Back to Journals » Journal of Inflammation Research » Volume 18
Succinate’s Dual Roles in Inflammatory Bowel Disease: A Narrative Review of Microbiota-Metabolism-Immune Crosstalk and Therapeutic Implications
Authors Zhao M, Zhou C, Wang D, Wu Q, Feng B
Received 25 August 2025
Accepted for publication 13 October 2025
Published 28 October 2025 Volume 2025:18 Pages 15017—15032
DOI https://doi.org/10.2147/JIR.S561871
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Nadia Andrea Andreani
Meihua Zhao,1 Chuan Zhou,1 Dandan Wang,1 Qiong Wu,2 Baisui Feng1
1Department of Gastroenterology, The Second Affiliated Hospital of Zhengzhou University, Zhengzhou, People’s Republic of China; 2Department of Pathology, The Third Affiliated Hospital of Zhengzhou University, Zhengzhou, People’s Republic of China
Correspondence: Baisui Feng, Department of Gastroenterology, The Second Affiliated Hospital of Zhengzhou University, No. 2 Courtyard, Jingba Road, Zhengzhou, Henan, People’s Republic of China, Email [email protected]
Abstract: Inflammatory bowel disease (IBD) is a chronic intestinal condition characterized by microbial dysbiosis, metabolic alterations, and immune dysregulation. Succinate, a key tricarboxylic acid (TCA) cycle intermediate and microbiota-derived metabolite, has emerged as a central regulator within the host microbiota-metabolism-immune axis in IBD. This narrative review delineates succinate metabolism and its dual signaling roles, operating both intracellularly and through extracellular receptors. We synthesize evidence on its context-dependent immunomodulatory functions, which can paradoxically drive both pro- and anti-inflammatory responses, and elucidate the specific factors that dictate these outcomes. Finally, we critically evaluate the translational potential of targeting the succinate pathway, outlining promising avenues for future research in IBD diagnosis and treatment.
Keywords: succinate, inflammatory bowel disease, immune regulation, microbiota-metabolism-immune axis, precision medicine
Introduction
Inflammatory bowel disease (IBD), which primarily includes Crohn’s disease (CD) and ulcerative colitis (UC), is a group of chronic, non-specific intestinal inflammatory disorders with an incompletely understood etiology.1 Clinically, it often presents with abdominal pain, diarrhea, fatigue, and weight loss, with some patients also exhibiting extraintestinal manifestations such as oral ulcers, osteoarticular lesions, dermatological manifestations, and ocular lesions. The course of IBD is characterized by alternating remission and active phases of varying durations, following a chronic relapsing and remitting pattern. Epidemiological studies show that approximately 0.3% to 0.5% of the global population suffers from IBD, and its incidence has been consistently rising in recent years.2 With disease progression, certain patients with IBD experience complications including intestinal stenosis, obstruction, hemorrhage, perforation, and occasionally malignant transformation, usually requiring surgical treatment, which significantly affects patient quality of life and imposes substantial economic burdens on families and society. Therefore, exploring novel diagnostic biomarkers and effective therapeutic strategies is of great significance.
The traditional view holds that the pathogenesis of IBD is primarily associated with genetic susceptibility, environmental factors, infection, and immune dysregulation. However, with the widespread application of high-throughput sequencing technologies, the role of the gut microbiota and its metabolites in the pathogenesis of IBD has gained increasing attention.3–5 Studies have demonstrated significant dysbiosis in the gut microbiota of IBD patients, marked by reduced microbial diversity and irregular shifts in certain bacterial populations.6–9 These alterations in microbial composition disrupt the intestinal metabolic microenvironment, resulting in reduced synthesis of short-chain fatty acids (SCFAs) and an abnormal buildup of metabolites like succinate. The abnormal changes in intestinal metabolites further establish a unique dynamic regulatory network of “microbiota-metabolism-immunity” by modulating mitochondrial function in intestinal epithelial cells, inducing metabolic reprogramming of immune cells, and affecting epigenetic modifications, ultimately profoundly influencing the pathophysiological progression of IBD.10–13
Recent studies have revealed that succinate plays a significant role in the development of various digestive system diseases, including IBD, as well as in cardiovascular diseases, metabolic disorders, and tumors.11,14–17 Present studies on IBD largely focus on the pro-inflammatory roles of succinate, while its anti-inflammatory effects remain underexplored. In this review, we systematically outline the metabolic features and signal transduction pathways of succinate and elucidate its dual immunomodulatory functions in IBD based on the emerging concept of the “microbiota-metabolism-immunity” axis. Lastly, we will focus on the clinical translation potential of succinate, discussing individualized therapeutic approaches targeting succinate and its applications in the diagnosis and precision medicine of IBD.
The Generation, Degradation, and Transport of Succinate
Succinate, a central metabolite in the gut microenvironment, functions as a critical nexus in the “microbiota-metabolism-immune” axis and significantly influences the onset and progression of IBD. It originates from two primary sources: the host’s intracellular tricarboxylic acid (TCA) cycle and the metabolic activities of the gut microbiota (Figure 1). This section will delineate the cellular and microbial origins of succinate, its subsequent metabolism, and its dynamic transport processes across cellular membranes.
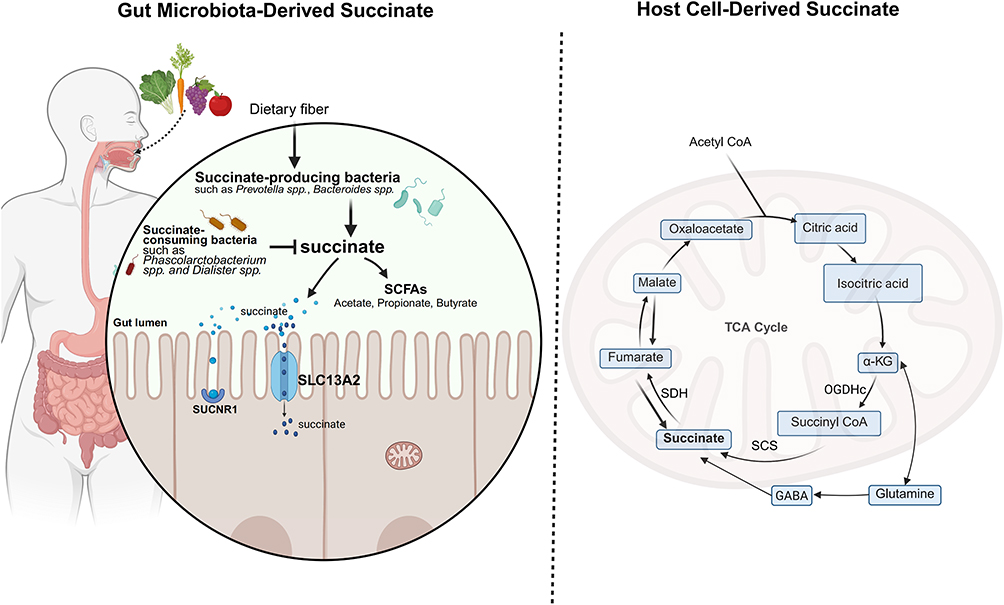 |
Figure 1 Dual Sources of Succinate in IBD. The diagram illustrates the key bacterial taxa, metabolic pathways, and transporters (SLC13A2) involved. In IBD, disruption of the balance between succinate-producing and consuming bacteria, coupled with host metabolic dysregulation, subsequently modulates intestinal immune responses. Created in BioRender. Zhao, M. (2025) https://BioRender.com/0fhmdfk. |
Host Cell-Derived Succinate
In host cells, succinate is predominantly generated through the classical TCA cycle.18 The TCA cycle, also known as the citric acid or Krebs cycle, is a key aerobic metabolic pathway. It occurs in the mitochondrial matrix and is essential for producing energy during cellular respiration. Through a series of enzymatic reactions, this cycle completely oxidizes metabolic products of carbohydrates, fats, and proteins into carbon dioxide and water, releasing substantial energy for Adenosine Triphosphate (ATP) synthesis.19 Within the cycle, α-ketoglutarate (α-KG) transforms into succinyl-CoA via the α-ketoglutarate dehydrogenase complex (OGDHc). Succinyl-CoA is then transformed into succinate by succinyl-CoA synthetase (SCS), and the resulting succinate undergoes further oxidation to fumarate by succinate dehydrogenase (SDH).20 Moreover, intracellular succinate can also be produced through the metabolism of glutamate via the γ-aminobutyric acid (GABA) shunt pathway.14
