")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: a review of the evidence to date
Authors Tuttolomondo A, Pecoraro R, Pinto A
Received 13 May 2014
Accepted for publication 8 August 2014
Published 7 November 2014 Volume 2014:8 Pages 2221—2239
DOI https://doi.org/10.2147/DDDT.S67655
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Antonino Tuttolomondo, Rosaria Pecoraro, Antonio Pinto
Biomedical Department of Internal and Specialistic Medicine, University of Palermo, Palermo, Italy
Abstract: The brain is very actively involved in immune-inflammatory processes, and the response to several trigger factors such as trauma, hemorrhage, or ischemia causes the release of active inflammatory substances such as cytokines, which are the basis of second-level damage. During brain ischemia and after brain trauma, the intrinsic inflammatory mechanisms of the brain, as well as those of the blood, are mediated by leukocytes that communicate with each other through cytokines. A neuroinflammatory cascade has been reported to be activated after a traumatic brain injury (TBI) and this cascade is due to the release of pro- and anti-inflammatory cytokines and chemokines. Microglia are the first sources of this inflammatory cascade in the brain setting. Also in an ischemic stroke setting, an important mediator of this inflammatory reaction is tumor necrosis factor (TNF)-α, which seems to be involved in every phase of stroke-related neuronal damage such as inflammatory and prothrombotic events. TNF-α has been shown to have an important role within the central nervous system; its properties include activation of microglia and astrocytes, influence on blood–brain barrier permeability, and influences on glutamatergic transmission and synaptic plasticity. TNF-α increases the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor density on the cell surface and simultaneously decreases expression of γ-aminobutyric acid receptor cells, and these effects are related to a direct neurotoxic effect. Several endogenous mechanisms regulate TNF-α activity during inflammatory responses. Endogenous inhibitors of TNF include prostaglandins, cyclic adenosine monophosphate, and glucocorticoids. Etanercept, a biologic TNF antagonist, has a reported effect of decreasing microglia activation in experimental models, and it has been used therapeutically in animal models of ischemic and traumatic neuronal damage. In some studies using animal models, researchers have reported a limitation of TBI-induced cerebral ischemia due to etanercept action, amelioration of brain contusion signs, as well as motor and cognitive dysfunction. On this basis, it appears that etanercept may improve outcomes of TBI by penetrating into the cerebrospinal fluid in rats, although further studies in humans are needed to confirm these interesting and suggestive experimental findings.
Keywords: tumor necrosis factor inhibitors, brain injury, stroke, TBI, traumatic brain injury
Introduction
Traumatic brain injury (TBI) and ischemic stroke are pathological events known to be significantly associated with a high rate of morbidity and mortality. These complex disorders are also characterized by two levels of damage that encompass primary and secondary injury pathological events.1
It is possible to schematize primary injury in these two clinical settings as follows:
- In TBI: damage due to mechanical factors synchronous with the time of trauma to neurons, axons, glia, and blood vessels, which can be considered a result of shearing, tearing, or stretching.
- In ischemic stroke: ischemic damage that occurs after a variable time interval following the initial ischemic event. Ischemic damage is also due to a wide range of delayed neurochemical or metabolic alterations at a cellular level.
Secondary injury mechanisms in both these clinical conditions include processes such as alteration of ionic homeostasis,2 increase of neurotransmitter levels (eg, glutamate-linked excitotoxicity mechanisms),3 neuronal apoptosis,4 lipid degradation,5 and immune-inflammatory activation.6 These neurochemical events involve inflammatory mediators, such as prostaglandins, oxidative metabolites, and inflammatory cytokines. This inflammatory activation causes a complex cascade of molecular events such as lipid peroxidation, blood–brain barrier (BBB) disruption, and cerebral edema.
Cerebral ischemia and TBI induces a cascade of inflammatory reactions that encompass genomic events as well as molecular and cellular alterations that occur in the central nervous system (CNS). In this series of inflammatory alterations, cytokines represent a central mediator of a stroke-linked immune-inflammatory cascade that leads to neuronal damage, inflammatory brain reactions linked to brain infarct size progression, and complex pathological events following a brain trauma.7
Recently, it became clear that inflammatory mediators such as cytokine release mediated by T cells and mononuclear/macrophage cells regulate many functions of some CNS cells such as macroglial and microglial cells. In particular, glial cells when activated can produce immunoregulatory factors that influence other cellular subsets such as mononuclear cells and glial cells.2
Cytokines such as tumor necrosis factor (TNF)-α, interferons, growth factors, colony-stimulating factors, and chemokines are pleiotropic proteins that express a wide range of processes, either physiological or pathological.
TNF-α plays an important role during cerebral ischemia: it exerts a chemotactic action toward leukocytes and induces the production of adhesion molecules in other cellular subtypes, such as several leukocyte subsets, endothelial cells, and other target cells, thus increasing inflammation events in the cerebral setting.8 Furthermore, TNF-α has a clear effect on enhancing thrombogenesis by increasing plasminogen-activating inhibitor-1 tissue factor and platelet-activating factor levels, and by inhibition of tissue plasminogen activator activity.8
On this basis, it is possible to suggest that, far from being an immunological sanctuary, brain is very actively involved in immune-inflammatory processes as a result of several trigger factors such as trauma and ischemia, and that these injuries to the brain are expressed by the release of mediators such as cytokines with their leading role in causing the so-called secondary neuronal damage.9
After focal brain ischemia, microglial cells gain an activated phenotype and they rush into the penumbra area and exacerbate and maintain neuronal injury at this site by several pathways of pathogenic events and mediators, such as poly(ADPribose) polymerase-1 activation and multiple matrix metalloproteinase (MMP) activation.4
In particular, MMP can influence the integrity of the BBB through damage of the endothelial function of this barrier, thus impairing the whole functional integrity of the so-called neurovascular unit. These pathological events may open a door for the entry of peripheral inflammatory cells into the brain, thus exposing neuronal cells to the attack of peripheral immune-competent cells.
After cerebral ischemia, it is possible to observe the induction of a series of inflammatory events such as infiltration of neutrophils and monocytes; activation of resident inflammatory cells such as microglia, astrocytes, and endothelial cells;8 and involvement of T cells and natural killer cells. All these inflammatory events are also initiated after a brain trauma by TNF-α, which is a pleiotropic protein involved in the regulation of numerous physiological and pathological processes, such as inflammation, cancer, autoimmunity, and infection.
Neuroinflammation after brain trauma
The immune-inflammatory activation after TBI is mediated by inflammatory mechanisms. Among the resident CNS cells, microglia represent a probable primary source of these inflammatory mediators in the CNS.2 Genetic studies in experimental models of TBI have shown that inflammatory genes are upregulated in the acute phase both after TBI and stroke.2–4
Additional studies analyzed genes of microglia inflammatory activation and their temporal-spatial localization after TBI, showing that chosen markers of microglial activation such as CD68, MHCII, or stress responses such as p22phox, heme oxygenase, or a gene related to chemokine activation such as CXCL10, CXCL6, are highly activated at a transcriptional level after TBI.5,6
TNF-α and other cytokines such as interleukin (IL)-1β and IL-6 have pleiotropic actions in the brain; for example, they regulate and control both systemic host responses to disease and local alterations on vessels, neurons, and endothelial cells caused by CNS inflammation.8,9
TNF-α, IL-1, and IL-6 are the main cytokines that have been reported as expressed by cultured brain cells in relation to various stimuli (Table 1). Astrocytes produce TNF-α in response to various stimuli such as lipopolysaccharide (LPS) and interferon-γ.10 Cultured cells of astrocytes also express high levels of other cytokines such as IL-6, IL-8, and granulocyte colony stimulating factor, in response to stimulation by IL-l or TNF-α.11
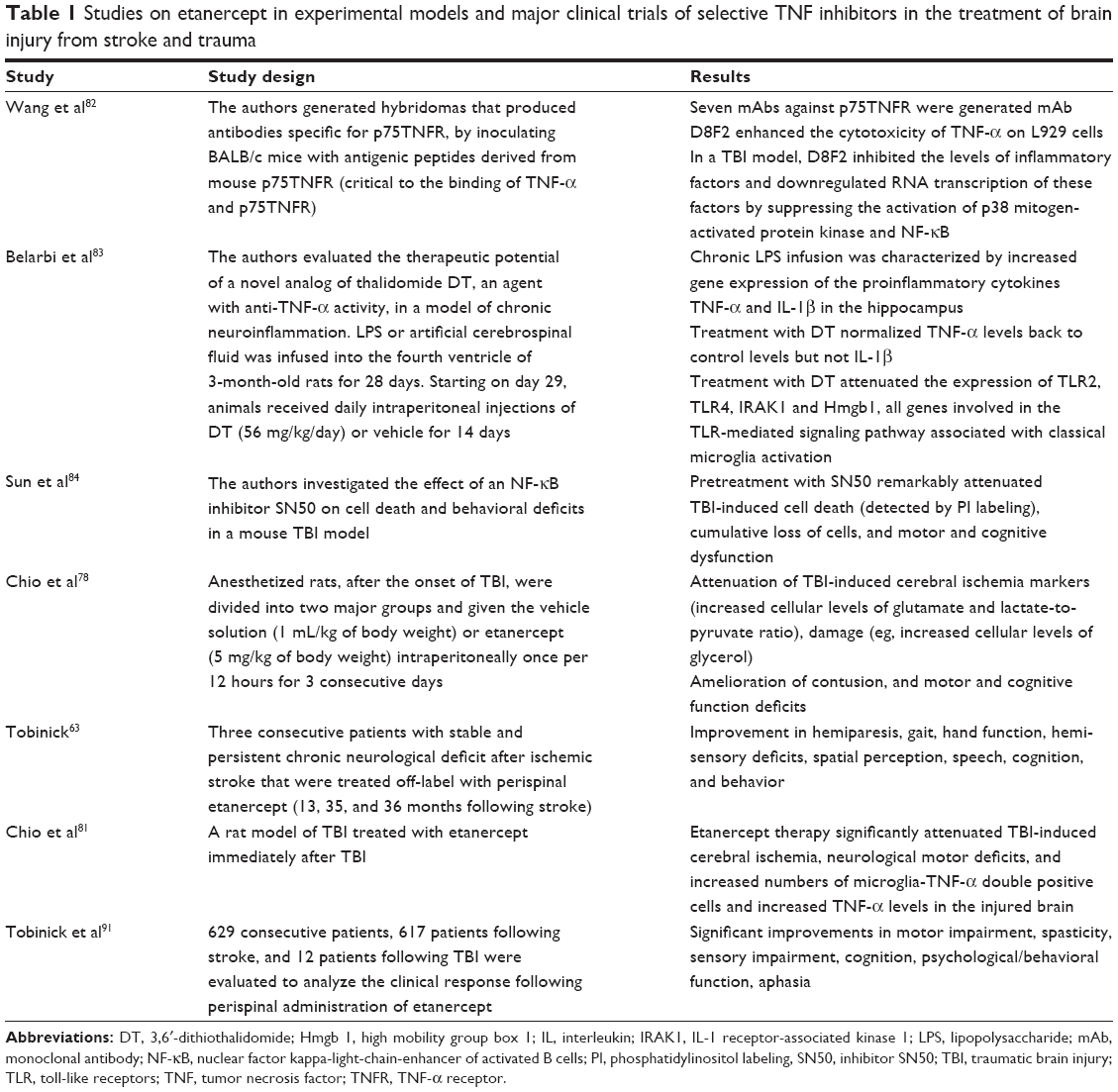 | Table 1 Studies on etanercept in experimental models and major clinical trials of selective TNF inhibitors in the treatment of brain injury from stroke and trauma |
Activated microglia regulate astrocyte proliferation, and this cell regulation is also expressed by means of the production of TNF-α.12 A complex cytokine network creates communication among stimulated brain cells. These cell relationships mediated by immune-inflammatory mediators represent the basis of an influence of each cell on the functional status of other inflammatory cells involved in this inflammatory network in the CNS setting.13
In this way, TNF-α achieves chemotaxis control action effects as a result of increased production of other chemokines. In addition, TNF-α may regulate other proteins called adhesion molecules, such as E-selectin, intercellular adhesion molecule (ICAM), vascular cell adhesion molecule (VCAM), and a monocyte adhesion protein.14,15
A higher degree of expression of adhesion molecules in astrocytes enhances a migration process of inflammatory cells, and it represents one of the possible pathogenic bases of inflammatory-mediated ischemic neuronal damage. ICAM-1 and VCAM-1 have been reported as highly expressed in cultured microglial cells after treatment with TNF-α.16 A high degree of expression of ICAM-1, VCAM-1, and E-selectin has also been shown in cultured endothelial cells from brain vessels after exposure to inflammatory stimuli such as TNF-α17 (Figure 1).
