Back to Journals » Drug Design, Development and Therapy » Volume 8
Sulforaphane reverses glucocorticoid-induced apoptosis in osteoblastic cells through regulation of the Nrf2 pathway
Authors Lin H, Wei B, Li G, Zheng J, Sun J, Chu J, Zeng R, Niu Y
Received 2 April 2014
Accepted for publication 10 May 2014
Published 19 July 2014 Volume 2014:8 Pages 973—982
DOI https://doi.org/10.2147/DDDT.S65410
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Hao Lin,1,* Bo Wei,1,* Guangsheng Li,1 Jinchang Zheng,1 Jiecong Sun,1 Jiaqi Chu,2 Rong Zeng,1 Yanru Niu2
1Department of Spinal Surgery, Affiliated Hospital of Guangdong Medical College, Zhanjiang, People’s Republic of China; 2Laboratory Institute of Minimally Invasive Orthopedic Surgery, Affiliated Hospital of Guangdong Medical College, Zhanjiang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Apoptosis of osteoblasts triggered by high-dose glucocorticoids (GCs) has been identified as a major cause of osteoporosis. However, the underlying molecular mechanisms accounting for this action remain elusive, which has impeded the prevention and cure of this side effect. Sulforaphane (SFP) is a naturally occurring isothiocyanate that has huge health benefits for humans. In this study, by using osteoblastic MC3T3-E1 cells as a model, we demonstrate the protective effects of SFP against dexamethasone (Dex)-induced apoptosis and elucidate the underlying molecular mechanisms. The results show that SFP could effectively inhibit the Dex-induced growth inhibition and release of lactate dehydrogenase in MC3T3-E1 cells. Treatment with Dex induced caspase-dependent apoptosis in MC3T3-E1 cells, as evidenced by an increase in the Sub-G1 phase, chromatin condensation, and deoxyribonucleic acid fragmentation, which were significantly suppressed by coincubation with SFP. Mitochondria-mediated apoptosis pathway contributed importantly to Dex-induced apoptosis, as revealed by the activation of caspase-3/-9 and subsequent cleavage of poly adenosine diphosphate ribose polymerase, which was also effectively blocked by SFP. Moreover, treatments of Dex strongly induced overproduction of reactive oxygen species and inhibited the expression of nuclear factor erythroid 2-related factor 2 (Nrf2) and the downstream effectors HO1 and NQO1. However, cotreatment with SFP effectively reversed this action of Dex. Furthermore, silencing of Nrf2 by small interfering ribonucleic acid significantly blocked the cytoprotective effects of SFP against Dex-induced apoptosis, which suggest the important role of Nrf2 signaling pathway and cell apoptosis induced by Dex. Taken together, this study provides a novel strategy for molecular intervention against Dex-induced osteoporosis using phytochemicals.
Keywords: osteoporosis, glucocorticoid, apoptosis, sulforaphane, Nrf2 pathway
Introduction
In the lives of vertebrates, bone is destroyed and reformed over and over in order to maintain their bone volume and calcium homeostasis. Osteoporosis, a prevalent health concern, is a bone disease characterized by a reduction in bone mineral density, disorder of bone architecture, and an increase in bone fragility.1 Osteoblasts and osteoclasts are two kinds of cells in charge of bone formation and resorption, respectively. No matter decrease in osteoblastic activity or increase in osteoclastic activity in the bone could result in osteoporosis.2 The function and differentiation of these have been found to play important roles during the occurrence of osteoporosis. Osteoblasts could build up the matrix of bone and enhance its mineralization. Many studies have found that cytokines, hormones, and some transcription factors contribute to the differentiation of osteoblasts. However, the underlying signaling mechanisms accounting for osteoporosis are still not well characterized.3,4
Glucocorticoids (GCs) have been widely used in clinics due to their anti-inflammatory and immunomodulatory effects. However, the therapeutic use of GCs is almost always limited by substantial adverse outcomes such as osteoporosis, diabetes, and obesity. These unwanted outcomes are a major dilemma for clinicians because improvements in the primary disorder seem to be achievable only by accepting substantial adverse effects that are often difficult to prevent or treat.4 Nowadays, GC-induced osteoporosis has been regarded as an important cause for osteoporosis and bone loss.5,6 For instance, long-term and high-dose intake of GCs, especially dexamethasone (Dex), has been identified as a major reason for human osteoporosis.7,8 Clinical studies have shown that over 30% of patients who receive overdosed GCs would develop bone fractures, and their bone displayed increased osteoblasts and osteocyte apoptosis.7,8 For the action mechanisms of GCs, Espina et al9 showed that Bim was an important regulator of osteoblast Dex-induced apoptosis in murine MBA-15.4 osteoblasts. O’Brien et al10 reported that Dex could directly induce apoptosis in osteoblasts and osteocytes and reduce the bone formation and architecture via regulation of 11β-hydroxysteroid dehydrogenase type 2. Studies also showed that inhibition of cytokines like interleukin 11 via interaction with AP-1 also contributed to the inhibitory effects of Dex on osteoblasts.6 Moreover, glycogen synthase kinase 3β and p38 mitogen-activated protein kinase were also activated in MC3T3-E1 cells by Dex.11 However, the underlying molecular mechanisms accounting for this action remain elusive, which has impeded the prevention and cure of this side effect.
