")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Study on the prostate cancer-targeting mechanism of aptamer-modified nanoparticles and their potential anticancer effect in vivo
Authors Wu X, Tai Z , Zhu Q, Fan W, Ding B , Zhang W, Zhang L, Yao C, Wang X, Ding X, Li Q, Li X, Liu G, Liu J, Gao S
Received 13 July 2014
Accepted for publication 9 September 2014
Published 21 November 2014 Volume 2014:9(1) Pages 5431—5440
DOI https://doi.org/10.2147/IJN.S71101
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Xin Wu,1,2,* Zongguang Tai,1,* Quangang Zhu,3,* Wei Fan,4 Baoyue Ding,5 Wei Zhang,1,6 Lijuan Zhang,1 Chong Yao,1 Xiaoyu Wang,1 Xueying Ding,2 Qin Li,2 Xiaoyu Li,2 Gaolin Liu,2 Jiyong Liu,1 Shen Gao1
1Department of Pharmaceutics, Changhai Hospital, Second Military Medical University, 2Department of Pharmaceutics, Shanghai First People’s Hospital, Shanghai Jiaotong University School of Medicine, 3Department of Pharmaceutics, Yueyang Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 4Department of Pharmaceutics, The 425th Hospital of PLA, Sanya, People’s Republic of China; 5Department of Pharmaceutics, Medical College of Jiaxing University, Jiaxing, People’s Republic of China; 6Department of Pharmaceutics, The 522nd Hospital of PLA, Luoyang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Ligand-mediated prostate cancer (PCa)-targeting gene delivery is one of the focuses of research in recent years. Our previous study reported the successful preparation of aptamer-modified nanoparticles (APT-NPs) in our laboratory and demonstrated their PCa-targeting ability in vitro. However, the mechanism underlying this PCa-targeting effect and their anticancer ability in vivo have not yet been elucidated. The objective of this study was to assess the feasibility of using APT-NPs to deliver micro RNA (miRNA) systemically to PCa cells, to testify their tumor-targeting efficiency, and to observe their biodistribution after systemic administration to a xenograft mouse model of PCa. In addition, the effect of APT depletion and endocytosis inhibitors on cellular uptake was also evaluated quantitatively in LNCaP cells to explore the internalization mechanism of APT-NPs. Finally, blood chemistry, and renal and liver function parameters were measured in the xenograft mouse model of PCa to see whether APT-NPs had any demonstrable toxicity in mice in vivo. The results showed that APT-NPs prolonged the survival duration of the PCa tumor-bearing mice as compared with the unmodified NPs. In addition, they had a potential PCa-targeting effect in vivo. In conclusion, this research provides a prototype for the safe and efficient delivery of miRNA expression vectors to PCa cells, which may prove useful for preclinical and clinical studies on the treatment of PCa.
Keywords: miRNA, aptamer, polyamidoamine, prostate-specific membrane antigen, targeted delivery, prostate cancer
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer and the second leading cause of cancer deaths in American males today.1 Novel and effective treatments for PCa, including gene therapy, are greatly desired.2 Gene therapy is the direct transfer of DNA or RNA into diseased cells for the purpose of therapy.3–5 In our earlier study,6 we constructed an efficient target gene delivery system mediated by the second-generation aptamer A10-3.2 (aptamer [APT]–polyethylene glycol [PEG]–polyamidoamine [PAMAM] [APT-PEG-PAMAM]) that could synergistically induce selective cell death of PCa cells by loading micro RNA (miRNA)-15a and miRNA-16-1, and found that APT-PEG-PAMAM could effectively deliver miRNA to PCa cells overexpressing prostate-specific membrane antigen (PSMA), resulting in tumoricidal efficacy.
PSMA is a 100 kDa membrane-bound glycoprotein that is upregulated in androgen-dependent PCa cells.7 Importantly, PSMA is continually recycled from the plasma membrane and constitutively endocytosed in PSMA-positive LNCaP cells, making it an attractive portal to deliver molecules intracellularly.8,9 The introduction of various biological ligands or antibodies into drug delivery systems has provided an opportunity for the targeted delivery of miRNA to PCa cells.10 Such ligands are recognized by PSMA on androgen-dependent PCa cell surfaces (LNCaP; 22Rv1), which later induce the cellular uptake of the ligand-decorated carriers via receptor-mediated endocytosis.11 Several studies on gene targeting therapy have demonstrated the strength of ligand-modified vehicles, such as antibody,12,13 peptide,7,14 and APT,6,15 in treating PSMA-positive PCa cells and xenografts. The high transfection efficiency and enhanced therapeutic effect indicate that ligand-decorated nanocarriers are potentially targeted vectors to PSMA-positive PCa cells for gene delivery.
APT-modified nanoparticles (NPs) (APT-PEG-PAMAM/miRNA) were a novel, efficient, PCa-targeting nonviral nanoscaled gene delivery system using the PAMAM dendrimer as the main macromolecular gene vector.6 APT was investigated as a PSMA-targeting ligand to modify PAMAM-based NPs to PCa cells, using PEG as a linker. APT-modified NPs have three important attributes: 1) high gene encapsulation ability using novel cationic macromolecular material, PAMAM, as the main gene vector; 2) enhanced target ability by targeting the PSMA in PCa cells (LNCaP; 22Rv1) via APT modification; and 3) multiple-target inhibitory effects on PCa and enhanced anticancer effects provided by the miRNA-15a and miRNA-16-1 system.
Although the PCa-targeting ability of APT-modified NPs has been demonstrated in our previous study,6 its underlying mechanism and in vivo anticancer ability remain elusive. The objective of this study was to validate the feasibility of systemic miRNA delivery to PCa cells by APT-PEG-PAMAM/miRNA, to testify its tumor-targeting efficiency, and to observe its biodistribution when it was administered systemically to a xenograft mouse model of PCa. The effect of APT depletion and endocytosis inhibitors on the cellular uptake of LNCaP cells were evaluated quantitatively to clarify the internalization mechanism of APT-PEG-PAMAM/miRNA. Finally, blood chemistry, and renal and liver function parameters in the xenograft mouse model of PCa were measured to see whether APT-PEG-PAMAM/miRNA had any demonstrable toxicity in vivo.