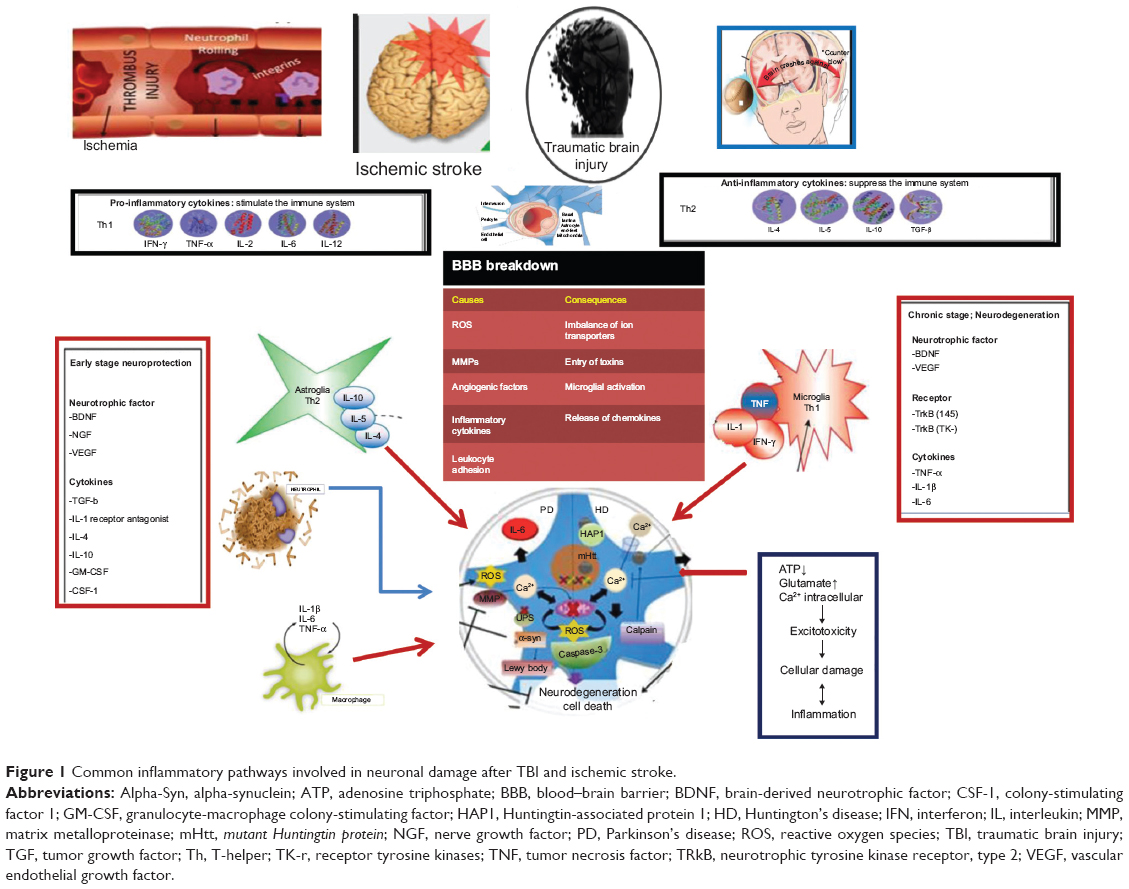 | Figure 1 Common inflammatory pathways involved in neuronal damage after TBI and ischemic stroke. |
Neuroinflammation after brain ischemia
An induction of messenger RNAs (mRNAs) of several cytokines such as IL-1β, IL-18, IL-6, and TNF-α after ischemic damage has been reported.18 High levels of TNF-α mRNA has been shown 1 hour after middle cerebral artery occlusion (MCAO), with a peak level at 12 hours, and persistently high levels for 5 days after experimental brain ischemia caused by arterial occlusion.19,20
A good example of neuroinflammation-mediated neuronal damage has been reported in heat stroke. In a study by Lin et al heat stroke, a reproducible model of brain injury, has been caused in rats exposed to a temperature of 42°C.22 This study reported that extracellular levels of some neurotransmitters and mediators such as dopamine, serotonin, or norepinephrine were upregulated in some brain areas such as the hypothalamus and corpus striatum in the course of the heat-stroke-induced cerebral ischemia and neuronal damage. The same authors showed high serum and brain levels of TNF-α induced by heat stroke and cerebral ischemic damage. They also demonstrated that depletion of brain dopamine or serotonin after an intracerebral injection of 6-hydroxydopamine or 5,7-dihydroxytryptamine ameliorated heat-stroke-induced cerebral ischemia. These findings indicate how increased brain levels of either dopamine or serotonin may represent important mediators for heat-stroke-induced cerebral ischemia and neuronal damage in experimental animal models.
Nevertheless, it is not easy to find a clear answer to the question of a possible source of inflammatory cytokines involved in ischemia-related neuronal damage. To answer this difficult question, some researchers such as Tarkowski et al23 evaluated serum and cerebrospinal fluid (CSF) levels of IL-lβ and IL-6 in patients with ischemic stroke. These authors showed higher levels of these cytokines in the CSF than in the serum. These findings suggest the possible existence of direct intracerebral production of these inflammatory cytokines strictly involved in the pathogenesis of ischemic neuronal damage.
Other authors such as Mathiesen et al reported a nonparallel increase of levels of IL-6 in the CSF and in the serum in subjects with subarachnoid hemorrhage.24 They also reported that increased levels of cytokines such as TNF-α were higher at day 6 and that these higher levels were more likely to be linked to the development of delayed ischemic neurological deficits.
Common pathways after TBI and ischemia
Higher degree of inflammatory activation in terms of TNF-α plasma levels has been observed in experimental models of brain ischemia as well as in patients with acute stroke23 and in patients with large cerebral infarct volume and poor stroke outcome.24
A neuroinflammatory cascade has also been described after a head trauma. A first event of this inflammatory cascade is microglia activation (MA) after either a traumatic or ischemic injury. After microglia involvement, it is possible to observe an increase in inflammatory cytokines such as IL-1β and TNF-α in both humans and rodents.25–28
TNF-α enhances the toxic effects of IL-1β in a synergistic way, thus both these inflammatory cytokines seem to regulate and induce neuroinflammation and brain damage after TBI29 and ischemic stroke.27,28 Nevertheless, the role of TNF-α in TBI-related neuronal damage remains less clear, with different findings in relation to studies conducted by evaluating acute and delayed stages of neuronal damage after TBI.30
Three different compounds have been reported as neuroprotective agents by means of a blocking action toward TNF-α. These substances are dexanabinol (HU-211), the TNF-binding protein, and pentoxifylline. Some researchers have reported that these substances are effective in offering a significant amelioration in neurological outcomes, probably also acting on BBB disruption and brain edema pathogenesis.31,32
It is possible to block inflammatory effects of cytokines and chemokines by several methods, including neutralizing antibodies, soluble cytokine/chemokine receptors, inhibitors of cytokine/chemokine synthesis, and anti-inflammatory cytokines.
Role of TNF-α in brain damage after ischemia
TNF-α levels progressively increase in ischemic brain damage after a TBI.33 High levels of TNF-α are strictly linked to BBB disruption34 and neuronal cell death after TBI and ischemic damage. TNF-α has been reported as a mediator in the mechanism of neuronal death in experimental models of neuronal damage caused by oxygen-glucose deprivation (OGD model) in vitro and in animal models of ischemic brain damage by means of brain artery occlusion or ligation.35 Some apoptotic pathways of neuron death showing peripheral aggregates have also been reported as linked to increased levels of TNF-α.36
A study37 explored the pathophysiological significance of brain TNF-α in the ischemic brain. The authors evaluated the effects of lateral cerebroventricular administration of exogenous TNF-α and tested agents that block the effects of TNF-α on focal stroke by administering TNF-α (2.5 or 25 pmol) intracerebroventricularly to spontaneously hypertensive rats 24 hours before MCAO. Animals were examined 24 hours later for neurological deficits and ischemic hemisphere necrosis and swelling. A neutralizing anti-TNF-α monoclonal antibody (mAb) (monoclonal hamster anti-murine TNF-α) was also administered in some of experimental animals (mAb intracerebroventricularly 30 minutes before exogenous TNF-α). They evaluated the direct neurotoxic effects of TNF-α in cultured rat cerebellar granule cells exposed to TNF-α by means of the evaluation of neurotransmitter release, glutamate toxicity, and oxygen radical toxicity, reporting that TNF-α increased after hemispheric infarct induced by MCAO. The toxic effects of TNF-α on brain, evaluated by means observation of swelling and neurological deficit after exogenous TNF-α, have been reported as reversed by preinjection of 60 pmol mAb.37 Blocking endogenous TNF-α also significantly reduced focal ischemic brain injury and reduced infarct size compared with control. These experimental experiences demonstrate that exogenous TNF-α exacerbates focal ischemic injury and that blocking endogenous TNF-α is neuroprotective. The specificity of the action(s) of TNF-α was demonstrated by antagonism of its effects with specific anti-TNF-α tools (ie, mAb and soluble TNF-α receptor 1 [sTNFR1]). TNF-α toxicity does not appear to be due to a direct effect on neurons or modulation of neuronal sensitivity to glutamate or oxygen radicals but apparently is mediated through nonneuronal cells. These data suggest that inhibiting TNF-α may represent a novel pharmacological strategy to treat ischemic stroke.