Recently, the search for naturally occurring phytochemicals from edible materials (especially fruits and vegetables) that can antagonize osteoporosis has gained more and more attention. Studies have found that tetrahydroxystilbene glucoside, dehydrocostus lactone, puerarin, and chemical constituents of the fruits of Prunus mume could reverse GC-induced apoptosis in osteoblasts by regulation of various signaling pathways.12–15 Nowadays, cruciferous vegetables such as broccoli are widely consumed all over the world. Epidemiological studies have revealed that consumption of a crucifers-rich diet could reduce the risk of chronic degenerative diseases.16 Evidence has demonstrated that glucosinolates are the major active component accounting for the protective effects of crucifers against chronic disease.17 Sulforaphane (SFP) is a naturally occurring isothiocyanate that has been identified as one of the most important components in vegetables that provide health benefits for humans.18 Xu et al19 demonstrated that SFP exhibited its antioxidant activity through regulation of nuclear factor erythroid 2-related factor 2 (Nrf-2) signaling pathways. Moreover, SFP was also identified as a significant inducer of Nrf2 that could rouse cells’ intrinsic defense system via regulation of cytoprotective genes.18 Based on this property, SFP also exhibited a potential protective phytochemical against neurodegenerative diseases by activating the Nrf2/antioxidant response element pathway.20 Studies also demonstrated the antigenotoxicity of SFP.21 Recently, Gross-Steinmeyer et al22 found that SFP and phenethyl isothiocyanate could inhibit aflatoxin B1-mediated genotoxicity in human hepatocytes through regulation of the GSTM1 genotype and CYP3A4 gene expression. Katoch et al23 also showed that SFP mitigated the genotoxicity induced by radiation and anticancer drugs in human lymphocytes. However, little information about the protective effects of SFP on GC-induced osteoporosis is available.
Therefore, in the present study, studies were carried out to clarify the protective effects of SFN to antagonize GC-induced apoptosis in osteoblasts and to examine the diversified signaling pathways accounting for this effect. Our results showed that SFP effectively reversed Dex-induced apoptosis in osteoblastic cells through regulation of the Nrf2 pathway. This study may provide a novel strategy for molecular intervention against Dex-induced osteoporosis by using phytochemicals.
Materials and methods
Materials
SFP, propidium iodide, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), JC-1, 4′,6-diamidino-2-phenylindole (DAPI), 2′,7′-dichlorofluorescin diacetate (DCFH-DA), and bicinchoninic acid kit for protein concentration measurement were purchased from Sigma-Aldrich, St Louis, MO, USA. The water used in this study was ultrapure Milli-Q® water (EMD Millipore, Billerica, MA, USA).
Cell culture
MC3T3-E1 preosteoblast (CRL-2593™) was purchased from American Type Culture Collection, Manassas, VA, USA. The cells were cultured in Alpha Minimum Essential Medium with ribonucleosides, deoxyribonucleosides, 2 mM L-glutamine, and 1 mM sodium pyruvate but without ascorbic acid (Life Technologies, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum in a humid incubator at 37°C (95% O2 and 5% CO2).
Drug treatments
MC3T3-E1 cells at a density of 3 × 103 cells/well were seeded in 96-well plates or Petri dishes for 24 hours. For protection experiments, the cells were pretreated with various concentrations of SFP for 2 hours, and then cultured in combination with 1 μM Dex for 24 hours. The cells were then subjected to biological analysis.
MTT assay
MC3T3-E1 cells at a density of 3 × 103 cells/well were seeded in 96-well plates for 24 hours and then treated with different concentrations of Dex and SFP for the indicated time. The cell viability was determined by MTT assay as previously described.24
TUNEL-DAPI costaining assay
Deoxyribonucleic acid (DNA) fragmentation and nucleus condensation induced by Dex at single cell level were investigated by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-DAPI staining assay according to the procedure of the assay kit (Hoffman-La Roche Ltd, Basel, Switzerland).25
Flow cytometric analysis
After exposure to different treatments of Dex and SFP, MC3T3-E1 cells were washed with phosphate buffered saline two times and then fixed with 75% ethanol at −20°C in dark for 24 hours. The fixed cells were stained with propidium iodide working solution (1.21 mg/mL Tris, 700 U/mL RNase, 50.1 μg/mL PI, pH 8.0) for 4 hours in dark, and then analyzed on a Coulter Epics XL flow cytometer (Beckman Coulter GmbH, Krefeld, Germany). Cell apoptosis was determined by quantifying the sub-G1 peak in the cell cycle distribution histogram.
Assessment of the caspase activity
The enzymatic activities of caspase-3, -8, and -9 in MC3T3-E1 cells treated with Dex and SFP were monitored by fluorometric method using specific caspase substrates (Ac-DEVD-AFC for caspase-3, Ac-IETD-AFC for caspase-8, and Ac-LEHD-AFC for caspase-9).25
Measurement of ROS generation
Intracellular reactive oxygen species (ROS) generation was determined by DCFH-DA fluorometric assay.24 The change in intracellular ROS levels of each group was determined by calculating ΔF = (F − F0)/F0, where F represents the fluorescence read at each time point and F0 the control fluorescence.
Western blot analysis
Treated MC3T3-E1 cells were harvested and collected as cell pellets, which were lysed in lysis buffer (Cell Signaling Technology, Inc., Beverly, MA, USA) on ice for 1 hour. The protein concentration was determined by bicinchoninic acid assay (Sigma-Aldrich) according to the manufacturer’s instructions. Equal proteins of each treatment were separated on 12% sodium dodecyl sulfate denaturing polyacrylamide gel and electrophoretically transferred to polyvinylidene difluoride membranes. After blocking with 5% nonfat milk, the membranes were incubated with primary antibodies (Cell Signaling Technology, Inc.) at 1:1,000 dilution overnight at 4°C. After washing, the membranes were incubated with corresponding secondary antibodies and visualized by Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA, USA).