Materials and methods
Materials
The materials used in this study were: PAMAM dendrimer (generation 5, 5% w/w solution in methyl alcohol, containing 128 surface primary amino groups with a molecular weight of 28,826); dithiothreitol (DTT; Sigma-Aldrich Co., St Louis, MO, USA); α-malemidyl-ω-N-hydroxysuccinimidyl polyethyleneglycol (NHS-PEG-MAL; molecular weight 3,400; Nektar Therapeutics, Huntsville, AL, USA); prostate carcinoma PC3 and LNCaP cell lines (American Type Culture Collection, Manassas, VA, USA); anti-PSMA APT (sequence 5′-GGGAGGACGAUGCGGAUCAGCCAUGUUUACGUCACUCCU-(CH2)6-S-S-(CH2)6-OH-3′ with 2′-fluoro pyrimidines) and nuclease-free water (Guangzhou RiboBio Co., Ltd., Guangzhou, People’s Republic of China); plasmid pre-miRNA-15a (sequence 5′-UAGCAGCACAUAAUGGUUUGUG-3′); plasmid pre-miRNA-16-1 (sequence 5′-UAGCAGCACGUAAAUAUUGGCG-3′) and plasmid prenegative control-miRNA (sequence 5′-UUCUCCGAACGUGUCACGUUTT-3′) (Shanghai YINGWEIXIN Information Technology Co. Ltd., Shanghai, People’s Republic of China); and micro-bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Male BALB/c mice (4–5 weeks old) weighing 19–20 g were provided by the Department of Experimental Animals of the Second Military Medical University (Shanghai, People’s Republic of China), and maintained under standard housing conditions. All animal experiments were carried out in accordance with the guidelines evaluated and approved by the ethics committee of the Second Military Medical University. All other materials were of reagent grade and obtained from commercial sources.
Synthesis of APT-modified vectors
APT, PEG, and PAMAM were successfully conjugated at 1:2:1 (mol/mol/mol), as described previously (designated as APT-PEG-PAMAM).6 Briefly, a 2:1 conjugate of NHS-PEG3400-MAL and PAMAM was prepared through a specific reaction between the NHS groups of the bifunctional PEG and the primary amino groups on the surface of a PAMAM derivative in phosphate buffered solution (PBS) (pH 8.0) for 15 minutes at 25°C. The resulting conjugate (designated as PEG-PAMAM) was purified by ultrafiltration using a 5 kDa molecular weight cutoff membrane. Then, the PEG-PAMAM was reacted with APT-SH (sulfhydryl) at 1:1 (mol/mol) in PBS (pH 7.0) for 24 hours at 25°C to obtain APT-PEG-PAMAM. The successful synthesis of APT-PEG-PAMAM was shown previously.6
Preparation of APT-modified NPs
APT-PEG-PAMAM/miRNA (miRNA-15-a:miRNA-16-1 =1:1) NPs were prepared by gently mixing 6 μg of miRNA/500 μL of distilled water with polymer stock solution at a N/P ratio of 15, into a final volume of 1 mL. The NPs were incubated for 30 minutes at 25°C prior to analysis. PEG-PAMAM/miRNA NPs were designated as unmodified NPs. APT-PEG-PAMAM/miRNA-cy3 and PEG-PAMAM/miRNA-cy3 NPs were designated in the same way as mentioned earlier.6 The size and zeta potential of APT-modified NPs formed in PBS (10 mM; pH 7.4) were measured in triplicate using a Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK). The morphology of the APT-modified NPs was studied by transmission electron microscopy (TEM) (JEOL JEM-1230; JEOL, Tokyo, Japan).
PCa cell LNCaP cellular uptake mechanism of APT-modified NPs
LNCaP cells were cultured in 24-well plates (Corning Incorporated, Corning, NY, USA) at a density of 1×104 cells/well for 48 hours, and examined for morphology and confluency under the microscope. The medium was removed, and the cells were incubated for 30 minutes with APT-PEG-PAMAM/NC-miRNA-cy3 (1 mM in medium) at 4°C or with APT-PEG-PAMAM/NC-miRNA-cy3 with and without the addition of 100 mM free APT at 37°C. After incubation, the wells were washed thrice with 1 mL of PBS (pH 7.4) and the cellular uptake was examined using a DM IL fluorescent microscope (Leica Microsystems, Wetzlar, Germany).16
LNCaP cells were cultured in 24-well plates (Corning Incorporated) at a density of 1×104 cells/well for 48 hours, and examined for morphology and confluency under the microscope. The medium was removed and the cells were pretreated with colchicine (10 mM), filipin (5 mg/mL), PhAsO (1 mM), excessive APT (100 mM), and polylysine (400 mg/mL) for 10 minutes at 37°C. Colchicine is an inhibitor of macropinocytosis,17 whereas cationic polylysine could inhibit the uptake of cationic NPs. Filipin is reported to block the caveolae-mediated process,18 and PhAsO is known to inhibit clathrin-dependent endocytosis.16,19 Excessive APT might induce both competitive and cationic inhibition. Subsequently, LNCaP cells were incubated with APT-PEG-PAMAM/NC-miRNA-cy3 and PEG-PAMAM/NC-miRNA-cy3 in a final concentration of 1 mM for 30 minutes at 37°C. The cells were washed thrice with PBS and visualized under a DM IL fluorescent microscope (Leica Microsystems). LNCaP-treated APT-PEG-PAMAM/NC-miRNA-cy3 and PEG-PAMAM/NC-miRNA-cy3 without any inhibition served as the control. LNCaP was treated as described earlier, incubated with Triton™ X-100 (1%, w/v) for cellular lysis, and centrifuged at 3,000 rpm for 1 minute for quantitative analysis. The fluorescence intensity of cy3 (excitation at 488 nm and emission at 570 nm) in the supernatant was assessed using a microplate fluorometer (LS 55; PerkinElmer Inc., Waltham, MA, USA). Protein concentrations of the cell lysates were determined using the Bradford colorimetric assay with bovine serum albumin (Thermo Fisher Scientific) as a protein standard. The relative uptake efficiency (RUE) was calculated as follows:
