")
Back to Journals » The Application of Clinical Genetics » Volume 16
Sturge-Weber Syndrome: A Review of Pathophysiology, Genetics, Clinical Features, and Current Management Approache
Authors Sánchez-Espino LF , Ivars M , Antoñanzas J , Baselga E
Received 2 November 2022
Accepted for publication 10 March 2023
Published 24 April 2023 Volume 2023:16 Pages 63—81
DOI https://doi.org/10.2147/TACG.S363685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Luis Fernando Sánchez-Espino,1 Marta Ivars,1 Javier Antoñanzas,2 Eulalia Baselga1
1Pediatric Dermatology Department, Barcelona Children’s Hospital Sant Joan de Dèu, Barcelona, Cataluña, Spain; 2Dermatology Department, Clínica Universidad de Navarra, Pamplona, Navarra, Spain
Correspondence: Eulalia Baselga, Department of Dermatology, Hospital Sant Joan de Deu, Passeig de Sant Joan de Déu, 2, Esplugues de Llobregat, Barcelona, 08950, Spain, Tel +34-686-68-9669, Email [email protected]
Abstract: Sturge-Weber syndrome (SWS) is a congenital, sporadic, and rare neurocutaneous disorder, characterized by the presence of a facial port-wine birthmark (PWB), glaucoma, and neurological manifestations including leptomeningeal angiomatosis and seizures. It is caused by a postzygotic, somatic, gain-of-function variant of the GNAQ gene, and more recently, the GNA11 gene in association with distinctive clinical features. Neuroimaging can help identify and stratify patients at risk for significant complications allowing closer follow-up; although no presymptomatic treatment has been demonstrated to be effective to date, these patients could benefit from early treatment and/or supportive interventions. Choroid plexus (CP) thickness measurements in brain magnetic resonance imaging (MRI) have a high sensitivity and specificity for early and incipient changes in SWS. In contrast, the absence of pathologic findings makes it possible to rule out associated neurological involvement and leads to periodical observation, with new imaging studies only in cases of new clinical signs/symptoms. Periodic ophthalmological examination is also recommended every 3 months during the first year and yearly afterwards to monitor for glaucoma and choroidal hemangiomas. Treatment for SWS depends on the extent and areas that are affected. These include laser surgery for PWB, anticonvulsants in the case of brain involvement, with either seizures or abnormal EEG, and medical treatment or surgery for glaucoma. Sirolimus has been used in a limited number of patients and appears to be a safe and potentially effective treatment for cutaneous and extra-cutaneous features, however controlled clinical studies have not been carried out. Better knowledge of GNAQ/GNA11 molecular pathways will help to develop future targeted treatments.
Keywords: Sturge-Weber-syndrome, port-wine stain, capillary malformation, targeted therapies, GNAQ/GNA11, sirolimus
Introduction
Sturge-Weber syndrome (SWS) is a congenital, sporadic, and rare neurocutaneous disorder, characterized by the presence of a facial port-wine birthmark (PWB), glaucoma, and leptomeningeal angiomatosis. Classically, it is caused by post-zygotic, somatic, gain-of-function variants in GNAQ but recently GNA11 variants have been described with subtle phenotypic particularities.1
SWS occurs at an estimated frequency of between 1:20,000 and 1:50,000 live births and affects males and females equally without racial predilection. It is the third most common neurocutaneous syndrome behind neurofibromatosis and tuberous sclerosis.2,3
Embryology
The exact reason for the vascular and neurologic involvements remains an ongoing debate; however, it is hypothesized that capillary malformations (CM) and leptomeningeal angiomatosis could be the result of the absent regression of a primitive cephalic plexus.2 The persistence of primordial sinusoidal vascular channels and the underdevelopment of the brains’ superficial venous drainage with compensatory deep dilated venous channels affect the capillaries as well.4
The embryological proximity of the ectoderm that will form the frontonasal skin and the contiguous neural folds (those involved in the primitive formation of the parieto-occipital lobes of the brain) could explain the association of PWB with leptomeningeal angiomatosis.5 The distribution of facial PWB in patients with SWS on the forehead area suggests a mutational event on the forebrain, from which the cerebral cortex and eye are also derived.6
Endothelial vessels in PWB co-express venous (EphB1) and arterial (EphrinB2) markers, demonstrating abnormal endothelial cell differentiation that results in venule-like vasculature and conforming areas of angiomatosis with abnormal flow.5 These areas of angiomatosis are described as capillary-venous-type vascular malformations, which result in an abnormally loose network of perivascular nerve fibers running longitudinally along the axis of the vessels. In fact, histological and imaging findings from brain angiomas in SWS have revealed tortuous and abnormal vascular structures in the thickened leptomeninges, ensuing in underlying brain tissue that may be atrophic, show neuronal loss, astrogliosis, dysgenic cortex, and/or calcification in cortical layers. There is a relative lack of superficial cortical veins, and the blood is shunted to the deep venous system by the enlarged medullary veins, which result in stasis and ischemic changes.
Genetics, Pathogenesis, and Genotype Correlation
Our knowledge and understanding of the pathogenesis of SWS, particularly its genetic origin and pathogenesis, has increased over recent years.
Shirley et al7 detected an activating somatic variant in the GNAQ gene (located in chromosome 9;locus 9q21.2), specifically, an arginine to glutamine substitution at 183 amino acid residues (c.548G→A, p.Arg183Gln) in 23 samples of affected tissue from 26 patients with SWS (88%), and in 12 of 13 patients with non-syndromic facial capillary malformations (92%). The variant allele frequency (VAF) was reported between 1% and 18%, confirming the mosaic nature of SWS.
Couto et al8 confirmed that CM is caused by somatic activating missense variants (VAF ranged from 2% to 11%) in GNAQ and that p.R183Q is the most common mutation. These authors identified two novel GNAQ variants in the same codon (p.R183L and p.R183G) and demonstrated that GNAQ mutations are enriched in the endothelial cell compartment.9 Familial cases of SWS due to a variant in this codon (R183Q) have been reported.10
GNAQ synthesizes Gαq (guanine nucleotide binding protein) - an alpha subunit of the heterotrimeric G protein that links G-protein–coupled receptors (GPCR) to activation of PLC (phospholipase C) β. The latter hydrolyzes phosphatidylinositol 4,5 biphosphate (PIP2) to generate diacylglycerol and inositol 1,4,5 triphosphate (IP3).11 G protein-coupled receptors (GPCRs) release guanosine diphosphate (GDP) and promote the binding of guanosine triphosphate (GTP) to GNAQ, causing its activation by the dissociation from the Gβγ complex.12 This effector process is stopped upon hydrolysis to its inactive form via the GTPase activating proteins (GAPs). The GTPase RAS-like domain (where the GDP/GTP binding and hydrolysis occur) is surrounded by an α-helical domain that protects it from binding to any effector nucleotides.13,14 The mutated R183 codon blocks the uncoupling function of Gαq with GDP, therefore promoting uncontrolled tumoral and vascular growth seen in cases of uveal melanoma as the second most common oncogenic mutation.10,15 Although no association between SWS and uveal melanoma has been reported to date, the fact that they share the same variant (also the reported isolated cases of uveal melanoma in phakomatosis pigmentovascularis) suggests that patients with SWS should likely undergo ophthalmic examination to evaluate features of ocular melanocytosis and potentially, yet unlikely, uveal melanoma.15
More recently, reports of SWS due to variants in the GNA11 gene have expanded our understanding of the SWS phenotypic spectrum.1,16,17 Patients with somatic GNA11 variants have been described with some distinctive clinical features (see clinical features section) from the classical SWS and are now to be considered within the phenotypic spectrum of this condition.17 The GNA11 gene, a paralog of GNAQ (Gα11 shares 90% amino acid affinity with Gαq), encodes the alpha subunits of the heterotrimeric G proteins, Gq and G11. Mutations in these proteins lead to dysregulation of several signaling pathways, including phospholipase C, and activation of the transcription factor YAP, leading to cellular proliferation via mitogen-activated protein kinase and abnormal gene transcription.1,18 The presence of GNAQ variants within blood vessels stimulates the activation of c-Jun N-terminal kinases (JNK) and extracellular signal regulated kinases (ERK).19
In SWS, the variant GNAQ/GNA11 gene causes abnormal gain-of-function signal enhancement that is further translated into active endothelial cells, proliferative capillary overgrowth, or differentiation-impaired endothelial cells with a progressive dilatation of immature venule-like vasculature.20 In a lentiviral construct to express p.R183Q in normal human endothelial colony forming cells showed constitutively activated PLC (phospholipase C) β3, a downstream effector of Gαq.21 Activated PLCβ3 has also been detected in human CM tissue sections. Bulk RNA sequencing of mutant endothelial colony forming cells showed constitutive activation of PKC (protein kinase C), NF-κB (nuclear factor kappa B), and calcineurin signaling. Increased expression of downstream targets in these pathways, ANGPT2 (angiopoietin-2) and DSCR (Down syndrome critical region protein) were confirmed by qPCR and immunostaining of human capillary malformation tissue sections.21 Mutant endothelial colony forming cells when injected subcutaneously into immunodeficient mice formed enlarged blood vessels, reminiscent of those found in human capillary malformation and suppression of ANGPT2 prevents the enlargement.21
Additionally, GNAQ and GNA11 genes are involved in calcium homeostasis. They exert functional effects on the endoplasmic reticulum and regulate the extracellular calcium (Ca2+e) levels by mediating calcium-sensing receptors (CaSR), a class C G-protein coupled receptor (GPCR).22 GNAQ/GNA11 variants cause increased activation of intracellular calcium signaling, which may explain low calcium serum values and abnormal brain calcifications seen in some patients with SWS.23 In fact, germline missense GNAQ/GNA11 variants or deletions have been associated with familial hypocalciuric hypercalcemia type-2 (FHH2) and autosomal dominant hypocalcemia type-2 (ADH2).22 It is believed that this may play a role in the pathogenesis of SWS due to the interplay of calcium with endothelial growth factors (VEGF) and endothelial receptors (VEGFR) (unpublished data), which are known promoters of extracellular growth via cytokine stimuli and growth factors interaction.20,23
Clinical Features
SWS is characterized by cutaneous, neurological, and ocular abnormalities. The characteristic vascular malformations are facial capillary malformation (PWB) and ocular and cerebral leptomeningeal capillary-venous malformation (or leptomeningeal angiomatosis), which are usually ipsilateral to facial PWB.
