Back to Journals » Journal of Pain Research » Volume 15
Study Design Characteristics and Endpoints for Enriched Enrollment Randomized Withdrawal Trials for Chronic Pain Patients: A Systematic Review
Authors Kopsky DJ ![]() , Szadek KM, Schober P, Vrancken AFJE, Steegers MAH
, Szadek KM, Schober P, Vrancken AFJE, Steegers MAH ![]()
Received 23 August 2021
Accepted for publication 6 January 2022
Published 17 February 2022 Volume 2022:15 Pages 479—496
DOI https://doi.org/10.2147/JPR.S334840
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Robert B. Raffa
David J Kopsky,1– 3 Karolina M Szadek,1 Patrick Schober,1 Alexander FJE Vrancken,3 Monique AH Steegers1
1Department of Anesthesiology, Amsterdam University Medical Center, Amsterdam, 1081 HV, the Netherlands; 2Institute for Neuropathic Pain, Amsterdam, 1056 SN, the Netherlands; 3Department of Neurology, Brain Centre University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands
Correspondence: David J Kopsky Institute for Neuropathic Pain, Vespuccistraat 64-III, Amsterdam, 1056 SN, the Netherlands Tel +31-6-28671847 Email [email protected]
Abstract: Enriched enrollment randomized withdrawal (EERW) pain trials are designed to include only responders with considerable pain relief without unacceptable side effects into the randomized phase. There are no recommendations for primary endpoints in such trials. Our objective was to propose recommendations based on assessment of trial characteristics, endpoints and effect sizes in EERW pain trials. We conducted a systematic review by searching electronic databases up to June 2020 for EERW trials comparing an analgesic with a placebo in adults suffering from chronic pain. A total of 28 trials met our criteria, involving 13662 patients in the open or single-blind phase and 7937 patients in the double-blind phase. As primary endpoint 18 trials used pain intensity measured with the visual analogue scale (VAS) or the 11-point numerical rating scale (NRS); 1 trial used a 4-point NRS. Loss of therapeutic response (LTR) was used in 1 trial and time to LTR was used in 8 trials as primary endpoint. Definitions of time to LTR differed considerably between trials. Only 2 out of 8 trials using time to LTR as primary endpoint reported the percentage of patients experiencing a minimum pain relief of 50%, compared to 14 out of 18 trials using NRS or VAS. Due to the complexity and diversity of time to LTR in EERW pain trials, we propose to use the NRS as primary endpoint with conservative imputation methods, and to use time to LTR as secondary endpoint.
Keywords: systematic review, pain, EERW trials, analgesics, endpoints
Introduction
Enriched enrollment randomized withdrawal (EERW) pain trials consist of an open or single-blind selection phase followed by the randomized double-blind phase (see Figure 1).1
 |
Figure 1 Design of enrichment enrolment withdrawal trials. |
The selection phase is often divided in 2 periods: 1) titration period, in which the analgesic drug is prescribed to the highest tolerable dose; 2) maintenance period to ensure a sustained pain-relieving effect at the optimal dose.1 The selection phase can also be used to switch trial patients from currently used analgesics to study analgesics.1
In the double-blind phase, responders tolerating the investigational drug are included and randomized into either the active or placebo arm.1 Responders are usually defined as patients experiencing a certain level of pain relief (often 30%) from baseline in the selection phase up to the start of the double-blind randomization phase.1 Patients randomized into the placebo arm are expected to experience pain aggravation, whereas patients randomized in the active arm are expected to maintain pain relief.1
The design of EERW trial ensures that only responders who experience considerable pain relief, and no or acceptable side effects on a predefined or titrated dose in the selection phase are included in the randomized double-blind phase. By this way patients not tolerating side effects drop out in the selection phase and do not affect the study results in the double-blind phase.1,2 Normally, physicians do not continue prescribing a drug that is not well tolerated. Therefore, EERW pain trials more closely resemble clinical practice because trial patients are not exposed to a non-tolerated treatment in the double-blind randomized phase.3 This design is also very useful in circumstances in which a placebo group is desirable, but prolonged exposure to placebo presents ethical or practical concerns as this design can incorporate escape to standard care or alternative treatments.4
In order to design a trial to detect a statistically significant difference between the active and placebo group, it is essential to choose the proper primary outcome and estimate a realistic effect size for a well-founded power calculation. In chronic pain trials, the following core outcome domains are recommended: pain intensity, interference in physical and emotional functioning, global improvement and satisfaction, and adverse events and patient disposition (including adherence to the treatment regimen and reasons for premature withdrawal from the trial).5,6 Regarding pain intensity, the 11-point numerical rating scale (NRS) is recommended as core outcome measure.5 Furthermore, additional analyses of minimal pain relief of 30% and 50% (MPR30 and MPR50) are recommended.5 MPR30 and MPR50 are seen as moderate and substantial clinical benefit.7
In EERW pain trials, mostly chosen endpoints are the following: 1) pain intensity measured with NRS or with the 10 cm visual analogue scale (VAS), 2) MPR30 and MPR50 compared to screenings baseline, and 3) time to loss of therapeutic response (LTR).2,8,9 Other descriptions of time to LTR are time to treatment failure, time to inadequate analgesic response, or time to exit.
Many factors can influence the effect size. For example, a systematic review on EERW chronic pain trials revealed that the standardized effect size decreased linearly by 0.13 for every 10 sites (P=0.037), from 0.75 to 0.36, until stabilization at 60 sites.9 The number of subjects required for 90% power to discriminate the difference between a study drug from placebo increased from 78 to 336 subjects, having 30 to 60 sites included. Another systematic review did not reveal a difference in effect size comparing enriched and non-enriched trials, whereas failure of blinding outcome assessor and failure to use intention to treat analysis were associated with the larger effect sizes.10 Other factors influencing the effect size negatively are longer duration of trials and more trial patients.11 Furthermore, these outcome differences likely reside in different patient populations, types of pain, drugs and administration routes. However, additional factors, related to the study design itself, may be related to the magnitude of the effect size. Identifying such factors is useful for the planning of future trials (eg, factors with respect to required sample size).
The purpose of this systematic review is to identify factors to design EERW pain trials optimally. With this systematic review, we 1) evaluate characteristics of the EERW pain trials with patients ≥18 years of age evaluating efficacy of analgesics compared to placebo 2) present the absolute effect size of difference in pain intensity, 3) correlate the absolute effect size with double-blind phase characteristics, ie, the stringency of inclusion criteria, duration of the double-blind phase and pain intensity at double-blind baseline and 4) propose recommendations for primary and secondary endpoints based on the qualitative analysis.
Methods
To perform this systematic literature review we followed the guidelines of Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.12
Protocol and Registration
The protocol of this systematic review was registered in advance through PROSPERO: CRD42020186453.
Eligibility Criteria
Our inclusion criteria were 1) EERW trial, 2) patients ≥18 years of age, 3) any type of chronic nonmalignant pain with a duration of at least 3 months and pain intensity as an outcome measure, 4) placebo-controlled trials with a double-blind phase of ≥4 weeks, 5) ≥10 patients per arm, 6) definition of a responder determined on a pain scale at the end of the selection phase, 7) presented power calculation. Exclusion criteria were 1) cross-over design in the double-blind randomization phase and 2) post-hoc analysis publications of EERW pain trials.