| (1) |
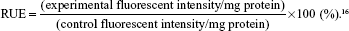
The PCa-targeting effect of APT-modified NPs in vivo
The in vivo PCa target effect of APT-modified NPs was evaluated by real-time fluorescence imaging analysis. Male athymic nude mice (BALB/c) were housed in standard microisolator conditions free of pathogens. LNCaP cells 5×106 were suspended in 0.2 mL of sterile PBS and injected subcutaneously via the left flank to the mice.20 The mouse model of human PCa was established when the tumor reached a mean diameter of 5–8 mm in about 2 weeks. APT-PEG-PAMAM/miRNA (APT-NPs) and PEG-PAMAM/miRNA (NPs) were labeled by cy7 (RuiCheng Industrial Co., Ltd., Shanghai, People’s Republic of China). In brief, for the synthesis of the cy7-labeled conjugate, PAMAM was first reacted with cy7-NHS in 100 mM of NaHCO3 for 12 hours at 4°C, purified and identified as described previously.6 The LNCaP tumor-bearing mice were injected with 0.2 mL of cy7-labeled APT-NPs or NPs via the tail vein. The mice were anesthetized with the intraperitoneal administration of 10% chloral hydrate and placed on an animal plate heated to 37°C. Fluorescent scan (from 620–900 nm) was performed at 0.5 hours, 2 hours, 6 hours, 12 hours, and 24 hours using an in vivo image system (Caliper Life Sciences; PerkinElmer Inc.).21 To compare tissue and tumor distributions of APT-NPs, the mice were sacrificed at 24 hours postinjection. The tumor and major organs including the heart, spleen, liver, lung, and kidney were dissected, washed with saline, and subjected to the in vivo image system to obtain the fluorescence images, as described previously.21,22
APT-modified NP toxicity study
Male athymic nude mice (BALB/c) were injected via the tail vein with either NaCl (0.9%) as the vehicle or APT-PEG-PAMAM/miRAN (miRNA-15-a/miRNA-16-1 =25 μg/25 μg) three times a week. Blood was collected at necropsy via cardiac puncture. The white blood cell (WBC) count was determined by the routine clinical laboratory technique.23,24 Plasma was obtained by centrifugation and used for the measurement of alanine aminotransferase (ALT) and creatinine (Cr) values by using a sequential multiple AutoAnalyzer system (Hitachi Ltd., Tokyo, Japan).24
The anticancer effect of APT-modified NPs in vivo
The anticancer effect of APT-NPs in vivo was investigated using the survival duration of the human PCa model mice. The mice were randomized into four groups and were respectively treated with 200 mL of NaCl (0.9%), APT-PEG-PAMAM/miRNA (miRNA-15-a/miRNA-16-1 =25 μg/25 μg), APT-PEG-PAMAM/NC-miRNA (50 μg), and PEG-PAMAM/miRNA (miRNA-15-a/miRNA-16-1 =25 μg/25 μg) at days 1, 3, 5, 7, and 11 after model establishment. The body weight was monitored every other day, and death was recorded as occurring after treatment. Tumors were excised and weighed after the mice died.
Statistical analysis
The data were presented as the mean ± standard deviation. For a comparison of individual time points, differences between the groups were tested by performing one-way analysis of variance. The Statistical Package for the Social Sciences (SPSS version 18.0; IBM Corporation, Armonk, NY, USA) was used for all statistical analyses. Statistical significance was defined as P<0.05.
Results
Characterization of APT-modified NPs
The successful synthesis of APT-PEG-PAMAM was shown previously.6 The mean diameter of APT-modified NPs was 177±17.5 nm (Figure 1A), with an acceptably good polydispersity index of <0.15, whereas the zeta potential value was 25.3±2.3 mV (Figure 1B). Owing to the enhanced permeability and retention (EPR) effect, APT-modified NPs may accumulate in the tumor more readily.6 The result of TEM showed that APT-modified NPs were moderately uniform particles with a spherical shape, whose size was in good agreement with that measured by the laser scattering technique (Figure 1C).
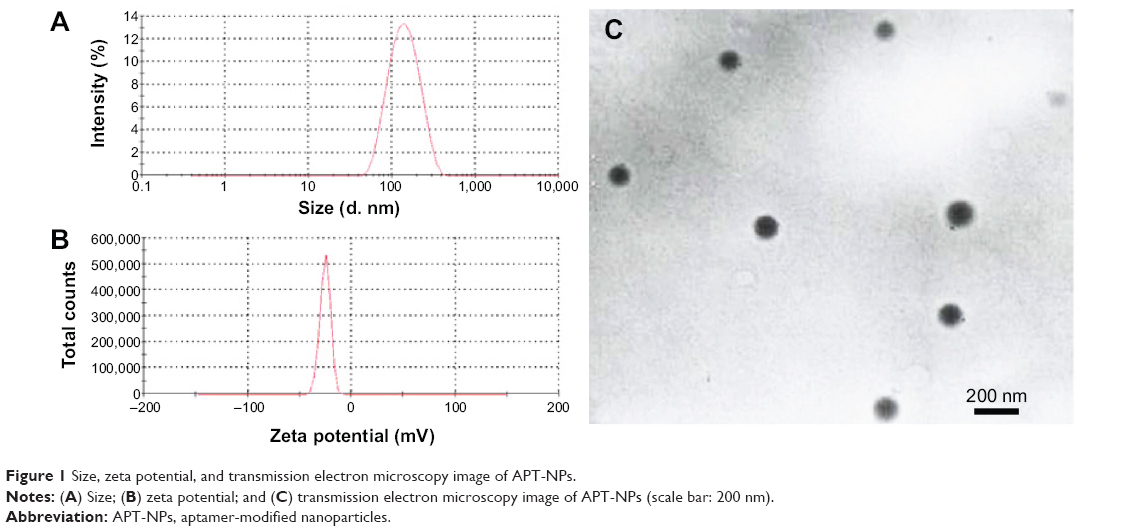 | Figure 1 Size, zeta potential, and transmission electron microscopy image of APT-NPs. |
Mechanism of cellular uptake of APT-modified NPs in LNCaP cells
The results of the cellular uptake of APT-modified NPs in LNCaP cells at different temperatures are shown in Figure 2. The cellular uptake of APT-NPs at 4°C was much lower than that at 37°C, and it was significantly inhibited by excessive free APT (Figure 2). APT-NPs were weakly internalized at 4°C when they were primarily associated with the plasma membrane (Figure 2A). The amount of cellular internalization of the APT-NPs was increased significantly under the same incubation conditions at 37°C (Figure 2B). In addition, both the cellular uptake of APT-NPs and the intracellular fluorescence decreased when APT-NPs were coincubated with excessive APT (Figure 2C).