Transient transfection with Nrf2 siRNA
Silencer® Select Pre-Designed siRNA (Thermo Fisher Scientific) (sense 5′-GCCCAUUGAUGUUUCUGAUTT-3′, antisense 5′-AUCAGAAACAUCAAUGGGCTT-3′) was used for knockdown of Nrf2. Briefly, MC3T3-E1 cells at 50%–60% confluence were transfected with siRNA–lipofectamine complex according to the manufacturer’s instructions. After incubation for 5 hours, the medium was replaced with complete Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific), and the cells were cultured for another 24 hours. The transfected cells were then subjected to treatments of Dex and SFP. The expression levels of Nrf2 and related proteins were determined by Western blotting.
Statistical analysis
In this study all the experiments were conducted at least in triplicate and the results were expressed as mean ± standard deviation. Significance in the difference among different treatments was analyzed by one-way analysis of variance at P<0.05 (*) or P<0.01 (**) level.
Results
SFP reverses Dex-induced cytotoxicity in osteoblasts through inhibition of cell apoptosis
In this study MC3T3-E1 cell line was used as a model to examine the cytotoxic effects of Dex on osteoblasts and the protective effects of SFP. As shown in Figure 1, the results of MTT assay revealed that Dex at the concentrations of 0.25–8.0 μM inhibited cell growth in a dose-dependent manner after treatment for 24 hours. In contrast, SFP alone was relatively nontoxic toward MC3T3-E1 cells at concentrations lower than 160 μM. Interestingly, the cytotoxic effect of Dex was significantly blocked by pretreatment of cells with SFP for 2 hours (Figure 2A). For instance, under the treatment of 1 μM Dex, SFP at 20 μM and 10 μM increased the viability of MC3T3-E1 cells from 73% to 92% and 86%, respectively. The cytotoxic effects of Dex were also effectively inhibited by SFP under cotreatment mode (Figure 2B). These results demonstrate that SFP is a novel agent to attenuate the toxicity of Dex to MC3T3-E1 cells.
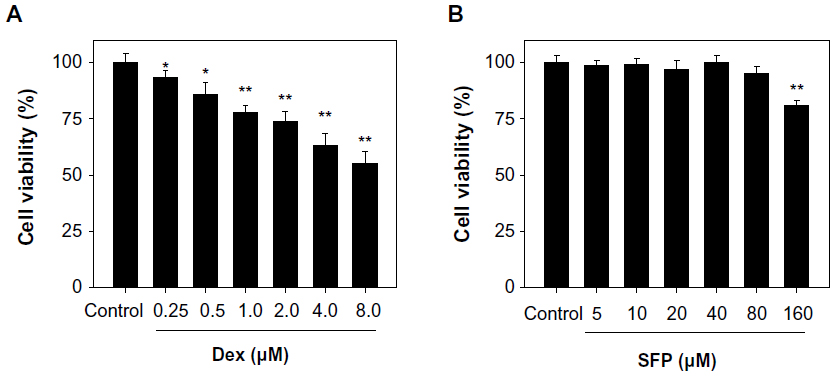 | Figure 1 Cytotoxic effects of dexamethasone (Dex) and sulforaphane (SFP) on MC3T3-E1 cells as determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The cells were treated with Dex and SFP for 24 hours, and the cell viability was examined by MTT assay. |
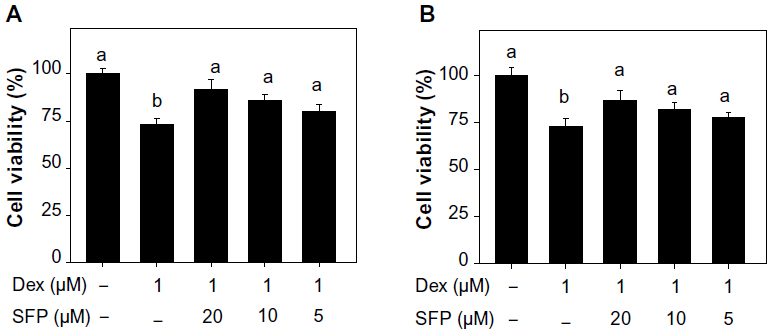 | Figure 2 Sulforaphane (SFP) rescues MC3T3-E1 cells from dexamethasone (Dex)-induced cytotoxicity. |
To understand the action mechanisms of Dex and SFP, we next examined the effects of Dex on the cell cycle distribution of MC3T3-E1 cells and the protection of SFP by DNA flow cytometric analysis. As shown in Figure 3A, treatment of the cells with Dex resulted in a remarkable increase in Sub-G1 cell population from 0.5% (control) to 31.8%, which suggests the induction of apoptosis induced by Dex. However, with cotreatment with SFP, the sub-G1 population was significantly decreased to 5.6%. Meanwhile, cells treated with SFP alone did not change the cell cycle distribution. The inhibition of Dex-induced apoptosis by SFP was further confirmed by TUNEL-DAPI costaining assay. As shown in Figure 3B, treatment with Dex led to the appearance of typical apoptotic features, including DNA fragmentation and chromatin condensation. However, pretreatment of the cells with SFP effectively reduced these changes induced by Dex. Taken together, SFP could reverse Dex-induced cytotoxicity in osteoblasts through inhibition of cell apoptosis.
