Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 13
Successful Anatomy Adapted Therapeutic Management and Genetic Profiling of Primary Pituitary Diffuse Large B-Cell Lymphoma
Authors Kimbrough EO ![]() , Gupta V
, Gupta V ![]() , Jiang L
, Jiang L ![]() , Tun HW
, Tun HW ![]()
Received 8 May 2023
Accepted for publication 13 July 2023
Published 28 July 2023 Volume 2023:13 Pages 25—32
DOI https://doi.org/10.2147/BLCTT.S420442
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wilson Gonsalves
ErinMarie O Kimbrough,1 Vivek Gupta,2 Liuyan Jiang,3 Han W Tun1
1Division of Hematology and Oncology, Mayo Clinic, Jacksonville, FL, USA; 2Department of Radiology, Mayo Clinic, Jacksonville, FL, USA; 3Department of Pathology, Mayo Clinic, Jacksonville, FL, USA
Correspondence: Han W Tun, Division of Hematology and Oncology, Mayo Clinic, 4500 San Pablo Road S., Jacksonville, FL, 32224, USA, Tel +1 904 953 2693, Fax +1 904 953 2315, Email [email protected]
Abstract: Primary pituitary diffuse large B-cell lymphoma (PPL) has been regarded as a subtype of primary central nervous system lymphoma (PCNSL); however, the pituitary gland is located outside the blood brain barrier (BBB) with neural and vascular connections to the brain. Given its unique anatomic location, a combination of non-central nervous system (CNS)-penetrating and CNS-penetrating therapeutic agents can be employed to treat PPL. We report a female patient with PPL who was successfully managed with anatomy-adapted therapy incorporating non-CNS penetrating chemoimmunotherapy [rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP)] alternating with CNS-penetrating chemoimmunotherapy [rituximab, high-dose methotrexate, and high-dose cytarabine (RMA)]. She received a total of eight cycles of treatment with four cycles of each regimen following partial transsphenoidal resection. She achieved a complete response after two cycles and has remained in complete remission for the last eight years. To our knowledge, this is the longest documented survival in a patient with PPL. Targeted genomic profiling with Next-Generation Sequencing (NGS) was recently performed on the lymphoma tissue. The genomic profile of PPL in this patient is quite different from the findings typically associated with PCNSL. We suggest that PPL may be biologically distinct from PCNSL and should be treated with an anatomy adapted approach. Additional research is necessary to confirm our findings.
Keywords: primary central nervous system lymphoma, pituitary gland, primary pituitary lymphoma, endocrine dysfunction
Introduction
Primary central nervous system lymphoma (PCNSL) is a lymphoma limited to the neuro-axis [brain, spinal cord, leptomeningeal compartment, cranial nerves, and eyes] without evidence of systemic disease.1–6 PCNSL is rare and accounts for 4–6% of all extranodal non-Hodgkin lymphomas with approximately 7 cases per 1,000,000 people per year in the US.2,3 It is more common among immunocompromised individuals; however, recently, there has been an increased incidence among immunocompetent individuals.2,5–7 Primary pituitary lymphoma (PPL) is currently classified as a rare subtype of PCNSL confined to the pituitary gland with or without extension into the suprasellar or parasellar regions.1,4–7 It has been suggested, however, that PPL is different from PCNSL given the embryonal origin and location of the pituitary gland outside of the blood brain barrier (BBB).4,8–10
PPL may be more common among women and presents in the 6th decade of life.1,4–6 Patients often present with headache, vision changes, cranial nerve (CN) palsies, and endocrinopathies and are less likely to endorse systemic symptoms.1,5,11 A combination of tissue biopsy and imaging with either computed tomography (CT), magnetic resonance imaging (MRI), or positron emission tomography-computed tomography (PET-CT) is useful in the diagnosis of PPL and helps exclude secondary pituitary involvement in the setting of widespread systemic disease.4,5,7,11 PPL is often mistaken for pituitary adenomas and is discovered after biopsy or surgical resection as part of the work-up for a pituitary mass.1,4–6 With less than 50 reported cases of PPL in immunocompetent patients, little is known about the genomic landscape, treatment, and outcomes of these patients.11
We report a case of PPL in an immunocompetent individual with the longest survival data available to our knowledge. We also provide a comprehensive genomic tumor profile and suggest a treatment strategy for PPL.