Encephalofacial angiomatosis, a flawed and outdated but persistent term, has been divided into three phenotypes recognizing variable distribution of facial capillary malformation to include isolated facial involvement versus associated leptomeningeal involvement.2,24 One of the initial clinical classifications of SWS, the Roach Scale classification, divided encephalofacial angiomatosis as follows: Type I: both facial and leptomeningeal malformations; may have glaucoma (classic SWS). Type II: facial capillary malformation alone; may have glaucoma. Type III: isolated leptomeningeal angiomatosis; usually no glaucoma.2 Nonetheless, this outdated classification incorrectly implies that all patients with a facial capillary malformation (without brain or eye involvement) have type II SWS. At present, we now know that the syndromic association to SWS and the risk of both brain and eye involvement can be more accurately stratified based on the extension and location of the facial capillary malformation (Figure 1).
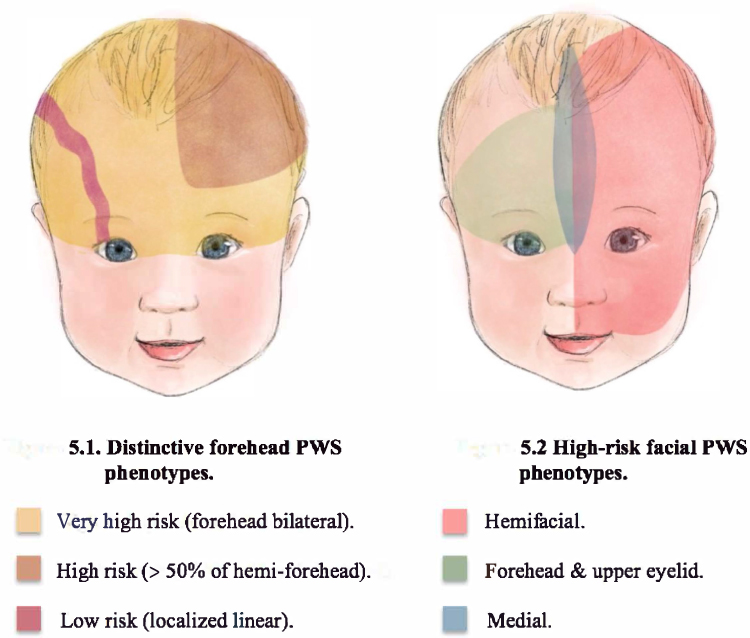 |
Figure 1 Risk stratification for the development of Sturge Weber Syndrome (SWS) based on distinct facial port-wine birthmark (PWB) phenotypes. |
Dermatological Manifestations
Capillary malformations are the most common type of vascular malformation, affecting about 0.3% of the general population.25 They range from pink and red to violaceous macules and patches that may be geographic or blotchier in nature. CM may affect the mucous membranes. In SWS, capillary malformation follows a distinctive anatomical distribution in the face, which is characteristic of these conditions (Figures 1 and 2). The associated risk of leptomeningeal or ocular involvement depends on the extent and location of the CM but it ranges from 7% to 28%.26
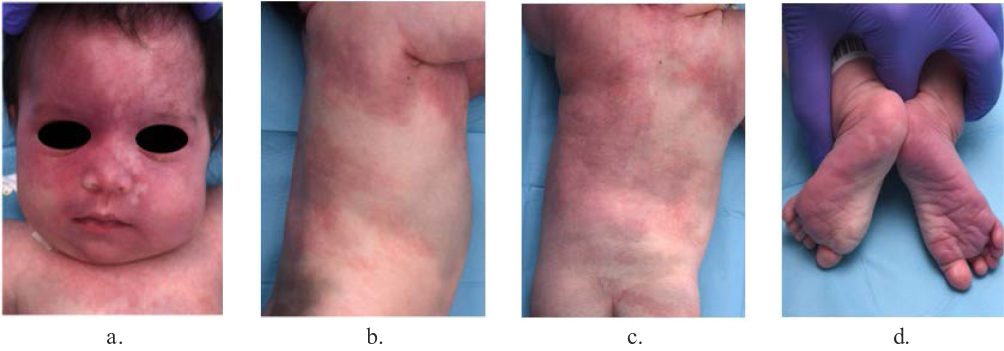 |
Figure 2 GNAQ SWS phenotype. Extensive facial Port-Wine birthmark with geographical and sharp borders (a). Presence of additional and extensive capillary malformations of the anterior, lateral and posterior trunk (b and c). Bilateral and symmetrical involvement of the plantar aspect of both feet (d). |
Historically, it was thought that the distribution of facial PWB followed the distribution of the sensory innervation branches of the trigeminal nerve (ophthalmologic [V1], maxillary [V2], and mandibular [V3]) and that children with involvement of the first branch (V1) were at highest risk of SWS.27–29 More recently, it has been demonstrated that the area of highest risk is the forehead, the area demarcated laterally and inferiorly by the outer margins of the canthus of the eye (including the upper eyelid) to the top of the ears.6 This area corresponds to the frontonasal prominence and the skin in the optic vesicle area. The skin in these areas is formed by the migration of neural crest cells from the prosencephalon (forebrain) and anterior mesencephalon (midbrain). Leptomeninges and the eye also derive from prosencephalon and therefore a somatic mutation in prosencephalon could affect the skin of the forehead, leptomeninges, and the eye.6,30
Aside from location, the risk of SWS is higher with increased extent of facial capillary malformation (Figure 1).30 Patients with extensive bilateral involvement in hemifacial forehead locations are at highest risk of SWS (Table 1).27,30–32 There is some debate as to whether median frontal location of PWB confers a higher risk than lateralized PWB. A retrospective study of 124 children reported that SWS was exclusively found in those with large and segmental PWB (at least half of a contiguous hemi forehead area), and this location had the highest specificity (72%) and positive predictive value (42%) for the risk of SWS compared to other forehead locations (Figure 1).30,33
 |
Table 1 Port Wine Stain Characteristics and Risk of CNS Involvement |
It is important to note that localized and minimal forehead involvement might still confer a small risk of SWS.33 Evaluation and screening of these patients should be tailored individually and based on additional supportive clinical findings.
Over time, facial skin lesions tend to darken, turning to a deep red to purple, or “port-wine” appearance. The affected skin might also grow thicker as the patient grows, and progressive vascular ectasias can lead to blebbing and nodularity on the surface of the PWB. There may be bony and soft tissue overgrowth, especially of the maxillary bone and of the lips when these areas are affected. This overgrowth is suspected to be triggered as well by the GNAQ/GNA11 activation. Soft tissue or bony hypertrophy occurs in approximately two-thirds of patients by the age of 50.34,35
Nodules are composed of either an aggregation of numerous thin-walled vessels whose lumina are of various calibers or authentic pyogenic granulomas. This may arise within capillary malformation both in the skin and mucosa.36,37 In some instances, these “angiomatous” nodules show a well-circumscribed proliferation of thick-walled muscle-containing blood vessels, lined by a single layer of endothelial cells resembling arteries, but lacking a well-formed elastic internal membrane and have been named acral arteriovenous tumors.38 It has been proposed that they can spontaneously grow from abnormal and microscopic arteriovenous anastomoses within the context of ectatic capillaries and veins.37 Dermal melanocytosis may be seen in patients with SWS and is called phakomatosis cessioflammea or phakomatosis pigmentovascularis.
The phenotypic spectrum of SWS recently expanded.17,39,40 Patients with a GNA11 variant have a slightly different clinical presentation compared to the classical SWS (Figure 3). In GNA11-SWS, the capillary malformation is pale and pink, and more reticulated in configuration (Figure 3). This variant may be accompanied with variable degree of other pigmentary changes in combination with capillary malformation, such as nevus anemicus (nevus vascularis mixtus), cafe-au-lait macules, and dermal melanocytosis. GNA11 capillary malformations tend to have progressive but slower purpuric darkening of the capillary stain over time compared to patients with GNAQ variants.17 Neurological manifestations tend to be milder and there are other distinctive extra-cutaneous features such as limb hyper or hypotrophy, renal anomalies, and hypertension (discussed below).1,16,17 There are only a few case reports and small cohorts of GNA11-SWS and therefore the distinct clinical phenotype needs further clarification.
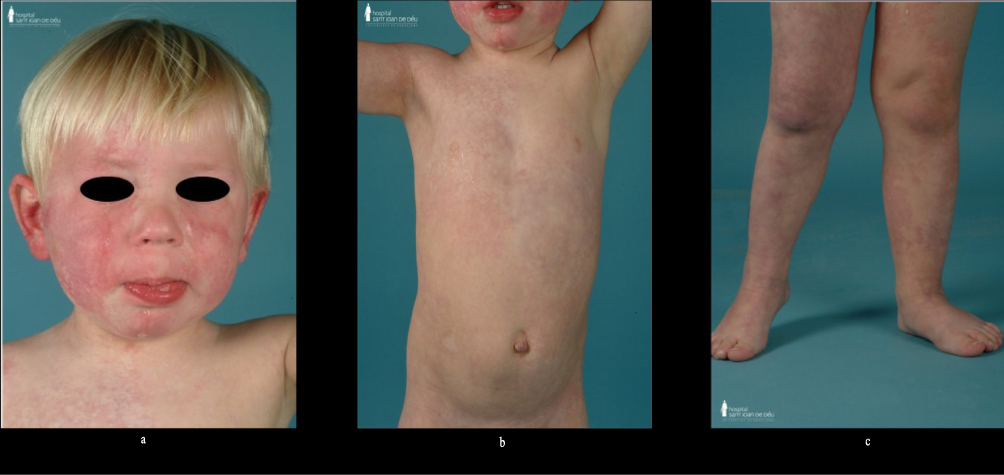 |
Figure 3 GNA11 SWS phenotype. Facial, truncal and appendicular extensive, reticulated and blotchy capillary malformation (a–c). |
Even though activating variants in GNAQ/GNA11 have potential oncogenic activity (as demonstrated in cases of uveal melanoma), PWB and SWS are not known to be associated with a significant risk of skin tumor development. However, several reports have been published describing basal cell carcinomas (BCC) arising within the PWB. Currently, there are reports of 36 patients and 79 BCCs arising within PWB, especially in patients who had undergone previous treatments with radiotherapy, or with occupational exposure to sunlight.41 These may be difficult to detect, mainly because over time the stain gradually darkens, becomes thicker, raised, and nodular, concealing potential malignant lesions. In most cases, simple excision of the tumor is sufficient, followed by skin grafting if needed. Local flaps must be considered for assessing the risk of flap complications due to surrounding tissues affected by PWB and compromising local vascular irrigation.
Therefore, it is important to encourage periodic follow-ups, and eventually, perform skin biopsies in areas of persistent scaling or ulceration within the PWB. This is specially recommended in patients previously treated with carcinogenic agents or with chronic exposure to sunlight.