Information Sources
To identify relevant studies, we performed a search in following databases: PubMed, PsychInfo and Scopus from the beginning of these databases up to June 2020. Furthermore, we have examined the references of systemic reviews about EERW chronic pain trials for potentially relevant trials. No limits were applied for language.
Search
We used the following search terms in all mentioned databases: withdrawal trial; pain. For more detailed information on the search strategy see Supplement 1. The search was conducted by DJK.
Study Selection
Two authors (DJK, KS) independently screened titles and abstracts imported into Rayyan database to select manuscripts meeting the inclusion criteria. If based on the title and abstract the article would be included, the full articles were screened. Any disagreement on separate preliminary decisions was resolved through discussions by the two authors (DJK, KS).
Data Collection Process and Items
One review author (DJK) extracted data into SPSS. The data was checked by two other authors (KS, PS). The following data were extracted: patients age, type of pain, pain intensity, investigational drug naïve or experienced patients, type of drug and application form, number of applications per day, allowance of rescue medication, number of research sites and countries, type of selection phase (ie, open or single-blind), inclusion criteria in selection and double-blind phase, number of patients in screening, selection and double-blind phase, number of drop-outs, duration of selection and double-blind phase, percentage of treatment emergent adverse events (TEAEs), primary and secondary endpoints, power calculation characteristics (ie, power, standard deviations, assumed effect size) absolute effect size and its confidence intervals, imputation methods for drop-outs, percentage of patients with MPR30 and MPR50 in double-blind phase compared to selection phase baseline pain score.
Assessment of Risk of Bias in Included Studies
Risk of bias and quality of reporting assessments were undertaken by the first and second authors (DJK, KS) independently with the revised version of the Cochrane tool, known as Risk of Bias 2 tool.13 The following risk of bias domains were assessed: bias arising from randomization process (random sequence generation, allocation concealment, baseline similarity between groups) bias due to deviations from intended interventions (blinding patients, carers and trial personnel, deviations due to intervention expectations, intention-to-treat analysis), bias due to missing outcome data, bias in measurement of the outcome (blinding the assessor), bias in selection of the reported result. The website of clinicaltrials.gov was accessed and data retrieved when full articles lacked information. These steps ensured the completeness of reporting and helped to determine the reliability and replicability of included trials. If any disagreements arose, consensus was reached through discussion between the two authors (DJK, KS).
We followed the definition of Risk of Bias tool regarding high-quality study with low risk of bias in all 5 domains; moderate quality study when some concerns arose in one of the domains; low quality of study when high risk of bias in one of the domains appeared.
Descriptive Analyses
The VAS measuring pain intensity can be reported in mm or cm. To be able to compare trial results using the VAS with trials using the 11-point NRS measuring pain intensity, we reported the former trials in cm, and named this outcome NRS for the sake of readability. The mean, standard deviations (SD) and range were used for continuous data with normal distribution. The difference between the active and placebo groups in their change in NRS pain intensity was measured from beginning until the end of the double-blind phase. A negative difference (negative effect size) implies an NRS increase in the placebo group, and thus an analgesic effect of the active treatment. Standardized effect size was calculated as follows: mean difference in pain intensity score between the active and placebo group divided by the SD. The Number Needed to Treat (NNT) is calculated using the MPR50 as follows: 100/(MPR50 active group – MPR50 placebo group).14
Additional Data Analyses
Explorative quantitative analyses were performed to assess the potential association between selected trial-level variables and the absolute effect size. To evaluate the potential influence of the enrichment stringency for the double-blind phase on the effect size, we grouped trials based on the presented inclusion criteria in categories with the following definitions: 1 = ≤4 NRS or ≥1 NRS pain relief at the end of the selection phase, 2 = ≤4.5 NRS or ≥1.5 NRS pain relief, 3 = ≥2NRS pain relief or ≥30% pain relief, and 4 = ≥50% pain relief. We also assessed the relationship between the baseline NRS at the double-blind phase and the effect size, as well as the relationship between the duration of the double-blind phase and the effect size. We hypothesized that 1) a more stringent enrichment, 2) a lower NRS at the double-blind baseline and 3) a longer double-blind treatment duration would be associated with a larger absolute effect size. These hypotheses were explored with meta-analysis and meta-regression using Stata 16.1 (StataCorp, College Station, TX, USA).15 Meta-analysis as well as meta-regression were specified as random effects models with restricted maximum likelihood estimation. Moreover, we hypothesized that stringent enrichment would result in a lower percentage of patients included in the double-blind phase (relative to the total number of patients included in the selection phase), and tested for a relationship between the enrichment stringency and the percentage of included patients in the double-blind phase with a Kruskal–Wallis test and meta-regression.16 We assessed publication bias using a funnel plot, in which the standard error of the effect size estimate is plotted against the effect size estimate. Asymmetry in the plot was determined by visual inspection as well as more formally with Egger’s test.
Results
Trial Selection
The primary search in all databases revealed 610 records of which 56 duplicates were removed. No additional records were found by screening the references in the 8 systemic reviews about EERW chronic pain trials.1,2,8,10,44–47 Twenty-eight trials met the inclusion criteria (see Figure 2).
 |
Figure 2 PRISMA flow diagram. Abbreviations: n, number; PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses. |
Trial Characteristics
The total number of patients in the selection and the double-blind phase were 13662 and 7937, respectively. The mean age of the patients in the trials was 55.1 years (SD: 6.2), ranging between mean 47.3 years (SD: 13.4),23 and 66.8 years (SD: 13.1),38 having included the youngest patient of 18 years,26,39,42 and the oldest of 93 years.37 The mean NRS at selection phase baseline and at double-blind baseline was 6.9 (SD: 0.5) and 3.0 (SD: 0.7), respectively. Eighteen trials used the 11-point NRS or VAS and one trial used a 4-point NRS as the primary endpoint (see Table 1).35 Eight trials used time to LTR with different definitions and one trial used treatment failure as a primary endpoint (see Table 2).43 In the 19 trials using the NRS or VAS as primary endpoint to measure pain intensity, the mean number of patients in the selection and double-blind phase was 522 (SD: 259, range: 37 to 1024) and 316 (SD: 149, range: 26 to 588), respectively. The number of screened patients was reported in only 14 out of 19 trials, with a mean of 997 (SD: 529, range: 51 to 1927). In the 9 trials using LTR or time to LTR as primary endpoint, the mean number of patients in the selection and double-blind phase is 416 (SD: 316, range: 67 to 1051) and 216 (SD: 168, range: 29 to 566), respectively. The number of screened patients was reported in 8 out of 9 trials, with a mean of 651 (SD: 558, range: 125 to 1777). No statistically significant difference in number of patients in both phases was found between trials having time to LTR and NRS or VAS as primary endpoint.
 |
Table 1 Characteristics of EERW Pain Trials with NRS or VAS as Primary Endpoint |
 |
Table 2 Characteristics of EERW Pain Trials with Time to LTR & Treatment Failure as Primary Endpoint |
The duration of the selection phase was in 14 trials up to 4 weeks;20,25,27–31,35,36,38,40,41,43 in 12 trials between 4 and 8 weeks;17–19,21–23,26,32 2 trials did limit this phase.33,34 Six trials used a single-blind design in the selection phase,24,27,30,37,39,41 of which 5 trials examined pregabaline.