Gut Microbiota-Derived Succinate
In the gut microenvironment, resident microbiota synthesize succinate by fermenting undigestible carbs like dietary fiber, a process that yields SCFAs. SCFAs are organic fatty acids containing 1 to 6 carbon atoms, with acetate, propionate, and butyrate as the primary forms. The total concentration of SCFAs in the colonic lumen is approximately 60–150 mmol/L.21 The biological functions of SCFAs are primarily mediated through activation of specific G protein-coupled receptors (GPCRs) and inhibition of histone deacetylase (HDAC) activity. In the pathogenesis of IBD, SCFAs play a protective role through several potential mechanisms, such as strengthening intestinal barrier integrity, regulating immune balance, and inhibiting excessive inflammatory responses. The main synthetic pathways of succinate in the intestinal lumen include the succinate-propionate pathway, succinate-acetate pathway, and succinate-butyrate pathway.22–24 As an example, taking the succinate-propionate pathway, certain gut bacteria first convert carbohydrates into phosphoenolpyruvate (PEP). In the presence of carbon dioxide (CO2), PEP is catalyzed by phosphoenolpyruvate carboxykinase (PEPCK) to form oxaloacetate (OAA). OAA is then sequentially reduced to succinate through enzymatic reactions involving malate dehydrogenase and fumarate reductase. Finally, with the participation of vitamin B12, succinate is converted through succinyl-CoA into methylmalonate (MMA), which is then further converted into propionate.22,23
Within the gut microbiota, Prevotella spp., Bacteroides spp., and members of the Veillonellaceae family are the primary producers of succinate.25–27 Conversely, Phascolarctobacterium spp. and Dialister spp. are key succinate-consuming bacteria. Importantly, succinate-producing and -consuming bacteria form a dynamic “succinate metabolic axis” via cross-feeding interactions to sustain gut homeostasis.28,29 Under normal physiological conditions, succinate levels in the gut lumen and feces are typically maintained at approximately 1 to 3 μM. Under inflammatory conditions like IBD, mitochondrial dysfunction in host cells, combined with gut microbiota dysbiosis, results in the abnormal accumulation of succinate both within host cells and the intestinal lumen. This leads to concentrations that are significantly elevated to 7–25 mM, which are thousands of times higher than those found in healthy individuals.20,30
The Dynamic Exchange of Succinate Inside and Outside the Cell
Succinate can dynamically exchange between intracellular and extracellular spaces; however, as a negatively charged dicarboxylic acid, it cannot freely diffuse across the cell membrane or mitochondrial inner membrane, and its transmembrane transport is strictly dependent on specific transport proteins.31–33 When succinate moves from the mitochondrial matrix to the extracellular space, it first enters the cytosol through the dicarboxylate carrier (DIC, SLC25A10), positioned within the mitochondrial inner membrane. Following this, the voltage-dependent anion channel (VDAC) enables the movement across the mitochondrial outer membrane into the cytoplasm, where succinate can be secreted into the extracellular fluid via organic anion transporters (OATs) or the monocarboxylate transporter 1 (MCT1). Extracellular succinate can then be taken up into cells via sodium-dependent dicarboxylate transporters (SLC13 family) or MCT1, thereby completing its dynamic intracellular-extracellular cycling.34,35 It is important to note that this SLC13-mediated transport underpins a critical transcellular pathway at the tissue level. Specifically, in the gut, luminal succinate derived from the microbiota is absorbed by epithelial cells via apical SLC13 transporters (eg, SLC13A2) and delivered to the lamina propria. There, macrophages enhance their uptake of succinate via the basolateral transporter SLC13A3, a process fine-tuned by SLC26A6 and SUCNR1. This microbiota-epithelium-macrophage succinate axis drives pro-inflammatory polarization in macrophages, positioning the SLC13 transport family as a central player in metabolic-immune communication, particularly in the context of inflammatory bowel diseases.31
The Signaling Pathways of Succinate
Succinate plays a pivotal role in the onset and progression of IBD. Its signaling functions are broadly categorized into intracellular and extracellular mechanisms, which, together, orchestrate a range of pro-inflammatory and anti-inflammatory responses in the intestinal microenvironment (Figure 2).
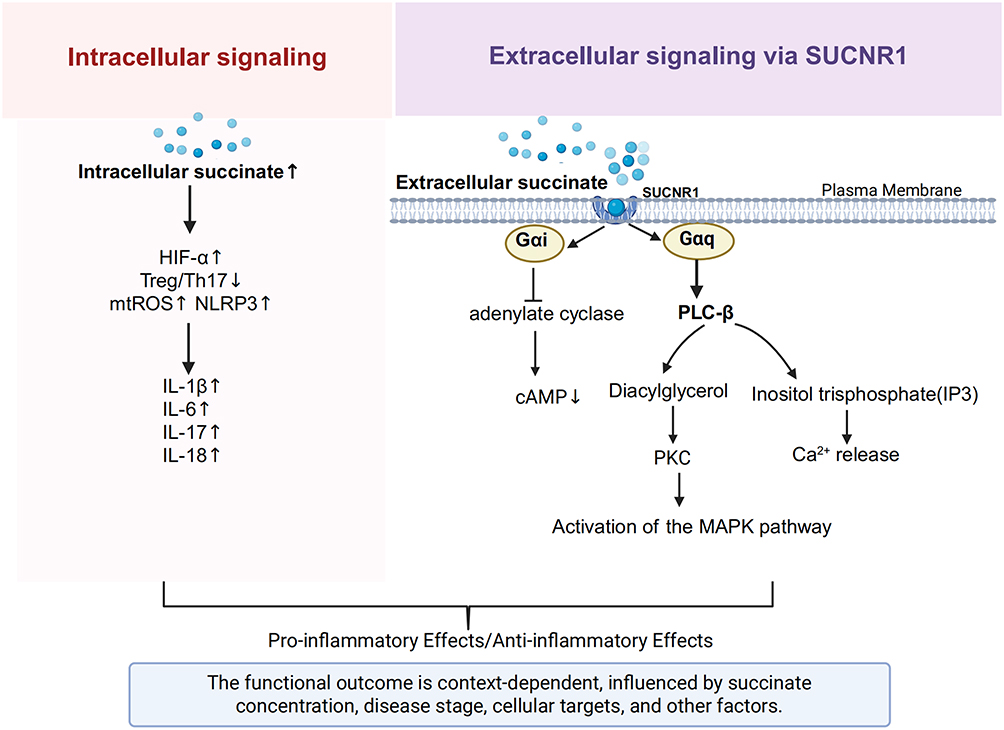 |
Figure 2 Intracellular and extracellular signaling pathways of succinate in IBD. Succinate modulates IBD inflammation by acting both inside and outside the cell. Intracellularly, it stabilizes HIF-1α, disrupts the Treg/Th17 balance, and promotes inflammasome activation. Extracellularly, by activating SUCNR1, it triggers Gαi and Gαq signaling, mobilizing PKC and MAPK effectors. The ultimate immune outcome is context-dependent. Created in BioRender. Zhao, M. (2025) https://BioRender.com/7bjvvwc. |
Intracellular Signaling of Succinate
Regarding intracellular mechanisms, succinate regulates several inflammation-associated signaling pathways via metabolic reprogramming, primarily involving the stabilization of the hypoxia-inducible factor-1α (HIF-1α) pathway, disrupting the equilibrium between regulatory T (Treg) cells and T helper 17 (Th17) cells, and inducing mitochondrial oxidative stress along with inflammasome activation. Specifically, intracellular succinate accumulation inhibits prolyl hydroxylase (PHD) activity, stabilizing HIF-1α and promoting its nuclear translocation.36 Activated HIF-1α subsequently boosts transcription of pro-inflammatory cytokines like interleukin-1β (IL-1β) and interleukin-6 (IL-6), promotes glycolysis, and disrupts the function of the intestinal mucosal barrier.37–39 Succinate within cells can also inhibit the expression of OGDHc, decreasing the synthesis of succinyl-CoA, leading to decreased succinylation of forkhead box protein P3 (FOXP3), the critical Treg cell transcription factor, thereby exposing its lysine residues and facilitating ubiquitin-dependent proteasomal degradation. FOXP3 deficiency leads to the loss of immunosuppressive function in Treg cells, causing them to shift towards a Th17-like phenotype with increased secretion of interleukin-17 (IL-17), which aggravates intestinal inflammation.40 Additionally, intracellular succinate accumulation competitively inhibits SDH activity, inducing mitochondrial dysfunction and increasing mitochondrial reactive oxygen species (mtROS) generation. These mtROS function as essential second messengers to trigger the activation of the NOD-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome, promoting maturation and secretion of pro-inflammatory cytokines including IL-1β and interleukin-18 (IL-18), thereby amplifying the inflammatory response.14,41
Extracellular Signaling of Succinate