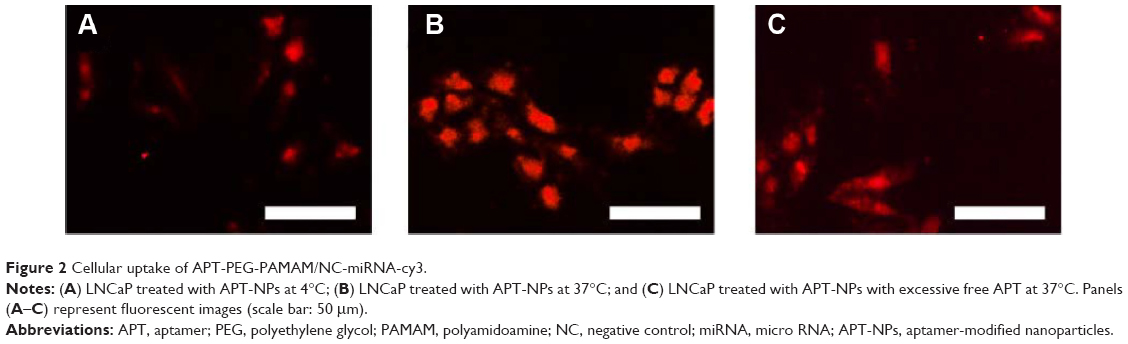 | Figure 2 Cellular uptake of APT-PEG-PAMAM/NC-miRNA-cy3. |
The effects of different inhibitors are shown in Figure 3. The cellular uptake of APT-NPs was inhibited by all the used inhibitors (Figures 3A–F), whereas the cellular uptake of unmodified NPs was inhibited by all but PhAsO, the inhibitor of clathrin-dependent endocytosis (Figures 3G–L). In addition, the RUE of colchicine, filipin, excessive APT, polylysine, and PhAsO on the cellular uptake of unmodified NPs was 55.94%, 39.46%, 69.01%, 24.38%, and 99.12%, respectively (Figure 3M). The RUE of colchicine, filipin, excessive APT, polylysine, and PhAsO on the cellular uptake of APT-NPs was 68.25%, 65.34%, 33.79%, 70.21%, and 35.11%, respectively (Figure 3M).
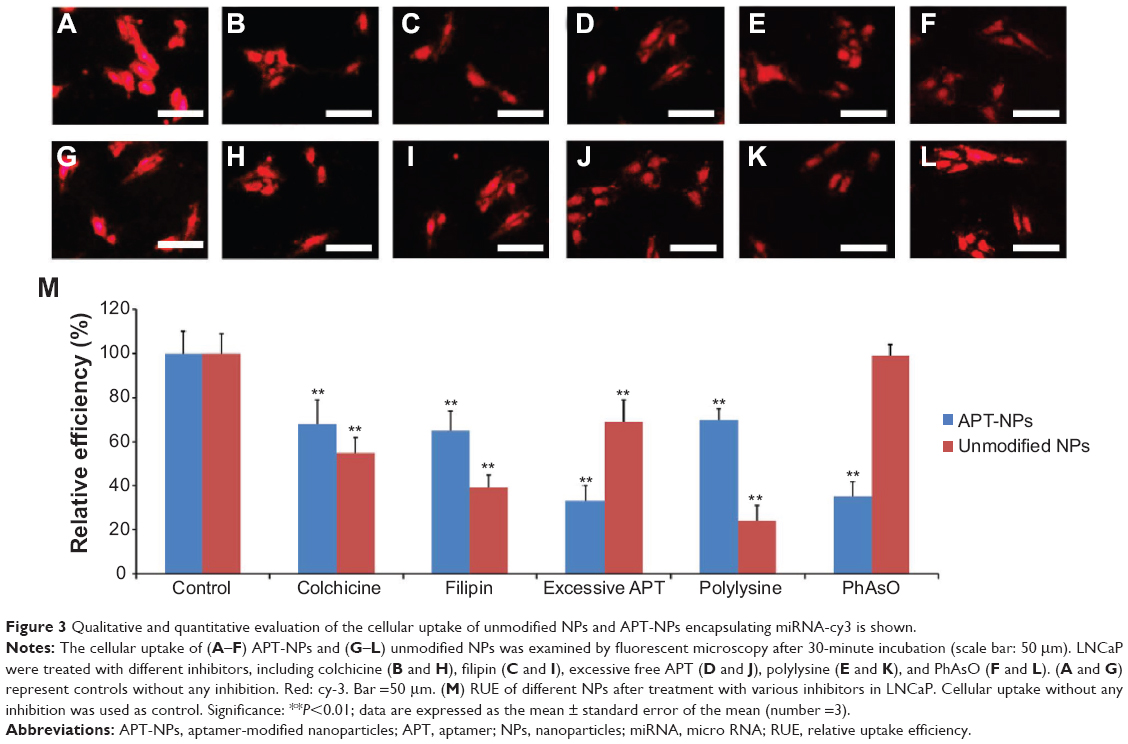 | Figure 3 Qualitative and quantitative evaluation of the cellular uptake of unmodified NPs and APT-NPs encapsulating miRNA-cy3 is shown. |
In vivo real-time imaging analysis of APT-modified NPs
Noninvasive near infrared fluorescence imaging showed that APT-NPs and cy7-labeled APT-NPs administered intravenously into the LNCaP tumor-bearing mice through the tail vein were distributed in a time-dependent manner. As shown in Figures 4A and B, the fluorescence signal was viewed in the tumor-bearing mice as early as 0.5 hours after injection, reaching the maximum at 2 hours after injection, and then decreasing gradually 24 hours after injection. Compared with the unmodified NP group, the fluorescence signal of the APT-NP group was significantly stronger at any postinjection time point ranging from 0.5–24 hours (Figures 4A and B).