Neurological and Neuroendocrine Manifestations
Neurologic complications are common and affect about 70–80% of patients with SWS. These are varied and include seizures, hemiparesis, stroke-like events, intellectual disability, and behavioral problems, amongst others.9 Seizures usually manifest as infantile spasms before the first year of age.2 Although rare, patients with their first seizure in adulthood have been reported.42,43 Seizures may be initially focal and may go unrecognized but often evolve to generalized seizures and status epilepticus. Leptomeningeal angiomatosis is not always symptomatic and up to 15–25% of affected children and young adults do not develop seizures.17,44,45
Leptomeningeal angiomatosis is usually unilateral, posterior, and ipsilateral to PWB; however, it may not always correlate with the distribution of facial birthmark.46 Also, the extent of facial lesion does not always predict the severity of brain lesion and intellectual disability.24,46 Bilateral intracranial involvement, reported in about 15% of patients, is associated with a higher incidence and earlier presentation of seizures and a poorer outcome.24
Seizures present in patients with SWS are the result of cortical irritability caused by cerebral vascular malformation, through mechanisms of hypoxia, ischemia, and gliosis.2 The underlying cause of these clusters remains unclear, but it may relate to underlying vascular or parenchymal changes, including dense calcifications near the leptomeningeal capillary malformations. Early onset of seizures, medical intractability, bilateral leptomeningeal involvement, and severe unilateral lesions are indicative of a poor prognosis.24 The latter can pose a therapeutic challenge due to its refractory nature and might respond poorly to multiple antiepileptic medications.
While epilepsy and mental retardation are the most common symptoms, migraine-like headaches have been recognized as an important feature of SWS. Twenty-eight percent of patients with SWS may experience headaches having clinical characteristics of migraines, which might also be debilitating to patients.24
“Stroke-like” episodes or transient/permanent hemiparesis episodes are seen with relative frequency in SWS.47 A retrospective cohort from the UK (n=102) reported that the frequency of these episodes ranges from 31% to 46% in SWS patients aged 2–22 years, similar data compared to a multi-center cohort study of the USA (n=277).47,48 These events tend to appear between 6 months to 5 years; however, when present <6 months of age, they could be long-lasting or permanent in nature. Its etiology and physiopathology has not been fully understood, but it is thought that these can be the result of either a transient ischemic event or seizures/epileptiform activity; however, due to the inconsistent epileptiform EEG and/or ischemic brain MRI changes during these events it continues to be subject of ongoing debate.9 A traumatic injury to the head (contusion or fall) should always be taken into consideration as a triggering event, as 1 of 5 patients report this association according to a previous report.49 The clinical variability amongst these episodes (degree of motor weakness) and broad time until recovery (1 min to 6 months) make the prognosis challenging. Low-dose aspirin and anti-epileptic drugs (read below in other treatments section) seem to delay the onset of seizures and frequency of stroke-like events,50 however, further studies are needed to better understand and characterize these events in order to provide accurate preventive strategies and potentially, effective treatments that might aid in a faster recovery and/or minimize sequelae.
Cognitive impairment is also common in patients with SWS. Attention, emotional, and behavioral problems, including a higher prevalence of depression, oppositional/defiant, and conduct disorders are also often described in this population.24 Social skills issues are more common in those children with seizures with cognitive deficits.51
Ophthalmological Manifestations
Up to 50% of patients with SWS have a variable degree of ocular abnormalities.52 The two most common manifestations include glaucoma and the presence of choroidal hemangiomas. Other eye vascular anomalies can affect the conjunctiva, the episclera, the retina, and/or the choroid, which can lead to optic atrophy and blindness.2
Glaucoma, the most common ocular problem in patients with SWS, is almost always ipsilateral to facial PWB or leptomeningeal angiomatosis. Glaucoma can be congenital or appear later in life.2,51 Its exact pathophysiology is unknown; however, it is thought to be secondary to abnormal angle development.52 Other suspected mechanisms include an increased episcleral venous pressure (witnessed in older children or young adults) and/or drainage abnormalities related to outlet resistance of the distal outflow pathway.2,52
Up to 30–70% of patients with SWS develop glaucoma, and 60% of them develop it during infancy.51 Glaucoma is challenging to manage and often refractory to medical and surgical therapy,53 and requires regular examinations due to its life-long risk.54 A study of 34 neonates with facial PWB who underwent ocular pressure measurements before 1 month of age noted that those with both frontal and malar involvement had the highest risk of glaucoma and correlated with the size of the capillary malformation in the malar area and involvement of the lower eyelid.55 Interestingly, in this report, none of the nine eyes lacking PWB eyelid involvement were diagnosed with glaucoma. Children with involvement of both eyelids were at highest risk, and in this study, involvement of the lower eyelid conferred higher risk than upper eyelid alone (83% vs 18%, respectively) (Table 2).55
 |
Table 2 Port Wine Stain Characteristics and Risk of Glaucoma |
Another study analyzed a cohort of 569 patients with facial PWB and stratified the risk of glaucoma based on specific features of facial capillary malformations.56 The prevalence of glaucoma in this study was 19.3% and correlated to a higher risk of developing glaucoma in bilateral and extensive PWB lesions involving the malar area. Also, male gender was associated with a higher risk for the development of early-onset glaucoma (defined as present <4 years of age) in this cohort.56
Choroidal hemangioma (CH), which is a benign vascular tumor, is prevalent in 40% to 50% of patients with SWS. It is more often diffuse in nature but can be circumscribed and is usually ipsilateral to the facial PWB.57,58 It is caused by aberrant and enlarged vessels and vascular channels that cause variable clinical features including reduced visual acuity, refractive errors, scotoma, or retinal detachment.52
Choroidal hemangioma gives an appearance of “tomato ketchup” fundus. Diagnostic suspicion can be confirmed by fundus fluorescein angiography, indocyanine green chorioangiography, B-scan ultrasound, OCT, and even MR. Management of CH depends on whether it is diffuse or circumscribed.
For diffuse CH causing vision loss, external beam radiotherapy, stereotactic radiotherapy, proton beam radiotherapy, and plaque brachytherapy may be used. Photodynamic therapy with verteporfin has become more popular recently. In circumscribed CH, argon lasers have been used in addition to the treatments used for diffuse choroidal hemangioma. Treatment may be challenging due to the anatomical regions that are affected (both juxta papillary and foveal regions).52
Other ocular abnormalities that present with variable frequency include vascular malformation, conjunctival affection (diffuse or localized), dilated retinal and persistent episcleral vessels, all of which have an ipsilateral presentation in relationship to facial PWB.52 Iris heterochromia, if present, is related to an increased risk (up to 45%) of glaucoma.52 Ocular melanocytosis, iris mammillations, cilioretinal occlusion, and hemiretinal occlusion have been reported in patients with SWS as well.52
Screening for ophthalmologic involvement is critical to promptly managing any complications in order to preserve vision.
Other Manifestations
Additionally, patients with SWS are at a higher risk of developing hormonal imbalances. An earlier publication reported that growth hormone deficiency can occur even in the absence of clear anatomical alterations of the pituitary or hypothalamus on brain imaging studies.59 Considering that patients with SWS are at a higher risk of hypothalamic–pituitary compromise, testing should be done based on the appearance of any suggestive signs/symptoms. Additionally, two studies reported central hypothyroidism in three patients, one of which was detected incidentally on a routine examination.60,61 We consider that a low threshold for endocrinology referral or evaluation is to be considered in patients with SWS.
Patients with SWS are at a higher risk of oral mucosal bleeding during regular dental procedures due to the presence of intra-oral angiomatosis exacerbated by the presence of soft-tissue overgrowth and gingival hyperplasia (triggered/worsened by the use of antiepileptic medications) which may complicate good dental hygiene in addition to compromising physiological hemostasis.62
Oral vascular lesions may range from localized vascular hyperplasia to severe angiomatous proliferation or pyogenic granulomas, making the affected mucosa friable to minimum trauma and minor dental interventions.63 These lesions are sharply demarcated in the midline and are ipsilateral to the facial lesion (Figure 4). For this reason, careful pre-operative planning should be carefully done prior to any dental surgery and a multidisciplinary approach is recommended in the hands of an experienced pediatric dentist and maxillofacial surgeon at tertiary centers rather than outpatient dental/surgical offices, which may offer limited support.64
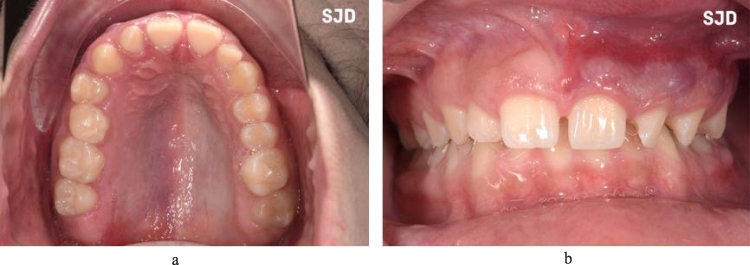 |
Figure 4 Palatal and gingival angiomatosis. Note the abrupt demarcation at the midline (a and b). |
Some additional features are to be considered and screened in patients with SWS (Table 3). Hyper and undergrowth have been reported in previous studies.16 An international cohort study of 45 patients reported that up to 9% of cases (where head circumference was measured) had macrocephaly. Growth asymmetry was reported in 35% of the cases in this cohort and renal hypertension was detected in a few patients with GNA11 mutation, therefore anthropometric measures are recommended to be screened thoroughly and blood pressure checks done regularly in these patients.17
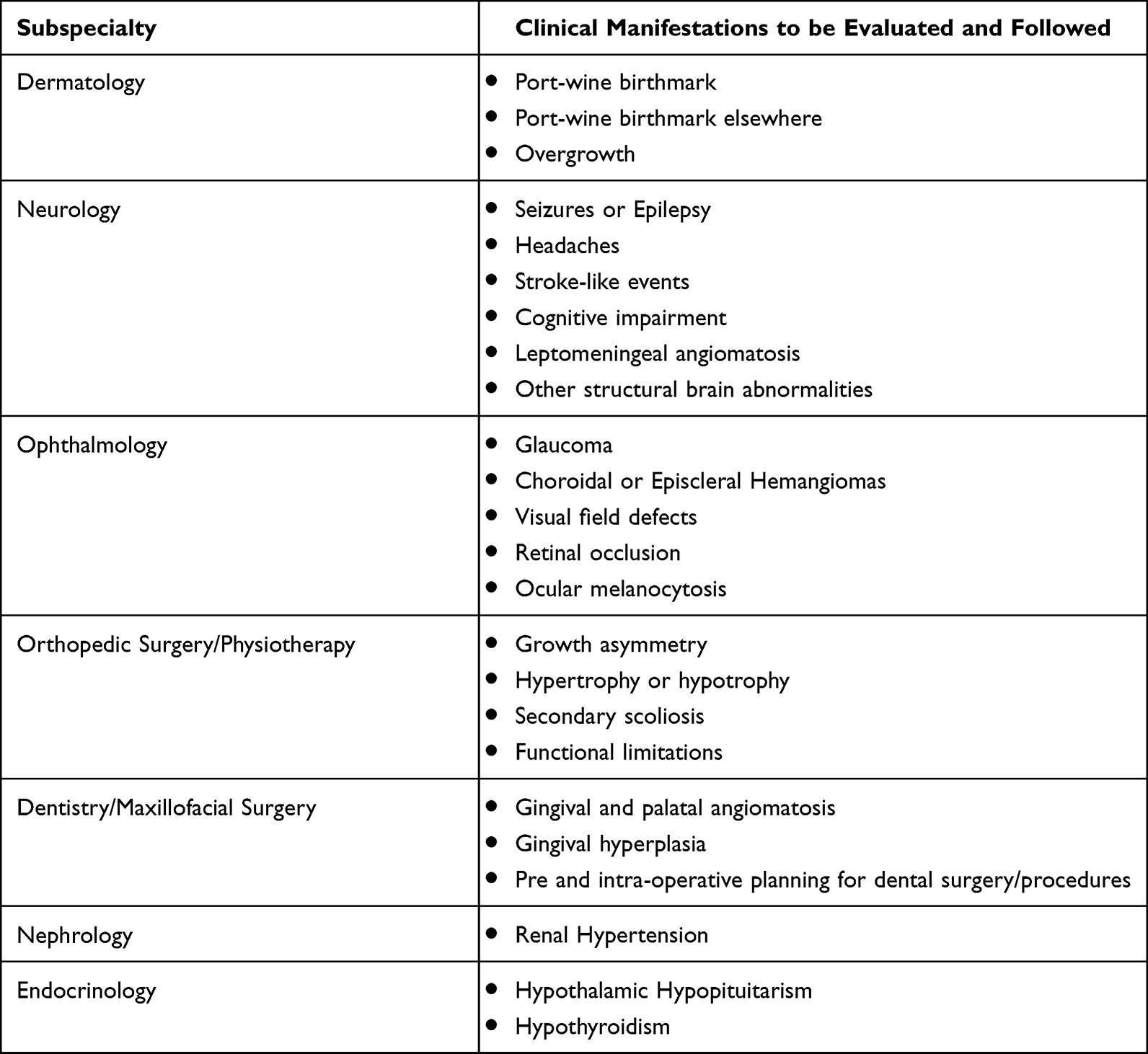 |
Table 3 Clinical manifestations in diferent organs to be evaluated and followed in patients with Sturge-Weber. |
Differential Diagnoses
Facial PWB may be confused early on with a precursor of hemangioma. Follow-up over time allows differentiation as hemangioma will proliferate, even if minimally and precursors of hemangioma usually develop a white peripheral halo and are more telangiectatic in nature.