Six trials had a relatively short double-blind phase of 4 or 5 weeks;27,30,35,36,41,43 20 trials had a double-blind phase of at least 12 weeks.17–26,28,29,31–34,37–40,42 One trial had a double-blind phase of 26 weeks.42 Only one trial was monocentric.27 The mean number of sites of the other trials (n=27) was 69 (SD: 46, range 9 to 258). Most of the trials were performed in 1 country (n=20). The trials were mainly performed in the United States (n=21), followed by Canada (n=7), South Africa (n=3) and Japan (n=3). A concise overview of data from these included trials is provided in Tables 1 and 2.
Types of Pain
Four trials included more types of pain (see Tables 1 and 2). Chronic low back pain was the most examined type of pain (n=14), followed by painful diabetic neuropathy (n=5), osteoarthritis (n=4), fibromyalgia (n=3), post herpetic neuralgia (n=3), trigeminal neuralgia (n=1), lumbosacral radiculopathy (n=1), complex regional pain syndrome (n=1), and post-operative pain (n=1).
Interventions
Most of the EERW trials examined the effect of opiates (n=19), mainly in extended-release formulations (see Tables 1 and 2), followed by pregabalin (n=6). Other interventions were a synthetic cannabinoid mimicking tetrahydrocannabinol (nabilone), a Nav 1.7 sodium channel blocker (raxatrigine) and a serotonin–norepinephrine reuptake inhibitor (milnacipran). Most of the trials examined an oral formulation (n=23). Other formulations consisted of patches (n=3) and buccal adhesive soluble film (n=2). Only one trial used a fixed dose in the selection and double-blind phase.43 All other trials used the selection phase for titrating medication up to the desired pain reduction to be able to enter the double-blind phase, or until side effects occurred. In most of the trials (n=17) patients took the investigational drug twice daily.17–22,25–27,29,30,32–34,36,40,42 In 6 trials patients took the investigational drug only once daily.23,31,37–39 One trial investigated a buprenorphine patch which had to be changed every week.28
Primary Endpoints
As primary endpoint, the 11-point NRS was used in 16 trials, the 4-point NRS in 1 trial, the VAS in 2 trials, the LTR in 1 trial, and the time to LTR in 8 trials (see tables 1 and 2). The MPR30 and MPR50 were never used as primary endpoints.
Use of Rescue Medication
In only 3 trials rescue medication was not allowed in the selection phase.28,33,35 In the double-blind phase, most of the trials allowed rescue medication, except for 2 trials.35,41 Two trials did not mention the use of rescue medication in both phases.27,43
Inclusion Criteria for the Selection Phase
For inclusion in the selection phase, most trials used the NRS or VAS (hereafter named as NRS) as one of the criteria measuring the intensity of pain. This inclusion criterion had different cut-off values:
- ≥4 last 24 hours,26,36
- daily mean of ≥4 during 1 week,24,27,37,41 with addition of having at least 4 days filled in,30,39,43 or with addition of ≥4 at 2 weekly screening visits,42
- ≥5 last 24 hours,22,32,38 daily mean of ≥5 during 3 days,25,29
- daily mean of ≥5 and ≤9 during 1 week having at least 4 days filled in,19
- daily mean of ≥5 during 2 weeks,23,28 with addition of <10,17 or
- ≥5 not further specified,33 with addition of ≤9,32
Other inclusion criteria were:
- ≥10 points as measured with the 24-point Roland-Morris Disability Questionnaire,17
- moderate to severe chronic low back pain not further specified,18,21
- pain despite the use of NSAIDs,35 the use of (opioid) analgesics,19 the use of milnacipran,40
- the use of between ≥60 and ≤320 mg daily morphine sulphate equivalent (MSE),31 ≤270 mg MSE (≤135 mg oxycodone),21,22 ≤200 mg MSE (≤100 mg oxycodone),23 ≤160 mg MSE,25,29 ≤120 mg MSE,36 ≤40 mg MSE,32 or ≥60mg MSE.34 In some trials, the selection phase was used for conversion from current opioid use to study opioids.19,23,26,31,34,36
In other trials, the screening phase was used to taper opioids to ≤30 mg MSE;18 or to wash-out prohibited analgesics.20,25,28,29,32
Most trials examining effect of opioids included both opioid naive and opioid experienced patients.19–23,29,32,35,36 Six opioid trials examined only opioid naive patients;17,25,28,33,38 three trials examined opioid experienced patients.18,26,31 Three trials evaluating other active pharmaceutical ingredients included experienced patients,37,39,40 three trials naive patients27,30,43 and one trial both naive and experienced patients.24
Selection Phase as Single-Blind
All 6 EERW trials defining the selection phase as single-blind, used this phase for titrating the optimal dose.24,27,30,37,39,41 Only one trial used this single-blind phase to exclude placebo responders taking placebo in the first week and experiencing ≥50% improvement in mean weekly pain score (n=14, 3.7%).41 Two articles stated that patients were permitted in this phase to titrate down to a lower dose.37,39 Two articles stated that patients were informed that they could receive both study medication (pregabalin) and placebo during the course of the trial.27,39
Inclusion Criteria for the Double-Blind Phase
Inclusion criteria for the double-blind phase varied considerably, mainly based on maximal pain score or minimal pain relief as measured with the NRS or VAS at the end of the selection phase compared to baseline pain score. The required pain reduction was measured from a one measurement moment up to the mean of the last 7 days. As a criterion for inclusion for the double-blind Phase 8 trials used ≤4 NRS or ≥1 NRS pain relief,19,21,22,25,29,31,33,34 ≤45 VAS or ≥15 VAS pain relief,38 ≥2 NRS pain relief or ≥30% pain relief17,18,20,23,24,26–28,30,32,36,37,41,43 or ≥50% pain relief.39,40,42 One trial defined ≤1 on a scale from 0 to 3 for several days as an inclusion criterion for the double-blind phase.35 The stringency of enrichment was significantly associated with the percentage of patients included in the double-blind phase (42.0% in the most stringent category 4 versus 64.9% in the least stringent category 1, Kruskal–Wallis P=0.036, meta-regression P<0.001).
Power Calculations of Trials with NRS or VAS as Primary Endpoints
One trial used the 4-point NRS.35 Two out of the other 18 EERW chronic pain trials used the VAS as the primary endpoint.33,34 Two out of the 19 EERW chronic pain trials using the NRS or VAS did not reach statistical significance between groups.22,24 Most of the trials calculated the number of patients needed with a power of 90%; other trials used 80%,27,33 88%,20 and 99%.31 The mean effect size on the NRS used in the power calculation of the 18 trials was 0.89 (SD: 0.47, range: 0.3 to 2). The mean standard deviation used in the power calculation was 1.91 (SD: 0.77, range: 0.6 to 3.0).