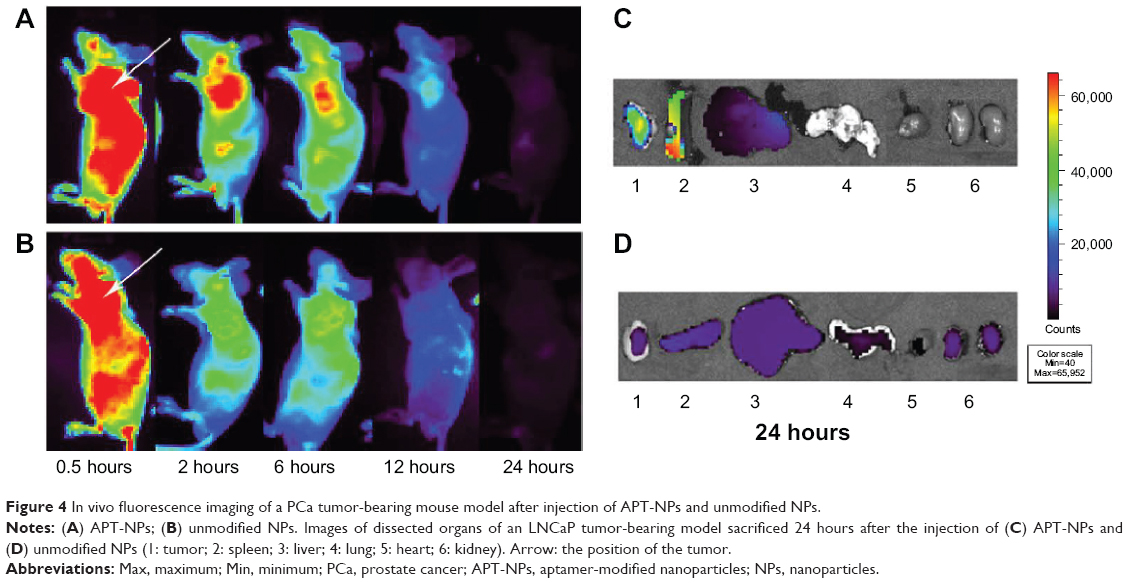 | Figure 4 In vivo fluorescence imaging of a PCa tumor-bearing mouse model after injection of APT-NPs and unmodified NPs. |
Ex vivo evaluation of the excised tumor and tissues from the heart, liver, spleen, lung, and kidney at 24 hours postinjection showed an obvious accumulation of APT-NPs in the tumor (Figures 4C and D). The fluorescence of unmodified NPs was located in the tumor, indicating that PEG-PAMAM/miRNA could accumulate in the PCa tumor via the EPR effect; more fluorescence of the APT-NPs was distributed in the PCa tumor, suggesting that PEG-PAMAM/miRNA modified with APT not only accumulated in the PCa tumor via the EPR effect, but it targeted the tumor via PSMA receptor-mediated endocytosis in the LNCaP cells. These findings suggest that APT-NPs had a potential PCa-targeting effect in vivo in the LNCaP tumor-bearing model.
Systemic toxicity in APT-modified NP-treated mice
The hematological toxicity of APT-NP-treated tumor-bearing mice was investigated by means of WBC count. There was no significant decrease in the WBC count after APT-NP treatment when compared to no treatment (control) (Table 1). In addition, blood biochemical assays showed that this treatment had no significant adverse effect on the liver and kidneys in terms of plasma ALT and Cr values (Table 1).
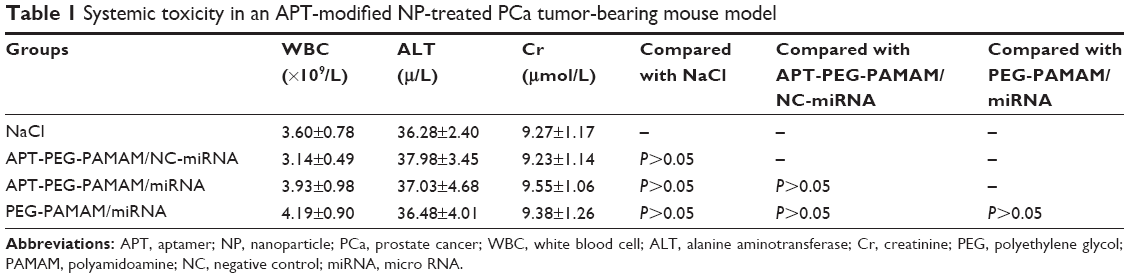 | Table 1 Systemic toxicity in an APT-modified NP-treated PCa tumor-bearing mouse model |
The in vivo anticancer effect of APT-modified NPs
In the human PCa mouse model, male nude mice that received 5×106 LNCaP cells subcutaneously into the left flank region were assigned to one of four groups (APT-PEG-PAMAM/miRNA, PEG-PAMAM/miRNA, APT-PEG-PAMAM/NC-miRNA, or NaCl). Comparisons of the anticancer effect between the four groups in terms of the survival duration are shown in Figure 5 and Table 2; the anticancer efficacy of APT-PEG-PAMAM/miRNA was superior to that of the other three groups. The mean survival duration of the NaCl, APT-PEG-PAMAM/NC-miRNA, PEG-PAMAM/miRNA, and APT-PEG-PAMAM/miRNA groups was 46 days, 45 days, 56 days, and 78 days, respectively. Log–rank analysis showed no significant difference in the survival duration between the NaCl and APT-PEG-PAMAM/NC-miRNA groups (P>0.05). Compared with the NaCl, APT-PEG-PAMAM/NC-miRNA, and PEG-PAMAM/miRNA groups, the survival duration in the APT-PEG-PAMAM/miRNA group was significantly longer (P<0.01). Although significant suppression of tumor growth was also observed in APT-PEG-PAMAM/miRNA and PEG-PAMAM/miRNA, the therapeutic effect on tumor growth was optimal with the treatment of APT-PEG-PAMAM/miRNA. Similar results were observed by the mean tumor weight. The suppressive effect of APT-PEG-PAMAM/miRNA on tumor growth was sustainable until the day of death, during which time the mean tumor weight grew to 0.36 g versus 1.21 g, 1.18 g, and 0.64 g in the NaCl, APT-PEG-PAMAM/NC-miRNA and PEG-PAMAM/miRNA groups, respectively (Figure 6 and Table 2).