Some authors reported that pretreatment with TNF-α in BALB/C mice prior to an experimental stroke induced by MCAO is associated with a significantly lower extension of brain infarct size,38 and to explain these findings the authors suggested a neuroprotective mechanism of ischemic preconditioning. Some CNS resident cells such as macrophages, astrocytes, and microglia synthesize TNF-α, which has inflammatory action in the CNS during the acute phase, but also immunosuppressive effects during the chronic phase.39
Several reports show that TNF-α is involved in several crucial pathways within the CNS (Figure 2).40–47 These pathways induce the activation of microglia and astrocytes, act on BBB integrity, enhance glutamatergic transmission, and regulate synaptic plasticity. Furthermore, a mechanism of scaling of excitatory synapses has been reported as linked to TNF-α promotion of amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and downregulation of γ-aminobutyric acid (GABA) receptors on the cell surface. These findings could suggest a possible role of TNF-α in the regulation of synaptic transmission at excitatory synapses, thus enhancing excitatory synaptic transmission and lowering inhibitory synaptic transmission.48–51
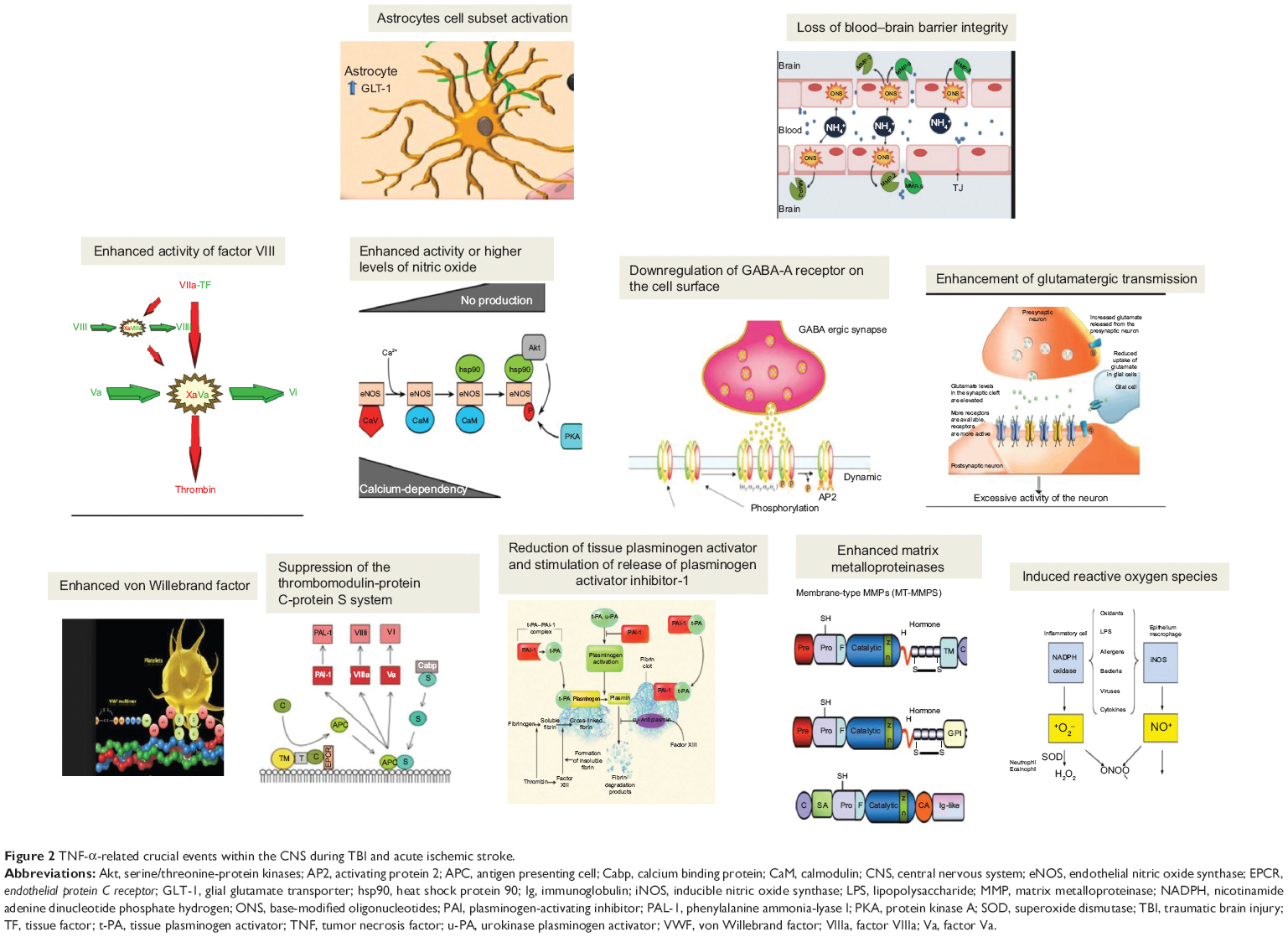 | Figure 2 TNF-α-related crucial events within the CNS during TBI and acute ischemic stroke. |
Some studies report that treatment with TNF-α receptor (TNFR) or anti-TNF-α antibody is able to reduce tetrodotoxin-induced promotion in the expression of AMPA receptors and increase spontaneous miniature excitatory synaptic current (mEPSC) amplitude, whereas they have been reported to decrease miniature inhibitory synaptic current amplitude.48,49
These mechanisms of regulation of synaptic scaling have also been demonstrated in some experimental cell models such as hippocampus cultures in TNF-α-deficient mice in which the authors did not report an increased mEPSC amplitude compared to wild-type cultures, thus underlying a TNF-α dependence on higher mEPSC amplitudes.48,49
Some research studies also reported how TNF-α is involved in the stimulation of tissue factor and adhesion molecule expression and in the induction of enhanced activity or higher levels of nitric oxide, factor VIII/von Willebrand factor, and platelet activating factor. In a procoagulant setting, TNF-α is also linked to endothelin, suppression of the thrombomodulin-protein C-protein S system, reduction of tissue plasminogen activator, and stimulation of release of plasminogen activator inhibitor-1. It also enhances MMP levels, thus actively acting on vascular permeability, which increases in response to TNF-α action. TNF-α can also induce reactive oxygen species synthesis on endothelial cells by means of an upregulation of some regulatory enzymes such as xanthine oxidase, cyclooxygenase, and nicotinamide adenine dinucleotide phosphate hydrogen oxidase, which are key enzymes in reactive oxygen species production pathways.50,51
All these cumulative effects increase the likelihood of local, inflammatory, and thrombotic micro events, thus inducing humeral and molecular backgrounds that predispose mechanisms of stroke initiation and induce the progression of ischemic neuronal damage after an ischemic stroke. TNF-α has an important role in the pathogenesis of atherosclerotic stroke through its role in promoting atherosclerotic plaque development and mechanisms that make these plaques unstable.52 TNF-α has a role in local endothelial prothrombotic and proinflammatory micro events.
Chemotaxis of lymphocytes and monocytes to atherosclerotic plaque has also been reported as directly stimulated by TNF-α, which enhances endothelial-cell adhesion molecule expression and elaboration of chemokines involved in chemotaxis mechanisms. TNF-α is involved in the accumulation of lipid-laden foam cells and smooth-muscle-cell proliferation, which are all key events in atherosclerosis development. One study on cultured macrophages from carotid plaques have reported the atherogenic properties of TNF-α on cultured macrophages from carotid plaques. These studies reported an increased degree of TNF-α, both serum and cell-linked, compared to the levels expressed by analyzing circulating control monocytes.51
The important role of TNF-α in ischemic stroke promotion has been demonstrated in reports of TNF-α activation in experimental brain ischemia models at both the mRNA and protein levels.53 Existing data on the time course of this cytokine after an acute ischemic stroke offer important information about the role of TNF-α in acute ischemic stroke in humans. A very important study by Sairanen et al evaluated the brain tissue distribution by analyzing its temporal kinetics and showed TNF-α brain tissue distribution pathways after an ischemic stroke (Figure 3).54 The study showed a typical neuronal TNF-α immunoreactivity pattern of cytoplasmic punctate staining with a swollen filament-like structure on the first day after ischemic stroke. On the second day, TNF-α immunoreactivity was reported only in a small number of neurons of the infarct core and in contralateral locations in dead subjects. Inflammatory cells showing TNF-α immunoreactivity were also observed in the infarct core on the second day, whereas a bilateral pattern of neuronal TNF-α immunoreactivity was reported after the second day.