Case Presentation
A 60-year-old female presented with a mild headache, nausea, vomiting, diarrhea, and lightheadedness of 1 day duration. She denied neurologic and B-symptoms (fevers, weight loss, night sweats). Her medical history was significant for osteoporosis and a recent acute exacerbation of her carpal tunnel syndrome for which hydrochlorothiazide and diclofenac were prescribed 2 days prior. On presentation, her physical exam was unremarkable including a neuro-ophthalmologic evaluation. Laboratory testing revealed severe, hypotonic hyponatremia with a sodium of 120 mmol/L (normal 135–145 mmol/L) and a serum osmolality of 242 mOsm/kg (normal 275–295 mOsm/kg). In addition, she had findings suggestive of hypothalamic-pituitary-adrenal dysfunction with low thyroid-stimulating hormone (TSH) <0.01 mIU/L (normal 0.30–4.20 mIU/L), low free T4 of 0.6 ng/dL (range 0.9–1.7 ng/dL), delayed response on Cortrosyn stimulation testing, and an elevated prolactin of 33.0 ng/mL (normal 4.8–23.3 ng/mL), see Table 1. She was started on levothyroxine and hydrocortisone for suspected partial anterior pituitary insufficiency. MRI of the brain demonstrated a 19×11 x 11 mm intrasellar mass without compression of the optic chiasm, Figure 1A. Surgical resection was recommended.
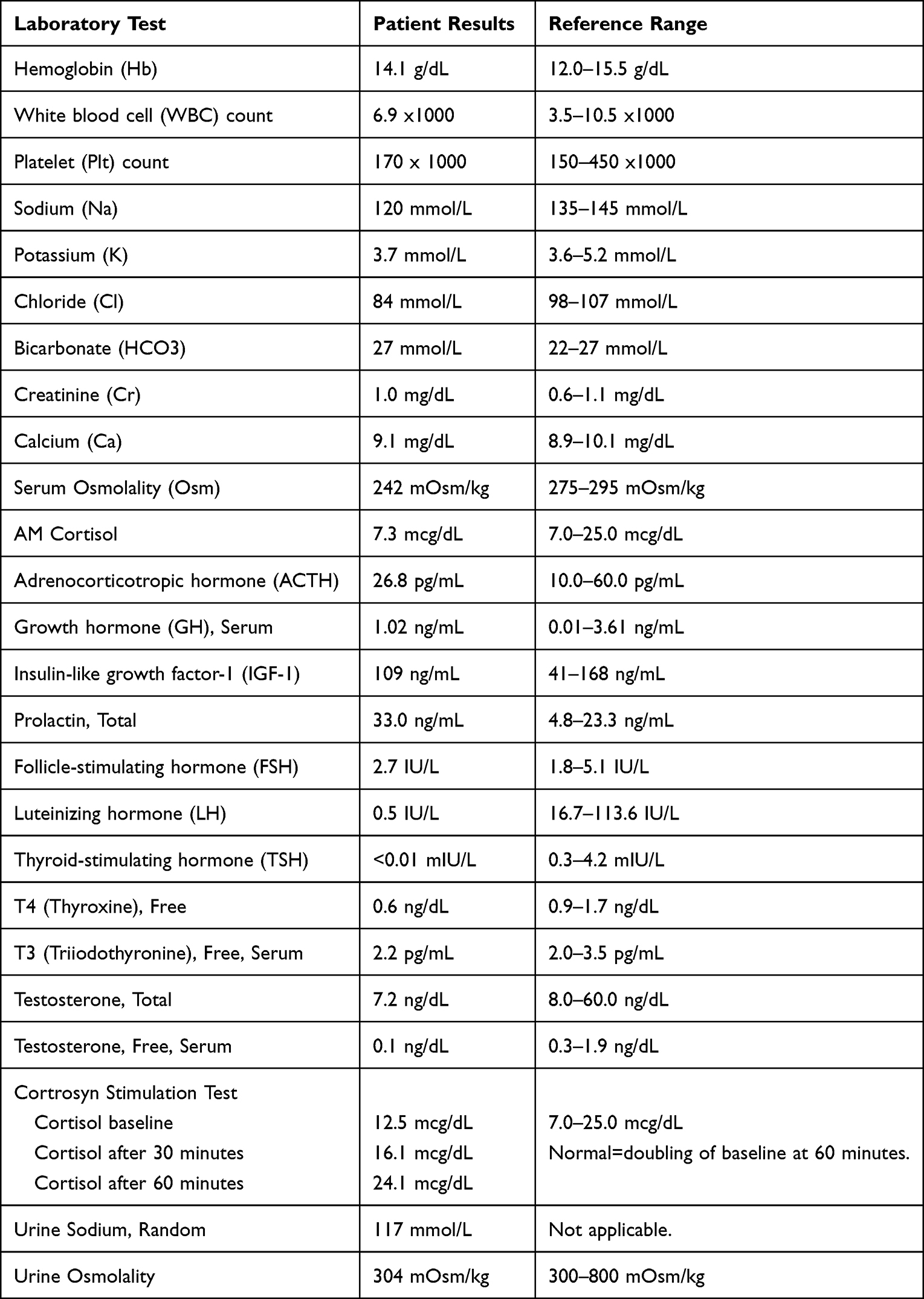 |
Table 1 Initial Laboratory Testing and Reference Ranges |
 |
Figure 1 MRI Brain Initial, 3 Month Surveillance Scan, Post-Surgery and Post-Treatment MRI. (A) Coronal enhanced T1-weighted MRI shows diffuse enlargement and diminished enhancement of the pituitary gland. (B) Follow up MRI 3 months later shows further enlargement of the pituitary (arrowhead) with suprasellar extension compressing the optic chiasm. (C) Residual tumor surrounding the surgical cavity (asterisk) following transsphenoidal resection. (D) MRI 10 weeks later upon completion of therapy showing complete resolution. Abbreviation: MRI, magnetic resonance imaging. |