Risk of Bias
Thirteen of the 28 included trials were of high quality (low risk of bias), 11 trials of moderate quality (some concerns of bias) and 4 trials of low quality (high risk of bias) (see Figure 3). Details of evaluation are presented in Supplement 2.
 |
Figure 3 Risk of bias summary. Abbreviations: L, chronic low back pain; LO, chronic low back pain and osteoarthritis; NP, neuropathic pain. |
Results of Individual Studies
In Table 3 the absolute and standardized effect sizes with its confidence intervals of the included EERW trials are presented. The absolute effect sizes ranged from −0.21 [95% CI: not reported]41 to −2.31 [95% CI: −3.78 to −0.83]41 on the NRS. The standardized effect size ranged from −0.248 [95% CI: −0.594 to −0.097]36 to −0.694 [95% CI: −0.976 to −0.412].33
 |
Table 3 Effect Sizes and Treatment-Emergent Adverse Events in the Double-Blind Phase |
Treatment-Emergent Adverse Events in the Double-Blind Phase
The percentage of patients experiencing treatment-emergent adverse events (TEAE) in the active and placebo groups is shown in Table 3. The mean percentage patients with TEAE in the active and placebo groups are 57.3% (SD: 14.1) and 50.6% (SD: 10.2), respectively, with a difference between groups of 6.7% (SD: 9.6). The difference in percentage patients with TEAE between active and placebo groups in trials with open label or single-blind selection phase is 7.0% (SD: 10.5) or 5.7% (SD: 6.5%), respectively.
Synthesis of Results
Because the patients, interventions, duration of trials and reported outcome measures varied markedly, we focused on describing the studies and their results on qualitative synthesis rather than meta-analysis.
MPR30 and MPR50 Calculations
To be able to compare trials with different endpoints, we evaluated the MPR30 and MPR50 at the end of the double-blind phase compared to the selection phase baseline (see Table 3). Only 2 out of 8 trials having time to LTR as primary endpoint reported the MPR30 and MPR50,37,39 compared to 14 out of 18 using the 11-point NRS or VAS as primary endpoint. Out of these 14 trials 12 on the MPR3017–20,23,25–27,29,31–33 and 8 on the MPR5018,20,23,25,26,29,31,33 reached statistically significant difference between active and placebo groups. Eleven opioid trials with 3992 patients in the double-blind phase entered the analysis.17–20,23,25,26,29,31–33 In total, the number of patients experiencing MPR30 and MPR50 in the active arms was 1219 and 878 (43.7%) out of 2010 patients, respectively, and in the placebo arms was 791 and 580 (29.3%) out of 1982 patients, respectively. Of the non-opioid trials, we entered 5 trials with 883 patients in the double-blind phase into the analysis.24,27,37,39,43 One trial did not report the MPR30.43 The MPR30 was reached in the active and placebo arms in 375 out of 431 patients and 327 out of 423 patients, respectively. The MPR50 was experienced in 317 out of 446 patients (71.1%) randomized in the active arm, and 248 out of 437 patients (56.8%) randomized into the placebo arm.
Drop-Out Rates of Opioid EERW Trials
Nineteen trials with 9626 patients were entered into the analysis. In total 3664 patients (38.1%) dropped-out in the selection phase (mean: 36.8%, SD: 5.9), Most of these drop-outs were due to adverse events (n=1419, 38.7%, mean: 41.6%, SD: 12.4). The mean drop-out due to lack of efficacy was 15.2% (SD: 9.7). The trial with highest drop-out rate in the selection phase (47.4%) examined oxycodone extended release.20 In the double-blind phase, the mean drop-out rate due to adverse events was for the active and placebo groups 8.4% (SD: 4.3) and 5.6% (SD: 2.6), respectively.
Drop-Out Rates of Non-Opioid EERW Trials
We entered nine trials with 4036 patients into the analysis. In total 2061 patients (51.1%), dropped-out in the selection phase (mean: 49.6%, SD: 12.3, range: 29.7–72.3%). Most of the drop-outs, extracted from 8 trials, were due to lack of efficacy (mean: 27.1%, SD: 24.5).24,27,30,37,39,41–43 The mean drop-out rate due to adverse events was 15.2% (SD: 11.0). The trial with highest drop-out rate in the selection phase (72.3%) examined pregabalin controlled release in fibromyalgia patients.39 In the double-blind phase, the mean drop-out rate due to adverse events was for the active and placebo groups 3.8% (SD: 5.0) and 3.3% (SD: 3.4), respectively.
Imputation Methods for Drop-Outs
Trials having time to LTR as a primary endpoint have no issues with drop-outs. In the trials using NRS or VAS as the primary endpoint, the way the drop-outs are handled can influence the effect size. Many imputation techniques used in EERW trials are presented hereunder.
Six trials handled missing data due to drop-outs in the primary analysis with last observation carried forward (LOCF) method.24,25,27,29,30,35 Two trials imputed drop-outs due to adverse events with screening observation carried forward (SOCF) and LOCF due to other reasons.23,28 Five trials handled drop-outs due to adverse events with SOCF, opioid withdrawal symptoms in placebo group with baseline observation carried forward (BOCF), and all other reasons with LOCF.26,31–34 One trial using the latter imputation techniques did not mention in which group patients should be when they discontinued due to opioid withdrawal symptoms,26 and one trial added an extra imputation method: SOCF when drop-out happened due to withdrawal symptoms in the active group.32 Two trials handled drop-outs as follows: SOCF due to adverse events, BOCF due to opioid withdrawal symptoms, LOCF due to lack of efficacy and multiple imputation approach due to other reasons.17,18 One trial used SOCF for adverse events and lack of efficacy drop-outs, BOCF for opioid withdrawal symptoms in the placebo group drop-outs and multiple imputation techniques for all other drop-out reasons.19 Two trials used multiple imputations for drop-outs,21,22 of which one imputed drop-outs in the active arm due to adverse events based on observed patient data in the placebo group.21 One article did not mention an imputation method in the primary analysis.20
Time to LTR Characteristics and Comparison with NRS
Time to LTR as Primary Endpoint
The primary endpoint time to LTR was in all 8 trials a composite endpoint of at least 2 criterions (see Table 2). Two out of 8 trials investigating the same drug (pregabalin controlled release), used the same relatively simple primary endpoint definition: <30% minimum pain relief compared to selection phase baseline pain score (<MPR30) or discontinuation due to adverse events or lack of efficacy.37,39 Two other trials investigating fentanyl patches had the same, though very complex primary endpoint definition (see Table 2, Arai 2015).38 Six out of 8 trials used <MPR30 as one of the primary endpoint criterions.36,37,39–42 Four out of these 6 trials defined <MPR30 more specifically: mean weekly pain score of <MPR30 at visit week 2, 4, 8 or 12,40 <MPR30 at 2 consecutive visits (week 2, 6, 10, 14, 18, 22, or 26) of which the first visit defined the time to LTR,42 <MPR30 (mean weekly pain score) and ≥1 on NRS,41 and <MPR30 or >3 on NRS for 3 consecutive days.36 Five out of 8 trials used “discontinuation” as one of the primary endpoint criterions without any specifications,41 or with specifications, eg, due to adverse events or lack of efficacy.37–39 Three out of 8 trials used as one of the primary endpoint criterions worsening of pain requiring an alternative treatment,40,42 or leading to dose increase, change or addition of analgesic, including rescue medication.36
Two out of 8 trials did not show statistical significance on the primary endpoint,38,41 compared to 2 out of 18 trials using the NRS or VAS as primary endpoint.22,24 One trial failed to show significance on the primary endpoint time to LTR, though showed a statistically significant difference between the 2 groups measured with the VAS at the end of the trial.38
Another trial having the LTR (thus not time to LTR) as primary endpoint did not show statistically significant difference between groups, though on the NRS as secondary endpoint there was a statistically significant difference between groups.43
Time to LTR as Secondary Endpoint
Twelve out of 19 trials having the NRS or VAS as primary endpoint reported time to LTR as secondary endpoint.19–22,24,26,28,30–34 These trials used different definitions for time to LTR (see Table 1). Three trials reached a statistically significant difference on the NRS or VAS, though did not show statistically significant difference on the endpoint time to LTR.21,28,30 On the other hand, 2 trials did not reach statistically significant difference on the NRS as primary endpoint, though reached statistically significant difference on time to LTR.22,24
Risk of Bias Across Studies
Visual inspection of the funnel plot (see Figure 4) as well as Egger’s test did not provide evidence for publication bias (P=0.694).