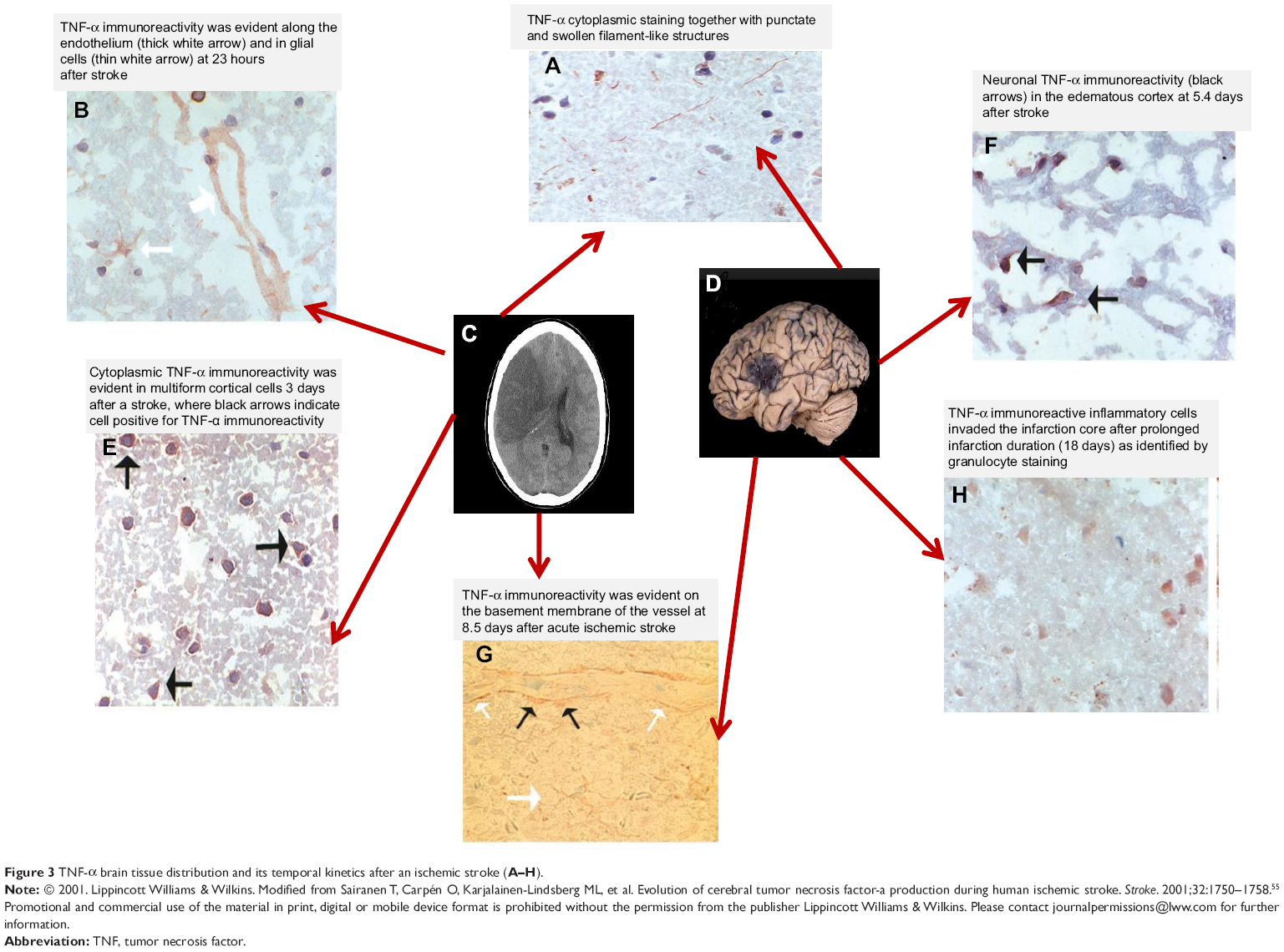 | Figure 3 TNF-α brain tissue distribution and its temporal kinetics after an ischemic stroke (A–H). |
On the third day, contralateral neuronal immunoreactivity decreased, whereas neuronal TNF-α immunoreactivity was demonstrable at 5.4 days in the infarct core and peri-infarct areas. A second wave of TNF-α immunoreactivity was also observed on astrocytes on the third day after an acute ischemic stroke. After 2 weeks, TNF-α immunoreactivity was shown, with inflammatory cells infiltrating the affected hemisphere in a chemotactic manner and resident astrocytes of controlateral brain areas only in a small amount.
The experimental findings of Sairanen et al54 offered a confirmation of the role of TNF-α production as a key element of neuronal damage after an acute ischemic stroke in humans. The long duration of kinetic distribution of TNF-α response observed in humans, with a peak between the second and the third day after an acute ischemic stroke and maintaining a high degree of TNF-α immunoreactivity at 2 weeks after stroke, fosters speculation about a possible therapeutic intervention in the immune-inflammatory background of the acute phase of ischemic stroke demonstrated by the progressive brain immunoreactivity of TNF-α.
Other researchers have suggested the possibility of a dual role of TNF-α in experimental models of ischemic stroke, such as a murine model in which the authors used LPS to induce LPS tolerance in the postischemia phase and in cell cultures with neuronal cells.55 These authors reported how LPS preconditioning resulted in a significant increase in circulating levels of TNF-α before experimental ischemic stroke induction by means of MCAO in mice; they also reported that the presence of TNF-α is necessary to induce a neuroprotective effect after experimental brain ischemia, and this issue has been supported by findings concerning the absence of neuroprotective effect after LPS injury in mice lacking TNF-α.55
Furthermore, these authors observed in LPS-preconditioned experimental mice a lowering of TNF-α, thus resulting in a subsequent neuronal modulation of a series of signaling molecules related to TNF-α such as TNFR1 and TNFR-associated death domain. Particularly interesting, it also appears that sTNFR1 levels increased after a stroke in LPS-preconditioned experimental mice, and this finding could explain a possible neutralizing effect toward TNF-α, thus ameliorating and lowering neuronal damage mechanisms mediated by TNF-α.
Role of TNF-α in brain damage after brain trauma
The role of TNF-α has also been evaluated in a more strictly traumatic setting. Some authors further examined the role of TNF-α in the acute pathophysiology of TBI.56 They analyzed TNF-α kinetics in terms of its expression, localization, and regulation in an experimental mouse model of head trauma. These authors observed higher TNF-α levels in brain cortex after TBI at 1 hour and 4 hours but not at 12 hours, 24 hours, or 72 hours. In early phases (between 1 hour and 4 hours after TBI), significantly higher levels of TNF-α have been reported as immunocytochemically detectable in neurons of the injured cerebral cortex, whereas positive immunochemistry staining for TNF-α has been reported in a small subset of astrocytes, ventricular cells, and microvessels, although this positive immunochemistry was not evaluated as TBI-dependent. Furthermore, these authors did not observe TNF-α expression in macrophages resident in the hemorrhagic zone along the external capsule and corpus callosum.
In the same research, intracerebroventricular administration of a selective TNF-α antagonist-sTNF-α receptor fusion protein in the early phase after TBI (at 15 minutes and at 1 hour after TBI) significantly ameliorated functional outcome in experimental mice, whereas intravenous administration of the same compound at the same time intervals did not result in the same findings. These results underline the fact that increased TBI-related TNF-α is responsible for many facets of subsequent neurological impairment.
Selective TNF inhibitors in the treatment of brain injury from ischemic stroke
Several possible mechanisms may control TNF-α activity during inflammatory activation. Endogenous inhibitors of TNF-α, such as prostaglandins and cyclic adenosine monophosphate, which inhibit TNF-α production, and glucocorticoids that upregulate in the presence of high TNF-α levels by means of activation of the hypothalamus pituitary adrenal axis, inhibit TNF-α production.57,58
Some authors55 evaluated neurons and microglial cells after treatment with IL-1β or IL-1 receptor agonist (ra) for 10 days in hippocampus slice cultures (OHSCs) from rats after an excitotoxic TBI by means of N-methyl-D-aspartate (NMDA). These researchers analyzed OHSCs by means of bright-field microscopy after hematoxylin staining and confocal laser scanning microscopy, after labeling of damaged neurons with propidium iodide and fluorescent staining of microglial cells. They reported that treatment with IL-1β in unlesioned OHSCs did not induce neuronal damage but induced an increase in the number of microglial cells, whereas excitotoxic NMDA-mediated lesions resulted in a higher density of microglial cells. Treatment of NMDA-lesioned OHSCs with IL-1β increased neuronal cell death and upregulated microglia cell density. In NMDA-lesioned culture, treatment with IL-1ra resulted in a significant amelioration of neuronal damage and reduced the number of microglial cells, whereas treatment with IL-1ra in unlesioned OHSCs has been reported as not inducing significant changes in the evaluated cell population.
On the basis of these facts, it is possible to suggest that: 1) IL-1β has a direct role in the pathogenesis of TBI-induced neuronal damage in the CNS setting; 2) IL-1β is responsible for microglia cell subset activation whereas it is not directly neurotoxic; 3) IL-1β is able to regulate excitotoxic neuronal damage and microglia upregulation; and 4) IL-1ra treatment inhibits neuronal cell death and downregulates microglia cell number after TBI by means of an excitotoxic damage.
Several anti-inflammatory mediators negatively regulate TNF-α action, such as IL-10, IL-13, and IL-4.51–53 Furthermore, other proteins are able to regulate TNF-α production such as the TNF-α converting enzyme (TACE) by cleaving the transmembrane form of TNF-α and generating its soluble form (sTNF-α). This enzyme can also modulate TNF receptors, generating sTNFRs able to bind circulatory sTNF.54–56,59
The vagus nerve has also been shown to inhibit TNF-α production by means of a cholinergic activation of muscarinic receptors.57 A series of proteins act as TNF-α inhibitors and have been called biologics, and their use has been used in the treatment of inflammatory and autoimmune disorders including rheumatoid and juvenile arthritis, ankylosing spondylitis, and Crohn’s disease. Among this class of drugs, infliximab, etanercept, and adalimumab act by binding soluble transmembrane form of TNF, thus inhibiting their interaction with TNF-α receptors. Infliximab, a chimeric bivalent Immunoglobulin G1 mAb with a human constant region and amurine variable regions; adalimumab, a bivalent mouse Immunoglobulin G1 mAb; and etanercept, a fusion protein of human IgG fused to a dimer of the extracellular regions of TNFR2, have been reported as active TNF-α inhibitor drugs.
Emerging evidence suggests that retrieved penumbra may be the site of ongoing inflammatory pathology, which includes extensive MA. This is a possible action area for TNF-α-blocking drugs. Etanercept has been reported to reduce MA in animal stroke models, and it has been reported as effective in reducing ischemic neuronal damage in murine models of brain ischemia and subsequent neuronal injury. Perispinal administration is a possible way to administer etanercept, and it has shown that perispinal administration is effective in Alzheimer’s disease,58,60,61 owing to the fact that this method of administration makes brain delivery of etanercept easier.
On this basis, a recent study evaluated the initial clinical response to perispinal etanercept in a first chronic stroke cohort.62 Researchers evaluated three patients with permanent postischemic neurological deficits with hemiparesis, in addition to other stroke deficits due to previous middle cerebral artery, brainstem (medulla), and left middle cerebral artery infarcts. These patients were treated with perispinal etanercept at 13 months, 35 months, and 36 months following acute brain ischemia, and the authors observed a significant amelioration following perispinal etanercept administration with clinical response in 10 minutes. Clinical improvement has been observed as a significant amelioration of motor deficit (hemiparesis), gait disturbances, sensory deficits, spatial perception, speech functions, cognitive performance, and behavioral disturbances. The authors reported further clinical improvement with a second perispinal etanercept dose 20 days after the first dose. These findings suggest that stroke may result in chronic TNF-mediated pathophysiology, which may be potentially suitable for therapeutic intervention even beyond an acute phase.