Prior to resection and 3 months after initial presentation, the patient developed blurred vision and left > right retro-orbital pain with associated nausea and vomiting. On exam, she had new onset left anisocoria with the left pupil measuring 5 mm and the right pupil measuring 3 mm. Visual fields were intact. She had decreased sensation on the right upper face, V1 distribution. These findings, consistent with partial third and fifth CN palsies, were concerning for tumor extension into the cavernous sinus. Repeat MRI brain demonstrated interval enlargement of the sellar mass with compression of the central and left optic chiasm and adjacent left optic tract with focal extension into the bilateral cavernous sinuses, Figure 1B. She underwent partial transsphenoidal tumor resection (Figure 1C) and pathologic examination revealed malignant lymphoma replacing the pituitary gland, Figure 2A. Immunohistochemistry showed the neoplastic lymphocytes were diffusely positive for CD20, CD79a, PAX5, MUM1, MYC (60%), and BCL2 (90%) and weakly positive for BCL6, Figure 2B–D. The neoplastic cells were negative for CD10. The proliferation rate by Ki-67 was high (>90%), Figure 2E. EBER in situ hybridization for Epstein-Barr virus (EBV) was completely negative. In addition, fluorescence in situ hybridization (FISH) testing was positive for BCL6 gene rearrangement in 100% of nuclei and was negative for MYC and BCL2 gene rearrangements. Extra copies of IGH, IGK, BCL2, IGL loci, and of chromosome 8 were also detected on FISH, Figure 2F. Given these findings, the lymphoma was best classified as “double-expressor” diffuse large B-cell lymphoma (DLBCL), non-germinal center phenotype by Hans algorithm. CT of the chest, abdomen, and pelvis was negative for lymphadenopathy. Bone marrow biopsy and cytology from a lumbar puncture were also negative for lymphoma.