 |
Figure 4 Funnel plot of effect sizes. Abbreviation: REML, restricted maximum likelihood. |
Additional Analyses: Correlations Between Absolute Effect Size and Double-Blind Phase Characteristics
A total of 20 trials reporting the effect size as defined above, along with data from which the standard error of the effect size could be calculated, were analyzed using meta-regression.17–21,23,25–30,32–34,36–38,43 No evidence was observed for a relationship between the effect size and either the stringency of enrichment (P=0.18) or the duration of the double-blind phase (P=0.96). However, lower NRS scores at double-blind baseline seem to be associated with larger absolute effect sizes (r=0.40, P=0.06, n=18) due to missing baseline NRS data in 2 trials (Figure 5).23,36
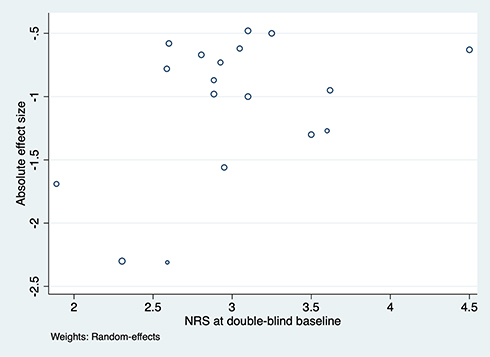 |
Figure 5 Relation between double-blind baseline NRS and absolute effect size. Abbreviation: NRS, 11-point numerical rating scale. |
Discussion
This systematic review on chronic pain EERW trials evaluated trial characteristics, absolute effect sizes and correlations between absolute effect sizes and 3 double-blind phase characteristics, ie, the stringency of inclusion criteria, duration and pain intensity at double-blind baseline. Furthermore, a qualitative comparison is made between time to LTR and NRS.
Most of the trials (n=18) used the 11-point NRS or VAS as primary endpoint. The definition for time to LTR as primary endpoint varied between the trials considerably (n=8), what makes comparison among trials on this endpoint impossible. Since only few EERW pain trials used time to LTR as a primary endpoint, there was no basis for the estimation of the standard deviation in the power calculation to rely on. The NRS in contrast to time to LTR is always defined the same way, which enables comparisons with trials having another design.
Regarding the definition of time to LTR most trials defined time to LTR as <MPR30 compared to screening baseline was one of the primary endpoint criterions. However, <MPR30 had a wide range of timeframe definitions, from reaching <MPR30 at the first timepoint to 2 consecutive visits with 1 month in between, the latter most probably chosen due to daily pain intensity fluctuations in many pain syndromes.48,49 To address these fluctuations, we suggest for the definition time to LTR as one criterion to use <MPR30 for 3 consecutive days counting the first day as time to LTR.
Imputation Methods
The only complicating aspect using the NRS as the primary endpoint in EERW pain trials is how to treat drop-outs. A potential risk of bias in favor of the active group is present when using only the LOCF imputation method in EERW pain trials with the NRS or VAS as primary endpoint. To minimize this potential risk, more conservative methods are used such as SOCF for drop-outs due to adverse events and BOCF in case of opioid withdrawal symptoms in the opioid group. On the other hand, using SOCF when drop-out due to lack of efficacy will inflate the treatment effect, as observed in one trial.19 The many imputation methods used in different EERW pain trials make comparison difficult. Sensitivity analyses testing the robustness of the outcome, may overcome bias due to imputation.
Remarkable Trial Characteristics
Most of trials (n=19) evaluated opioids, most probably to filter out the patients with too much TEAEs in the selection phase. Systematic reviews evaluating EERW and non-EERW opioid trials revealed that non-EERW trials reported more types and higher percentage of TEAEs than EERW trials in the double-blind phase.45,47
All but one trial used the selection phase to titrate the optimal dose. Six trials designed the selection phase in a single-blind fashion, most probably not to unblind patients in the double-blind phase due to the already experienced TEAEs. Surprisingly, the percentage of patients with TEAEs in the active and placebo groups was comparable when stratifying on trials with single-blind and open phase. Patients with TEAEs in the open label phase would most probably continue to experience TEAEs in the double-blind phase when randomized to the active group. On the other hand, patients with TEAEs randomized to the placebo group, might continue to experience side effects in the double-blind phase due to priming. Thus, the single-blind phase is most likely not needed in the selection phase to reduce the risk of bias due to deviations from intended interventions, though might be used to exclude placebo responders in the double-blind phase. Only 1 of the 6 trials with a single-blind selection phase, filtered out placebo responders in the first week (3.7%).41
MPR30 and MPR50
MPR50 and sometimes the MPR30 is used to calculate the NNT. To our knowledge, no chronic pain trial has used the MPR50 as primary endpoint. This might be due to some controversy using a dichotomous outcome which could result in diminished statistical power.50 Also, the broad range in MPR50 difference between the active and placebo groups, reflected in NNT (see Table 3) might be the reason to use MPR50 only as a secondary endpoint. The NNT should be interpreted cautiously due to distortions in calculation as placebo effects approach treatment effects, with the possibility of infinite values; difficulties in estimating the NNT’s confidence interval; and difficulties in interpretation.51 The NNT also requires selecting cutoffs of the original variable for dichotomization, with the NNT often changing depending on the cut-off value.51 Lastly, the NNT calculated from the EERW pain trials is probably too low compared to parallel trials, due to not including the non-responders in the open phase.