Salmon patches can be confused with PWB, especially when they are intense in color. Salmon patches can be distinguished by their characteristic locations such as bilateral upper eyelids, glabella, nape, and scalp. PWB is commonly lateralized to one side of the face, but on rare occasions PWB can present at the medial forehead location.26,65 Differentiation in these cases may be impossible from a clinical point of view and close follow-up or even brain imaging may be considered.
Arteriovenous malformations in their quiescent phase present as a “capillary” stain that may be confused with a PWB. The precursors of an arteriovenous malformation are usually warmer to palpation, more heterogeneous in color, and have archipelago-like borders. A Doppler ultrasound may help in differentiation.
Megalencephaly capillary malformation syndrome (MCAP), a PIK3CA overgrowth syndrome, may be confused with SWS as this condition is also associated with facial (medial forehead location) and truncal reticulated capillary malformations (similar to the GNA11 SWS phenotype), partial overgrowth, and macrocephaly. However, the more extensive overgrowth; toe abnormalities, the frequent involvement of the philtrum, and the lack of leptomeningeal angiomatosis or glaucoma in MCAP help in differentiation. Genetic studies also help in differentiation.
Other conditions within the PIK3CA-related overgrowth syndromes (PROS) spectrum can also present with a facial capillary malformation but lack leptomeningeal angiomatosis or glaucoma and have a more pronounced (and often lipomatous) overgrowth of subcutaneous tissue. A few patients with SWS with more extensive capillary malformations and mild overgrowth have been reported as having a SWS-Klippel Trenaunay syndrome (KTS) overlap. In our opinion, these patients lack the more characteristic venous anomalies and the degree of overgrowth seen in KTS and fit more into the SWS definition.66–70 When performed, genetic studies demonstrate overlap the GNAQ R183 variant in these patients with clinical overlap.71,72
Radiological Management and Neuroimaging
Gadolinium enhanced MRI is the main imaging modality for the diagnosis of SWS brain involvement. It can detect leptomeningeal enhancement, abnormal venous drainage, cortical atrophy, and hypermyelination underneath the leptomeningeal angiomatosis, as well as calcifications. There is extensive controversy regarding imaging studies of presymptomatic infants at risk, and despite a recent expert panel recommendation, the debate has not been settled26 (Figure 5).
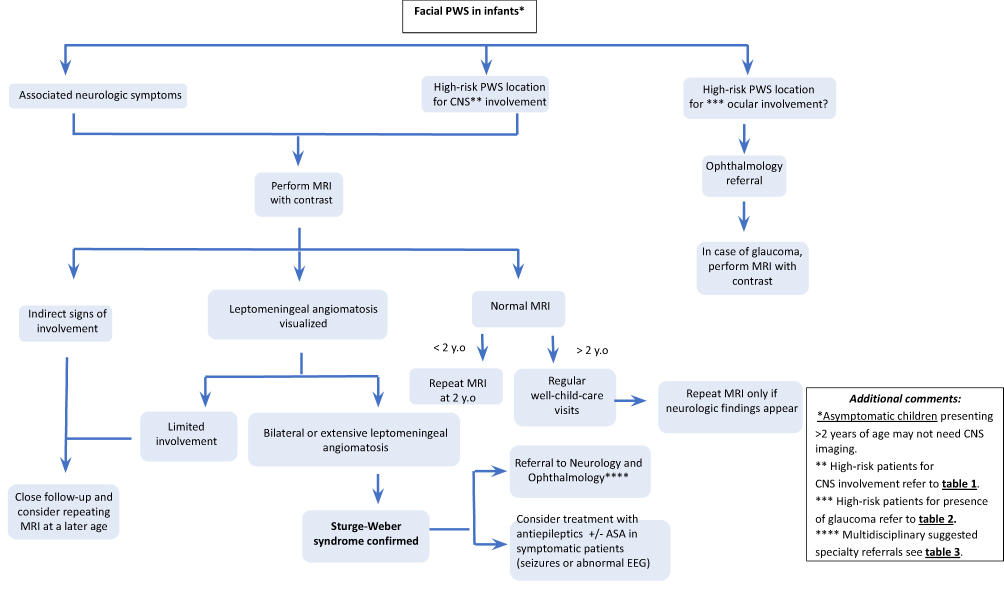 |
Figure 5 Management algorithm of patients with a facial port-wine birthmark (PWB). |
Those against presymptomatic imaging argue that early MRI requires anesthesia in young infants and may give false-negative results as leptomeningeal angiomatosis may be difficult to visualize early on. There is a current push to raise awareness of subtle and incipient radiological findings that could be supportive of the diagnoses of SWS from an earlier age.73 One study showed that choroid plexus (CP) thickness and CP thickness ratio (cut-off value of 5.6 mm of the affected side) correlated with a sensitivity and specificity of 91.7% and 100%, respectively, for the diagnostic ratification of SWS.
Other indirect findings, such as focal areas of abnormal white matter signal, cortical atrophy, and CP asymmetry, can also be helpful to prove the diagnosis in more challenging or subtle cases.74 Another study reported that susceptibility-weighted imaging (SWI) and contrast-enhanced fluid-attenuated inversion recovery (FLAIR) provided the best sensitivity for the detection of pial vascular abnormalities in SWS.16
Bar et al74 evaluated the diagnostic yield of early asymptomatic screening via brain MRI in infants with SWS before 3 months of age for the detection of leptomeningeal angiomatosis, documenting a sensitivity and specificity of 85% and 94%, respectively. Additional use of indirect MRI signs increased the sensitivity to 100%.74 However, another publication mentioned that, in the absence of contrast administration, the sensitivity decreased to 35% (using the same diagnostic criteria of Bar et al74), as it compromised subtle and indirect signs.73 For this reason, careful and cautious evaluations should be taken into consideration, particularly in non-contrast weighted imaging modalities.
Earlier detection could potentially be proven beneficial by identifying high-risk patients that are likely to present symptoms who may benefit from closer multidisciplinary monitoring. On the other hand, a normal MRI, even considering a small percentage of false-negatives, is reassuring, and these low-risk patients may save some appointments. However, the risks and benefits of sedation before the age of 2 years need to be weighted on a patient-to-patient basis. In cases where sedation might not be desired, alternative non-sedative imaging modalities might provide insightful clinical data.75 After 2 years of age, imaging studies are debatable in asymptomatic patients, considering that most of the patients with brain involvement will have presented with seizures before 1 year of age.2
The preferred post-symptomatic imaging modality is pre- and post-contrast magnetic resonance imaging (MRI) sequences focusing on parenchymal and vascular anomalies, however, this is subject to individual hospital policies and procedures. Susceptibility weighted imaging (SWI) and contrast-enhanced FLAIR (fluid attenuated inversion recovery) images are particularly important to obtain as it may complement conventional contrast-enhanced T1 weighted MRI in characterizing brain abnormalities of Sturge-Weber syndrome.76,77
Routine follow-up MRIs are not recommended in SWS children with stable clinical symptoms, controlled seizures, and no neuro-cognitive decline, due to the potential risks delineated above. An additional MRI may be indicated in select patients with known SWS, eg, in the case of new or progressive neurological symptom(s), uncontrolled or worsening seizures, change in seizure semiology, new stroke-like symptoms with prolonged neurological deficit, new-onset migraine, or other clinical symptoms that raise concerns of progressive changes in brain structure and/or function.54
In the case of stroke-like episodes, a repeated MRI (after the event) is controversial considering that the radiological changes (diffusion restriction abnormalities and increased edema) are self-resolving in nature. Also, event is suspected to occur at the microangiopathic level, where the use of a cerebral angiogram has been questioned in previous studies.78
The primary goal of pre-surgical MRI in children with drug-resistant epilepsy is to determine the extent and severity of brain involvement (frontal lobes, eloquent cortical areas, unilateral versus bilateral involvement). Imaging in the context of pre-surgical evaluation could be of benefit for tailored planning for a surgical approach (ie, epilepsy surgery), both of which should be performed in an experienced neurosurgical and neuroradiological center.26
Non-invasive (and non-sedative) imaging modalities like MRI with the feed and wrap technique or the transcranial Doppler (TCD) merit consideration in the evaluation of SWS. The “feed and wrap” technique is a modified MRI technique, which is practical, easily reproducible, and when done properly, it can provide useful radiological information in infants under 3 months of age.79 It can avoid unnecessary periods of fasting, insertion of intravenous lines, and the use of sedatives and/or general anesthesia in newborns and infants. Nevertheless, many centers do not use contrast for non-sedated cases, making this technique much less informative. To our knowledge, no studies have addressed the use of the “feed and wrap” technique in SWS. This is to be explored further, as earlier, or incipient brain imaging findings can be detected at a younger age in SWS (as discussed above) safely without the risks of sedation or anesthesia, as it is widely performed in Sickle Cell disease.