 |
Figure 2 Excisional Biopsy, Pituitary Tumor and FISH Testing. The excisional biopsy of the pituitary lesion revealed a diffuse proliferation of large lymphocytes with open chromatin and prominent nuclei; mitoses are frequent (A, H&E x 40). IHC studies showed the lymphocytes are positive for CD20, BCL2, MYC with a high proliferative rate by Ki-67 (B–E, x 20). The lymphocytes are negative for CD10 and EBER ISH. FISH study revealed BCL6 gene rearrangement (F, green and red arrows indicate separation of break-apart gene probes) and multiple copies of MYC, BCL2, IGL, and IGK. Abbreviations: EBER ISH, EBER in situ hybridization for Epstein-Barr virus; FISH, fluorescence in-situ hybridization; IHC, immunohistochemistry. |
She was diagnosed with PPL, DLBCL subtype. She was treated with rituximab (R) 375 mg/m2 and high-dose (HD) methotrexate (MTX) 3.5 g/m2 on day 1 and high-dose cytarabine (HiDAC) 2.0 g/m2 every 12 hours on days 2–3 (RMA) of each odd cycle alternating with rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2, vincristine 2 mg on day 1 and prednisone 100 mg days 1–5 (R-CHOP) for a total of eight cycles with four cycles of each regimen. She was in complete remission (CR) after 2 cycles (Figure 1D) and has remained in CR for the past 8 years. She continues to follow with endocrinology for management of secondary adrenal insufficiency and central hypothyroidism and takes hydrocortisone and levothyroxine. She has not experienced any significant long-term impact on her quality of life related to her lymphoma treatment.
Recently, a targeted genomic profile of the pituitary tumor tissue was performed through the Genomic Testing Cooperative Hematology Plus (GTC-Hematology Plus) platform. GTC-Hematology Plus utilizes a combination of next-generation sequencing (NGS), Sanger Sequencing, and fragment length analysis to assess genes (DNA of 179 genes and RNA in 1408 genes) frequently found in hematologic malignancies. The results are included in Table 2.
 |
Table 2 Genomic Profiling of the Pituitary Tumor |
Discussion
PPL is rare with less than 50 cases reported in immunocompetent individuals in the literature.11 PPL involves the sella with or without suprasellar or parasellar extension and no other systemic involvement.1,4–7 The mean age at presentation is 59 and PPL may be more common in females.1,11
The etiology of PPL remains uncertain, but the proposed risk factors differ depending on the immune status of the patient.1,5,6,11 In immunosuppressed patients, those with human immunodeficiency virus infection or prior transplant on chronic immunosuppressive therapy, viral reactivation with EBV is thought to be a driver of central nervous system (CNS) lymphoma.1,5,6 In immunocompetent patients, chronic inflammation in the setting of infection or lymphocytic hypophysitis (reported in 7% of cases), prior chemotherapy or radiation to the CNS, and concomitant pituitary adenoma (reported in 11% of cases) may predispose to PPL.1,5,6,11–13 Secretory adenomas are thought to enhance lymphoma cell proliferation as these malignant cells can express endocrine hormone receptors and may be stimulated by prolactin or growth hormone.7,11
Presenting symptoms vary depending on the size and location of the mass.1,5,11 The symptoms can include headache, vision changes, double vision, CN palsies, fatigue, and rarely B-symptoms.1,5,11 Pituitary dysfunction is common and has been reported in as many as 89.7% of cases.11 Anterior pituitary dysfunction is the most common, but panhypopituitarism, posterior pituitary failure (diabetes insipidus), and hyperprolactinemia as a result of stalk effect have also been reported.1,5,11 Anterior pituitary dysfunction was reported in 70% of patients and diabetes insipidus in 36% of patients in a study by Tarabay et al.1