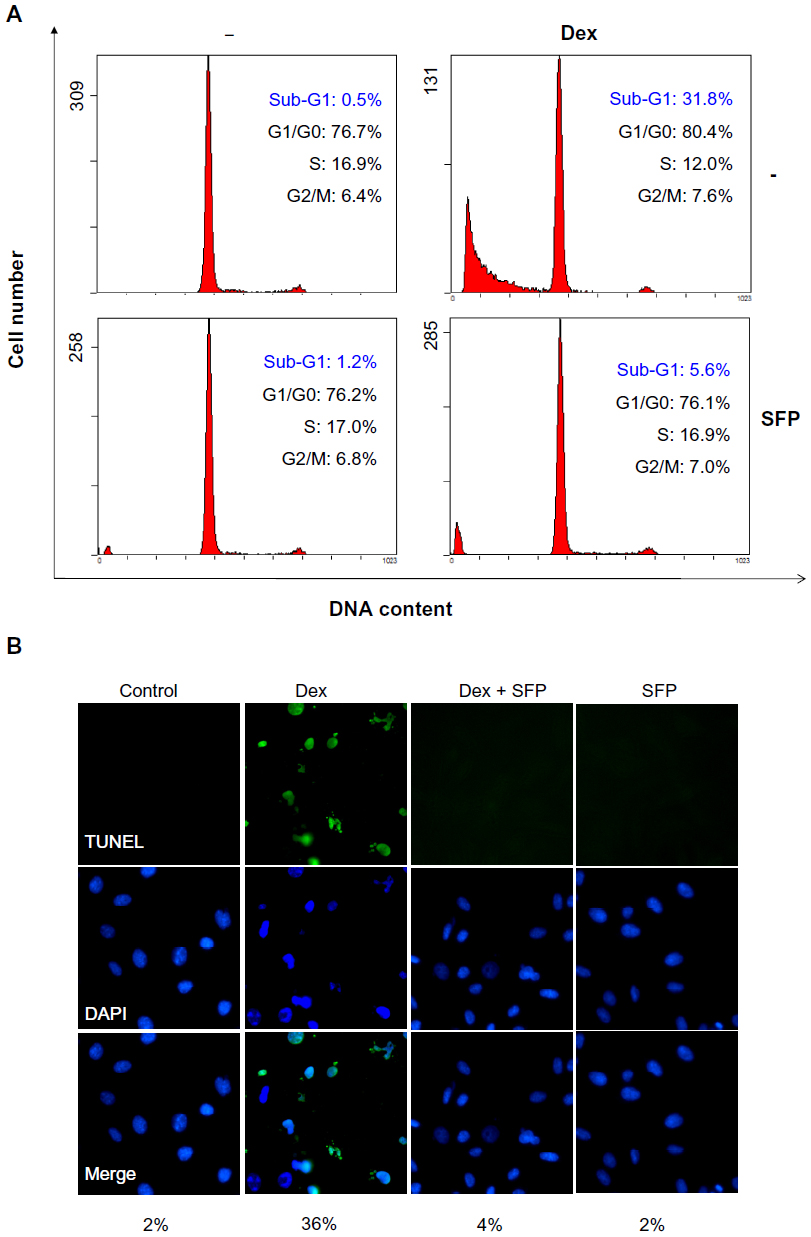 | Figure 3 Protective effects of sulforaphane (SFP) on MC3T3-E1 cells against dexamethasone (Dex)-induced apoptosis as examined by flow cytometric analysis (A) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) 4′,6-diamidino-2-phenylindole (DAPI) (TUNEL-DAPI) costaining assay (B). |
SFP suppresses Dex-induced activation of caspase and poly adenosine diphosphate ribose polymerase
Caspase is a family of endoproteases that control inflammation and cell apoptosis, while poly adenosine diphosphate ribose polymerase (PARP) is a downstream effecter of caspase family proteins in the apoptotic pathways.26 Nowadays, cleavage of PARP after activation of caspase has been identified as a biochemical hallmark of cell apoptosis. In this study, to examine the possible roles of caspases in apoptosis induced by Dex, we have examined the changes in the activities of various initiator and effector caspases. As shown in Figure 4, treatment of the cells with Dex resulted in a significant increase in the activities of caspase-3, -8, and -9. The higher activation degree of caspase-9 than caspase-8 suggests the more important role of the mitochondria-mediated apoptotic pathway in Dex-induced apoptosis. Moreover, the addition of SFP at different concentrations effectively reduced the caspase-3, -8, and -9 activities in a dose-dependent manner. Moreover, as shown in Figure 4D, PARP activity in cells exposed to Dex was increased to 190% compared with the control group. However, cotreatment with SFP at the concentrations of 20 μM, 10 μM, and 5 μM effectively inhibited it to 161%, 132%, and 117%, respectively. Taken together, these results indicate that SFP suppresses Dex-induced apoptosis through inhibition of caspase activation and PARP cleavage, and the intrinsic mitochondrial apoptotic pathway plays an important role in the action of Dex and the protection by SFP.