Chen et al performed a qualitative and quantitative analysis of a rapid (5-min sequence) and high-resolution adapted MRI sequence, including susceptibility weighted imaging (SWI) and quantitative susceptibility mapping (QSM) modalities in a pediatric SWS cohort minimizing the time and degree of sedation.80 The authors were able to obtain high-quality data for accurate imaging reading interpretation and analysis, however, the authors mention that larger and prospective studies are required to evaluate the feasibility of this modified technique versus the traditional MR sequences in SWS.80
Transcranial Doppler as a non-invasive imaging modality was investigated in a cohort of 14 patients with confirmed SWS, who had unilateral brain involvement confirmed with MRI comparing them with age-matched controls.75 Overall, subjects with SWS had lower peak systolic velocity (PSV) and end diastolic velocity (EDV) scores in the median, posterior, and anterior cerebral arteries compared to healthy controls (both in affected and unaffected sides). Patients with the lowest mean flow velocities of the middle cerebral artery (MCA) had the worst neurological scores. This study suggested that TCD might be a valuable non-invasive tool with predictive value of disease monitoring and progression in SWS.75
Perfusion imaging by either positron emission tomography (PET) or single photon emission computed tomography (SPECT) may be important to monitor disease progression in SWS.81 PET can detect subtle changes in the degree and asymmetry of glucose activity/levels in the brain, which are sensitive markers for cognitive decline and seizure activity in SWS. In a study, the use of SPECT concomitantly used with MRI demonstrated a “steal” phenomenon of the affected brain areas during seizure activity leading to significant remote ischemia.82 As this is another mechanism that can provoke brain injury in children with SWS, it urges for aggressive seizure control and prompt management early on.
Other imaging modalities include computed tomography with or without contrast, which can detect calcification, cortical atrophy, enlarged choroid plexus, and enlarged ventricles.
Electroencephalography (EEG) is a cost-effective and minimally invasive diagnostic tool that can aid in the detection of cortical affection and epilepsy in SWS. Subclinical seizure activity in SWS might manifest as rhythm and voltage asymmetry and might be seen more frequently than epileptiform abnormalities.27,83 Subclinical seizure activity usually reflects brain injury from prior insult (seizure or calcification). There is discrepancy as to when the initial EEG should be performed in SWS; however, according to some reports it can detect background voltages and rhythm asymmetry starting at 3–6 months of age.27,46 Nonetheless, subtle or incipient changes might go unrecognized. The use of quantitative EEG for the prompt detection of asymptomatic patients remains attractive, although guidelines and recommendations for the standardized use of EEG in SWS are lacking, and its clinical relevance remains to be evaluated in larger and higher-powered studies. Finally, EEG is an important test to perform for SWS patients with a febrile illness and altered mental status (unexplained confusion or unresponsiveness), as they are at risk for non-convulsive status epilepticus. Prolonged video EEG may be needed in some cases of episodic mental status changes.54
Neurodevelopmental outcomes depend on optimal seizure control and early detection of neurological red flags.27 For this reason, we recommend a low threshold for the performance of EEG (regardless of imaging considerations) and the referral to a pediatric neurologist should always be considered for regular monitoring, screening, and prompt treatment when required in patients with extensive facial PWB.27
Biomarkers for Disease Monitoring
Sreenivasan et al84 published on the utility of urine vascular biomarkers in SWS. They analyzed the urine levels of Matrix Metalloproteinases (MMP) MMP-2 and MMP-9, Vascular Endothelial Growth Factor (VEGF), and basic Fibroblast Growth Factor (b-FGF) and correlated them with disease progression. The results suggested that MMP-2 and MMP-9 levels may be useful as non-invasive methods for assessing SWS progression, but it could also show which patients might benefit from earlier and aggressive treatment, while b-FGF levels may be useful in judging the efficacy of neurologic treatment in SWS.
Laser Therapy
Sabeti et al65 published consensus guidelines for the treatment and management of PWB in SWS. PDL is considered the first line and gold standard amongst light-based instruments.65,85 Treatment response varies depending on the depth of the vessels;86 the initial PWB color; patients phototype; and the affected anatomical site, where frontal lesions have the greatest degree of clearance compared to malar and perioral lesions.2,65 Based on expert observations and limited studies, treatment of PWBs at an earlier age, particularly in the first year of life, results in better outcomes.65 Several PDL sessions (between 7 and 15) are usually needed to achieve the desired therapeutic response; however, the capillary stains do not clear completely. Complete to near-complete clearance was reported in only 3% of patients in a multicentric, retrospective, cohort study of patients with hypertrophic.34 Less than 50% improvement was reported in 67.2% of these patients. Of note, the median age of starting laser therapy in this study was 6 years with a range of 3 months to 58 years in 67.2% of these patients.34
Laser treatments are usually scheduled every 6 weeks. More frequent treatments are safe but do not show a better therapeutic response.85 There is no evidence that laser treatment can prevent hypertrophy and PWB may become thicker even if early treatment is performed. After initial lightening there may be some redarkening over time and additional treatments may be needed at a later age. PDL is considered safe in children and has a low incidence of complication when used appropriately. Treatment of the periocular areas, with adequate eye shields, does not worsen existing glaucoma or precipitate seizures.
It is believed that partial or non-responders might suffer from ongoing regeneration and/or revascularization of the photocoagulated vessels. Intense pulse light may also be used with success if PDL is not available. Longer laser wavelengths such as those of Alexandrite laser and NdYag are commonly employed for hypertrophic, nodular lesions or deeper/larger vessels, or in PDL resistant stain.87 Other laser modalities such as ablative lasers like CO2, Erbium:YAG, and Erbium:Glass have been used in combination with PDL but will inevitably leave a scar.85,88
Alternative therapies for PWB that do not respond to conventional laser therapy include adjuvant medications (see below), photodynamic therapy (intravenous administration of photosensitizer), surgery, or corrective cover-up.65
The adjuvant use of topical agents with the use of PDL for the treatment of PWB has been studied recently.
A Phase II, randomized, double-blind, intraindividual placebo-controlled, clinical trial demonstrated that the use of topical rapamycin (RPM) was more effective than the use of PDL treatment alone in the treatment of facial PWS in an adult cohort.89 Nevertheless, treatment quadrants were non-randomly assigned, so a cautious interpretation of the results is recommended considering the study design. Musalem et al90 reported on the response to dual therapy by adding topical RPM to subsequent laser sessions where PDL was ineffective alone in an observational cohort study, suggesting that dual therapy can increase clearance rates in some resistant cases. However, based on a review of the literature, these findings have not been replicated consistently in other publications.91 The use of an enhanced drug delivery system using non-thermal resurfacing technology has shown promising results without significant side effects.
Nonetheless, further prospective, head-to-head, and comparative studies are required to rigorously evaluate the efficacy and long-term safety of the combination of topical RPM with PDL.
Other adjuvant combination topical agents in PDL-resistant cases have been used. Timolol and topical Imiquimod in combination with the use of PDL have been studied without conclusive results.91–93 A study evaluated the concomitant use of PDL and topical axitinib (an inhibitor of MEK/ERK), a medication with angiogenic properties, using animal models.94 The treatment combination was shown to prevent vessel regrowth, although efficacy in patients has yet to be demonstrated. One small-scale pilot study evaluated topical bosentan (endothelin receptor antagonist) 1 day before PDL and for 14 days after the session in PDL-resistant PWB and showed partial efficacy in one of four PWB patients treated.95
Photodynamic therapy, which consists of the administration (topical, oral, or intravenous) of photosensitizing agents that create a photochemical reaction targeting and destroying specific cells (ie cancer cells), has been used for the treatment of refractory choroidal hemangiomas in PWB.96 The use of 5-aminolevulinic acid (ALA) in combination with PDL was evaluated in one study and noted mild-to-moderate improvement of recalcitrant PWB.97
Li et al98 reported on the effective and safe use of photodynamic therapy with hematoporphyrin monomethyl ether (HMME) for the treatment of patients with SWS and large/segmental PWB. However, undesirable reactions such as facial swelling, infection, and skin necrosis have been reported so this modality of treatment should be used cautiously and in an experienced center.99
As laser therapy is associated with discomfort, pain management should be discussed with parents and patients before therapy. Pain can be minimized with topical anesthetics, local anesthetics, and nerve blocks. For younger children, these methods may not be feasible or may be intolerable and general anesthesia may be needed. The risk of multiple general anesthesia events should be weighed against the benefits from early treatment. There are studies on the safety of a single anesthesia episode in infants; however, there are no studies addressing the long-term safety, neurocognitive, neuropsychological, and behavioral outcomes of multiple anesthesia events in young children.100–102
Pulsed dye laser treatment without general anesthesia may be preferred in very young children, particularly before 12 months of age. There are several positive considerations in favor: the laser procedure only lasts a few seconds, the treatment area in infants is smaller (compared to older children and adults); eye shields can be placed easily in infants, and they are more easily restrained from motion than older children. Non-medical interventions are commonly used to minimize discomfort, anxiety, and pain, including soothers, sucrose, distraction, and the company of the parents or caregivers (depending on the laser surgeon’s comfort).