Sample Sizes
An earlier systematic review reported in a post hoc analysis of 8 EERW pain trials that the required sample size to achieve 80% or 90% power for time to LTR was the same or lower than the required sample size for the mean pain intensity endpoint.2 However, no statistical comparison of sample sizes between time to LTR and NRS was presented, and our analysis did not show any statistically significant difference in mean sample sizes between trials using time to LTR and NRS as primary endpoint. Furthermore, the argument that time to LTR is relatively immune to imputation problems compared to the NRS could be overruled having a consensus on a conservative imputation method.
Correlations Between Enrichment Stringency, Double-Blind Baseline Score and Absolute Effect Size
No direct relationship was found between stringency of enrichment and the absolute effect size, which is likely explained by the fact that stringency is difficult to quantify when performing an analysis on a group of heterogenic trials. One EERW neuropathic pain trial examined the stringency of responders entering the double-blind phase, stratifying responders in three groups: pain decrease of 1) ≥30%, 2) ≥10% to <30%, and 3) <10%.52 Although the first group numerically had the highest sensitivity, the combined first and second group excluded fewer patients and achieved a balance between assay sensitivity and rate of enrollment.52
We found an apparent relationship between low NRS score at the double-blind baseline and the absolute effect size (r=0.40, P=0.06), suggesting that lower NRS score may be associated with larger absolute effect sizes. While this observation failed to reach statistical significance at the traditional 0.05 threshold, it is important to note that the threshold is arbitrary. We share the view expressed by the renowned statistician Altman that P-values just above or under the threshold should lead to very similar conclusions, not diametrically opposed ones.53 The statistical power of our analysis is limited by the available number of EERW pain trials. While the evidence for a relationship is thus not particularly strong, it supports our hypothesis that aiming at low NRS scores at the end of the enrichment period may increase the likelihood of observing a larger treatment effect. This observation is also plausible from the theoretical perspective. Nonetheless, this hypothesis needs to be confirmed with future EERW pain trials, allowing to test for associations between study-level covariates and effect sizes with higher statistical power.
Numbers Needed to Treat
The presented NNTs in Table 3 are in daily practice higher, than calculated from EERW pain trials, because non-responders or most of the drop-outs due to unbearable adverse events are already filtered out in the selection phase. Nonetheless, NNT is a viable measurement to compare EERW pain trials.
Risk of Bias Due to Deviations of the Intended Interventions
In EERW trials, unblinding in the double-blind phase might occur when TEAEs remain in the selection phase. Unfortunately, none of the included trials have reported the transience of the TEAEs. When patients still experiencing TEAEs when entering the double-blind phase, the chance is there that they will be unblinded by disappearance when randomized in the placebo group. On the other hand, due to priming, TEAEs might continue in the placebo group. Our systematic review reveals that there is little difference between the active and placebo groups in the percentage of patients experiencing TEAEs. Thus, the risk of bias due to deviations of the intended interventions is low in the selected EERW pain trials.
Advantages and Disadvantages of EERW Pain Trials
Advantages and disadvantages of EERW pain trials compared with the common parallel design RCTs are discussed in previous papers and are summarized hereunder.1,2,8,27,54 Advantages of EERW chronic pain designs are that 1) all patients can try the active treatment which resembles more clinical practice, 2) selection phase provides estimates of analgesic response and adverse events for all patients, 3) selection phase can be used for titration mimics clinical practice, 4) minimization of patients receiving an ineffective of poorly tolerated treatment in the double-blind phase, 5) greater ease in discerning pain increase than pain reduction, 6) assay sensitivity of the double-blind phase can be increased and 7) FDA acceptation for registration of drug treatments.
The only systematic review comparing 8 EERW pain trials with 39 non-EERW pain trials examining opioids did not find a statistically significant difference in effect size,45 thus in assay sensitivity. However, the small number of included EERW trials may have lacked sufficient power to detect a statistically significant difference in effect size.
Possible disadvantages of EERW chronic pain designs are that 1) this design is prone to criticism on generalizability of the double-blind data, 2) comparisons with other trial designs are more complex, especially when using time to LTR as primary endpoint. An EERW trial could be used as a confirmatory trial, including patients from prospective parallel placebo-controlled trials.
Limitations
Due to the limited number of EERW pain trials available, the results of the quantitative analysis are only of exploratory nature. For quality assessment, we relied solely on reported data and followed previous systematic review not to contact authors to avoid “response” bias.46 This might have underestimated the quality of the included trials. Because limited number of trials reported the MPR50, a full comparison between trials using the NRS and time to LTR could not be made.
Conclusions and Recommendations
In EERW chronic pain trials many composite definitions are used for time to LTR which makes comparison between trials on this endpoint impossible. We recommend to use the 11-point NRS as primary endpoint in EERW chronic pain trials, using the following predefined conservative imputation methods, based on theoretical considerations, literature and our expert consensus.55
- Impute missing data due to adverse event drop-outs with SOCF.17–19,23,26,28,31–34 This imputation method will reduce the absolute effect size. Since most of the adverse event drop-outs occur in the selection phase and only a small percentage of adverse event drop-outs occur in the double-blind phase, usually equally divided between groups, this imputation method will not have a big impact on the absolute effect size.
- Impute missing data due to withdrawal symptoms drop-outs with BOCF.17–19,26,31–34 Withdrawal symptom drop-outs are mainly expected in the placebo group of opioid trials will reduce the absolute effect size. A taper period sufficient in length in the double-blind phase for the placebo group can prevent this drop-out.
- Impute missing data due to lack of efficacy drop-outs or other reasons with LOCF.26,31–34 These types of drop-outs are mainly expected in the placebo group, experiencing a rise in the NRS. To correct for daily fluctuations, impute these drop-outs with the mean NRS of the last 3 consecutive days.
- Test with sensitivity analyses, so that drop-outs will not inflate the absolute effect size.
We recommend to use time to LTR as a secondary endpoint with a simple uniform definition to be able to compare trials. The proposed definition of time to LTR is <MPR30 for 3 consecutive days counting the first day as time to LTR, or discontinuation. Furthermore, we recommend to present MPR30 and MPR50 so that the NNT can be calculated.
Disclosure
David J Kopsky reports pending patents “Topical phenytoin for use in the treatment of peripheral neuropathic pain (WO2018106107)” and “Topical pharmaceutical composition containing phenytoin and a (co-)analgesic for the treatment of chronic pain (WO2018106108)”. This systematic review is outside the scope of these patent applications. Maybe in the future a topical might be evaluated by an EERW trial. The authors have no other conflicts of interest related to this work.