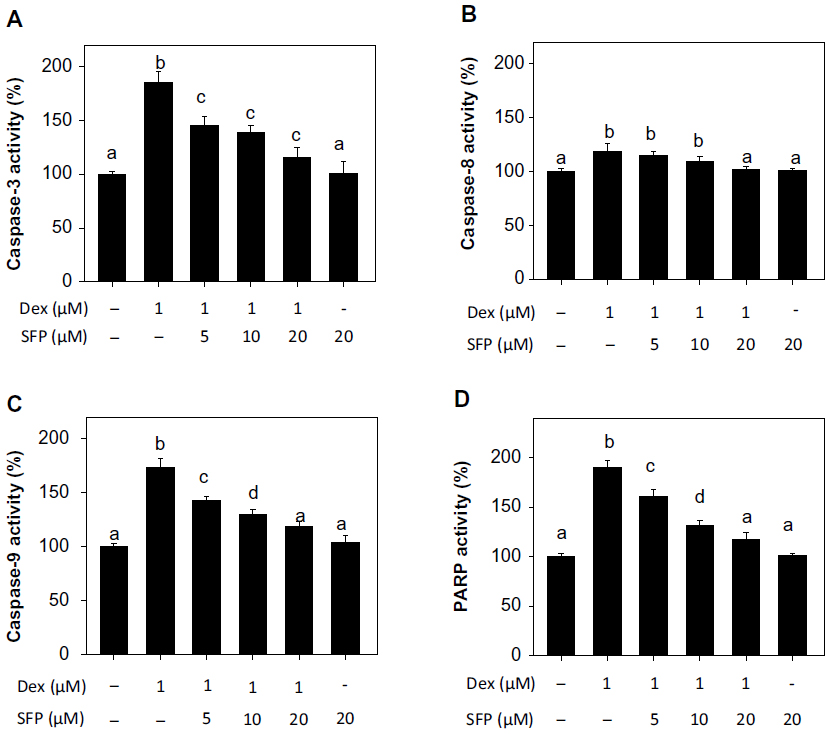 | Figure 4 Sulforaphane (SFP) suppresses dexamethasone (Dex)-induced caspase activation (A, B, and C) and poly adenosine diphosphate ribose polymerase (PARP) cleavage (D) in MC3T3-E1 cells. |
To further evaluate the roles of different caspases in Dex-induced apoptosis, we next examined the effects of various caspase inhibitors, including general caspase inhibitor z-VAD-fmk, caspase-3/7 inhibitor z-DEVD-fmk, caspase-8 inhibitor Z-IETD-fmk, and caspase-9 inhibitor Z-LEHD-fmk, on the cell apoptosis induced by 1 μM Dex. As shown in Figure 5, z-VAD-fmk and z-DEVD-fmk effectively suppressed the Dex-induced sub-G1 apoptotic peak from 32.0% to 6.8% and 9.4%, respectively, indicating that Dex-induced apoptosis occurs in a caspase-dependent manner. Moreover, addition of caspase-9 inhibitor (Z-LEHD-fmk) effectively inhibited cell apoptosis to 13.9%, while caspase-8 inhibitor (Z-IETD-fmk) only slightly decreased cell apoptosis to 27.8%. These results further confirm that mitochondria-mediated apoptosis plays the major role in Dex-induced cell apoptosis, while the contribution of the death receptor-mediated extrinsic apoptosis pathway is insignificant.
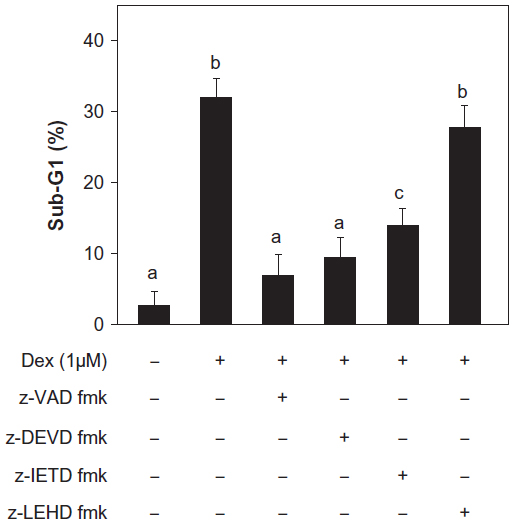 | Figure 5 Effects of various caspase inhibitors on apoptosis induced by dexamethasone (Dex). |
SFP recovers Dex-induced inhibition on osteocalcin, Runx2, and osterin
Osteocalcin is an important bone matrix protein that could be upregulated and activated by Runx227and osterix.28,29 Runx2 and osterix are transcription active factors that play important roles in osteoblastic differentiation and bone formation.27 In this study, to examine the effects of Dex and SFP on the function of MC3T3-E1 cells, we have determined the changes in the expression levels of osteocalcin, Runx2, and osterin in cells exposed to Dex and SFP. As shown in Figure 6, treatment of the cells with 1 μM Dex markedly reduced the expression levels of osteocalcin. Interestingly, this effect of Dex was significantly attenuated by the addition of SFP. Moreover, Dex also significantly downregulated the levels of Runx2 and osterix, and protective effects were also observed by the addition of SFP. Taken together, SFP could recover Dex-induced inhibition on osteocalcin, Runx2, and osterin in MC3T3-E1 cells.
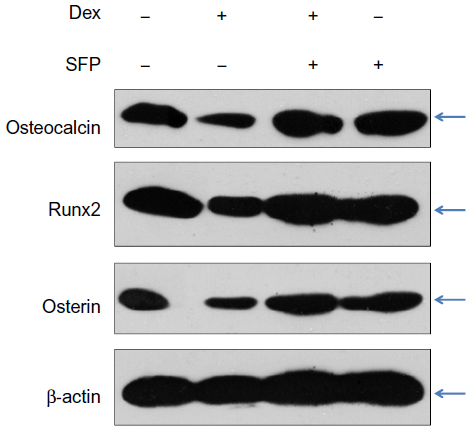 | Figure 6 Effects of dexamethasone (Dex) and sulforaphane (SFP) on the expression levels of osteocalcin, Runx2, and osterin. |
SFP suppresses Dex-induced ROS overproduction by regulation of Nrf2 pathways
Many studies have shown that ROS plays an important role in the signaling pathways triggered by GCs.12–15,30 Therefore, in this study we examined the intracellular levels of ROS in cells exposed to Dex by DCFH-DA fluorescence assay. As shown in Figure 7A, cells treated with 1 μM Dex displayed a rapid increase in intracellular ROS generation. However, cotreatment with SFP at different concentrations effectively reduced the intracellular ROS level. Likewise, the levels of superoxide in cells treated with Dex were also significantly higher than those treated in combination with SFP. Taken together, these results suggest that SFP suppresses Dex-induced ROS overproduction in MC3T3-E1 cells.