On the extracellular signaling front, succinate mediates regulation via its specific receptor, succinate receptor 1 (SUCNR1), also referred to as G protein-coupled receptor 91 (GPR91).42 SUCNR1 is extensively expressed across different tissues and cells, including the kidney, liver, spleen, adipose tissue, and intestine, as well as in specialized cells such as intestinal epithelial cells, macrophages, dendritic cells, and T cells.43,44 Notably, SUCNR1 expression is markedly increased in the intestinal tissues of patients with CD who have penetrating lesions (B3 type), particularly in areas surrounding the fistula.45 As a G protein-coupled receptor, SUCNR1 can differentially activate Gαq and Gαi signaling pathways, with its signal output highly dependent on the cell’s metabolic state and receptor subcellular localization. Stimulation of the Gαq pathway triggers phosphoinositide hydrolysis mediated by phospholipase Cβ (PLCβ), leading to the release of intracellular calcium ions (Ca²⁺). This subsequently initiates the protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) signaling cascades. In contrast, Gαi pathway activation inhibits adenylate cyclase, reducing cyclic adenosine monophosphate (cAMP) concentrations.42,44 Furthermore, recent studies indicate that SUCNR1 signaling can also exert key immunoregulatory roles by activating the interleukin-4 receptor α subunit (IL-4Rα)/HIF-1α axis on the surface of myeloid cells.46 Succinate activates intestinal lamina propria macrophages and dendritic cells through SUCNR1 binding, upregulating the CCL2/CCR2 chemokine axis to promote monocyte recruitment to sites of inflammation.46 In intestinal epithelial cells, succinate engagement of SUCNR1 activates Wnt/β-catenin signaling. This activation drives epithelial-mesenchymal transition (EMT), a process strongly implicated in CD-associated fistula pathogenesis. Simultaneously, the succinate-SUCNR1 signaling axis can also directly promote intestinal fibroblast proliferation and collagen deposition, collectively mediating IBD-associated intestinal fibrosis.15,45 Moreover, gut microbiota-derived succinate activates T helper 9 cells (Th9) through the SUCNR1/HIF-1α axis, stimulating substantial IL-9 secretion, which disrupts the intestinal epithelial barrier function, ultimately exacerbating colonic inflammation.29
Interactions Between Succinate and Other Intestinal Metabolites
Intestinal metabolites are diverse, and succinate, as one of them, exhibits complex interactions with SCFAs, secondary bile acids (SBAs), tryptophan metabolites, and others, forming an intricately regulated metabolic network.47,48 Research indicates that SCFAs like butyrate play a key role in immune regulation by stimulating the development of Treg cells and enhancing epithelial barrier function. This occurs through the activation of G protein-coupled receptors 41 and 43 (GPR41/43), as well as the suppression of HDAC activity. As a result, butyrate helps counteract some of the inflammatory responses triggered by succinate.49 There is a notable correlation between succinate and secondary bile acids. In IBD patients, the levels of both succinate and deoxycholic acid (DCA) are significantly increased. Succinate signals through its receptor SUCNR1, whereas DCA activates G protein-coupled bile acid receptor 1 (GPBAR1/TGR5) and farnesoid X receptor (FXR), jointly promoting Th17 differentiation and intensifying pro-inflammatory responses. Conversely, lithocholic acid (LCA) and its metabolite 12-keto lithocholic acid (12-KLCA) significantly inhibit secretion of interleukin-17A (IL-17A) by colonic group 3 innate lymphoid cells (ILC3s) through activating vitamin D receptor (VDR) and pregnane X receptor (PXR), thereby antagonizing succinate’s pro-inflammatory effect.50 Moreover, the tryptophan metabolite kynurenine is also significantly elevated in patients with active IBD and, similar to succinate, can suppress Treg cell function. However, indole derivatives produced by probiotic metabolism of tryptophan, such as indole-3-aldehyde (I3A), enhance epithelial barrier function and induce anti-inflammatory interleukin-22 (IL-22) expression through the activation of the aryl hydrocarbon receptor (AhR), thus exacerbating profibrotic effects via the succinate-SUCNR1 pathway.11 The complex interaction network among these metabolites provides an important theoretical basis for developing combination therapies for IBD, especially the joint use of probiotics and SUCNR1 antagonists.27
The Dual Role of Succinate in IBD
The role of succinate in IBD is complex and context-dependent, with substantial evidence supporting its pro-inflammatory actions, while emerging research reveals specific scenarios where it may facilitate anti-inflammatory and repair processes (Table 1).
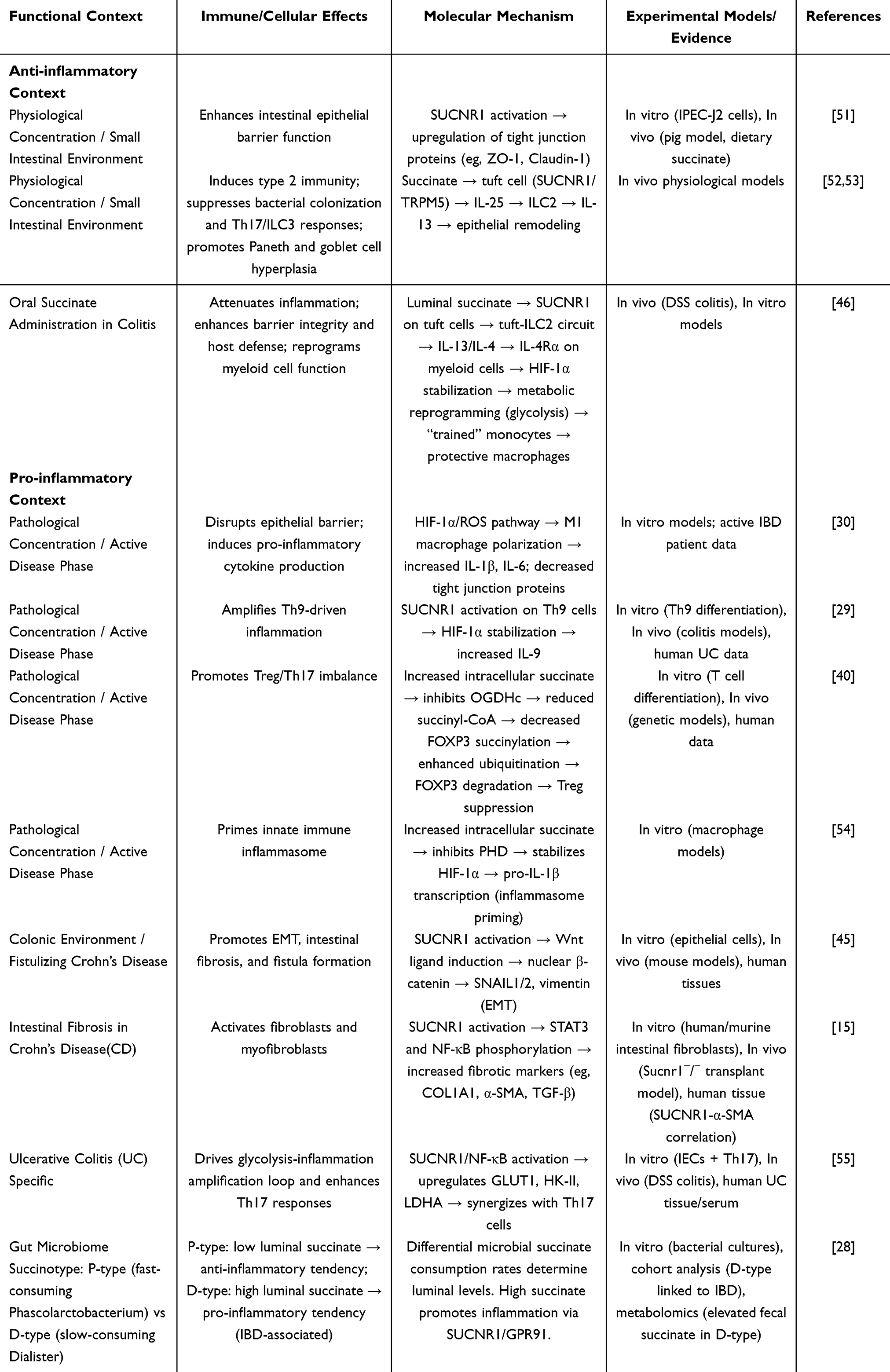 |
Table 1 Summary of the Dual Immunoregulatory Roles of Succinate in IBD |
The Pro-Inflammatory Role
Substantial evidence from clinical and preclinical studies establishes succinate as a potent pro-inflammatory driver in IBD. Elevated fecal and serum succinate levels are observed in IBD patients compared to healthy individuals, correlating positively with clinical disease activity, as demonstrated in clinical studies.29,31,56 In vivo studies directly demonstrate succinate’s pro-inflammatory properties. In the dextran sulfate sodium (DSS)-induced mouse colitis model, intrarectal administration of exogenous succinate significantly exacerbates disease severity, as evidenced by greater weight loss, severe colonic shortening, higher histopathology scores, and significantly elevated levels of pro-inflammatory cytokines, including IL-6 and TNF-α, in colon tissue. Notably, in this model, transplantation of succinate-consuming bacteria or administration of SUCNR1 antagonists effectively reduces colonic succinate levels, alleviates intestinal inflammation, and is accompanied by suppression of Th9 cell-related inflammatory pathways.29,46 Beyond acute inflammation, the succinate-SUCNR1 axis is critically implicated in the pathogenesis of intestinal fibrosis, a major complication of Crohn’s disease. SUCNR1 expression is significantly upregulated in the intestinal tissue and isolated fibroblasts of CD patients. Activation of SUCNR1 by succinate drives fibroblast activation in a dose-dependent manner, upregulating fibrotic markers (COL1A1, α-SMA) and pro-fibrotic mediators (TGF-β), and activating STAT3 and NF-κB signaling pathways. Conversely, genetic deletion of SUCNR1 in mice confers protection against both TNBS-induced colitis and a heterotopic transplant model of intestinal fibrosis, characterized by attenuated collagen deposition, reduced expression of pro-inflammatory cytokines, and a shift in macrophage polarization towards an anti-inflammatory M2 phenotype.15 In vitro studies further confirm the pro-inflammatory role of succinate; treatment of human CD4⁺ T cells with 5–10 mmol/L succinate promotes differentiation of Th17 cells, while concurrently mediating degradation of FOXP3 protein via the ubiquitin-proteasome pathway, thereby significantly impairing the immunosuppressive function of Treg cells.40 Succinate activates pro-inflammatory signaling networks through multiple pathways, among which the SUCNR1-Gq-PLC-IP3 pathway and the HIF-1α pathway are core drivers.38,57,58
The Anti-Inflammatory Role