Several studies have indicated how TNF-α production is regulated at both transcriptional and translational levels.63 Potentially effective classes of drugs include calphostin C, protein kinase C inhibitors of microbial origin,64 for example, which are able to block LPS-stimulated TNF-α production either from human monocytes in vitro65 or LPS and virus-stimulated TNF-α increase in astrocytic cell lines.66
Another possible application of a TNF-α blocking strategy has been offered by a blocking treatment against some key enzymes involved in TNF biosynthesis pathways such as p38 and other monoaminophosphate kinases. Soluble TNF-α is catalyzed by the membrane-bound Zn-metalloprotease, TACE, which has been cloned using a pro-TNF-α peptide cleavage assay.67
One study suggested a crucial role for TACE as the major physiological TNF-α processing enzyme, and this activity has been shown to be susceptible to a metalloprotease inhibitor that significantly reduces LPS-induced TNF release in cells.68
TACE has been shown to have an important role in the CNS both in physiological and pathological conditions,69 and some authors have reported that, after a cerebral ischemia, it is possible to observe a significant upregulation of gelatinase MMP-2 and MMP-9 activities, as well as signs of neuronal damage at hippocampus sites.70 In these experiments, it has been reported that treatment with BB-94 before and soon after a transient global ischemia induces a significant reduction in signs of hippocampus neuronal damage, producing a significant lowering of hippocampus gelatinase activities. Furthermore, Yang et al have shown that the TACE/MMP-9 inhibitor KB-R7785 significantly reduces brain damage.71 In this study, the administration of KB-R7785 was reported to induce a significant lowering of infarct volume after MCAO in mice. All these studies demonstrate how TNF-α production is a target of therapeutic interventions owing to the fact that its production is regulated at both transcriptional and translational levels (Figure 4).72
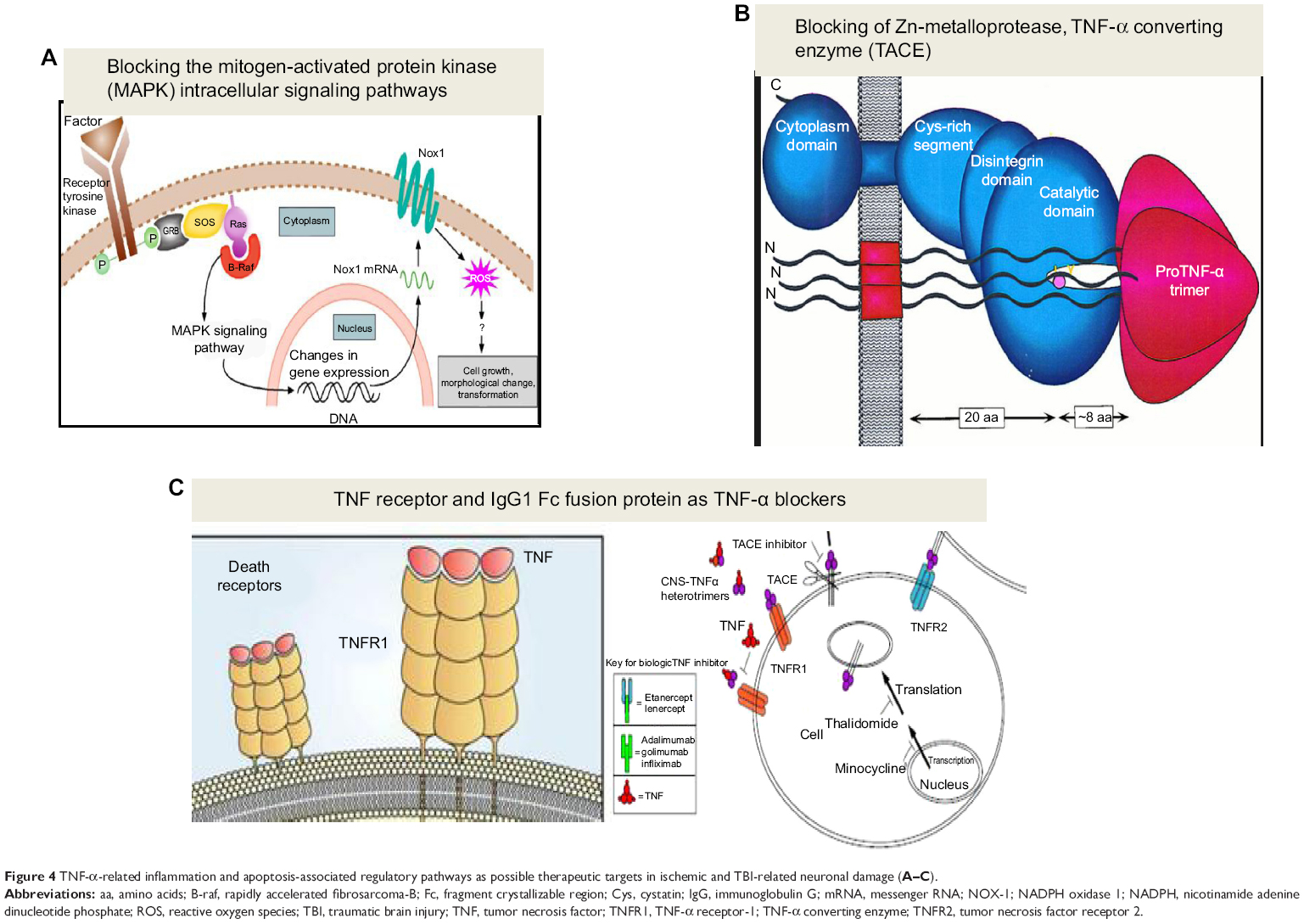 | Figure 4 TNF-α-related inflammation and apoptosis-associated regulatory pathways as possible therapeutic targets in ischemic and TBI-related neuronal damage (A–C). |
Inflammation- and apoptosis-associated regulatory pathways are other possible therapeutic targets in TBI-related neuronal damage. On this basis, some researchers analyzed the therapeutic strategy of blocking the mitogen-activated protein kinase (MAPK) intracellular signaling pathways. Three distinct MAPK pathways have been reported. For example, in stress-activated MAPK, p38, and c-Jun N terminal kinase have been described as having an important role in mediating signals by transcription factors and inflammatory cytokines.73
In a study by Chio et al, experimental rats after TBI (Table 1) were randomized to vehicle solution or etanercept (5 mg/kg of body weight) intraperitoneally administered once every 12 hours for 3 consecutive days.74 Etanercept was reported as effective in ameliorating cerebral ischemia surrogate markers such as increased cellular levels of glutamate, lactate-to-pyruvate ratio, and increased cellular levels of glycerol. Etanercept also ameliorated some brain edema and functional (motor and cognitive) signs. TBI-induced neuronal damage is mediated by neuronal, glia, and astrocytic apoptosis induction expressed by a higher degree of terminal deoxynucleotidyl transferase αUTP nick-end labeling and neuronal-specific nuclear protein double-positive cells, a higher degree of terminal deoxynucleotidyl transferase αUTP nick-end labeling and glia fibrillary acidic protein double-positive cells, and increased numbers of glia fibrillary acidic protein positive cells. Other surrogate markers of TBI-induced cell damage are MA activation expressed by some markers, such as a higher degree of ionized calcium-binding adapter molecule 1-positive cells, TNF-α, IL-1β, and IL-6, and all these post-TBI inflammatory markers have been reported as significantly reduced by treatment with etanercept.
Some experimental evidence underlines that post-TBI inflammation may persist longer than the duration of acute ischemia. Some studies have reported how MA in the ischemic core, the peri-infarct zone, and the contralateral hemisphere may persist for 30 days.75–77 Excess TNF released by activated glia may perpetuate inflammation and produce a cycle of continued glia activation. Thus, etanercept represents a possible anti-inflammatory and neuroprotective therapy for TBI (Figure 5).
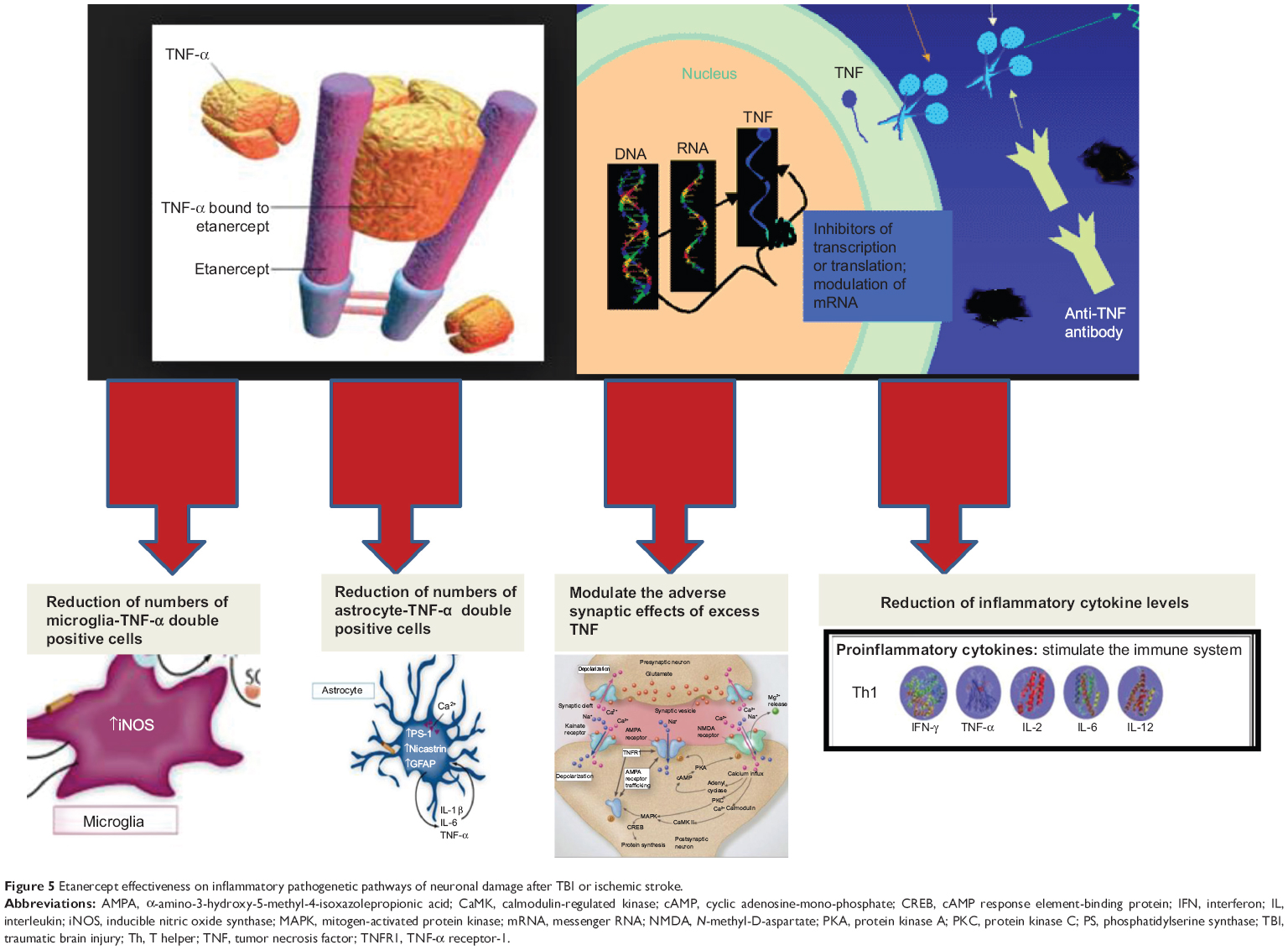 | Figure 5 Etanercept effectiveness on inflammatory pathogenetic pathways of neuronal damage after TBI or ischemic stroke. |
Monoclonal neutralizing anti-TNF-α antibody administered intracerebroventricularly (or into the brain cortex in the experimental animals with MCAO significantly decreases focal ischemic brain injury. A recent study74 has been conducted to evaluate whether inhibiting the action of TNF-α attenuates brain injury and reduces inflammatory responses in murine models of ischemic stroke. Mice underwent MCAO for 1 hour and monoclonal neutralizing anti-murine TNF-α antibody (mAb) was administrated intraventricularly in the mouse with temporary MCAO. The authors reported that the infarct volume in mice treated with anti-TNF-α mAb was significantly smaller than that in the control group.