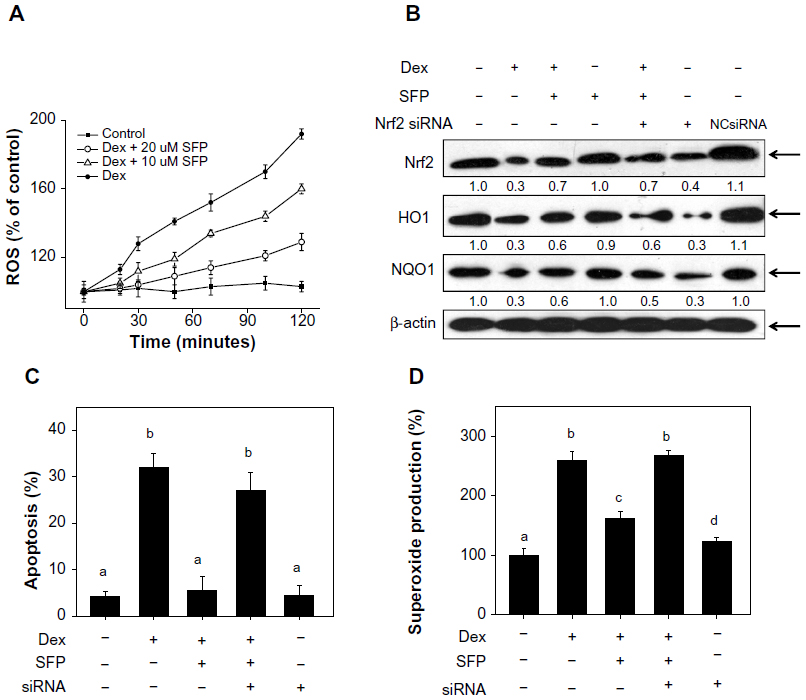 | Figure 7 Knockdown of nuclear factor erythroid 2-related factor 2 (Nrf2) blocks the reactive oxygen species (ROS) generation and cytoprotective effects of sulforaphane (SFP) on dexamethasone (Dex)-induced apoptosis. |
The Keap1-Nrf2 pathway is an important cytoprotective regulator in mammalian cells in response to endogenous and exogenous stresses.31 Previous studies have demonstrated the important role of Nrf2 signaling in the action of SFP.18–20 Therefore, we have examined the effects of Dex and SFP on the expression level of Nrf2 in MC3T3-E1 cells by Western blotting. As shown in Figure 7B, Dex significantly inhibited the Nrf2 signaling pathway, as revealed by the decrease in the expression level of Nrf2 and its downstream effectors NQO1 and HO1, but SFP alone did not alter the expression levels of these proteins. However, cotreatment of the cells with SFP in combination with Dex strongly suppressed the inhibitory effects of Dex on the Nrf2 signaling pathway, as demonstrated by the increase in the expression levels of Nrf2, NQO1, and HO1. The results suggest that the Nrf2 pathway may be involved in Dex-induced apoptosis and the protection of SFP. To clarify the involvement of the Nrf2 pathway in Dex-induced apoptosis, we silenced this gene by using a small interfering ribonucleic acid (siRNA) technique and examined its effects on the cytoprotection of SFP against Dex-induced apoptosis. As shown in Figure 7B, knockdown of Nrf2 significantly reduced the protein level of Nrf2 elevated by SFP. It also inhibited the upregulation of NQO1 and HO1 by SFP in Dex-treated cells. These results indicate that Nrf2 is the key factor in the cellular oxidative stress induced by Dex.
To confirm the importance of Nrf2 to the protective effects of SFP against Dex-induced apoptosis, we further examined the effects of Nrf2 knockdown on cell apoptosis. As shown in Figure 7C, knockdown of Nrf2 almost completely blocked the cytoprotective effects of SFP on Dex-induced apoptosis. For instance, SFP suppressed Dex-induced apoptotic cell death from 32% to 5%, which was recovered to 27% by Nrf2 knockdown. Similarly, SFP suppressed Dex-induced superoxide production from 259% to 162%, which was recovered to 268% by Nrf2 knockdown (Figure 7D). Taken together, these results suggest that Nrf2 is crucial for the protective effects of SFP against Dex-induced apoptosis.
Discussion
Osteoporosis is a very common human bone disease affecting the life quality of millions of people all over the world. GCs are widely used for their unsurpassed anti-inflammatory and immunomodulatory effects. However, the therapeutic use of GCs is almost always limited by substantial adverse outcomes such as osteoporosis, diabetes, and obesity. These unwanted outcomes are a major dilemma for clinicians because improvements in the primary disorder seem to be achievable only by accepting substantial adverse effects that are often difficult to prevent or treat.4 GC-induced osteoporosis is regarded as an important cause of osteoporosis.5,6 The search for naturally occurring phytochemicals from edible materials (especially fruits and vegetables) that can antagonize osteoporosis has gained more and more attention. Studies have found that tetrahydroxystilbene glucoside, dehydrocostus lactone, puerarin, and chemical constituents of the fruits of Prunus mume could reverse GC-induced apoptosis in osteoblasts by regulation of various signaling pathways.12–15 In this study we found that SFP, a naturally occurring isothiocyanate that has huge health benefits for humans, could reverse GC-induced apoptosis in osteoblastic cells through regulation of the Nrf2 pathway. This study may provide a novel strategy for molecular intervention against Dex-induced osteoporosis by using phytochemicals.