In contrast to the prevailing pro-inflammatory paradigm, certain conditions unveil a protective role for succinate. In the DSS-induced experimental colitis mouse model, supplementation of succinate (300 mM) in drinking water significantly alleviates disease severity, as evidenced by attenuation of colon shortening, reduction of histopathological damage, reduced expression of critical inflammatory mediators TNF-α and IL-6, and enhancement of intestinal barrier function. Mechanistically, succinate in the intestinal lumen exerts its effects mediated by the receptor SUCNR1, chiefly through the IL-4Rα/HIF-1α signaling pathway. This signaling axis drives metabolic reprogramming of Ly6Chi monocytes, promoting their differentiation into intestinal macrophages with tissue repair functions while retaining pro-inflammatory response potential. This process ultimately restores intestinal barrier integrity and suppresses pro-inflammatory cytokine expression, thereby significantly alleviating DSS-induced colitis.46
Determinants of Succinate’s Dual Immunoregulatory Role in IBD
The net immunoregulatory effect of succinate in IBD, spanning pro-inflammatory to anti-inflammatory outcomes, is orchestrated by a complex interplay of factors. These factors include gut microbiota composition, local succinate concentration, disease stage and location, target cell types, host genetics, and dietary influences, which collectively determine its context-dependent functions (Figure 3).
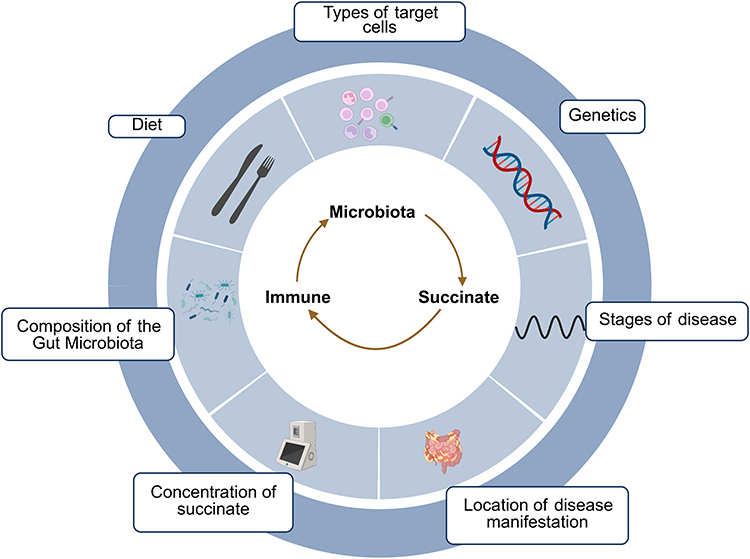 |
Figure 3 The Microbiota-Succinate-Immune Axis in IBD and Its Regulatory Factors. As a key hub molecule of the “microbiota-metabolism-immune” axis, succinate exhibits complex, dual immunoregulatory effects in IBD, which can both promote and inhibit inflammation. The final effect is determined by various critical factors, including gut microbiota composition, local succinate concentration, disease location, disease stage, target cell types, genetics, diet, and other factors. Created in BioRender. Zhao, M. (2025) https://BioRender.com/avpf4ul. |
Gut Microbiota Composition and External Influences
Microbiota play a critical role in maintaining the succinate balance in the gut. Succinate-producing bacteria are enriched in IBD patients, primarily generating succinate through the fumarate reductase pathway by metabolizing dietary fibers. This process leads to their excessive proliferation, which is positively associated with colitis severity. In contrast, key succinate-consuming bacteria, including Phascolarctobacterium and Dialister, are often deficient or diminished in IBD patients. These bacteria utilize the succinyl-CoA transferase to methylmalonyl-CoA decarboxylase pathway to transform succinate into propionate. Significantly, these two genera of succinate consumers demonstrate pronounced, mutually exclusive colonization within the intestine, forming distinct “succinotypes”. In fact, experimental in vitro findings demonstrate that Phascolarctobacterium consumes succinate at an average rate of 63.7 mM/h/OD, about double that of Dialister’s 30.7 mM/h/OD. This inherent difference in succinate consumption efficiency between the two genera is a major factor driving inter-individual variation in intestinal succinate levels and may be directly linked to IBD susceptibility.28
Diet and antibiotics are two critical external factors that regulate microbial composition and succinate metabolism. Regarding diet, the specific dietary composition directly influences microbial functions and succinate accumulation by altering the availability of fermentable substrates in the gut. For instance, in mouse studies, consumption of purified fructooligosaccharides elevated cecal succinate levels, while supplementation with refined inulin has been shown to induce abnormal succinate accumulation, which aggravates colitis and potentially facilitates tumor development. On the other hand, a low-calorie Mediterranean diet combined with physical activity can increase the abundance of succinate-consuming bacteria, optimize the production-consumption ratio of succinate, and reduce its overall level in the gut. The use of antibiotics severely disrupts the equilibrium of the gut microbiota and markedly alters succinate metabolism. While eliminating pathogenic bacteria, antibiotics also selectively deplete key succinate-consuming microbes, disrupting metabolic homeostasis. This disruption leads to succinate accumulation, which can trigger inflammatory responses or facilitate opportunistic infections such as Clostridioides difficile.27,59
Concentration of Succinate
Succinate exhibits a significant concentration-dependent bidirectional regulatory effect on intestinal inflammation. At physiological concentrations (1–3 mM), succinate triggers SUCNR1 signaling in both intestinal epithelial and immune cell populations, including macrophages and tuft cells, mediating type 2 immune responses, promoting monocyte differentiation into tissue-repairing macrophages, inducing a trained immunity-like phenotype, enhancing intestinal barrier integrity, and significantly alleviating experimental colitis. The underlying protective mechanisms include activation of the IL-4Rα/HIF-1α signaling axis, increased host antibacterial capability, and decreased intestinal mucosal permeability.46 Conversely, at pathological high concentrations of succinate (>5 mM), excessive activation of SUCNR1 leads to Wnt/β-catenin signaling-dependent EMT, which promotes intestinal fibrosis and fistula formation.45 This concentration-dependent “metabolic switch” effect enables succinate at physiological levels to exert mucosal protective functions, while excessive concentrations may drive inflammatory cascade responses.27
Location of Disease Manifestation
The immunomodulatory effects of succinate display marked regional heterogeneity along the gastrointestinal tract. Under physiological conditions in the small intestine, succinate activates the SUCNR1 receptors on tuft cells, leading to IL-25 secretion and subsequent activation of the type 2 innate lymphoid cell (ILC2)/interleukin-13 (IL-13) axis. This in turn promotes Paneth cell hyperplasia and reshapes antimicrobial peptide expression, effectively inhibiting mucosal bacterial colonization and improving barrier integrity, thereby suppressing excessive inflammation via GATA3⁺ cell expansion and IL-4/IL-13-driven Th2 immune polarization. Such coordinated responses help maintain intestinal homeostasis.52,53 In contrast, within the colon, where the elevated microbiota density and reduced oxygen tension restrict SDH activity, there is abnormal succinate accumulation in this segment of the gut. As a result of this metabolic shift, elevated succinate levels activate SUCNR1, inducing Wnt1/4/10A ligand expression, triggering β-catenin nuclear translocation, and upregulating SNAIL1/2 and vimentin-mediated EMT, thus facilitating fibrosis and fistula development.45 Additionally, in the specific context of ulcerative colitis, succinate accumulation activates the SUCNR1/NF-κB pathway, synergizing with Th17 cells to drive a pathogenic glycolytic switch in intestinal epithelial cells. This UC-specific metabolic-immune cascade, characterized by upregulation of GLUT1, HK-II, and LDHA, establishes a pro-inflammatory feed-forward loop that perpetuates mucosal inflammation.55