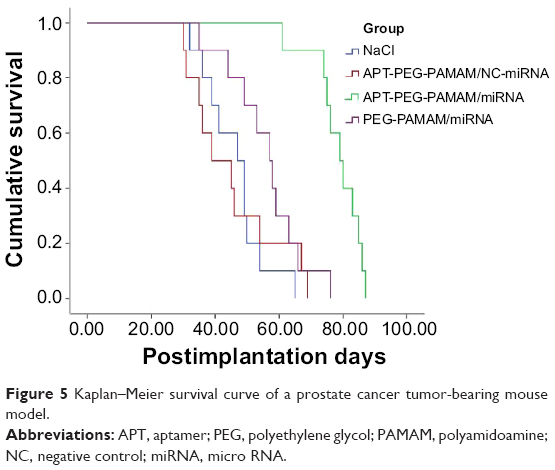 | Figure 5 Kaplan–Meier survival curve of a prostate cancer tumor-bearing mouse model. |
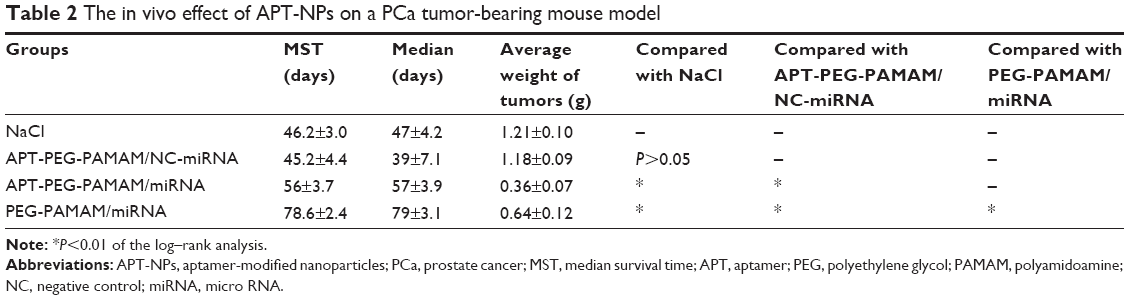 | Table 2 The in vivo effect of APT-NPs on a PCa tumor-bearing mouse model |
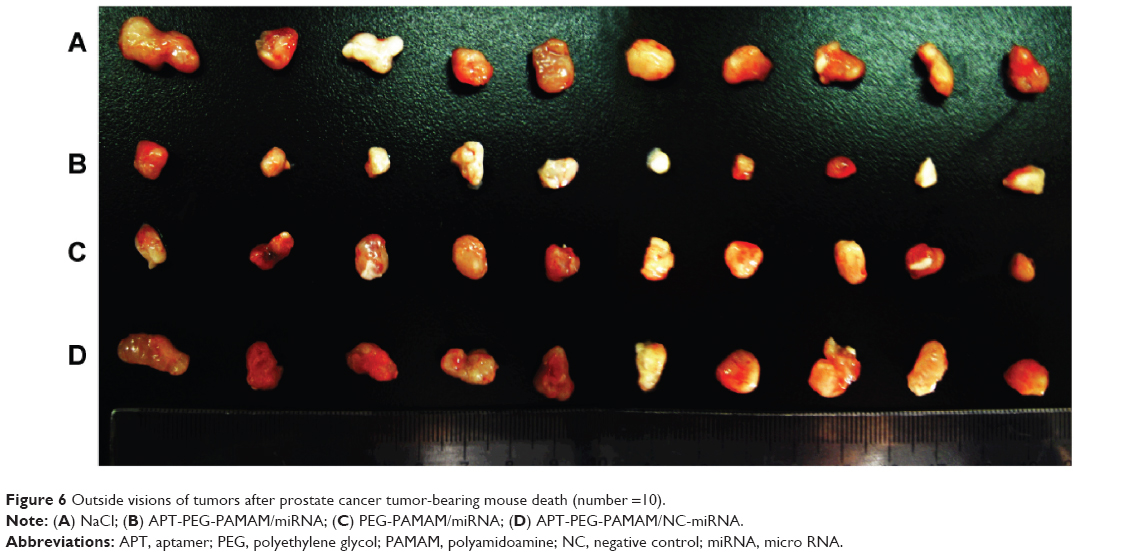 | Figure 6 Outside visions of tumors after prostate cancer tumor-bearing mouse death (number =10). |
To evaluate the safety of these NPs, body weight was monitored as a marker of overall toxicity. The change in body weight was recorded at 21 days. As shown in Figure 7, the mice in the APT-PEG-PAMAM/miRNA, APT-PEG-PAMAM/NC-miRNA, and PEG-PAMAM/miRNA groups gained weight during the first 15 days after treatment, as did those in the NaCl group, implying that APT-NPs were safe. However, there was a weight loss 15 days after treatment in all four groups, probably because the tumors grew more rapidly after 15 days. Thus, the APT-NPs could be a safe gene delivery system for PCa treatment. Similar results were also observed in the previous section (Systemic toxicity in APT-modified NP-treated mice).