Apoptosis of osteoblasts triggered by high-dose GCs has been identified as a major cause of osteoporosis. However, the underlying molecular mechanisms accounting for this action remain elusive, which has impeded the prevention and cure of this side effect. In this study we found that SFP effectively antagonized Dex-induced apoptosis of osteoblastic cells. Firstly, we analyzed the underlying mechanisms accounting for these effects. From the results of MTT assay, cotreatment of the cells with SFP in combination with Dex can reduce the cytotoxicity of Dex in MC3T3-E1 cells. The results of flow cytometric analysis and DAPI-TUNEL staining assay proved that SFP blocked Dex-induced apoptosis in MC3T3-E1 cells with the inhibition of caspase activation and PARP cleavage. Caspase-3 was considered to be a central mediator of cell apoptosis pathway, while caspase-8 and -9 act as initiators of extrinsic (death receptor-mediated) and intrinsic (mitochondria-mediated) apoptotic pathways, respectively. The higher activation degree of caspase-9 than caspase-8 suggests the more important role of the mitochondria-mediated apoptotic pathway in Dex-induced apoptosis. Moreover, cotreatment of the cells with different concentrations of SFP effectively reduced the caspase-3, -8, and -9 activities in a dose-dependent manner. These results suggest that mitochondria-mediated apoptosis plays an important role in the protective action of SFP. To further evaluate the roles of the different caspases in Dex-induced apoptosis, we next examined the effects of various caspase inhibitors. The results showed that Dex-induced apoptosis occurs in a caspase-dependent manner. Moreover, mitochondria-mediated apoptosis plays the major role in Dex-induced cell apoptosis, while the contribution of the death receptor-mediated extrinsic apoptosis pathway is insignificant.
To examine the effects of Dex and SFP on the function of MC3T3-E1 cells, we determined the changes in the expression levels of osteocalcin, Runx2, and osterin. The results showed that treatment of the cells with Dex markedly reduced the expression levels of osteocalcin, which was significantly attenuated by SFP. Interestingly, Dex also significantly downregulated the levels of Runx2 and osterix, and protective effects were also observed by the addition of SFP. These data suggest that SFP may block cell apoptosis induced by GCs and recover the function of MC3T3-E1 cells.
Oxidative stress is a result of overproduction of ROS, which could induce apoptosis and decrease activities of osteoblasts. Bone mass is kept balanced by bone remodeling via regulation of bone formation and resorption.32 A recent study showed that ROS could enhance bone resorption through promotion of osteoclast formation and an increase in their activities.33 Moreover, ROS also induced apoptosis of osteoblasts and led to a reduction of osteoblast differentiation and bone formation.30 Therefore, administration of antioxidants that could scavenge excess intracellular ROS could be a potential approach for treatment of osteoporosis. In this study we found that SFP could effectively suppress Dex-induced cytotoxicity in osteoblastic cells through inhibition of intracellular ROS generation and regulation of the Nrf2 pathway.
Previous studies have reported that Nrf2 could protect human cells from oxidative and electrophilic damage.27,28 In this study, due to the detection of ROS in MC3T3-E1 cells by Dex, the changes in the expression level of Nrf2 and two representative target genes NQO1 and HO1 in cells exposed to Dex and SFP were examined by Western blotting. Dex significantly inhibited the Nrf2 signaling pathway, as revealed by the decrease in the expression level of Nrf2 and its downstream effectors NQO1 and HO1. However, cotreatments of the cells with SFP strongly suppressed the inhibitory effects of Dex on the Nrf2 signaling pathway. To further clarify the importance of Nrf2, we examined the effects of Nrf2 knockdown on the cytoprotection of SFP against Dex-induced apoptosis. The results showed that knockdown of Nrf2 almost completely blocked the cytoprotective effects of SFP on Dex-induced apoptosis and ROS overproduction. Overall, we conclude that Nrf2 plays an important role in the protective effects of SFP against Dex-induced apoptosis.
Conclusion
In summary, the present study demonstrates the protective effects of SFP against Dex-induced cytotoxicity, and elucidates the underlying molecular mechanisms. Specifically, treatment of the cells with SFP attenuated Dex-induced caspase activation, PARP cleavage, mitochondrial dysfunction, and DNA damage. These effects were mainly mediated by regulation of the Nrf2 pathway to inhibit the production of ROS. Taken together, this study provides a novel strategy for molecular intervention against Dex-induced osteoporosis using phytochemicals.
Acknowledgments
This study was supported by grants from the Natural Science Foundation of Guangdong Province, People’s Republic of China (No S2013020012866), the Science and Technology Planning Project of Guangdong Province, People’s Republic of China (No 2011B031800212), the Foundation for Youth Scholars of Guangdong Medical College, People’s Republic of China (No XQ1322), and the Science and Technology Development Planning Project of Zhanjiang, People’s Republic of China (No 2012C3101026).
Disclosure
The authors report no conflicts of interest in this work.