Selective TNF inhibitors in the treatment of brain injury from TBI
Etanercept has been reported as possibly effective in reducing TBI-induced cerebral edema, motor and cognitive impairment, and other candidate markers of TBI-linked neuronal damage such as upregulation of astrocytes and microglial cells, and to modulate other inflammation mediators. It seems that etanercept’s effectiveness could be related to the fact that this drug is able to reach TBI-lesioned brain tissue and to lower TBI-induced cognitive and motor impairment by promoting a neurogenesis-involved pathway such as doublecortin (DCX), a microtubule-associated protein. DCX is upregulated in neuronal precursors of the developing brain, thus representing a candidate target marker of neuronal regeneration achieved by blocking TNF-α. DCX expression has been reported as higher within areas of the subventricular zone of the lateral ventricles and the subgranular zone at the dentate gyrus/hilus interface of hippocampus10,11 in the adult brain. In a post-TBI neuronal regeneration setting, an increased number of divided cells in the subventricular zone and subgranular zone is expressed by immature neurons simultaneously with DCX expression. Several experimental models suggest that glia TNF-α produces neurotoxicity and that TNF-α-linked neurotoxicity may be reversible by the blockade of TNF-α.78
Etanercept may improve functional outcomes of TBI owing to the fact that it reaches the CSF in rats, thus reaching damaged brain areas. Some authors assessed etanercept’s effectiveness on acute neurological and motor injury in rats 1 day prior to and 7 days after surgery.79 These authors used the degree of the co-localizations of 5-bromodeoxyuridine and DCX on damaged areas evaluated by immunofluorescence staining as markers of brain contusion and brain injury. They reported detectable brain levels of etanercept in the damaged brain 7 days after systemic administration of etanercept, and also reported a lowering action of this drug on neurological and motor deficits signs, cerebral contusion markers, and degree of expression of brain TNF-α. Furthermore, they reported a higher degree of neurogenesis markers such as 5-bromodeoxyuridine- and DCX-specific markers in the contused brain tissues caused by TBI after treatment with etanercept, thus showing that etanercept may reach the TBI-damaged brain tissues, ameliorating outcomes of TBI by a broad anti-inflammatory action and a specific promotion of neurogenesis.
A study was conducted that aimed to evaluate the role of etanercept after TBI on TNF-α-related markers and on astrocyte and neuron damage surrogate markers.80 The authors used a rat model and, after a mechanical TBI, performed a systemic measure of the neurological severity score and motor function before injury and at day 3 after etanercept administration. They reported a higher degree of microglia-TNF-α double-positive cells after TBI but not TNF-α neurons or TNF-α astrocyte double-positive cells in the injured brain areas. Etanercept administration significantly lowered all the surrogate markers of TBI neuronal damage, such as numbers of microglia-TNF-α double-positive cells, and increased TNF-α levels in the injured brain and ameliorated several signs of neurological motor deficits. This study further underlined how an early microglia overproduction of TNF-α in the injured brain region after TBI could represent a main contributor to cerebral ischemia and neurological motor deficits, thus representing a candidate therapeutic target of etanercept action.
TBI patients do not show clear structural brain defects, but in general they frequently suffer from long-lasting cognitive, behavioral, and emotional difficulties. Owing to the central role of TNF-α in the regulation of systemic inflammatory processes, and in regulating inflammatory-mediated neuronal and glia toxicity, it may represent a promising drug target even after the early stages of TBI. Some researchers reported that TNF-α levels increase after TBI and may also be responsible for later secondary damage to brain tissue.80
Wang et al81 evaluated the effects of the activation of the p75TNFR signaling pathway in mice with TBI in an experimental model of hybridomas that produced antibodies specific for p75TNFR, which is critical to the binding of TNF-α and p75TNFR. The authors detected the levels of inflammatory factors in C57BL/6 mice with TBI, and then they injected the mice with either saline (control) or p75TNFR agonist to analyze the effects of p75TNFR agonist mAb on p38MAPK and nuclear factor-κB (NF-κB) signals. They showed that mAb D8F2 was able to inhibit posttraumatic inflammatory responses effectively, offering an agonist anti-mouse p75TNFR mAb which may be used for the clinical treatment of inflammation after brain trauma.
Belarbi et al82 conducted a study (Table 1) to evaluate the therapeutic potential of a analog of thalidomide, namely 3,6′-dithiothalidomide (DT), an agent with anti-TNF-α activity, in a model of chronic neuroinflammation. They showed that chronic LPS infusion was characterized by increased gene expression of the proinflammatory cytokines TNF-α and IL-1β in the hippocampus and that treatment with DT normalized TNF-α levels but not IL-1β. The authors also showed that treatment with DT ameliorated cognitive function in LPS-infused animals, demonstrating that the TNF-α synthesis inhibitor DT can significantly reverse hippocampus-dependent cognitive impairment induced by chronic neuroinflammation.
NF-κB plays important roles in the regulation of apoptosis and inflammation. A recent study83 evaluated the effect of an NF-κB inhibitor SN50 on cell death and behavioral deficits in mice with TBI models. This study showed that pretreatment with SN50 significantly lowered TBI-induced cell death and motor and cognitive dysfunction and also that SN50 lowered TNF-α expression.
Another recent study evaluated the efficacy of the experimental TNF-α synthesis inhibitor DT on neurological deficits after a TBI due to a rapid induction of high levels of brain TNF-α.84 These authors reported a significant amelioration of neurological functional impairment in mice that received a one-time administration of DT, thus indicating a possible role of DT as a neuroprotective drug against TBI-related neuronal damage.
More than a decade after the introduction of biologic TNF-α inhibitors into clinical practice, there is substantial evidence that excess TNF-α plays a key role in brain dysfunction and also that physiological levels of TNF-α are involved in normal brain physiology.85–87 TNF-α also acts as a glia transmitter of glial cell production and has been reported to regulate synaptic communication.88,89
A noninvasive method of delivery of etanercept, through perispinal extrathecal administration, was developed for the treatment of neuroinflammatory disorders.89 Stroke patients and patients with TBI, owing to the reported pathogenic role of TNF-α and other cytokines in this clinical context, could represent a possible target for these drugs. Furthermore, the long interval (years) between the stroke event and rapid clinical improvement that has been described in these patients is consistent with experimental evidence that TNF-α generation persists in the CSF for very much longer (10 months) than in the serum (gone in 6 hours), thus offering a long-term opportunity for therapeutic intervention.85
These findings permitted researchers to also evaluate the role of etanercept in late stages of post-TBI neuronal damage because the interesting findings on experimental mice suggested the possible role of selective TNF-α inhibition in ameliorating neuroinflammation-related neuronal damage after TBI, also in humans.
Selective TNF inhibitors in the treatment of brain injury from stroke and from TBI
Since brain injury from stroke and TBI may result in a persistent neuroinflammatory response in the injury penumbra, and owing to the fact that this inflammatory response may include MA and excess levels of TNF, and that previous experimental data suggest etanercept, a selective TNF inhibitor, has the ability to ameliorate MA and modulate the adverse synaptic effects of excess TNF, it has been hypothesized that perispinal administration may enhance etanercept delivery across the blood–CSF barrier.
Perispinal administration of etanercept may facilitate the delivery of the drug into the brain. Tobinick et al conducted a study to evaluate perispinal etanercept administration (Table 1) in patients with chronic ischemic stroke, enrolling three consecutive patients with chronic neurological impairment after acute ischemic stroke not resolved after rehabilitation.90 In all these patients treated with etanercept, the authors observed a significant clinical improvement that was evident within 10 minutes of perispinal injection. They also observed a significant amelioration of motor dysfunction, gait disorder, hemi-neglect disorders, speech, cognitive dysfunction, and behavioral disorders. After a second perispinal etanercept dose, they noticed additional clinical improvement.
Furthermore, Tobinick et al conducted another study (Table 1) to evaluate the effectiveness of intraspinal etanercept administration in patients with postischemic and post-TBI neurological impairment, enrolling 629 consecutive patients.91 The authors observed a significant improvement in motor impairment, spasticity, sensory impairment, cognition, psychological/behavioral function, and aphasia, and also an amelioration of pseudobulbar symptoms and urinary incontinence. These findings were also observed in patients treated even in the late phase of their poststroke neurological dysfunction. In this subgroup of patients, the authors also observed a significant improvement of motor symptoms and spasticity. This study suggested how stroke and TBI may lead to delayed neuroinflammatory response in the brain, which could be a possible therapeutic target for selective inhibition of TNF, even a long time after an acute injury. These findings further permit us to conclude that high brain and serum levels of TNF-α may represent a possible pathogenic basis of chronic neurological and neuropsychiatric impairment after stroke and TBI.