PPL is often mistaken for pituitary macroadenomas or other more common CNS lesions.1,5 It should be considered in the differential of patients presenting with a sellar mass, particularly if the lesion is growing quickly or behaving in an atypical manner.1,5 If upfront surgical management in deferred, as in our case, serial imaging is critical to monitor rate of growth and assess for impending neurologic compromise.5 Imaging with CT, MRI, or PET-CT can help identify sellar pathology; however, transsphenoidal tissue biopsy or resection is necessary for definitive diagnosis.1,4,5,11 Once PPL is suspected, it should be distinguished from disseminated lymphoma with secondary CNS involvement.5 Work-up should include ophthalmologic evaluation, cerebrospinal fluid analysis, bone marrow biopsy, and evaluation of the chest, abdomen, and pelvis with either CT or PET-CT.1,5,12
B-cell lymphomas, in particular DLBCLs, account for the majority of PPL cases.1,5,7,11 T-cell and NK/T-cell lymphomas have also been reported.1 Given the rarity of the cancer, little is known about the genomic profile of these tumors, and to our knowledge we provide the first comprehensive genomic profiling of PPL. Comprehensive genomic analysis of our patient’s tumor was most consistent with DLBCL, non-germinal center subtype. Interestingly, the mutation profile did not resemble that of PCNSL. There was no evidence of MYD88, CDKN2A, or CD79A alterations which are commonly found in PCNSL.14 This suggests the biology and pathogenesis of PPL may differ from other PCNSL. This could be explained by the anatomic location of the pituitary outside of the BBB and its unique embryologic origin—the anterior pituitary originates from oral ectoderm (epithelial origin) and the posterior pituitary arises from neural ectoderm.8–10
Patients with PPL are treated similarly to other patients with PCNSL.1,4,11 Treatment regimens have included surgical resection, radiation, chemotherapy, or a combination of these therapies.1,4,6,11 Surgical tumor resection is thought to be an inadequate stand-alone therapy due to the infiltrative nature of lymphoma; however, partial or complete surgical resection helps to alleviate immediate symptoms as a result of tumor compression or invasion of surrounding structures that can cause CN palsies.1,3,5,6,11 Chemotherapy alone has resulted in CR and is thought to cause less long-term neurologic toxicity than combined chemoradiation.1,3,5 Therapies utilizing HD or intrathecal MTX, in regimens like lenalidomide, rituximab, methotrexate (R2-MTX) and R-CHOP with HD-MTX have resulted in CR.1,3–5,11
PPL is different from PCNSL in that the pituitary gland is outside the BBB while having direct vascular and neural connections to the brain. As such, it develops outside the BBB with risk of infiltrating and spreading to the brain.1,4,8,9,11 The therapeutic management of PPL should take into consideration the unique anatomy of the pituitary and include both CNS-penetrating and non-CNS penetrating therapeutic agents, like HD-MTX and R-CHOP regimens previously utilized.11
We adopted an anatomy-adapted therapeutic approach for our patient, alternating non-CNS penetrating systemic chemoimmunotherapy (R-CHOP) with CNS-targeted chemoimmunotherapy (RMA). She received a total of eight treatments with four cycles of each administered in an alternating fashion. This approach worked very well for our patient as evidenced by the longest continuous complete remission (~ eight years) reported in the literature. In contrast, a mean overall survival of 14.4 months has been reported.1,4,5
Conclusion
Although PPL has been regarded as a rare subtype of PCNSL, we believe it should be considered as a distinct entity given its unique anatomic localization outside the BBB and its proximity to and direct neural and vascular connections with the brain increasing the risk for cerebral infiltration by lymphoma. In addition, the genomic alternations found in our patient’s tumor are different from those commonly seen in PCNSL and also suggest this may be a distinct entity.
We suggest that an anatomy-adapted therapeutic approach should be considered in the management of PPL, employing both CNS-penetrating and non-CNS penetrating therapeutic agents. This approach helps address the unique anatomic localization of the pituitary. Additional research is necessary to further elucidate the biological mechanisms underpinning PPL and the most optimal therapeutic approach.