The potential impact of painful procedures early in life must be carefully weighed against the risk of general anesthesia and the advantages of having more rapid vascular clearing with significant cosmetic improvement before school age. Joint decision-making with parents before moving forward with laser under general anesthesia is strongly recommended. Quality of life (QoL), self-esteem, and related psychosocial issues should not be overlooked and should be taken into consideration when deciding and discussing the timing for laser surgery.65
Targeted Therapies, a Novel Approach in SWS
To date, there is no known targeted treatment for SWS. Treatment with sirolimus has shown some benefit, but the effect on the GNAQ pathway may be indirect. Sirolimus binds to FKBP-12, an intracellular protein, forming an intracellular complex that inhibits mTOR (target of rapamycin), a regulatory kinase. It inhibits T-lymphocyte activation and proliferation via antigenic and cytokine stimulation, later inhibiting antibody production.103 This mechanism makes Sirolimus an attractive therapeutic choice for downstream signaling pathway like PI3K/AKT/mTOR (mammalian target of rapamycin).20,104 However, GNAQ has not been shown to signal directly through this pathway.
In 2021, a systematic review evaluated the efficacy and safety of sirolimus, showing that it can improve the prognosis of vascular tumors (especially those with severe coagulopathy), venous and lymphatic malformations.105 These results, taken together with the importance of mTOR in cell growth and proliferation, have motivated its use in other vascular anomalies, including CM (Capillary Malformations) associated with SWS.
The effectiveness of topical rapamycin in SWS was evaluated in phase II, randomized, double-blind, intraindividual placebo-controlled, clinical trial.1 The use of topical rapamycin in combination with PDL proved to be the most effective intervention in both subjective and objective measurements (including photographs, clinical evaluation, and histopathology) compared to either topical or laser therapy alone in post-hoc paired analysis in this study. This treatment modality was postulated as presumably most effective in the pediatric population,89 where earlier tampering of the expression of proangiogenic factors that contribute to ongoing angiogenesis in SWS could potentially inhibit the formation of larger ectatic vessels that are challenging to treat later in adulthood.
Rapamycin in combination with aspirin has been given orally in an infant with bilateral facial and extensive leptomeningeal involvement prophylactically.106 The patient was prophylactically started on oral rapamycin (0.8 mg/m2/dose) in combination with aspirin (10 mg/kg/day) at 3-weeks of age and remained seizure free at 23-months; the patient also underwent 10 sessions of PDL (starting at 2 months of age) that were considered significantly effective, showing notable lightening of the PWB.
Sun et al reported on the beneficial use of sirolimus on a small cohort of patients, in whom their epilepsy was gradually controlled and had sustained effective remission for 26 months and without any other antiepileptic medications.44 Neurologic development was reported as stable. Moreover, the associated hemihypertrophy and color of the capillary stain improved with treatment as well. Another study showed that cognitive functioning, recovery after stroke-like events, and quality of life were improved with the use of sirolimus in SWS.45
Soft-tissue overgrowth, which is seen in some patients with SWS, can be significantly distressing to patients and their caregivers, as it can lead to progressive cosmetic disfigurement and functional limitations such as limited eye and mouth opening. The use of rapamycin was shown to be beneficial with a reduction in the associated overgrowth within the PWB of an 11-year-old girl 8-months into treatment, without the use of other adjuvant therapies.107 Furthermore, attenuation of capillary discoloration and reduction of ocular pressure were seen as added treatment benefits.
Sirolimus was considered safe and well tolerated in all the reports discussed above. Adverse events were mild and classified as grade I (CTCAE classification), reporting hypertriglyceridemia, decreased HDL, mouth sores, headaches, increased LFTs, and personality changes (worsening behavior).45
There are only a few other studies evaluating other potential targeted therapy agents in SWS. A recent study by Feng et al108 has demonstrated that the presence of oncogenic GNAQ mutations can cause an overactivation of FAK-mediated YAP activation, which transcriptionally activates the TEAD transcription factor family and downstream pro-growth and pro-survival genes. No specific YAP inhibitors are currently in clinical use; however, Truong et al109 demonstrated that combined therapy with trametinib (MEK1/2 inhibition) and the lysosome inhibitor chloroquine, increased cytotoxicity while indirectly decreasing YAP nuclear localization and transcriptional activity. This could be of interest in the treatment of uveal melanoma and, potentially, also SWS. Additional in vivo and in vitro complex studies are needed to advance our understanding of PWB and SWS.
Other Treatments
The mainstay of treatment for seizures remains antiepileptic medications. From a multicenter registry of 268 patients with SWS, the most frequently used medications were levetiracetam (48.1%), low-dose aspirin (44.8%), oxcarbazepine (39.9%), and phenobarbital (14.9%). In this study, the presence of family history of seizures, bilateral brain involvement, and no anti-epileptic medication use was associated with a higher incidence of neurosurgery.110 It is currently advisable to warn parents and caregivers about the risk of seizure clusters and to encourage the development of individualized emergency plans that include the use of rescue benzodiazepine therapy. Some providers recommend the use of intermittent benzodiazepine therapy during febrile illnesses. A similar strategy, as used for the prevention of status epilepticus in Dravet syndrome, may be considered.111
Low-dose aspirin (3–5 mg/kg/day) can be used as concomitant therapy (in addition to antiepileptic drugs), as it decreases both the frequency and severity of headaches, seizures, and stroke-like events in patients with SWS.50 Aspirin was demonstrated to be safe and well tolerated in a pediatric cohort study.112 In this study, patients were introduced to the drug starting as early as 1 month of age.112 Patients with anatomical brain abnormalities detected on imaging that are asymptomatic may particularly benefit from treatment.50,112 Speculation regarding the concomitant relief of glaucoma (via endothelial antiaggregant effect) of aspirin has been proposed; however, this requires further study.
Use of (CBD) in patients with SWS and refractory epilepsy has been reported.113 Doses of 2–25 mg/kg/day of CBD were employed in this study and although adverse effects were frequent, they were transient, mild, and resolved with dosage adjustments or transient discontinuation of the CBD.113
Conclusion
In summary, SWS is a neurocutaneous disorder caused by GNAQ and GNA11 somatic variants that lead to progressive capillary vessel dilation and dysfunction in skin, eye, and brain tissue with negative impact on patients’ quality of life.
Our understanding and knowledge about SWS have improved over recent years and will continue to evolve rapidly. However, a better understanding of the pathophysiological basis of the disease is needed in order to identify accurate biomarkers capable of monitoring disease progression. The identification of the activating somatic GNAQ mutation and more recently, in the GNA11 gene are of significant interest for future and more specific targeted therapies.
Although the number of cases is still limited, sirolimus seems to be effective for both cutaneous and extra-cutaneous features in SWS and is alluring for prophylactic therapy. Robust multicenter, blinded, and prospective clinical trials with long-term follow-up are required to demonstrate the effectiveness and safety of sirolimus and other targeted therapeutic agents in SWS.
Abbreviations
ADH2, Autosomal dominant hypocalcemia type-2; B-FGF, Fibroblast Growth Factor; Ca2+e, Extracellular calcium; CaSR, Calcium-sensing receptor; CH, Choroidal hemangioma; CM, Capillary malformation; CNS – Central nervous system; CP, Choroid plexus; CTCAE, Common Terminology Criteria for Adverse Events; EEG, Electroencephalography; EDV, End Diastolic Velocity; FHH2, Familial hypocalciuric hypercalcemia type-2; FLAIR- Contrast-enhanced fluid-attenuated inversion recovery; GPCR, Class C G-protein coupled receptor; LFT, Liver function tests; NGS, Next-generation sequencing; MAF, Mutant Allelic Frequencies; MCA, Middle Cerebral Artery; MCAP, Megalencephaly-capillary malformation syndrome; MMP, Metalloproteinases; PDL, Pulse Dye Laser; PET, Positron emission tomography; PG, Pyogenic granulomas; PSV, Peak Systolic Velocity; PWS, Port Wine Stain; SWI, Susceptibility weighted imaging; SWS, Sturge-Weber Syndrome; TCD, Transcranial Doppler; VEGF – Vascular Endothelial Growth Factor; VEGFR, Vascular Endothelial receptors; WES, Whole-exome sequencing.
Acknowledgments
We would like to thank Beatriz Trigo González, graphic designer, for her support in the creation and design of the facial PWS images/phenotypes used in this paper.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Thorpe J, Frelin LP, McCann M, et al. Identification of a mosaic activating mutation in GNA11 in atypical Sturge-Weber syndrome. J Invest Dermatol. 2021;141:685–688.
2. Higueros E, Roe E, Granell E, Baselga E. Sturge-Weber syndrome: a review. Actas Dermo Sifiliográficas. 2017;108(5):407–417.
3. Singh AK, Keenaghan M. Sturge-Weber syndrome; 2017.
4. Singh AK, Keenaghan M. Sturge-Weber syndrome; 2022.
5. Comi AM. Sturge--Weber syndrome. Handb Clin Neurol. 2015;132:157–168.
6. Waelchli R, Aylett SE, Robinson K, Chong WK, Martinez AE, Kinsler VA. New vascular classification of port-wine stains: improving prediction of Sturge--Weber risk. Br J Dermatol. 2014;171(4):861–867.
7. Shirley MD, Tang H, Gallione CJ, et al. Sturge--Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971–1979.
8. Couto JA, Huang L, Vivero MP, et al. Endothelial cells from capillary malformations are enriched for somatic GNAQ mutations. Plast Reconstr Surg. 2016;137(1):77e.
9. Comi AM. Presentation, diagnosis, pathophysiology and treatment of the neurologic features of Sturge-Weber Syndrome. Neurologist. 2011;17(4):179.
10. Huang Z, Li Y, Zhao Z, et al. GNAQ mutation R183Q as a potential cause of familial Sturge-Weber syndrome: a case report. Oncol Lett. 2017;13(4):2665–2669.
11. Asadi S. The role of genetic mutations in gene GNAQ in Sturge-Weber syndrome. J Cell Signal Damage Assoc Mol Patterns. 2020;1:15–19.
12. Martins L, Giovani PA, Rebouças PD, et al. Computational analysis for GNAQ mutations: new insights on the molecular etiology of Sturge-Weber syndrome. J Mol Graph Model. 2017;76:429–440.
13. Syrovatkina V, Alegre KO, Dey R, Huang XY. Regulation, signaling, and physiological functions of G-proteins. J Mol Biol. 2016;428(19):3850–3868.
14. Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat Struct Mol Biol. 2006;13(9):772–777.
15. Fry MV, Williams JBK, Kim HJ, Di Nicola M. Choroidal melanoma in phakomatosis pigmentovascularis with overlapping Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Retin Cases Br Reports. 2022. doi:10.1097/ICB.0000000000001154
16. Dompmartin A, van der Vleuten CJM, Dekeuleneer V, et al. GNA11-mutated Sturge--Weber syndrome has distinct neurological and dermatological features. Eur J Neurol. 2022;29(10):3061–3070.