Positron emission tomographic brain imaging and pathological examination showed how a chronic neuroinflammatory response lasting for years after a single brain injury is present in humans and how TNF is fully involved in this neuroinflammatory cascade. Very recently, Tobinick et al91 reported that a single dose of perispinal etanercept produced an amelioration of aphasia, speech apraxia, and left hemiparesis in patients with acute ischemic stroke for more than 3 years after acute brain injury. These findings could suggest that acute brain-injury-induced pathologic levels of TNF may provide a therapeutic target that can be addressed years after injury.
Conclusion
Neuronal damage after an ischemic stroke or TBI has a complex pathogenesis with several inflammatory pathways that have been reported as involved. Several sources of evidence indicate that an inflammatory-immunologic cascade is involved in the pathogenesis of cerebral ischemia. Circulatory inflammatory cells such as neutrophils and macrophages reach the damaged area of the ischemic brain in some experimental models of ischemic stroke and also in human subjects with cerebral ischemia. Brain resident cells such as astrocytes, microglia, or endothelia have also been reported to be involved in this immune-inflammatory activation subsequent to a cerebral injury, either mechanical (TBI) or ischemic. These cells, which are involved in this inflammatory activation, become able to communicate with one another by producing inflammatory mediators such as cytokines and adhesion molecules. These molecules appear to be responsible for the accumulation of inflammatory cells in the injured brain, and the resulting immunologic-inflammatory cascade produces an environment that may affect the survival of neurons subjected to ischemic injury.
Clinical studies have reported a higher degree of proinflammatory cytokine and adhesion molecule serum levels in the peripheral blood and CSF of patients with ischemic stroke. Among the cytokines involved in the pathogenesis of ischemic stroke, TNF-α has been reported as higher in experimental ischemia at both the mRNA and protein levels. Furthermore, increased levels of cytokines, such as IL-1β, TNF-α, and IL-6, as well as adhesion molecules, such as ICAM-1 have been observed after experimental brain ischemia. High IL-6 concentrations in CSF and plasma have been associated with larger infarct size, neurological deterioration, and poor outcome, independently of the stroke subtype. TNF-α originating from a variety of cell types in either the periphery or the brain itself can act on the brain to regulate neural–immune interactions as well as other brain functions. TNF-α derived from peripheral immune cells can act in a hormone-like fashion through the hypothalamus–pituitary–adrenal axis and the vagus nerve to stimulate and affect several CNS responses.
Several sources of clinical, genetic, and epidemiologic evidence indicate a central role of high levels of TNF-α in the pathogenesis of ischemic stroke and of TBI, suggesting how TNF-α overexpression could be a therapeutic target. High levels of TNF-α have been reported as responsible for chronic neurologic, neuropsychiatric, and clinical impairment in stroke and TBI clinical settings.98,99 On this basis, perispinal administration of etanercept induces significant clinical improvement in patients with poststroke and TBI motor, sensory, and neuropsychiatric chronic clinical impairment, and this extension of the therapeutic time window exists even up to 10 years after stroke and TBI.100 Nevertheless, randomized clinical trials will be necessary to further quantify and characterize the clinical response.
However, ischemic stroke is a heterogeneous disease and is not unique, owing to the fact that five subtypes of ischemic stroke exist,92,101,102 and some studies report that every subtype shows a different degree of immune-inflammatory activation in terms of cytokines such as TNF-α in the acute phase.93–95 On this basis, it is possible to hypothesize differential effectiveness for selective TNF inhibitors in the treatment of brain injury from stroke in relation to clinical subtypes of ischemic stroke (atherothromboembolic, cardioembolic, or lacunar) that express different degrees of immune-inflammatory activation in the acute phase in terms of cytokine serum levels, as reported in some studies.96,97
Disclosure
The authors report no conflicts of interest in this work.
References
Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. | ||
Gentile NT, McIntosh TK. Antagonists of excitatory amino acids and endogenous opioid peptides in the treatment of experimental central nervous system injury. Ann Emerg Med. 1993;22:1028–1034. | ||
Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244(4906):798–800. | ||
Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J Neurotrauma. 1997;14:23–34. | ||
Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. | ||
Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. | ||
Morrison B 3rd, Eberwine JH, Meaney DF, McIntosh TK. Traumatic injury induces differential expression of cell death genes in organotypic brain slice cultures determined by complementary DNA array hybridization. Neuroscience. 2000;96(1):131 -139. | ||
Liu T, Clark RK, McConnell PC, et al. Tumor necrosis factor in ischemic neurons. Stroke. 1994;25:148–148. | ||
Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2006;184:53–68. | ||
Rothwell NJ, Stroemer P, Lawrence C, Davies C. Actions of IL1β in neurodegeneration. In: Kriegelstein J, editor. Pharmacology of Cerebral Ischaemia. Stuttgart: Medpharm; 1996:125–129. | ||
Benveniste EN, Sparacio SM, Norris JG, Grenett HE, Fuller GM. Induction and regulation of interleukin-6 gene expression in rat astrocytes. J Neuroimmunol. 1990;30(2–3):201–212. | ||
Beamer NB, Coull BM, Clark WM, et al. Focal cerebral ischemia in the rat, temporal profile of neutrophil responses. Neurosci Res Commun. 1992;11:179–186. | ||
Aloisi F, Carè A, Borsellino G, et al. Production of hemolymphopoietic cytokines (IL-6, IL-8, colonystimulating factors) by normal human astrocytes in response to ILl/l and tumor necrosis factor-a. J Immunol. 1992;149:2358–2366. | ||
Frei K, Nohava K, Malipiero UV, Schwerdel C, Fontana A. Production of macrophage colony-stimulating factor by astrocytes and brain macrophages. J Neuroimmunol. 1992;40:189–195. | ||
Lee SC, Liu W, Roth P, Dickson DW, Berman JW, Brosnan CF. Macrophage colony-stimulating factor in human fetal astrocytes and microglia. Differential regulation by cytokines and lipopolysaccharide, and modulation of class II MHC on microglia. J Immunol. 1993;150(2):594–604. | ||
Gautam SC, Noth CJ, Niewenhuis LM, Janakiraman N, Kim JS, Chopp M. Transforming growth factor beta-1 (TGF-beta 1) potentiates IL1 alpha-induced IL6 mRNA and cytokine protein production in a human astrocytoma cell line. Oncol Res. 1993;5:423–432. | ||
Sebire G, Hery C, Peudenier S, Tardieu M. Adhesion proteins on human microglial cells and modulation of their expression by IL-l p and TNFcr. Res Virol. 1993;144:47–52. | ||
Wong D, Dorovini-Zis K. Upregulation of intercellular adhesion molecules-l (ICAM-1) expression in primary cultures of human microvessel endothelial cells by cytokines and lipopolysaccharide. J Neuroimmunol. 1992;39:11–21. | ||
Minami M, Kuraishi Y, Yabuuchi K, Yamazaki K, Satoh M. Induction of interleukin-lb mRNA in rat brain after transient forebrain ischaemia. J Neurochem. 1992;58:390–392. | ||
Buttini M, Sauter A, Boddeke HWGM. Induction of interleukin-1b mRNA after focal cerebral ischaemia in the rat. Mol Brain Res. 1994;23:126–134. | ||
Morganti-Kossmann MC, Hans VH, Lenzlinger PM, et al. TGF-beta is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood–brain barrier function. J Neurotrauma. 1999;16:617–628. | ||
Lin MT. Heatstroke-induced cerebral ischemia and neuronal damage. Involvement of cytokines and monoamines. Ann N Y Acad Sci. 1997;15(813):572–580. | ||
Tarkowski E, Rosengren L, Blomstrand C, et al. Early intrathecal production of interleukin-6 predicts the size of brain lesion in stroke. Stroke. 1995;26:1393–1398. | ||
Matbiesen T, Anderson B, Loftenius A, von Holst H. Increased interleukin-6 levels in cerebrospinal fluid following subarachnoid hemorrhage. J Neurosurg. 1993;78:562–567. | ||
Fassbender K, Rossol S, Kammer T, et al. Proinflammatory cytokines in serum of patients with acute cerebral ischemia, kinetics of secretion and relation to the extent of brain damage and outcome of disease. J Neurol Sci. 1994;122:135–139. | ||
Beamer NB, Coull BM, Clark WM, et al. Persistent inflammatory response in stroke survivors. Neurology. 1997;50:1722–1728. | ||
Tarkowski E, Rosengren L, Blomstrand C, Jensen C, Ekholm S, Tarkowski A. Intrathecal expression of proteins regulating apoptosis in acute stroke. Stroke. 1999;30(2):321–327. | ||
Fan L, Young PR, Barone FC, Feuerstein GZ, Smith DH, McIntosh TK. Experimental brain injury induces expression of interleukin-1 beta mRNA in the rat brain. Brain Res Mol Brain Res. 1995;30:125–130. | ||
Lu D, Goussev A, Chen J, et al. Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma. 2004;21:21–32. | ||
Rothwell N. Interleukin-1 and neuronal injury: mechanisms, modification, and therapeutic potential. Brain Behav Immun. 2003;17:152–157. | ||
Basu A, Krady JK, O’Malley M, Styren SD, DeKosky ST, Levison SW. The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J Neurosci. 2002;22:6071–6082. | ||
Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun. 1995;9:355–365. | ||
Ross SA, Halliday MI, Campbell GC, Byrnes DP, Rowlands BJ. The presence of tumour necrosis factor in CSF and plasma after severe head injury. Br J Neurosurg. 1994;8:419–425. | ||
Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72:169–177. | ||
Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, et al. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999;19:6248–6256. | ||
Liu T, Clark RK, McDonnell PC, et al. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25(7):1481–1488. | ||
Barone FC, Arvin B, White RF, et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28(6):1233–1244. | ||
Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood–brain barrier opening after reperfusion in rat brain. Stroke. 1998;29(10):2189–2195. | ||