References
1. Moore RA, Wiffen PJ, Eccleston C, et al. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. Pain. 2015;156(8):1382–1395. doi:10.1097/j.pain.0000000000000088
2. Katz N. Enriched enrollment randomized withdrawal trial designs of analgesics: focus on methodology. Clin J Pain. 2009;25(9):797–807. doi:10.1097/AJP.0b013e3181b12dec
3. Campbell J, King NB. ‘Unsettling circularity’: clinical trial enrichment and the evidentiary politics of chronic pain. BioSocieties. 2017;12:191–216. doi:10.1057/biosoc.2016.7
4. Dworkin RH, Turk DC, Peirce-Sandner S, et al. Research design considerations for confirmatory chronic pain clinical trials: IMMPACT recommendations. Pain. 2010;149(2):177–193. doi:10.1016/j.pain.2010.02.018
5. Dworkin RH, Turk DC, Farrar JT, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain. 2005;113(1–2):9–19. doi:10.1016/j.pain.2004.09.012
6. Turk DC, Dworkin RH, Allen RR, et al. Core outcome domains for chronic pain clinical trials: IMMPACT recommendations. Pain. 2003;106(3):337–345. doi:10.1016/j.pain.2003.08.001
7. Dworkin RH, Turk DC, Wyrwich KW, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9(2):105–121. doi:10.1016/j.jpain.2007.09.005
8. Meske DS, Lawal OD, Elder H, Langberg V, Paillard F, Katz N. Efficacy of opioids versus placebo in chronic pain: a systematic review and meta-analysis of enriched enrollment randomized withdrawal trials. J Pain Res. 2018;11:923–934. doi:10.2147/JPR.S160255
9. Meske DS, Vaughn BJ, Kopecky EA, Katz N. Number of clinical trial study sites impacts observed treatment effect size: an analysis of randomized controlled trials of opioids for chronic pain. J Pain Res. 2019;12:3161–3165. doi:10.2147/JPR.S201751
10. Yamato TP, Maher CG, Saragiotto BT, et al. Comparison of effect sizes between enriched and nonenriched trials of analgesics for chronic musculoskeletal pain: a systematic review. Br J Clin Pharmacol. 2017;83(11):2347–2355. doi:10.1111/bcp.13350
11. Finnerup NB, Haroutounian S, Baron R, et al. Neuropathic pain clinical trials: factors associated with decreases in estimated drug efficacy. Pain. 2018;159(11):2339–2346. doi:10.1097/j.pain.0000000000001340
12. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535. doi:10.1136/bmj.b2535
13. Higgins JPT, Savović J, Page MJ, et al. Chapter 8: assessing risk of bias in a randomized trial. In: Higgins JPT, Thomas J, Chandler J, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 6.0. Cochrane; 2019.
14. Cook RJ, Sackett DL. The number needed to treat: a clinically useful measure of treatment effect. BMJ. 1995;310(6977):452–454. doi:10.1136/bmj.310.6977.452
15. Schober P, Vetter TR. Meta-analysis in clinical research. Anesth Analg. 2020;131(4):1090–1091. doi:10.1213/ANE.0000000000005001
16. Schober P, Vetter TR. Nonparametric statistical methods in medical research. Anesth Analg. 2020;131(6):1862–1863. doi:10.1213/ANE.0000000000005101
17. Rauck RL, Potts J, Xiang Q, Tzanis E, Finn A. Efficacy and tolerability of buccal buprenorphine in opioid-naive patients with moderate to severe chronic low back pain. Postgrad Med. 2016;128(1):1–11. doi:10.1080/00325481.2016.1128307
18. Gimbel J, Spierings EL, Katz N, Xiang Q, Tzanis E, Finn A. Efficacy and tolerability of buccal buprenorphine in opioid-experienced patients with moderate to severe chronic low back pain: results of a Phase 3, enriched enrollment, randomized withdrawal study. Pain. 2016;157(11):2517–2526. doi:10.1097/j.pain.0000000000000670
19. Rauck RL, Hale ME, Bass A, et al. A randomized double-blind, placebo-controlled efficacy and safety study of ALO-02 (extended-release oxycodone surrounding sequestered naltrexone) for moderate-to-severe chronic low back pain treatment. Pain. 2015;156(9):1660–1669. doi:10.1097/j.pain.0000000000000230
20. Katz N, Kopecky EA, O’Connor M, Brown RH, Fleming AB. A phase 3, multicenter, randomized, double-blind, placebo-controlled, safety, tolerability, and efficacy study of Xtampza ER in patients with moderate-to-severe chronic low back pain. Pain. 2015;156(12):2458–2467. doi:10.1097/j.pain.0000000000000315
21. Hale ME, Zimmerman TR, Eyal E, Malamut R. Efficacy and safety of a hydrocodone extended-release tablet formulated with abuse-deterrence technology in patients with moderate-to-severe chronic low back pain. J Opioid Manag. 2015;11(6):507–518. doi:10.5055/jom.2015.0304
22. Hale ME, Laudadio C, Yang R, Narayana A, Malamut R. Efficacy and tolerability of a hydrocodone extended-release tablet formulated with abuse-deterrence technology for the treatment of moderate-to-severe chronic pain in patients with osteoarthritis or low back pain. J Pain Res. 2015;8:623–636. doi:10.2147/JPR.S83930
23. Wen W, Sitar S, Lynch SY, He E, Ripa SR. A multicenter, randomized, double-blind, placebo-controlled trial to assess the efficacy and safety of single-entity, once-daily hydrocodone tablets in patients with uncontrolled moderate to severe chronic low back pain. Expert Opin Pharmacother. 2015;16(11):1593–1606. doi:10.1517/14656566.2015.1060221
24. Raskin P, Huffman C, Toth C, et al. Pregabalin in patients with inadequately treated painful diabetic peripheral neuropathy: a randomized withdrawal trial. Clin J Pain. 2014;30(5):379–390. doi:10.1097/AJP.0b013e31829ea1a1
25. Vinik AI, Shapiro DY, Rauschkolb C, et al. A randomized withdrawal, placebo-controlled study evaluating the efficacy and tolerability of tapentadol extended release in patients with chronic painful diabetic peripheral neuropathy. Diabetes Care. 2014;37(8):2302–2309. doi:10.2337/dc13-2291
26. Rauck RL, Nalamachu S, Wild JE, et al. Single-entity hydrocodone extended-release capsules in opioid-tolerant subjects with moderate-to-severe chronic low back pain: a randomized double-blind, placebo-controlled study. Pain Med. 2014;15(6):975–985. doi:10.1111/pme.12377
27. Toth C, Mawani S, Brady S, et al. An enriched-enrolment, randomized withdrawal, flexible-dose, double-blind, placebo-controlled, parallel assignment efficacy study of nabilone as adjuvant in the treatment of diabetic peripheral neuropathic pain. Pain. 2012;153(10):2073–2082. doi:10.1016/j.pain.2012.06.024
28. Steiner DJ, Sitar S, Wen W, et al. Efficacy and safety of the seven-day buprenorphine transdermal system in opioid-naïve patients with moderate to severe chronic low back pain: an enriched, randomized, double-blind, placebo-controlled study. J Pain Symptom Manage. 2011;42(6):903–917. doi:10.1016/j.jpainsymman.2011.04.006
29. Schwartz S, Etropolski M, Shapiro DY, et al. Safety and efficacy of tapentadol ER in patients with painful diabetic peripheral neuropathy: results of a randomized-withdrawal, placebo-controlled trial. Curr Med Res Opin. 2011;27(1):151–162. doi:10.1185/03007995.2010.537589
30. Gilron I, Wajsbrot D, Therrien F, Lemay J. Pregabalin for peripheral neuropathic pain: a multicenter, enriched enrollment randomized withdrawal placebo-controlled trial. Clin J Pain. 2011;27(3):185–193. doi:10.1097/AJP.0b013e3181fe13f6
31. Hale M, Khan A, Kutch M, Li LS. Once-daily OROS hydromorphone ER compared with placebo in opioid-tolerant patients with chronic low back pain. Curr Med Res Opin. 2010;26(6):1505–1518. doi:10.1185/03007995.2010.484723
32. Katz N, Hale M, Morris D, Stauffer J. Morphine sulfate and naltrexone hydrochloride extended release capsules in patients with chronic osteoarthritis pain. Postgrad Med. 2010;122(4):112–128. doi:10.3810/pgm.2010.07.2179
33. Katz N, Rauck R, Ahdieh H, et al. A 12-week, randomized, placebo-controlled trial assessing the safety and efficacy of oxymorphone extended release for opioid-naive patients with chronic low back pain. Curr Med Res Opin. 2007;23(1):117–128. doi:10.1185/030079906X162692
34. Hale ME, Ahdieh H, Ma T, Rauck R. Efficacy and safety of OPANA ER (oxymorphone extended release) for relief of moderate to severe chronic low back pain in opioid-experienced patients: a 12-week, randomized, double-blind, placebo-controlled study. J Pain. 2007;8(2):175–184. doi:10.1016/j.jpain.2006.09.011
35. Caldwell JR, Hale ME, Boyd RE, et al. Treatment of osteoarthritis pain with controlled release oxycodone or fixed combination oxycodone plus Acetaminophen added to nonsteroidal antiinflammatory drugs: a double blind, randomized, multicenter, placebo controlled trial. J Rheumatol. 1999;26(4):862–869.