17. Polubothu S, Al-Olabi L, Del Boente MC, et al. GNA11 mutation as a cause of Sturge-Weber syndrome: expansion of the phenotypic spectrum of G$α$/11 mosaicism and the associated clinical diagnoses. J Invest Dermatol. 2020;140(5):1110.
18. Formisano M, Di Pippo MC, Scuderi L, Abdolrahimzadeh S. Current concepts on diffuse choroidal hemangioma in Sturge Weber syndrome. Ophthalmic Genet. 2021;42(4):375–382.
19. Tan W, Nadora DM, Gao L, Wang G, Mihm MC, Nelson JS. The somatic GNAQ mutation (R183Q) is primarily located within the blood vessels of port wine stains. J Am Acad Dermatol. 2016;74(2):380–383.
20. Nguyen V, Hochman M, Mihm JMC, Nelson JS, Tan W. The pathogenesis of port wine stain and Sturge Weber syndrome: complex interactions between genetic alterations and aberrant MAPK and PI3K activation. Int J Mol Sci. 2019;20(9):2243.
21. Huang L, Bichsel C, Norris AL, et al. Endothelial GNAQ p. R183Q increases ANGPT2 (Angiopoietin-2) and drives formation of enlarged blood vessels. Arterioscler Thromb Vasc Biol. 2022;42(1):e27–e43.
22. Gluck AK, Stevenson M, Falcone S, et al. OR07-06 the roles of GNAQ and GNA11 in calcium-sensing receptor (CaSR) signalling. J Endocr Soc. 2020;4(Supplement_1):OR07–OR106.
23. Zecchin D, Knoepfel N, Gluck A, et al. Aberrant calcium signalling and systemic hypocalcaemia in GNAQ/11 mosaic diseases. Br J Dermatol. 2022;187:92.
24. Sudarsanam A, Ardern-Holmes SL. Sturge--Weber syndrome: from the past to the present. Eur J Paediatr Neurol. 2014;18(3):257–266.
25. Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58(2):218–222.
26. Sabeti S, Ball KL, Bhattacharya SK, et al. Consensus statement for the management and treatment of Sturge-Weber syndrome: neurology, neuroimaging, and ophthalmology recommendations. Pediatr Neurol. 2021;121:59–66.
27. Zallmann M, Leventer RJ, Mackay MT, Ditchfield M, Bekhor PS, Su JC. Screening for Sturge-Weber syndrome: a state-of-The-art review. Pediatr Dermatol. 2018;35(1):30–42.
28. Garzon MC, Huang JT, Enjolras O, Frieden IJ. Vascular malformations: part II: associated syndromes. J Am Acad Dermatol. 2007;56(4):541–564.
29. Ch’ng S, Tan ST. Facial port-wine stains--clinical stratification and risks of neuro-ocular involvement. J Plast Reconstr Aesthetic Surg. 2008;61(8):889–893.
30. Dutkiewicz AS, Ezzedine K, Mazereeuw-Hautier J, et al. A prospective study of risk for Sturge-Weber syndrome in children with upper facial port-wine stain. J Am Acad Dermatol. 2015;72(3):473–480.
31. Kaseka ML, Bitton JY, Décarie JC, Major P. Predictive factors for epilepsy in pediatric patients with Sturge-Weber syndrome. Pediatr Neurol. 2016;64:52–58. doi:10.1016/j.pediatrneurol.2016.08.009
32. Dymerska M, Kirkorian AY, Offermann EA, Lin DD, Comi AM, Cohen BA. Size of facial port-wine birthmark may predict neurologic outcome in Sturge-Weber syndrome. J Pediatr. 2017;188:205–209.
33. Boos MD, Bozarth XL, Sidbury R, et al. Forehead location and large segmental pattern of facial port-wine stains predict risk of Sturge-Weber syndrome. J Am Acad Dermatol. 2020;83(4):1110–1117.
34. Passeron T, Salhi A, Mazer JM, et al. Prognosis and response to laser treatment of early-onset hypertrophic port-wine stains (PWS). J Am Acad Dermatol. 2016;75(1):64–68.
35. Savas JA, Ledon JA, Franca K, Chacon A, Nouri K. Pulsed dye laser-resistant port-wine stains: mechanisms of resistance and implications for treatment. Br J Dermatol. 2013;168(5):941–953.
36. Mopagar VP, Choudhari S, Subbaraya DK, Peesapati S. Sturge-Weber syndrome with pyogenic granuloma. Contemp Clin Dent. 2013;4(3):360.
37. Rancan A, Boscarelli A, Codrich D, Berti I, Guida E, Schleef J. Pyogenic granuloma arising within capillary malformations in children: a case report and literature review. Dermatol Report. 2021;13(2):9115.
38. Carrasco L, Pastor A, Fariña C, Martn L, Manzarbeitia F, Requena L. Acral arteriovenous tumor developed within a nevus flammeus in a patient with Sturge-Weber syndrome. Am J Dermatopathol. 2003;25(4):341–345.
39. Jordan M, Carmignac V, Sorlin A, et al. Reverse phenotyping in patients with skin capillary malformations and mosaic GNAQ or GNA11 mutations defines a clinical spectrum with genotype-phenotype correlation. J Invest Dermatol. 2020;140(5):1106–1110.
40. Thomas AC, Zeng Z, Rivière JB, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136(4):770–778.
41. Marangi GF, Segreto F, Alessandri-Bonetti M, et al. Basal cell carcinoma arising within port-wine stain. Int J Dermatol. 2021. doi:10.1111/ijd.15944
42. Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10(1):49–58.
43. Ishikawa H, Ii Y, Niwa A, Matsuura K, Maeda M, Tomimoto H. A case of 55-year-old man with first-ever generalized seizure diagnosed with Sturge-Weber syndrome type III by characteristic MRI findings. Rinsho shinkeigaku. 2017;57(5):214–219.
44. Sun B, Han T, Wang Y, Gao Q, Cui J, Shen W. Sirolimus as a potential treatment for Sturge-Weber syndrome. J Craniofac Surg. 2021;32(1):257–260.
45. Sebold AJ, Day AM, Ewen J, et al. Sirolimus treatment in Sturge-Weber syndrome. Pediatr Neurol. 2021;115:29–40.
46. Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, Viaño J. Sturge-Weber syndrome. Study of 55 patients. Can J Neurol Sci. 2008;35(3):301–307.
47. Tillmann RP, Ray K, Aylett SE. Transient episodes of hemiparesis in Sturge Weber syndrome-causes, incidence and recovery. Eur J Paediatr Neurol. 2020;25:90–96.
48. Day AM, McCulloch CE, Hammill AM, et al. Physical and family history variables associated with neurological and cognitive development in Sturge-Weber syndrome. Pediatr Neurol. 2019;96:30–36.
49. Juhász C. Toward a better understanding of stroke-like episodes in Sturge-Weber syndrome. Eur J Paediatr Neurol EJPN off J Eur Paediatr Neurol Soc. 2020;25:3.
50. Comi A. Current therapeutic options in Sturge-Weber syndrome. In: Seminars in Pediatric Neurology. Vol. 22. WB Saunders; 2015:295–301.
51. Alejandro J, Luat AF, Juhász C, et al. A multidisciplinary consensus for clinical care and research needs for Sturge-Weber syndrome. Pediatr Neurol. 2018;84:11–20.
52. Hassanpour K, Nourinia R, Gerami E, Mahmoudi G, Esfandiari H. Ocular manifestations of the Sturge--Weber syndrome. J Ophthalmic Vis Res. 2021;16(3):415.
53. Silverstein M, Salvin J. Ocular manifestations of Sturge--Weber syndrome. Curr Opin Ophthalmol. 2019;30(5):301–305.
54. Mantelli F, Bruscolini A, La Cava M, Abdolrahimzadeh S, Lambiase A. Ocular manifestations of Sturge--Weber syndrome: pathogenesis, diagnosis, and management. Clin Ophthalmol. 2016;10:871.
55. Ha A, Kim JS, Baek SU, et al. Facial port-wine stain phenotypes associated with glaucoma risk in neonates. Am J Ophthalmol. 2020;220:183–190.
56. Wu Y, Yu RJ, Chen D, et al. Glaucoma in patients with eyes close to areas affected by port-wine stain has lateral and gender predilection. Chin Med J. 2017;130(24):2922–2926.
57. Formisano M, Abdolrahimzadeh B, Mollo R, Bruni P, Malagola R, Abdolrahimzadeh S. Bilateral diffuse choroidal hemangioma in Sturge Weber syndrome: a case report highlighting the role of multimodal imaging and a brief review of the literature. J Curr Ophthalmol. 2019;31(2):242–249.
58. Le Guin CHD, Metz KA, Kreis SH, et al. GNAQ Q209R mutations are highly specific for circumscribed choroidal hemangioma. Cancer. 2019;11(7):1031.
59. Miller RS, Ball KL, Comi AM, Germain-Lee EL. Growth hormone deficiency in Sturge--Weber syndrome. Arch Dis Child. 2006;91(4):340–341.
60. Saroj G, Gangwar A, Dhillon JK. Hypothyroidism and Sturge-Weber syndrome associated with bilateral port-wine nevus. Int J Clin Pediatr Dent. 2016;9(1):82.
61. Comi AM, Bellamkonda S, Ferenc LM, Cohen BA, Germain-Lee EL. Central hypothyroidism and Sturge-Weber syndrome. Pediatr Neurol. 2008;39(1):58–62.
62. Mapara PN, Taur SM, Hadakar SG, et al. Sturge--Weber syndrome: roots to a cure a nightmare in pediatric dentistry. Int J Clin Pediatr Dent. 2021;14(1):145.
63. Tripathi AK, Kumar V, Dwivedi R, Saimbi CS. Sturge-Weber syndrome: oral and extra-oral manifestations. Case Reports. 2015;2015:bcr2014207663.
64. Carvalho VA, Dallazen E, Statkievicz C, et al. Oral surgery in patients with Sturge-Weber syndrome. J Craniofac Surg. 2021;32(1):e85–e88.
65. Sabeti S, Ball KL, Burkhart C, et al. Consensus statement for the management and treatment of port-wine birthmarks in Sturge-Weber syndrome. JAMA Dermatol. 2021;157(1):98–104.
66. Kentab AY. Klippel-Trenaunay and Sturge-Weber overlapping syndrome in a Saudi boy. Sudan J Paediatr. 2016;16(2):86.
67. Chhajed M, Pandit S, Dhawan N, Jain A. Klippel-Trenaunay and Sturge-Weber overlap syndrome with phakomatosis pigmentovascularis. J Pediatr Neurosci. 2010;5(2):138.
68. Barros FS, Marussi VHR, Amaral LLF, et al. The rare neurocutaneous disorders: update on clinical, molecular, and neuroimaging features. Top Magn Reson Imaging. 2018;27(6):433–462.