Abbreviations
BBB, blood brain barrier; CN, cranial nerve; CNS, central nervous system; CR, complete remission; CT, computed tomography; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein-Barr virus; FISH, fluorescence in situ hybridization; GTC, Genomic Testing Cooperative; HD, high-dose; HiDAC, high-dose cytarabine; MRI, magnetic resonance imaging; MTX, methotrexate; NGS, next-generation sequencing; PCNSL, primary central nervous system lymphoma; PET-CT, positron emission tomography-computed tomography; PPL, primary pituitary lymphoma; R, rituximab; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; RMA, rituximab, high-dose methotrexate, high-dose cytarabine; R2-MTX, lenalidomide, rituximab, methotrexate; TSH, thyroid-stimulating hormone.
Consent for Publication
The study participant has given written informed consent to participate as well as written informed consent to publish the data, case details, and images. Institutional approval was not required to publish this case.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tarabay A, Cossu G, Berhouma M, Levivier M, Daniel RT, Messerer M. Primary pituitary lymphoma: an update of the literature. J Neurooncol. 2016;130(3):383–395. doi:10.1007/s11060-016-2249-z
2. Shiels MS, Pfeiffer RM, Besson C, et al. Trends in primary central nervous system lymphoma incidence and survival in the US. Br J Haematol. 2016;174(3):417–424. doi:10.1111/bjh.14073
3. Grommes C, DeAngelis LM. Primary CNS lymphoma. J Clin Oncol. 2017;35(21):2410–2418. doi:10.1200/JCO.2017.72.7602
4. Duan L, Liu J, Zhang Y, et al. Primary pituitary lymphoma in immunocompetent patients: a report on two case studies and the review of literature. Front Endocrinol. 2020;11:562850. doi:10.3389/fendo.2020.562850
5. Giustina A, Gola M, Doga M, Rosei EA. Clinical review 136: primary lymphoma of the pituitary: an emerging clinical entity. J Clin Endocrinol Metab. 2001;86(10):4567–4575. doi:10.1210/jcem.86.10.7909
6. Rainsbury P, Mitchell-Innes A, Clifton N, Khalil H. Primary lymphoma of the pituitary gland: an unusual cause of hemianopia in an immunocompetent patient. JRSM Short Rep. 2012;3(8):55. doi:10.1258/shorts.2012.012067
7. Ravindra VM, Raheja A, Corn H, et al. Primary pituitary diffuse large B-cell lymphoma with somatotroph hyperplasia and acromegaly: case report. J Neurosurg. 2017;126(5):1725–1730. doi:10.3171/2016.5.JNS16828
8. Nussey S, Whitehead S. Endocrinology: An Integrated Approach. CRC Press; 2001.
9. Dash P Chapter 11: blood brain barrier and cerebral metabolism. McGovern medical school at UTHealth; 2022. https://nba.uth.tmc.edu/neuroscience/m/s4/chapter11.html.
10. Larkin S, Ansorge O. Development and microscopic anatomy of the pituitary gland. In: Feingold KR, Anawalt B, Boyce A, editors. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000.
11. Caputo M, Prencipe N, Bisceglia A, et al. Primary pituitary lymphoma as rare cause of a pituitary mass and hypopituitarism in adulthood. Endocr Pract. 2020;26(11):1337–1350. doi:10.4158/EP-2020-0286
12. Ren S, Lu Q, Xiao Y, et al. Coexistence of pituitary adenoma and primary pituitary lymphoma: a case report and review of the literature. Front Surg. 2022;9:842830. doi:10.3389/fsurg.2022.842830
13. Huang YY, Lin SF, Dunn P, Wai YY, Hsueh C, Tsai JS. Primary pituitary lymphoma presenting as hypophysitis. Endocr J. 2005;52(5):543–549. doi:10.1507/endocrj.52.543
14. Radke J, Ishaque N, Koll R, et al. The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat Commun. 2022;13(1):2558. doi:10.1038/s41467-022-30050-y
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.