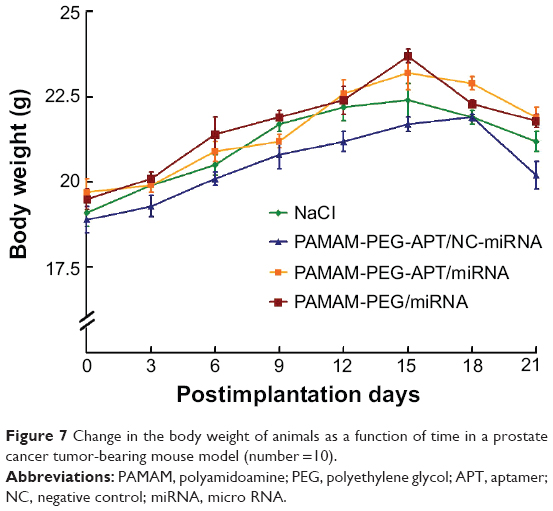 | Figure 7 Change in the body weight of animals as a function of time in a prostate cancer tumor-bearing mouse model (number =10). |
Discussion
Nanoscaled PCa-targeting drug delivery systems have been used for small molecules, proteins, and genes.25 Owing to the elucidation of the exact targeting mechanisms, nanoscaled PCa-targeting drug delivery systems were designed efficiently. The high transfection efficiency and enhanced therapeutic effect of these drug delivery systems indicate that ligand-decorated nanocarriers are potentially targeted vectors for PCa treatment. In our previous study,26 we identified that APT was a novel PSMA-targeting ligand, and we used it as a gene delivery vector to target PCa cells successfully. To the best of our knowledge, this study is the first systematic investigation on the PCa-targeting mechanism of APT-modified NPs and their in vivo anticancer efficacy.
The APT-modified NPs designed in this study mainly consist of the ligand APT and the dendritic PAMAM. It has been indicated that APT can bind to the specific PSMA-receptors in the androgen-dependent PCa cells.27–29 In addition, PAMAM is positively charged with primary amino groups on the surface.30 Therefore, the synthesized APT-modified NPs should also be positively charged. It was found in this study that the cellular uptake of APT-modified NPs was energy dependent and inhibited by excessive APT (Figure 2). In addition, the uptake of APT-modified NPs by LNCaP was related to the endocytic processes, including macropinocytosis, caveolae-mediated endocytosis, and clathrin-dependent endocytosis, with regards to the cationic properties of PAMAM and APT (Figure 3). These results suggest that both receptor and adsorptive-mediated mechanisms might contribute to the cellular uptake of APT-modified NPs.
To further verify the PCa-targeting effect of APT-NPs in vivo, LNCaP tumor-bearing nude mice were used to investigate the distribution of cy7-labeled NPs. The results from in vivo and ex vivo imaging photos of the tumor-bearing mice showed that APT modification enhanced the accumulation of the NPs in the PCa tumor. The fluorescence of unmodified NPs was located on the tumor, indicating that PEG-PAMAM/miRNA could accumulate in the PCa tumor via the EPR effect, while more fluorescence of the APT-NPs distributed on the PCa tumor, suggesting that PEG-PAMAM/miRNA modified with APT not only accumulated in the PCa tumor via the EPR effect, but that it could target the tumor via PSMA receptor-mediated endocytosis in LNCaP cells. The accumulation of APT-NPs and unmodified NPs in the liver and spleen suggested that both APT-NPs and unmodified NPs were scavenged predominantly by the liver and spleen. In the presence of PEG on the surface of APT-NPs, the rate of reticuloendothelial system uptake of the NPs was greatly reduced, thus allowing the APT-NPs to have an increased chance to distribute to the tumor. This may be the mechanism underlying the potential PCa-targeting effect of APT-NPs in the LNCaP tumor-bearing model in vivo.
Our data concerning the systemic toxicity of APT-NPs show that APT-NPs were well tolerated by the mice. In addition, no significant sign of toxicity on the major organs was observed in terms of changes in body weight (Figure 7), WBC, ALT, and Cr values (Table 1), suggesting that APT-NPs do not induce functional impairment to the normal and vital organs and tissues. These results also imply that APT-NPs can be used as a safe and effective miRNA delivery system for PCa treatment.
A previous study31 has demonstrated that miRNA-15a and miRNA-16-1 are effective anti-PCa molecules. The present study demonstrated that APT-NPs had an anticancer effect in terms of the survival time and mean tumor weight in the human PCa mouse model (Figures 5 and 6; Table 2). Unlike the delivery of short hairpin RNAs or small interfering RNA, which are usually designed to target single genes, miRNAs can concurrently target multiple genes and pathways involved in PCa, thus potentially enhancing the anti-PCa activity. Compared with unmodified NPs, APT-NPs also had a potential PCa-targeting effect in vivo, thus prolonging the survival time of the mice in the human PCa tumor-bearing model.
Conclusion
To the best of our knowledge, this is the first study focusing on the PCa-targeting mechanism of APT-modified NPs and their anticancer ability in vivo. The uptake of APT-modified NPs by LNCaP was related to endocytic processes, including macropinocytosis, caveolae-mediated endocytosis, and clathrin-dependent endocytosis, with regard to the cationic properties of vectors and APT. The results from in vivo and ex vivo imaging photos of the human PCa tumor-bearing mice indicate that APT modification could enhance the accumulation of NPs in the PCa tumor. Our data suggest that APT-NPs do not induce functional impairment to normal tissues and vital organs. APT-NPs may prove to be a safe and effective miRNA delivery system for PCa treatment. Compared with unmodified NPs, APT-NPs could prolong the survival time of the mice in the human PCa tumor-bearing model; they also exhibited a potential PCa-targeting effect in vivo. In conclusion, this research provides a prototype for the safe and efficient delivery of miRNA expression vectors to PCa cells, which could be widely used in preclinical and, eventually, clinical arenas of PCa treatment.