Stages of Disease
The immunoregulatory function of succinate evolves with disease progression. During the active phase of the disease, pro-inflammatory effects predominate. In patients with active UC, dysbiosis is characterized by an increased relative abundance of succinate-producing bacteria and a reduction of succinate-consuming bacteria, leading to succinate accumulation in the intestinal lumen. The accumulated succinate activates intestinal Th9 cells via SUCNR1, promoting secretion of IL-9 and other factors, indirectly recruiting neutrophils and amplifying the inflammatory cascade.29 Additionally, through the HIF-1α/ROS pathway, succinate facilitates M1 macrophage polarization, which elevates the production of pro-inflammatory cytokines including IL-1β and IL-6, and suppresses expression of epithelial tight junction proteins like occludin and claudin-1, thereby worsening the damage to the intestinal barrier.30 In active IBD patients, clinical data reveal a strong positive correlation (r=0.78, p<0.001) between fecal succinate concentrations and fecal calprotectin levels.28 Conversely, during disease remission, succinate shows protective functions. In the DSS-induced colitis recovery model, exogenous succinate activates intestinal tuft cell SUCNR1 receptors, stimulating ILC2 to secrete interleukin-4 (IL-4) and IL-13, thereby promoting macrophage polarization to the reparative M2 type and facilitating intestinal tissue repair.46
Types of Target Cells
The biological effects of succinate are highly cell-type specific within the intestinal microenvironment. In innate immune cells, succinate directly activates dendritic cells and macrophages via its receptor SUCNR1, promoting NLRP3 inflammasome assembly and IL-1β release.54,60 In adaptive immune cell subsets, high concentrations of succinate significantly inhibit the immunosuppressive function of Treg cells by inducing degradation of the key transcription factor FOXP3, while promoting differentiation of Th9 and Th17 cells, thereby driving IL-9 and IL-17-mediated intestinal inflammatory responses.29,40 In intestinal epithelial cells, physiological concentrations of succinate upregulate the expression of tight junction proteins ZO-1 and Claudin-1 via the SUCNR1 signaling pathway, helping to maintain intestinal barrier integrity.51 However, at pathological concentrations, succinate induces mitochondrial fission, disrupting energy metabolic homeostasis in epithelial cells, which exacerbates intestinal barrier dysfunction. Notably, butyrate can effectively antagonize this mitochondrial damage process by activating free fatty acid receptor 3 (FFAR3).61 In intestinal stromal cells, succinate activates SUCNR1 on fibroblasts, initiating the transforming growth factor beta (TGF-β) signaling cascade, promoting collagen deposition and activation of α-smooth muscle actin (α-SMA)-positive myofibroblasts, thereby accelerating intestinal fibrosis progression and potentially leading to intestinal stricture formation.15 In conclusion, succinate performs complex and varied biological roles via multiple signaling pathways in various cell types.
The Potential Application of Succinate in Clinical Diagnosis of IBD
Numerous studies have demonstrated that succinate levels in urine and feces can serve as important biomarkers reflecting intestinal microbiota dysbiosis and disease activity.62 Clinically, the detection of succinate primarily utilizes techniques such as gas chromatography-mass spectrometry (GC-MS), liquid chromatography-tandem mass spectrometry (LC-MS/MS), capillary electrophoresis (CE), and enzymatic assays. Each method, however, presents notable limitations: GC-MS requires complex derivatization procedures; LC-MS/MS, while offering high sensitivity and specificity, is costly and susceptible to matrix effects; CE suffers from poor reproducibility; and enzymatic assays lack specificity, typically yielding only semi-quantitative results. These technical challenges, therefore, currently hinder the standardization and high-throughput application of succinate measurement in routine IBD diagnostics.29,63
The relationship between succinate levels and IBD pathophysiology is increasingly clear. In CD patients, reduced urinary succinate correlates with gut metabolic dysregulation, whereas heightened fecal succinate indicates pathogenic bacterial expansion and inflammatory activation.26,56 Furthermore, the “succinotype” classification strategy based on gut microbiota metabolic features provides a novel dimension for precise IBD subtype classification.28 Clinical cohort research confirms that IBD patients characterized by a Dialister-dominant D-type succinotype have higher rates of disease relapse and more severe mucosal injury observed endoscopically than those with a Phascolarctobacterium-dominant P-type succinotype.64 Additionally, future approaches may integrate fecal succinate levels, urinary metabolomics, and microbiota sequencing to develop non-invasive, personalized dynamic monitoring systems, enabling more accurate assessment of IBD disease activity.56,65
The Potential Application of Succinate in Clinical Treatment of IBD
The immunomodulatory function of succinate in IBD is notably bidirectional, and its final biological outcomes are intricately governed by multidimensional networks such as the intestinal microbiota composition. Based on this regulatory network, potential targeted therapeutic strategies mainly include three aspects.
Targeting the Gut Microbiota Structure
Regulating the gut microbiota “succinotype” by supplementing probiotics or succinate-consuming bacteria, and implementing personalized dietary plans to adjust the consumption/production ratio of succinate. Preliminary studies indicate that oral administration of Phascolarctobacterium faecium can significantly reduce fecal succinate levels in UC patients and improve clinical symptoms via the succinate-to-propionate metabolic pathway.66 Supplementing fermentable dietary fibers such as oligofructose can promote the proliferation of succinate-consuming bacteria, while the use of refined fibers like inulin additives should be avoided to prevent dysbiosis and abnormal succinate accumulation.25
Targeting Succinate Signaling and Metabolic Pathways
Succinate mainly mediates multiple immune pathways via its receptor SUCNR1; therefore, developing SUCNR1 antagonists is a core strategy. Specifically, SUCNR1 antagonists can be used to block pro-inflammatory signals during active IBD, whereas during remission, succinate or its precursors can be supplemented to promote mucosal repair. Although SUCNR1 antagonists for IBD treatment have defined targets and lead compounds such as TUG-2465, clinical translation still faces challenges including species differences, insufficient tissue selectivity, and the bidirectionality of succinate/SUCNR1 signaling, all of which require humanized animal models and more precise delivery strategies to overcome.67 Moreover, the development of drugs targeting key succinate metabolism enzymes like SDH and SCS could hold considerable potential.
