma. Ann Oncol. 2011;22(2):295–300. | ||
Cella D, Michaelson MD, Bushmakin AG, et al. Health-related quality of life in patients with metastatic renal cell carcinoma treated with sunitinib vs interferon-alpha in a phase III trial: final results and geographical analysis. Br J Cancer. 2010;102(4):658–664. | ||
Abdel-Rahman O, Fouad M. Efficacy and toxicity of sunitinib for non clear cell renal cell carcinoma (RCC): a systematic review of the literature. CritRev Oncol Hematol. 2015;94(2):238–250. | ||
Zama IN, Hutson TE, Elson P, et al. Sunitinib rechallenge in metastatic renal cell carcinoma patients. Cancer. 2010;116(23):5400–5406. | ||
Bjarnason GA, Khalil B, Hudson JM, et al. Outcomes in patients with metastatic renal cell cancer treated with individualized sunitinib therapy: correlation with dynamic microbubble ultrasound data and review of the literature. Urol Oncol. 2014;32(4):480–487. | ||
Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol. 2014;191(3):611–618. | ||
Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer. 2014;50(6):1084–1089. | ||
Lee JL, Kim MK, Park I, et al. RandomizEd phase II trial of Sunitinib four weeks on and two weeks off versus Two weeks on and One week off in metastatic clear-cell type REnal cell carcinoma: RESTORE trial. Ann Oncol. 2015;26(11):2300–2305. | ||
Bracarda S, Iacovelli R, Boni L, et al. Sunitinib administered on 2/1 schedule in patients with metastatic renal cell carcinoma: the RAINBOW analysis. Ann Oncol. 2015;26(10):2107–2113. | ||
Motzer RJ, Hutson TE, Olsen MR, et al. Randomized phase II trial of sunitinib on an intermittent versus continuous dosing schedule as first-line therapy for advanced renal cell carcinoma. J Clin Oncol. 2012;30(12):1371–1377. | ||
Hutson TE, Bukowski RM, Rini BI, et al. Efficacy and safety of sunitinib in elderly patients with metastatic renal cell carcinoma. Br J Cancer. 2014;110(5):1125–1132. | ||
Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338. | ||
Demetri GD, Garrett CR, Schoffski P, et al. Complete longitudinal analyses of the randomized, placebo-controlled, phase III trial of sunitinib in patients with gastrointestinal stromal tumor following imatinib failure. Clin Cancer Res. 2012;18(11):3170–3179. | ||
Demetri GD, Heinrich MC, Fletcher JA, et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res. 2009;15(18):5902–5909. | ||
Reichardt P, Kang YK, Rutkowski P, et al. Clinical outcomes of patients with advanced gastrointestinal stromal tumors: safety and efficacy in a worldwide treatment-use trial of sunitinib. Cancer. 2015;121(9):1405–1413. | ||
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. | ||
Prior JO, Montemurro M, Orcurto MV, et al. Early prediction of response to sunitinib after imatinib failure by 18F-fluorodeoxyglucose positron emission tomography in patients with gastrointestinal stromal tumor. J Clin Oncol. 2009;27(3):439–445. | ||
Rutkowski P, Bylina E, Klimczak A, et al. The outcome and predictive factors of sunitinib therapy in advanced gastrointestinal stromal tumors (GIST) after imatinib failure – one institution study. BMC Cancer. 2012;12:107. | ||
George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45(11):1959–1968. | ||
Kulke MH, Lenz HJ, Meropol NJ, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26(20):3403–3410. | ||
Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513. | ||
de Mestier L, Walter T, Brixi H, Lombard-Bohas C, Cadiot G. Sunitinib achieved fast and sustained control of VIPoma symptoms. Eur J Endocrinol. 2015;172(1):K1–K3. | ||
Chen J, Wang C, Han J, et al. Therapeutic effect of sunitinib malate and its influence on blood glucose concentrations in a patient with metastatic insulinoma. Expert Rev Anticancer Ther. 2013;13(6):737–743. | ||
Carr LL, Mankoff DA, Goulart BH, et al. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res. 2010;16(21):5260–5268. | ||
Bikas A, Kundra P, Desale S, et al. Phase 2 clinical trial of sunitinib as adjunctive treatment in patients with advanced differentiated thyroid cancer. Eur J Endocrinol. 2016;174(3):373–380. | ||
Thornton K, Kim G, Maher VE, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res. 2012;18(14):3722–3730. | ||
Viola D, Cappagli V, Elisei R. Cabozantinib (XL184) for the treatment of locally advanced or metastatic progressive medullary thyroid cancer. Future Oncol. 2013;9(8):1083–1092. | ||
McFarland DC, Misiukiewicz KJ. Sorafenib in radioactive iodine-refractory well-differentiated metastatic thyroid cancer. OncoTargets Ther. 2014;7:1291–1299. | ||
Nair A, Lemery SJ, Yang J, et al. FDA approval summary: lenvatinib for progressive, radio-iodine-refractory differentiated thyroid cancer. Clin Cancer Res. 2015;21(23):5205–5208. | ||
Lee KW, Park SR, Oh DY, et al. Phase I study of sunitinib plus capecitabine/cisplatin or capecitabine/oxaliplatin in advanced gastric cancer. Invest New Drugs. 2013;31(6):1547–1558. | ||
Sweeney CJ, Chiorean EG, Verschraegen CF, et al. A phase I study of sunitinib plus capecitabine in patients with advanced solid tumors. J Clin Oncol. 2010;28(29):4513–4520. | ||
Ready NE, Pang HH, Gu L, et al. Chemotherapy With or Without Maintenance Sunitinib for Untreated Extensive-Stage Small-Cell Lung Cancer: A Randomized, Double-Blind, Placebo-Controlled Phase II Study-CALGB 30504 (Alliance). J Clin Oncol. 2015;33(15):1660–1665. | ||
Kao J, Chen CT, Tong CC, et al. Concurrent sunitinib and stereotactic body radiotherapy for patients with oligometastases: final report of a prospective clinical trial. Target Oncol. 2014;9(2):145–153. | ||
Chen HM, Ma G, Gildener-Leapman N, et al. Myeloid-Derived Suppressor Cells as an Immune Parameter in Patients with Concurrent Sunitinib and Stereotactic Body Radiotherapy. Clin Cancer Res. 2015;21(18):4073–4085. | ||
Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015;4(3):e989764. | ||
Farsaci B, Donahue RN, Coplin MA, et al. Immune consequences of decreasing tumor vasculature with antiangiogenic tyrosine kinase inhibitors in combination with therapeutic vaccines. Cancer Immunol Res. 2014;2(11):1090–1102. | ||
Horowitz JR, Rivard A, van der Zee R, et al. Vascular endothelial growth factor/vascular permeability factor produces nitric oxide-dependent hypotension. Evidence for a maintenance role in quiescent adult endothelium. Arterioscler Thromb Vasc Biol. 1997;17(11):2793–2800. | ||
Lee WJ, Lee JL, Chang SE, et al. Cutaneous adverse effects in patients treated with the multitargeted kinase inhibitors sorafenib and sunitinib. Br J Dermatol. 2009;161(5):1045–1051. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.