References
Hsu WL, Chen CY, Tsauo JY, Yang RS. Balance control in elderly people with osteoporosis. J Formos Med Assoc. 2014;113(6):334–339. | |
Reid IR. Anti-resorptive therapies for osteoporosis. Semin Cell Dev Biol. 2008;19(5):473–478. | |
Salam SN, Eastell R, Khwaja A. Fragility fractures and osteoporosis in CKD: pathophysiology and diagnostic methods. Am J Kidney Dis. 2014;S0272–6386(14):499–495. | |
Seibel MJ, Cooper MS, Zhou H. Glucocorticoid-induced osteoporosis: mechanisms, management, and future perspectives. Lancet Diabetes Endocrinol. 2013;1(1):59–70. | |
Brennan-Speranza TC, Henneicke H, Gasparini SJ, et al. Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. J Clin Invest. 2012;122(11):4172–4189. | |
Rauch A, Seitz S, Baschant U, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010;11(6):517–531. | |
den Uyl D, Bultink IE, Lems WF. Advances in glucocorticoid-induced osteoporosis. Curr Rheumatol Rep. 2011;13(3):233–240. | |
Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365(1):62–70. | |
Espina B, Liang M, Russell RG, Hulley PA. Regulation of Bim in glucocorticoid-mediated osteoblast apoptosis. J Cell Physiol. 2008;215(2):488–496. | |
O’Brien CA, Jia D, Plotkin LI, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145(4):1835–1841. | |
Yun SI, Yoon HY, Jeong SY, Chung YS. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3beta. J Bone Miner Metab. 2009;27(2):140–148. | |
Choi EM, Kim GH, Lee YS. Protective effects of dehydrocostus lactone against hydrogen peroxide-induced dysfunction and oxidative stress in osteoblastic MC3T3-E1 cells. Toxicol In Vitro. 2009;23(5):862–867. | |
Wang Y, Wang WL, Xie WL, et al. Puerarin stimulates proliferation and differentiation and protects against cell death in human osteoblastic MG-63 cells via ER-dependent MEK/ERK and PI3K/Akt activation. Phytomedicine. 15, 2013;20(10):787–796. | |
Yan XT, Lee SH, Li W, et al. Evaluation of the antioxidant and anti-osteoporosis activities of chemical constituents of the fruits of Prunus mume. Food Chem. 2014;156:408–415. | |
Zhang JK, Yang L, Meng GL, et al. Protective effect of tetrahydroxystilbene glucoside against hydrogen peroxide-induced dysfunction and oxidative stress in osteoblastic MC3T3-E1 cells. Eur J Pharmacol. 2012;689(1–3):31–37. | |
Wiseman M. The second World Cancer Research Fund/American Institute for Cancer Research expert report. Food, nutrition, physical activity, and the prevention of cancer: a global perspective. Proc Nutr Soc. 2008;67(3):253–256. | |
Gupta P, Kim B, Kim SH, Srivastava SK. Molecular targets of isothiocyanates in cancer: recent advances. Mol Nutr Food Res. Epub February 10, 2014. | |
Houghton CA, Fassett RG, Coombes JS. Sulforaphane: translational research from laboratory bench to clinic. Nutr Rev. 2013;71(11):709–726. | |
Xu T, Ren D, Sun X, Yang G. Dual roles of sulforaphane in cancer treatment. Anticancer Agents Med Chem. 2012;12(9):1132–1142. | |
Tarozzi A, Angeloni C, Malaguti M, Morroni F, Hrelia S, Hrelia P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxid Med Cell Longev. 2013;2013:415078. | |
Barcelo S, Gardiner JM, Gescher A, Chipman JK. CYP2E1-mediated mechanism of anti-genotoxicity of the broccoli constituent sulforaphane. Carcinogenesis. 1996;17(2):277–282. | |
Gross-Steinmeyer K, Stapleton PL, Tracy JH, Bammler TK, Strom SC, Eaton DL. Sulforaphane- and phenethyl isothiocyanate-induced inhibition of aflatoxin B1-mediated genotoxicity in human hepatocytes: role of GSTM1 genotype and CYP3A4 gene expression. Toxicol Sci. 2010;116(2):422–432. | |
Katoch O, Kumar A, Adhikari JS, Dwarakanath BS, Agrawala PK. Sulforaphane mitigates genotoxicity induced by radiation and anticancer drugs in human lymphocytes. Mutat Res. 2013;758(1–2):29–34. | |
Fan C, Chen J, Wang Y, et al. Selenocystine potentiates cancer cell apoptosis induced by 5-fluorouracil by triggering reactive oxygen species-mediated DNA damage and inactivation of the ERK pathway. Free Radic Biol Med. 2013;65:305–316. | |
Su J, Lai H, Chen J, et al. Natural borneol, a monoterpenoid compound, potentiates selenocystine-induced apoptosis in human hepatocellular carcinoma cells by enhancement of cellular uptake and activation of ROS-mediated DNA damage. PLoS One. 2013;8(5):e63502. | |
McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. | |
Xu ZS, Wang XY, Xiao DM, et al. Hydrogen sulfide protects MC3T3-E1 osteoblastic cells against H2O2-induced oxidative damage-implications for the treatment of osteoporosis. Free Radic Biol Med. 2011;50(10):1314–1323. | |
Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. | |
Komori T. Regulation of osteoblast differentiation by transcription factors. J Cell Biochem. 2006;99(5):1233–1239. | |
Arai M, Shibata Y, Pugdee K, Abiko Y, Ogata Y. Effects of reactive oxygen species (ROS) on antioxidant system and osteoblastic differentiation in MC3T3-E1 cells. IUBMB Life. 2007;59(1):27–33. | |
Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1(1):45–49. | |
Seeman E, Delmas PD. Bone quality: the material and structural basis of bone strength and fragility. N Engl J Med. 2006;354(21):2250–2261. | |
Lee NK, Choi YG, Baik JY, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005; 106(3):852–859. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.