Targeting the Transporters of Succinate
The distribution of succinate between intracellular and extracellular environments depends on specific transport proteins such as DIC, OATs, MCTs, and members of the SLC13 family. Regulating the activity of these transporters could influence the local microenvironment’s succinate levels and its accessibility to SUCNR1, thus modulating downstream signaling cascades. Therefore, the further application of this approach requires the elucidation of the precise concentration thresholds and tissue-specific effects through which succinate mediates its distinct immune regulatory roles.
Conclusion and Prospects
This review comprehensively summarizes the dual immunomodulatory role of succinate in IBD and highlights its central position in the microbiota-metabolism-immune axis. Research shows that the function of succinate exhibits significant context dependence: it can mediate immune cell activation and pro-inflammatory responses through both SUCNR1-dependent and independent pathways, while simultaneously contributing to anti-inflammatory and tissue repair processes in specific microenvironments. This duality of action indicates that the final biological effects of succinate are not solely determined by its intrinsic properties, but rather depend on its concentration, spatial distribution, and the complex interactions between local metabolism and immune status in the gut. Therefore, understanding the dynamic balance between these opposing roles is crucial for elucidating its overall impact on intestinal homeostasis.
Despite continuous basic research revealing the key role of succinate in the pathogenesis of IBD, its clinical translation still faces significant challenges. Although numerous studies have deepened our understanding of succinate-mediated inflammatory pathways, translating these findings into effective therapies remains difficult. For example, strategies to regulate the gut “succinotype” through methods such as fecal microbiota transplantation (FMT) face issues such as lack of precision and limited success rates.68 In addition, such interventions often fail to achieve stable and long-term modulation of intestinal metabolites, further limiting their clinical applicability. Moreover, the systemic application of SUCNR1 antagonists presents additional obstacles, as their widespread receptor distribution may result in unintended off-target effects and potential liver or kidney toxicity. Taken together, these limitations underscore the urgent need to develop novel, precise intervention strategies that can selectively modulate succinate signaling within the intestinal microenvironment while minimizing systemic risks.
Future research should focus on the following directions. Firstly, there is a need to develop more targeted intervention tools, such as live biological therapies based on engineered probiotics or succinate-consuming bacteria,69,70 microenvironment-specific SUCNR1 antagonists, and smart nanoparticle drug delivery systems,67,71 aimed at improving intervention efficiency and safety. Secondly, it is essential to integrate multi-omics data, such as host genetic polymorphisms, gut microbiota metabolic functions, and dynamic metabolomics, to construct comprehensive and predictive models for the precise stratification of IBD patients, thereby providing a solid basis for personalized succinate-related treatments.72–74 Furthermore, it will be crucial to strengthen longitudinal and translational studies that link molecular mechanisms to clinical outcomes, so as to ensure that findings from laboratory research can be effectively applied in clinical contexts.
It should be noted that the immunomodulatory mechanisms of succinate discussed in this review are primarily derived from preclinical models and thus require direct validation in human tissues and clinical cohorts. Therefore, bridging this gap between experimental and clinical research will be essential to confirm the translational relevance of current findings. As research into IBD immunometabolism continues to deepen, the integration of multi-dimensional analyses of the succinate functional network with the rational design of precise and personalized intervention strategies is expected to transform these mechanistic insights into effective clinical applications, ultimately opening up new paths for the diagnosis and treatment of IBD.
Abbreviations
IBD, Inflammatory Bowel Disease; CD, Crohn’s Disease; UC, Ulcerative Colitis; SCFAs, short-chain fatty acids; TCA, tricarboxylic acid; ATP, Adenosine Triphosphate; α-KG, α-ketoglutarate; OGDHc, α-ketoglutarate dehydrogenase complex; SCS, succinyl-CoA synthetase; SDH, succinate dehydrogenase; GABA, γ-aminobutyric acid; GPCRs, G protein-coupled receptors; HDAC, histone deacetylase; PEP, phosphoenolpyruvate; CO2, carbon dioxide; PEPCK, phosphoenolpyruvate carboxykinase; OAA, oxaloacetate; MMA, methylmalonate; DIC, dicarboxylate carrier; VDAC, voltage-dependent anion channel; OATs, organic anion transporters; MCT1, monocarboxylate transporter 1; HIF-1α, hypoxia-inducible factor-1α; Treg, regulatory T cells; Th17, T helper 17 cells; PHD, prolyl hydroxylase; IL-1β, interleukin-1β; IL-6, interleukin-6; FOXP3, forkhead box protein P3; IL-17, interleukin-17; IL-17A, interleukin-17A; mtROS, mitochondrial reactive oxygen species; NLRP3, NOD-like receptor pyrin domain-containing protein 3; IL-18, interleukin-18; SUCNR1, succinate receptor 1; GPR91, G protein-coupled receptor 91 (succinate receptor 1); PLCβ, phospholipase Cβ; PKC, protein kinase C; MAPK, mitogen-activated protein kinase; cAMP, cyclic adenosine monophosphate; IL-4Rα, interleukin-4 receptor α subunit; CCL2, C-C Motif Chemokine Ligand 2; CCR2, C-C Motif Chemokine Receptor 2; EMT, epithelial-mesenchymal transition; Th9, T helper 9 cells; SBAs, secondary bile acids; GPR41, G protein-coupled receptor 41; GPR43, G protein-coupled receptor 43; DCA, deoxycholic acid; GPBAR1, G protein-coupled bile acid receptor 1; FXR, farnesoid X receptor; LCA, lithocholic acid; 12-KLCA, 12-keto lithocholic acid; ILC3s, group 3 innate lymphoid cells; VDR, vitamin D receptor; PXR, pregnane X receptor; I3A, indole-3-aldehyde; IL-22, interleukin-22; AhR, aryl hydrocarbon receptor; DSS, dextran sulfate sodium; TNF-α, tumor necrosis factor-α; ILC2, type 2 innate lymphoid cell; IL-13, interleukin-13; IL-4, interleukin-4; FFAR3, free fatty acid receptor 3; TGF-β, transforming growth factor-β; α-SMA, α-smooth muscle actin; FMT, fecal microbiota transplantation.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analysed in this study.
Acknowledgments
Graphical abstracted was created in BioRender. zhao, M. (2025) https://BioRender.com/xeillqz.
Author Contributions
Meihua Zhao: Conceptualization, Writing - original draft, Writing - review & editing, Visualization, Formal analysis. Chuan Zhou: Investigation, Writing - review & editing, Validation, Data curation. Dandan Wang: Investigation, Writing - review & editing, Methodology. Qiong Wu: Writing - review & editing, Validation, Formal analysis. Baisui Feng: Writing - review & editing, Project administration, Resources. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Henan Province Medical Science and Technology Research Plan Joint Construction Project (LHGJ20210419), the Nature Sciences Foundation of Henan Province (242300421282), and the Medical Science and Technology Project of Henan Province (SBGJ202102178).
Disclosure
The authors have no conflicts of interest to declare in this work.