Martin-Villalba A, Hahne M, Kleber S, et al. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001;8(7):679–686. | ||
Robertson J, Beaulieu JM, Doroudchi MM, Durham HD, Julien JP, Mushynski WE. Apoptotic death of neurons exhibiting peripherin aggregates is mediated by the proinflammatory cytokine tumor necrosis factor-alpha. J Cell Biol. 2001;155(2):217–226. | ||
Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1997;17(5):483–490. | ||
Pober JS, Cotran RS. Cytokines and endothelial cell biology. Physiol Rev. 1990;70:427–451. | ||
Tansey MG, Wyss-Coray T. Cytokines in CNS inflammation and disease. In: Lane TE, Carson M, Bergmann C, Wyss-Coray T, editors. Central Nervous System Diseases and Inflammation. New York, NY: Springer; 2008:59–106. | ||
Selmaj KW, Farooq M, Norton WT, Raine CS, Brosnan CF. Proliferation of astrocytes in vitro in response to cytokines. A primary role for tumor necrosis factor. J Immunol. 1990;144:129–135. | ||
Sedgwick JD, Riminton DS, Cyster JG, Korner H. Tumor necrosis factor: a master-regulator of leukocyte movement. Immunol Today. 2000;21:110–113. | ||
Leon LR. Invited review: cytokine regulation of fever: studies using gene knockout mice. J Appl Physiol. 2002;92:2648–2655. | ||
Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNF-alpha. Science. 2002;295:2282–2285. | ||
Pickering M, Cumiskey D, O’Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. | ||
Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-[alpha]. Nature. 2006;440:1054–1059. | ||
Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential Regulation of AMPA Receptor and GABA Receptor Trafficking by Tumor Necrosis Factor-{alpha}. J Neurosci. 2005;25:3219–3228. | ||
Weber C, Erl W, Pietsch A, Ströbel M, Ziegler-Heitbrock HW, Weber PC. Antioxidants inhibit monocyte adhesion by suppressing nuclear factor-B mobilization and induction of vascular cell adhesion molecule-1 in endothelial cells stimulated to generate radicals. Arterioscler Thromb. 1994;14:1665–1673. | ||
Tipping PG, Hancock WW. Production of tumor necrosis factor and interleukin-1 by macrophages from human atheromatous plaques. Am J Pathol. 1993;142:1721–1728. | ||
Uno H, Matsuyama T, Akita H, Nishimura H, Sugita M. Induction of tumor necrosis factor-a in the mouse hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab. 1997;17:491–499. | ||
Sairanen T, Carpén O, Karjalainen-Lindsberg ML, et al. Evolution of cerebral tumor necrosis factor-a production during human ischemic stroke. Stroke. 2001;32:1750–1758. | ||
Hailer NP, Vogt C, Korf HW, Dehghani F. Interleukin-1beta exacerbates and interleukin-1 receptor antagonist attenuates neuronal injury and microglial activation after excitotoxic damage in organotypic hippocampal slice cultures. Eur J Neurosci. 2005;21(9):2347–2360. | ||
Perlstein RS, Whitnall MH, Abrams JS, Mougey EH, Neta R. Synergistic roles of interleukin-6, interleukin-1, and tumor necrosis factor in the adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology. 1993;132:946–952. | ||
Borovikova LV, Ivanova S, Zhang M, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. | ||
Seckinger P, Isaaz S, Dayer JM. A human inhibitor of tumor necrosis factor alpha. J Exp Med. 1988;167:1511–1516. | ||
Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95(1–2):115–125. | ||
Seckinger P, Isaaz S, Dayer JM. Purification and biologic characterization of a specific tumor necrosis factor alpha inhibitor. J Biol Chem. 1989;264:11966–11973. | ||
Novick D, Engelmann H, Wallach D, Rubinstein M. Soluble cytokine receptors are present in normal human urine. J Exp Med. 1989;170:1409–1414. | ||
Tobinick E. Rapid improvement of chronic stroke deficits after perispinal etanercept: three consecutive cases. CNS Drugs. 2011;25(2):145–155. | ||
Gerard C, Bruyns C, Marchant A, et al. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J Exp Med. 1993;177:547–550. | ||
de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;74:1209–1220. | ||
Tracey KJ, Cerami A. Tumor necrosis factor, an updated review of its biology. Crit Care Med. 1993;21(10 suppl):S415–S422. | ||
Semmler J, Wachtel H, Endres S. The specific type IV phosphodiesterase inhibitor rolipram suppresses tumor necrosis factor-alpha production by human mononuclear cells. Int J Immunopharmacol. 1993;15(3):409–413. | ||
Takahashi I, Kobayashi E, Nakano H, et al. Potent selective inhibition of 7-O-methyl UCN-01 against protein kinase C. Pharmacol Exp Ther. 1990;255(3):1218–1221. | ||
Prabhakar U, Lipshutz D, Pullen M, et al. Protein kinase C regulates TNF-alpha production by human monocytes. Eur Cytokine Netw. 1993;4(1):31–37. | ||
Lieberman AP, Pitha PM, Shin ML. Poly(A) removal is the kinase-regulated step in tumor necrosis factor mRNA decay. J Biol Chem. 1992;267(4):2123–2126. | ||
Moss ML, Jin SL, Milla ME, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385(6618):733–736. | ||
Yang Y, Jalal FY, Thompson JF, et al. Tissue inhibitor of metalloproteinases-3 mediates the death of immature oligodendrocytes via TNF-α/TACE in focal cerebral ischemia in mice. J Neuroinflammation. 2011;8:108. | ||
Moro MA, Hurtado O, Cárdenas A, et al. Expression and function of tumour necrosis factor-alpha-converting enzyme in the central nervous system. Neurosignals. 2003;12(2):53–58. | ||
Lee SR, Tsuji K, Lee SR, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24(3):671–678. | ||
Chio CC, Lin JW, Chang MW, et al. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J Neurochem. 2010;115(4):921–929. | ||
Kyriakis JM, Avruch J. Sounding the alarm, protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271(40):24313–24316. | ||
Kummer JL, Rao PK, Heidenreich KA. Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J Biol Chem. 1997;272(33):20490–20494. | ||
Yang GY, Gong C, Qin Z, Ye W, Mao Y, Bertz AL. Inhibition of TNF-alpha attenuates infarct volume and ICAM-1 expression in ischemic mouse brain. Neuroreport. 1998;9:2131–2134. | ||
Jiang X, Namura S, Nagata I. Matrix metalloproteinase inhibitor KB-R7785, attenuates brain damage resulting from permanent focal cerebral ischemia in mice. Neurosci Lett. 2001;305(1):41–44. | ||
Cheong CU, Chang CP, Chao CM, Cheng BC, Yang CZ, Chio CC. Etanercept attenuates traumatic brain injury in rats by reducing brain TNF- α contents and by stimulating newly formed neurogenesis. Mediators Inflamm. 2013;2013:620837. | ||
Chio CC, Chang CH, Wang CC, et al. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-α. BMC Neurosci. 2013;15(14):33. | ||
Wang YX, You Q, Su WL, et al. Ruan CPA study on inhibition of inflammation via p75TNFR signaling pathway activation in mice with traumatic brain injury. J Surg Res. 2013;182(1):127–133. | ||
Belarbi K, Jopson T, Tweedie D, et al. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J Neuroinflammation. 2012;9:23. | ||
Sun YX, Dai DK, Liu R, et al. Therapeutic effect of SN50, an inhibitor of nuclear factor-κB, in treatment of TBI in mice. Neurol Sci. 2013;34(3):345–355. | ||
Baratz R, Tweedie D, Rubovitch V, et al. Tumor necrosis factor-α synthesis inhibitor, 3,6′-dithiothalidomide, reverses behavioral impairments induced by minimal traumatic brain injury in mice. J Neurochem. 2011;118(6):1032–1042. | ||
Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28(9):2221–2230. | ||
Immonen RJ, Kharatishvili I, Niskanen JP, Gröhn H, Pitkänen A, Gröhn OH. Distinct MRI pattern in lesional and perilesional area after traumatic brain injury in rat: 11 months follow-up. Exp Neurol. 2009;215(1):29–40. | ||
Price CJ, Wang D, Menon DK, et al. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke. 2006;37(7):1749–1753. | ||
Jiang H, Hampel H, Prvulovic D, et al. Elevated CSF levels of TACE activity and soluble TNF receptors in subjects with mild cognitive impairment and patients with Alzheimer’s disease. Mol Neurodegener. 2011;6:69. | ||
Rossi D, Martorana F, Brambilla L. Implications of gliotransmission for the pharmacotherapy of CNS disorders. CNS Drugs. 2011;25(8):641–658. | ||
Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs. 2012;26(12):1051–1070. | ||
Tobinick E, Rodriguez-Romanacce H, Levine A, Ignatowski TA, Spengler RN. Immediate neurological recovery following perispinal etanercept years after brain injury. Clin Drug Investig. 2014;34(5):361–366. | ||
Adams HP Jr, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993;24(1):35–41. | ||
Davì G, Tuttolomondo A, Santilli F, et al. CD40 ligand and MCP-1 as predictors of cardiovascular events in diabetic patients with stroke. J Atheroscler Thromb. 2009;16(6):707–713. | ||
Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Inflammation in ischemic stroke subtypes. Curr Pharm Des. 2012;18(28):4289–4310. | ||
Tuttolomondo A, Pinto A, Corrao S, et al. Immuno-inflammatory and thrombotic/fibrinolytic variables associated with acute ischemic stroke diagnosis. Atherosclerosis. 2009;203(2):503–508. | ||
Licata G, Tuttolomondo A, Corrao S, et al. Immunoinflammatory activation during the acute phase of lacunar and non-lacunar ischemic stroke: association with time of onset and diabetic state. Int J Immunopathol Pharmacol. 2006;19(3):639–646. | ||
Tuttolomondo A, Di Raimondo D, Forte GI, et al. Single nucleotide polymorphisms (SNPs) of pro-inflammatory/anti-inflammatory and thrombotic/fibrinolytic genes in patients with acute ischemic stroke in relation to TOAST subtype. Cytokine. 2012;58(3):398–405. | ||
Cheng X, Yang L, He P, Li R, Shen Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J Alzheimers Dis. 2010;19(2):621–630. | ||
Tweedie D, Ferguson RA, Fishman K, et al. Tumor necrosis factor-alpha synthesis inhibitor 3,60-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J Neuroinflammation. 2012;9:106. | ||
Tobinick E. Perispinal etanercept for neuroinflammatory disorders. Drug Discov Today. 2009;14(3–4):168–177. | ||
Albanese A, Tuttolomondo A, Anile C, et al. Spontaneous chronic subdural hematomas in young adults with a deficiency in coagulation factor XIII. Report of three cases. J Neurosurg. 2005;102(6):1130–1132. | ||
Pinto A, Di Raimondo D, Tuttolomondo A, Fernandez P, Arnao V, Licata G. Twenty-four hour ambulatory blood pressure monitoring to evaluate effects on blood pressure of physical activity in hypertensive patients. Clin J Sport Med. 2006;16(3):238–243. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.