69. John PR. Klippel-Trenaunay syndrome. Tech Vasc Interv Radiol. 2019;22(4):100634.
70. Setty BA, Wusik K, Hammill AM. How we approach genetics in the diagnosis and management of vascular anomalies. Pediatr Blood Cancer. 2022;69:e29320.
71. Minami Y, Okamoto T, Hirotsu Y, et al. Phakomatosis pigmentovascularis type IIb with Klippel-Trenaunay syndrome: association with GNAQ mutation in vascular endothelial cells. J Dermatol. 2022;49(12):e444–e445.
72. He R, Liao S, Yao X, et al. Klippel--Trenaunay and Sturge--Weber overlap syndrome with KRAS and GNAQ mutations. Ann Clin Transl Neurol. 2020;7(7):1258–1264.
73. Catsman-Berrevoets CE, Koudijs SM, Buijze MSJ, de Laat PCJ, Pasmans SGMA, Dremmen MHG. Early MRI diagnosis of Sturge Weber Syndrome type 1 in infants. Eur J Paediatr Neurol. 2022;38:66–72.
74. Bar C, Pedespan JM, Boccara O, et al. Early magnetic resonance imaging to detect presymptomatic leptomeningeal angioma in children with suspected Sturge--Weber syndrome. Dev Med Child Neurol. 2020;62(2):227–233.
75. Offermann EA, Sreenivasan A, DeJong MR, et al. Reliability and clinical correlation of transcranial Doppler ultrasound in Sturge-Weber syndrome. Pediatr Neurol. 2017;74:15–23.e5. doi:10.1016/j.pediatrneurol.2017.04.026
76. Juhasz C, Haacke EM, Hu J, et al. Multimodality imaging of cortical and white matter abnormalities in Sturge-Weber syndrome. Am J Neuroradiol. 2007;28(5):900–906.
77. Hu J, Yu Y, Juhasz C, et al. MR susceptibility weighted imaging (SWI) complements conventional contrast enhanced T1 weighted MRI in characterizing brain abnormalities of Sturge-Weber syndrome. J Magn Reson Imaging an off J Int Soc Magn Reson Med. 2008;28(2):300–307.
78. Kumar KR, Hon K, Schultz D, Agzarian MJ, Jones DN, Thyagarajan D. Transient changes on brain magnetic resonance imaging in a patient with Sturge-Weber syndrome presenting with hemiparesis. Neurologist. 2009;15(6):351–354.
79. Antonov NK, Ruzal-Shapiro CB, Morel KD, et al. Feed and wrap MRI technique in infants. Clin Pediatr. 2017;56(12):1095–1103.
80. Chen Y, Xuan Y, Juhasz C, Hu J, Haacke EM. Strategically Acquired Gradient Echo (STAGE) imaging as a means for multi-contrast quantitative pediatric neuroimaging with minimized sedation: a pilot study in Sturge-Weber syndrome; 2019.
81. Comi AM. Advances in Sturge-Weber syndrome. Curr Opin Neurol. 2006;19(2):124–128.
82. Namer IJ, Battaglia F, Hirsch E, Constantinesco A, Marescaux C. Subtraction ictal SPECT co-registered to MRI (SISCOM) in Sturge--Weber syndrome. Clin Nucl Med. 2005;30(1):39–40.
83. Ewen JB, Kossoff EH, Crone NE, et al. Use of quantitative EEG in infants with port-wine birthmark to assess for Sturge--Weber brain involvement. Clin Neurophysiol. 2009;120(8):1433–1440.
84. Sreenivasan AK, Bachur CD, Lanier KE, et al. Urine vascular biomarkers in Sturge--Weber syndrome. Vasc Med. 2013;18(3):122–128.
85. Updyke KM, Khachemoune A. Port-wine stains: a focused review on their management. J Drugs Dermatology JDD. 2017;16(11):1145–1151.
86. Kwiek B, Rożalski Michałand Sieczych J, Paluch Ł, Kowalewski C, Ambroziak M. Predictive value of dermoscopy for the treatment of port-wine stains with large spot 532 nm laser. Lasers Surg Med. 2019;51(7):569–583.
87. Carlsen BC, Wenande E, Erlendsson AM, Faurschou A, Dierickx C, Haedersdal M. A randomized side-by-side study comparing alexandrite laser at different pulse durations for port wine stains. Lasers Surg Med. 2017;49(1):97–103.
88. Toren KL, Marquart JD. Fractional thermoablation using an erbium-doped yttrium aluminum garnet fractionated laser for the treatment of pulsed dye laser--resistant port wine stain birthmarks. Dermatologic Surg. 2011;37(12):1791–1794.
89. Marqués L, Núñez-Córdoba JM, Aguado L, et al. Topical rapamycin combined with pulsed dye laser in the treatment of capillary vascular malformations in Sturge-Weber syndrome: phase II, randomized, double-blind, intraindividual placebo-controlled clinical trial. J Am Acad Dermatol. 2015;72(1):151–158.e1. doi:10.1016/J.JAAD.2014.10.011
90. Musalem HM, Alshaikh AA, Tuleimat LM, Alajlan S. Outcome with topical sirolimus for port wine stain malformations after unsatisfactory results with pulse dye laser treatment alone. Ann Saudi Med. 2018;38(5):376–380.
91. Lipner SR. Topical adjuncts to pulsed dye laser for treatment of port wine stains: review of the literature. Dermatologic Surg. 2018;44(6):796–802.
92. Passeron T, Maza A, Fontas E, et al. Treatment of port wine stains with pulsed dye laser and topical timolol: a multicenter randomized controlled trial. Br J Dermatol. 2014;170(6):1350–1353.
93. Tremaine AM, Armstrong J, Huang YC, et al. Enhanced port-wine stain lightening achieved with combined treatment of selective photothermolysis and imiquimod. J Am Acad Dermatol. 2012;66(4):634–641.
94. Gao L, Nadora DM, Phan S, et al. Topical axitinib suppresses angiogenesis pathways induced by pulsed dye laser. Br J Dermatol. 2015;172(3):669–676.
95. Taquin H, Lacour JP, Le Duff F, Chiaverini C, Passeron T. Treatment of resistant port-wine stains with bosentan and pulsed dye laser: a pilot prospective study. J Eur Acad Dermatol Venereol JEADV. 2015;30(8):1432–1434.
96. Nugent R, Lee L, Kwan A. Photodynamic therapy for diffuse choroidal hemangioma in a child with Sturge-Weber syndrome. J Am Assoc Pediatr Ophthalmol Strabismus. 2015;19(2):181–183.
97. Liu S, Yang C, Yang S, Wang Z, Luo D, Zhang X. Topical application of 5-aminolevulinic acid followed by 595-nm pulsed dye laser irradiation for the treatment of recalcitrant port-wine stains: a primary study. J Cosmet Laser Ther. 2012;87(6):189–192.
98. Li X, Diao P, Liu L, et al. Hematoporphyrin monomethyl ether photodynamic therapy for the treatment of Sturge--Weber syndrome and large segmental facial port-wine stain. Dermatol Ther. 2022;35(5):e15404.
99. Chen Y, Zhang L, Zhang C, Pan L, Wu S. Skin necrosis due to post-treatment care failure after photodynamic therapy of facial port-wine stain in Sturge-Weber Syndrome-A case report. Photodiagnosis Photodyn Ther. 2021;36:102546.
100. McCann ME, De Graaff JC, Dorris L, et al. Neurodevelopmental outcome at 5 years of age after general anaesthesia or awake-regional anaesthesia in infancy (GAS): an international, multicentre, randomised, controlled equivalence trial. Lancet. 2019;393(10172):664–677.
101. Sun LS, Li G, Miller TLK, et al. Association between a single general anesthesia exposure before age 36 months and neurocognitive outcomes in later childhood. JAMA. 2016;315(21):2312–2320.
102. Warner DO, Zaccariello MJ, Katusic SK, et al. Neuropsychological and behavioral outcomes after exposure of young children to procedures requiring general anesthesia: the Mayo Anesthesia Safety in Kids (MASK) study. Anesthesiology. 2018;129(1):89–105.
103. Sehgal SN. Sirolimus: its discovery, biological properties, and mechanism of action. In: Transplantation Proceedings. Vol. 35. Elsevier; 2003:S7–S14.
104. Queisser A, Seront E, Boon LM, Vikkula M. Genetic basis and therapies for vascular anomalies. Circ Res. 2021;129(1):155–173.
105. Freixo C, Ferreira V, Martins J, et al. Efficacy and safety of sirolimus in the treatment of vascular anomalies: a systematic review. J Vasc Surg. 2020;71(1):318–327.
106. Triana Junco PE, Sánchez-Carpintero I, López-Gutiérrez JC. Preventive treatment with oral sirolimus and aspirin in a newborn with severe Sturge-Weber syndrome. Pediatr Dermatol. 2019;36(4):524–527.
107. Giacaman A, Salinas Sanz JA, Navarro Noguera S, Lastra Rodriguez J, Montis Palos MC, Mart\’\in-Santiago A. Facial hemihypertrophy in a girl with Sturge-Weber syndrome: treatment with oral sirolimus. Pediatr Dermatol. 2021;38(2):469–471.
108. Feng X, Arang N, Rigiracciolo DC, et al. A platform of synthetic lethal gene interaction networks reveals that the GNAQ uveal melanoma oncogene controls the Hippo pathway through FAK. Cancer Cell. 2019;35(3):457–472.
109. Truong A, Yoo JH, Scherzer MT, et al. Chloroquine Sensitizes GNAQ/11-mutated Melanoma to MEK1/2 InhibitionChloroquine and MEK1/2 Inhibition in GNAQ/11-mutant Melanoma. Clin Cancer Res. 2020;26(23):6374–6386.
110. Smegal LF, Sebold AJ, Hammill AM, et al. Multicenter research data of epilepsy management in patients with Sturge-Weber syndrome. Pediatr Neurol. 2021;119:3–10.
111. Van Trigt WK, Kelly KM, Hughes CCW. GNAQ mutations drive port wine birthmark-associated Sturge-Weber syndrome: a review of pathobiology, therapies, and current models; 2022.
112. Lance EI, Sreenivasan AK, Zabel TA, Kossoff EH, Comi AM. Aspirin use in Sturge-Weber syndrome: side effects and clinical outcomes. J Child Neurol. 2013;28(2):213–218.
113. Kaplan EH, Offermann EA, Sievers JW, Comi AM. Cannabidiol treatment for refractory seizures in Sturge-Weber syndrome. Pediatr Neurol. 2017;71:18–23.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.