36. Kawamata M, Iseki M, Kawakami M, et al. Efficacy and safety of controlled-release oxycodone for the management of moderate-to-severe chronic low back pain in Japan: results of an enriched enrollment randomized withdrawal study followed by an open-label extension study. J Pain Res. 2019;12:363–375. doi:10.2147/JPR.S179110
37. Huffman CL, Goldenberg JN, Weintraub J, et al. Efficacy and safety of once-daily controlled-release pregabalin for the treatment of patients with postherpetic neuralgia: a double-blind, enriched enrollment randomized withdrawal, placebo-controlled trial. Clin J Pain. 2017;33(7):569–578. doi:10.1097/AJP.0000000000000445
38. Arai T, Kashimoto Y, Ukyo Y, Tominaga Y, Imanaka K. Two placebo-controlled, randomized withdrawal studies to evaluate the fentanyl 1 day patch in opioid-naive patients with chronic pain. Curr Med Res Opin. 2015;31(12):2207–2218. doi:10.1185/03007995.2015.1092127
39. Arnold LM, Arsenault P, Huffman C, et al. Once daily controlled-release pregabalin in the treatment of patients with fibromyalgia: a Phase III, double-blind, randomized withdrawal, placebo-controlled study. Curr Med Res Opin. 2014;30(10):2069–2083. doi:10.1185/03007995.2014.928275
40. Clauw DJ, Mease PJ, Palmer RH, Trugman JM, Wang Y. Continuing efficacy of milnacipran following long-term treatment in fibromyalgia: a randomized trial. Arthritis Res Ther. 2013;15(4):R88. doi:10.1186/ar4268
41. Baron R, Freynhagen R, Tölle TR, et al. The efficacy and safety of pregabalin in the treatment of neuropathic pain associated with chronic lumbosacral radiculopathy. Pain. 2010;150(3):420–427. doi:10.1016/j.pain.2010.04.013
42. Crofford LJ, Mease PJ, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain. 2008;136(3):419–431. doi:10.1016/j.pain.2008.02.027
43. Zakrzewska JM, Palmer J, Morisset V, et al. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 2017;16(4):291–300. doi:10.1016/S1474-4422(17)30005-4
44. McQuay HJ, Derry S, Moore RA, Poulain P, Legout V. Enriched enrolment with randomised withdrawal (EERW): time for a new look at clinical trial design in chronic pain. Pain. 2008;135(3):217–220. doi:10.1016/j.pain.2008.01.014
45. Furlan A, Chaparro LE, Irvin E, Mailis-Gagnon A. A comparison between enriched and nonenriched enrollment randomized withdrawal trials of opioids for chronic noncancer pain. Pain Res Manag. 2011;16(5):337–351. doi:10.1155/2011/465281
46. Sommer C, Klose P, Welsch P, Petzke F, Häuser W. Opioids for chronic non-cancer neuropathic pain. An updated systematic review and meta-analysis of efficacy, tolerability and safety in randomized placebo-controlled studies of at least 4 weeks duration. Eur J Pain. 2020;24(1):3–18. doi:10.1002/ejp.1494
47. Petzke F, Klose P, Welsch P, Sommer C, Häuser W. Opioids for chronic low back pain: an updated systematic review and meta-analysis of efficacy, tolerability and safety in randomized placebo-controlled studies of at least 4 weeks of double-blind duration. Eur J Pain. 2020;24(3):497–517. doi:10.1002/ejp.1519
48. Schneider S, Junghaenel DU, Keefe FJ, Schwartz JE, Stone AA, Broderick JE. Individual differences in the day-to-day variability of pain, fatigue, and well-being in patients with rheumatic disease: associations with psychological variables. Pain. 2012;153(4):813–822. doi:10.1016/j.pain.2012.01.001
49. Bartley EJ, Robinson ME, Staud R. Pain and fatigue variability patterns distinguish subgroups of fibromyalgia patients. J Pain. 2018;19(4):372–381. doi:10.1016/j.jpain.2017.11.014
50. Gilron I, Carr DB, Desjardins PJ, Kehlet H. Current methods and challenges for acute pain clinical trials. Pain Rep. 2019;4(3):e647. doi:10.1097/PR9.0000000000000647
51. Katz N, Paillard FC, Van Inwegen R. A review of the use of the number needed to treat to evaluate the efficacy of analgesics. J Pain. 2015;16(2):116–123. doi:10.1016/j.jpain.2014.08.005
52. Hewitt DJ, Ho TW, Galer B, et al. Impact of responder definition on the enriched enrollment randomized withdrawal trial design for establishing proof of concept in neuropathic pain. Pain. 2011;152(3):514–521. doi:10.1016/j.pain.2010.10.050
53. Altman DG. Practical Statistics for Medical Research. Boca Raton, FL: Chapman & Hall/CRC; 1999.
54. Gilron I. Methodological issues associated with clinical trials in neuropathic pain. Expert Rev Clin Pharmacol. 2016;9(11):1399–1402. doi:10.1080/17512433.2016.1240029
55. National Research Council (US) Panel on Handling Missing Data in Clinical Trials. The prevention and treatment of missing data in clinical trials. Washington (DC): National Academies Press (US); 2010.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.