References
1. Muzammil MA, Fariha F, Patel T, et al. Advancements in inflammatory bowel disease: a narrative review of diagnostics, management, epidemiology, prevalence, patient outcomes, quality of life, and clinical presentation. Cureus. 2023;15(6):e41120. doi:10.7759/cureus.41120
2. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–2778. doi:10.1016/s0140-6736(17)32448-0
3. Nagayama M, Gogokhia L, Longman RS. Precision microbiota therapy for IBD: premise and promise. Gut Microbes. 2025;17(1):2489067. doi:10.1080/19490976.2025.2489067
4. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146(6):1489–1499. doi:10.1053/j.gastro.2014.02.009
5. Sharma B, Agriantonis G, Twelker K, et al. Gut microbiota serves as a crucial independent biomarker in inflammatory bowel disease (IBD). Int J Mol Sci. 2025;26(6):2503. doi:10.3390/ijms26062503
6. Wang X, Peng J, Cai P, et al. The emerging role of the gut microbiota and its application in inflammatory bowel disease. Biomed Pharmacother. 2024;179:117302. doi:10.1016/j.biopha.2024.117302
7. Lee M, Chang EB. Inflammatory Bowel Diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology. 2021;160(2):524–537. doi:10.1053/j.gastro.2020.09.056
8. Ma HQ, Yu TT, Zhao XJ, Zhang Y, Zhang HJ. Fecal microbial dysbiosis in Chinese patients with inflammatory bowel disease. World J Gastroenterol. 2018;24(13):1464–1477. doi:10.3748/wjg.v24.i13.1464
9. Andoh A, Nishida A. Alteration of the gut microbiome in inflammatory bowel disease. Digestion. 2023;104(1):16–23. doi:10.1159/000525925
10. Zhang Y, Zhang J, Duan L. The role of microbiota-mitochondria crosstalk in pathogenesis and therapy of intestinal diseases. Pharmacol Res. 2022;186:106530. doi:10.1016/j.phrs.2022.106530
11. Gasaly N, de Vos P, Hermoso MA. Impact of bacterial metabolites on gut barrier function and host immunity: a focus on bacterial metabolism and its relevance for intestinal inflammation. Front Immunol. 2021;12:658354. doi:10.3389/fimmu.2021.658354
12. Fu Y, Lyu J, Wang S. The role of intestinal microbes on intestinal barrier function and host immunity from a metabolite perspective. Front Immunol. 2023;14:1277102. doi:10.3389/fimmu.2023.1277102
13. Alula KM, Dowdell AS, LeBere B, et al. Interplay of gut microbiota and host epithelial mitochondrial dysfunction is necessary for the development of spontaneous intestinal inflammation in mice. Microbiome. 2023;11(1):256. doi:10.1186/s40168-023-01686-9
14. Zhang W, Lang R. Succinate metabolism: a promising therapeutic target for inflammation, ischemia/reperfusion injury and cancer. Front Cell Dev Biol. 2023;11:1266973. doi:10.3389/fcell.2023.1266973
15. Macias-Ceja DC, Ortiz-Masiá D, Salvador P, et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol. 2019;12(1):178–187. doi:10.1038/s41385-018-0087-3
16. Atallah R, Olschewski A, Heinemann A. Succinate at the crossroad of metabolism and angiogenesis: roles of SDH, HIF1α and SUCNR1. Biomedicines. 2022;10(12):3089. doi:10.3390/biomedicines10123089
17. Chen H, Jin C, Xie L, Wu J. Succinate as a signaling molecule in the mediation of liver diseases. Biochim Biophys Acta Mol Basis Dis. 2024;1870(2):166935. doi:10.1016/j.bbadis.2023.166935
18. Murphy MP, O’Neill LAJ. Krebs cycle reimagined: the emerging roles of succinate and itaconate as signal transducers. Cell. 2018;174(4):780–784. doi:10.1016/j.cell.2018.07.030
19. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277(34):30409–30412. doi:10.1074/jbc.R200006200
20. Connors J, Dawe N, Van Limbergen J. The role of succinate in the regulation of intestinal inflammation. Nutrients. 2018;11(1):25. doi:10.3390/nu11010025
21. Deleu S, Machiels K, Raes J, Verbeke K, Vermeire S. Short chain fatty acids and its producing organisms: an overlooked therapy for IBD? EBioMedicine. 2021;66:103293. doi:10.1016/j.ebiom.2021.103293
22. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41. doi:10.1111/1462-2920.13589
23. Krautkramer KA, Fan J, Bäckhed F. Gut microbial metabolites as multi-kingdom intermediates. Nat Rev Microbiol. 2021;19(2):77–94. doi:10.1038/s41579-020-0438-4
24. Kwong WK, Zheng H, Moran NA. Convergent evolution of a modified, acetate-driven TCA cycle in bacteria. Nat Microbiol. 2017;2:17067. doi:10.1038/nmicrobiol.2017.67
25. Tian S, Paudel D, Hao F, et al. Refined fiber inulin promotes inflammation-associated colon tumorigenesis by modulating microbial succinate production. Cancer Rep. 2023;6(11):e1863. doi:10.1002/cnr2.1863
26. Lin H, Chen Y, Zhou M, et al. Comprehensive analysis of faecal metagenomic and serum metabolism revealed the role of gut microbes and related metabolites in detecting colorectal lateral spreading tumours. Virulence. 2025;16(1):2489154. doi:10.1080/21505594.2025.2489154
27. Fernández-Veledo S, Grau-Bové C, Notararigo S, Huber-Ruano I. The role of microbial succinate in the pathophysiology of inflammatory bowel disease: mechanisms and therapeutic potential. Curr Opin Microbiol. 2025;85:102599. doi:10.1016/j.mib.2025.102599
28. Anthamatten L, von Bieberstein PR, Menzi C, et al. Stratification of human gut microbiomes by succinotype is associated with inflammatory bowel disease status. Microbiome. 2024;12(1):186. doi:10.1186/s40168-024-01897-8
29. Dalal R, Sadhu S, Batra A, et al. Gut commensals-derived succinate impels colonic inflammation in ulcerative colitis. NPJ Biofilms Microbiomes. 2025;11(1):44. doi:10.1038/s41522-025-00672-3
30. Wei YH, Ma X, Zhao JC, Wang XQ, Gao CQ. Succinate metabolism and its regulation of host-microbe interactions. Gut Microbes. 2023;15(1):2190300. doi:10.1080/19490976.2023.2190300
31. Fremder M, Kim SW, Khamaysi A, et al. A transepithelial pathway delivers succinate to macrophages, thus perpetuating their pro-inflammatory metabolic state. Cell Rep. 2021;36(6):109521. doi:10.1016/j.celrep.2021.109521
32. Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447(5):689–709. doi:10.1007/s00424-003-1099-7
33. Bisbach CM, Hass DT, Thomas ED, Cherry TJ, Hurley JB. Monocarboxylate Transporter 1 (MCT1) mediates succinate export in the retina. Invest Ophthalmol Vis Sci. 2022;63(4):1. doi:10.1167/iovs.63.4.1
34. Reddy A, Winther S, Tran N, et al. Monocarboxylate transporters facilitate succinate uptake into brown adipocytes. Nat Metab. 2024;6(3):567–577. doi:10.1038/s42255-024-00981-5
35. Yang Q, Guo C, Zhang L. The role of metabolite sensors in metabolism-immune interaction: new targets for immune modulation. Clin Transl Med. 2025;15(4):e70294. doi:10.1002/ctm2.70294
36. Taylor CT, Scholz CC. The effect of HIF on metabolism and immunity. Nat Rev Nephrol. 2022;18(9):573–587. doi:10.1038/s41581-022-00587-8
37. Solanki S, Shah YM. Hypoxia-induced signaling in gut and liver pathobiology. Annu Rev Pathol. 2024;19:291–317. doi:10.1146/annurev-pathmechdis-051122-094743
38. Huang H, Li G, He Y, et al. Cellular succinate metabolism and signaling in inflammation: implications for therapeutic intervention. Front Immunol. 2024;15:1404441. doi:10.3389/fimmu.2024.1404441
39. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
40. Wang H, Hu D, Cheng Y, et al. Succinate drives gut inflammation by promoting FOXP3 degradation through a molecular switch. Nat Immunol. 2025;26(6):866–880. doi:10.1038/s41590-025-02166-y
41. Lee H, Jeon JH, Kim ES. Mitochondrial dysfunctions in T cells: focus on inflammatory bowel disease. Front Immunol. 2023;14:1219422. doi:10.3389/fimmu.2023.1219422