Acknowledgments
This research work was supported by the funds provided by the National Natural Science Foundation of China (numbers 81372762, 81302212, 81373896, 81201809, 81101658, 81100800); the Foundation of Shanghai Science and Technology Committee (numbers 11NM0405600, 13NM1401900, 14JC1491300); the Hainan Provincial Natural Science Foundation (number 814392); the Zhejiang Provincial Natural Science Foundation (number LQ12H30005); and the Science and Technology Projects of Jiaxing (number 2012AY1075-3).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Yang YF, Xue SY, Lu ZZ, et al. Antitumor effects of oncolytic adenovirus armed with PSA-IZ-CD40L fusion gene against prostate cancer. Gene Ther. 2014;21(8):723–731. | ||
Azab BM, Dash R, Das SK, et al. Enhanced prostate cancer gene transfer and therapy using a novel serotype chimera cancer terminator virus (Ad.5/3-CTV). J Cell Physiol. 2014;229(1):34–43. | ||
Jiang Z, Sun C, Yin Z, et al. Comparison of two kinds of nanomedicine for targeted gene therapy: premodified or postmodified gene delivery systems. Int J Nanomedicine. 2012;7:2019–2031. | ||
Xue HY, Wong HL. Solid lipid-PEI hybrid nanocarrier: an integrated approach to provide extended, targeted, and safer siRNA therapy of prostate cancer in an all-in-one manner. ACS Nano. 2011;5(9):7034–7047. | ||
Wu X, Ding B, Gao J, et al. Second-generation aptamer-conjugated PSMA-targeted delivery system for prostate cancer therapy. Int J Nanomedicine. 2011;6:1747–1756. | ||
Xiang B, Dong DW, Shi NQ, et al. PSA-responsive and PSMA-mediated multifunctional liposomes for targeted therapy of prostate cancer. Biomaterials. 2013;34(28):6976–6991. | ||
Zhou J, Rossi JJ. Aptamer-targeted cell-specific RNA interference. Silence. 2010;1(1):4. | ||
Su Y, Yu L, Liu N, et al. PSMA specific single chain antibody-mediated targeted knockdown of Notch1 inhibits human prostate cancer cell proliferation and tumor growth. Cancer Lett. 2013;338(2):282–291. | ||
Civenni G, Malek A, Albino D, et al. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res. 2013;73(22):6816–6827. | ||
Yao V, Berkman CE, Choi JK, O’Keefe DS, Bacich DJ. Expression of prostate-specific membrane antigen (PSMA), increases cell folate uptake and proliferation and suggests a novel role for PSMA in the uptake of the non-polyglutamated folate, folic acid. Prostate. 2010;70(3):305–316. | ||
Milowsky MI, Nanus DM, Kostakoglu L, et al. Vascular targeted therapy with anti-prostate-specific membrane antigen monoclonal antibody J591 in advanced solid tumors. J Clin Oncol. 2007;25(5):540–547. | ||
Moffatt S, Cristiano RJ. PEGylated J591 mAb loaded in PLGA-PEG-PLGA tri-block copolymer for targeted delivery: in vitro evaluation in human prostate cancer cells. Int J Pharm. 2006;317(1):10–13. | ||
Barve A, Jin W, Cheng K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J Control Release. 2014;187:118–132. | ||
Dassie JP, Hernandez LI, Thomas GS, et al. Targeted inhibition of prostate cancer metastases with an RNA aptamer to prostate-specific membrane antigen. Mol Ther. Epub 2014 Jun 23. | ||
Huang R, Ke W, Han L, et al. Brain-targeting mechanisms of lactoferrin-modified DNA-loaded nanoparticles. J Cereb Blood Flow Metab. 2009;29(12):1914–1923. | ||
Liu J, Shapiro JI. Endocytosis and signal transduction: basic science update. Biol Res Nurs. 2003;5(2):117–128. | ||
Kim HR, Gil S, Andrieux K, et al. Low-density lipoprotein receptor-mediated endocytosis of PEGylated nanoparticles in rat brain endothelial cells. Cell Mol Life Sci. 2007;64(3):356–364. | ||
Visser CC, Stevanović S, Heleen Voorwinden L, et al. Validation of the transferrin receptor for drug targeting to brain capillary endothelial cells in vitro. J Drug Target. 2004;12(3):145–150. | ||
Schwarzenböck S, Sachs D, Souvatzoglou M, et al. [[11C]choline as a pharmacodynamic marker for docetaxel therapy. Response assessment in a LNCaP prostate cancer xenograft mouse model]. Nuklearmedizin. 2013;52(4):141–147. German. | ||
Xin H, Jiang X, Gu J, et al. Angiopep-conjugated poly(ethylene glycol)-co-poly(ε-caprolactone) nanoparticles as dual-targeting drug delivery system for brain glioma. Biomaterials. 2011;32(18):4293–4305. | ||
Xin H, Chen L, Gu J, et al. Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(ε-caprolactone) nanoparticles: In vitro and in vivo evaluation. Int J Pharm. 2010;402(1–2):238–247. | ||
Jiang T, Zhou C, Gu J, et al. Enhanced therapeutic effect of cisplatin on the prostate cancer in tumor-bearing mice by transfecting the attenuated Salmonella carrying a plasmid co-expressing p53 gene and mdm2 siRNA. Cancer Lett. 2013;337(1):133–142. | ||
Fang J, Sawa T, Akaike T, Greish K, Maeda H. Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int J Cancer. 2004;109(1):1–8. | ||
Cabral H, Miyata K, Kishimura A. Nanodevices for studying nano-pathophysiology. Adv Drug Deliv Rev. 2014;74C:35–52. | ||
Dassie JP, Liu XY, Thomas GS, et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27(9):839–849. | ||
Hao Z, Fan W, Hao J, et al. Efficient delivery of micro RNA to bone-metastatic prostate tumors by using aptamer-conjugated atelocollagen in vitro and in vivo. Drug Deliv. Epub 2014 Jun 3. | ||
Taghdisi SM, Danesh NM, Sarreshtehdar Emrani A, et al. Targeted delivery of Epirubicin to cancer cells by PEGylated A10 aptamer. J Drug Target. 2013;21(8):739–744. | ||
Min K, Jo H, Song K, et al. Dual-aptamer-based delivery vehicle of doxorubicin to both PSMA (+) and PSMA (−) prostate cancers. Biomaterials. 2011;32(8):2124–2132. | ||
Markowicz-Piasecka M, Luczak E, Chałubiński M, Broncel M, Mikiciuk-Olasik E, Sikora J. Studies towards biocompatibility of PAMAM dendrimers – Overall hemostasis potential and integrity of the human aortic endothelial barrier. Int J Pharm. 2014;473(1–2):158–169. | ||
Bonci D, Coppola V, Musumeci M, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14(11):1271–1277. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.