42. Gilissen J, Jouret F, Pirotte B, Hanson J. Insight into SUCNR1 (GPR91) structure and function. Pharmacol Ther. 2016;159:56–65. doi:10.1016/j.pharmthera.2016.01.008
43. Liebing AD, Rabe P, Krumbholz P, et al. Succinate receptor 1 signaling mutually depends on subcellular localization and cellular metabolism. Febs J. 2025;292(8):2017–2050. doi:10.1111/febs.17407
44. Krzak G, Willis CM, Smith JA, Pluchino S, Peruzzotti-Jametti L. Succinate receptor 1: an emerging regulator of myeloid cell function in inflammation. Trends Immunol. 2021;42(1):45–58. doi:10.1016/j.it.2020.11.004
45. Ortiz-Masiá D, Gisbert-Ferrándiz L, Bauset C, et al. Succinate activates EMT in intestinal epithelial cells through SUCNR1: a novel protagonist in fistula development. Cells. 2020;9(5):1104. doi:10.3390/cells9051104
46. Liang L, Dang B, Ouyang X, et al. Dietary succinate supplementation alleviates DSS-induced colitis via the IL-4Rα/Hif-1α Axis. Int Immunopharmacol. 2025;152:114408. doi:10.1016/j.intimp.2025.114408
47. Zheng L. New insights into the interplay between intestinal flora and bile acids in inflammatory bowel disease. World J Clin Cases. 2022;10(30):10823–10839. doi:10.12998/wjcc.v10.i30.10823
48. Lavelle A, Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(4):223–237. doi:10.1038/s41575-019-0258-z
49. Xiao J, Guo X, Wang Z. Crosstalk between hypoxia-inducible factor-1α and short-chain fatty acids in inflammatory bowel disease: key clues toward unraveling the mystery. Front Immunol. 2024;15:1385907. doi:10.3389/fimmu.2024.1385907
50. Li N, Ma P, Li Y, et al. Gut microbiota-derived 12-ketolithocholic acid suppresses the IL-17A secretion from colonic group 3 innate lymphoid cells to prevent the acute exacerbation of ulcerative colitis. Gut Microbes. 2023;15(2):2290315. doi:10.1080/19490976.2023.2290315
51. Li X, Mao M, Zhang Y, Yu K, Zhu W. Succinate modulates intestinal barrier function and inflammation response in pigs. Biomolecules. 2019;9(9):486. doi:10.3390/biom9090486
52. Banerjee A, Herring CA, Chen B, et al. Succinate produced by intestinal microbes promotes specification of tuft cells to suppress ileal inflammation. Gastroenterology. 2020;159(6):2101–2115.e5. doi:10.1053/j.gastro.2020.08.029
53. Fung C, Fraser LM, Barrón GM, et al. Tuft cells mediate commensal remodeling of the small intestinal antimicrobial landscape. Proc Natl Acad Sci U S A. 2023;120(23):e2216908120. doi:10.1073/pnas.2216908120
54. O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15–23. doi:10.1084/jem.20151570
55. Huo L, Chen Q, Jia S, et al. Gut microbiome promotes succinate-induced ulcerative colitis by enhancing glycolysis through SUCNR1/NF-κB signaling pathway. Am J Physiol Cell Physiol. 2025;329(2):C440–c454. doi:10.1152/ajpcell.00411.2025
56. Baskaran K, Moshkovich M, Hart L, et al. The role of urine metabolomics in the diagnosis and management of adult and pediatric Crohn’s disease and ulcerative colitis. Biomarkers. 2025;30(1):104–113. doi:10.1080/1354750x.2024.2438734
57. Astorga J, Gasaly N, Dubois-Camacho K, et al. The role of cholesterol and mitochondrial bioenergetics in activation of the inflammasome in IBD. Front Immunol. 2022;13:1028953. doi:10.3389/fimmu.2022.1028953
58. Yuan Y, Ni S, Zhuge A, Li L, Li B. Adipose-derived mesenchymal stem cells reprogram M1 macrophage metabolism via PHD2/HIF-1α pathway in colitis mice. Front Immunol. 2022;13:859806. doi:10.3389/fimmu.2022.859806
59. Li X, Huang G, Zhang Y, et al. Succinate signaling attenuates high-fat diet-induced metabolic disturbance and intestinal barrier dysfunction. Pharmacol Res. 2023;194:106865. doi:10.1016/j.phrs.2023.106865
60. Bauset C, Lis-Lopez L, Coll S, et al. SUCNR1 Mediates the Priming Step of the Inflammasome in Intestinal Epithelial Cells: relevance in Ulcerative Colitis. Biomedicines. 2022;10(3):532. doi:10.3390/biomedicines10030532
61. Hamed SA, Mohan A, Navaneetha Krishnan S, et al. Butyrate reduces adherent-invasive E. coli-evoked disruption of epithelial mitochondrial morphology and barrier function: involvement of free fatty acid receptor 3. Gut Microbes. 2023;15(2):2281011. doi:10.1080/19490976.2023.2281011
62. Čipčić Paljetak H, Barešić A, Panek M, et al. Gut microbiota in mucosa and feces of newly diagnosed, treatment-naïve adult inflammatory bowel disease and irritable bowel syndrome patients. Gut Microbes. 2022;14(1):2083419. doi:10.1080/19490976.2022.2083419
63. Kaczmarczyk O, Dąbek-Drobny A, Woźniakiewicz M, et al. Fecal levels of lactic, succinic and short-chain fatty acids in patients with ulcerative colitis and Crohn disease: a pilot study. J Clin Med. 2021;10(20):4701. doi:10.3390/jcm10204701
64. O’Sullivan J, Patel S, Leventhal GE, et al. Host-microbe multi-omics and succinotype profiling have prognostic value for future relapse in patients with inflammatory bowel disease. Gut Microbes. 2025;17(1):2450207. doi:10.1080/19490976.2025.2450207
65. Kaz AM, Venu N. Diagnostic methods and biomarkers in inflammatory bowel disease. Diagnostics. 2025;15(11):1303. doi:10.3390/diagnostics15111303
66. Ikeyama N, Murakami T, Toyoda A, et al. Microbial interaction between the succinate-utilizing bacterium Phascolarctobacterium faecium and the gut commensal Bacteroides thetaiotaomicron. Microbiologyopen. 2020;9(10):e1111. doi:10.1002/mbo3.1111
67. Ciba M, Dibnah B, Hudson BD, Rexen Ulven E. Development and characterization of potent succinate receptor fluorescent tracers. J Med Chem. 2023;66(13):8951–8974. doi:10.1021/acs.jmedchem.3c00552
68. Yadegar A, Bar-Yoseph H, Monaghan TM, et al. Fecal microbiota transplantation: current challenges and future landscapes. Clin Microbiol Rev. 2024;37(2):e0006022. doi:10.1128/cmr.00060-22
69. Huang Y, Peng S, Zeng R, Yao H, Feng G, Fang J. From probiotic chassis to modification strategies, control and improvement of genetically engineered probiotics for inflammatory bowel disease. Microbiol Res. 2024;289:127928. doi:10.1016/j.micres.2024.127928
70. Sang G, Wang B, Xie Y, Chen Y, Yang F. Engineered probiotic-based biomaterials for inflammatory bowel disease treatment. Theranostics. 2025;15(8):3289–3315. doi:10.7150/thno.103983
71. Yasmin F, Najeeb H, Shaikh S, et al. Novel drug delivery systems for inflammatory bowel disease. World J Gastroenterol. 2022;28(18):1922–1933. doi:10.3748/wjg.v28.i18.1922
72. Fiocchi C, Dragoni G, Iliopoulos D, Katsanos K, Ramirez VH, Suzuki K. Results of the seventh scientific workshop of ECCO: precision medicine in IBD-what, why, and how. J Crohns Colitis. 2021;15(9):1410–1430. doi:10.1093/ecco-jcc/jjab051
73. Gudiño V, Bartolomé-Casado R, Salas A. Single-cell omics in inflammatory bowel disease: recent insights and future clinical applications. Gut. 2025. doi:10.1136/gutjnl-2024-334165
74. Cannarozzi AL, Latiano A, Massimino L, et al. Inflammatory bowel disease genomics, transcriptomics, proteomics and metagenomics meet artificial intelligence. United Eur Gastroenterol J. 2024;12(10):1461–1480. doi:10.1002/ueg2.12655
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Awareness and Predictors of the Use of Bioinformatics in Genome Research in Saudi Arabia
Alomair L, Abolfotouh MA
International Journal of General Medicine 2023, 16:3413-3425
Published Date: 11 August 2023