Back to Journals » Drug Design, Development and Therapy » Volume 20
Structure-Based Discovery of Glyoxalase-I Inhibitors: Evaluating the Role of Active Site Water Molecules
Authors Anaam R ![]() , Isawi IH
, Isawi IH ![]() , Al-Balas Q
, Al-Balas Q ![]()
Received 18 October 2025
Accepted for publication 7 April 2026
Published 23 April 2026 Volume 2026:20 575242
DOI https://doi.org/10.2147/DDDT.S575242
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Tamer Ibrahim
Reema Anaam, Israa H Isawi, Qosay Al-Balas
Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan
Correspondence: Israa H Isawi, Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan, Email [email protected]
Background: Glyoxalase-I (GLO-I) is a zinc-dependent metalloenzyme and a promising target for anticancer drug discovery. It catalyzes the detoxification of methylglyoxal, a cytotoxic byproduct of glycolysis, a metabolic shift commonly observed in cancer cells. GLO-I overexpression in tumor cells promotes multidrug resistance and tumor progression. However, the role of conserved water molecules within the GLO-I active site remains insufficiently explored, and understanding their influence on ligand binding may improve structure-based inhibitor design.
Purpose: This study aimed to identify potential GLO-I inhibitors by examining the effect of conserved active site water molecules on ligand binding and activity predictions using a structure-based drug design approach.
Materials and Methods: Three human GLO-I crystal structures were used to generate structure-based pharmacophore models under two conditions: with and without crystallographic active site water molecules. The models were applied to virtually screen the OTAVA Lead-Like library (commercial lead-like compound library). Molecular docking of the resulting hits was then performed under both hydration conditions to evaluate effects on ligand binding affinity and pose orientation. Top-ranked compounds were purchased and evaluated in vitro for GLO-I inhibition. The most active hit was further evaluated by 1000 ns molecular dynamics (MD) simulations (± crystallographic waters), including analysis of pose stability and binding-site water behavior.
Results: Among the 22 compounds tested in vitro, five showed inhibitory activity, with IC50 values ranging from 12.07 to 25.36 μM. The most potent compound (hit 19) demonstrated an IC50 of 12.07 ± 0.31 μM and 85.63% inhibition at 50 μM. Docking analysis indicated that including crystallographic water molecules often increased docking scores but could distort binding orientations, whereas docking without conserved active site water molecules more consistently produced plausible poses and better agreement with experimental activity trends.
Conclusion: For GLO-I, docking without conserved active site water molecules provided more accurate results and may represent a more reliable approach for studying ligand binding and guiding inhibitor design.
Keywords: glyoxalase-I, anticancer, pharmacophore modeling, molecular docking, crystallographic water effect
Introduction
Cancer continues to pose a substantial global health challenge, ranking as one of the leading causes of mortality worldwide.1 According to the National Cancer Institute, an alarming increase can be expected in the incidence of cancer cases by 2050, reaching an estimated number of 35.3 million new cases and 18.5 million deaths annually.1 A characteristic feature of cancer is uncontrolled cell growth and proliferation, partly driven by a unique metabolic shift referred to as the “Warburg effect”.2 This metabolic rewiring allows cancer cells to depend predominantly on glycolysis for energy production, even in the presence of oxygen, and thereby promotes their proliferation and survival.2
As a critical outcome of this altered metabolism, cancer cells accumulate toxic byproducts, especially methylglyoxal (MG), a highly reactive dicarbonyl derived from glycolysis.3 In mitigating this toxicity, cells depend on the glyoxalase system, consisting of two enzymes: Glyoxalase I (GLO-I) and Glyoxalase II (GLO-II). Figure 1 depicts that the first step in the metabolism of MG involves the non-enzymatic reaction with glutathione (GSH) to form a hemithioacetal intermediate; GLO-I then converts this intermediate into S-D-lactoylglutathione, which, in turn, is hydrolyzed by GLO-II into D-lactate and regenerating GSH, completing the detoxification cycle.3,4 Because MG is cytotoxic, efficient glyoxalase activity provides a survival advantage to highly glycolytic cancer cells.
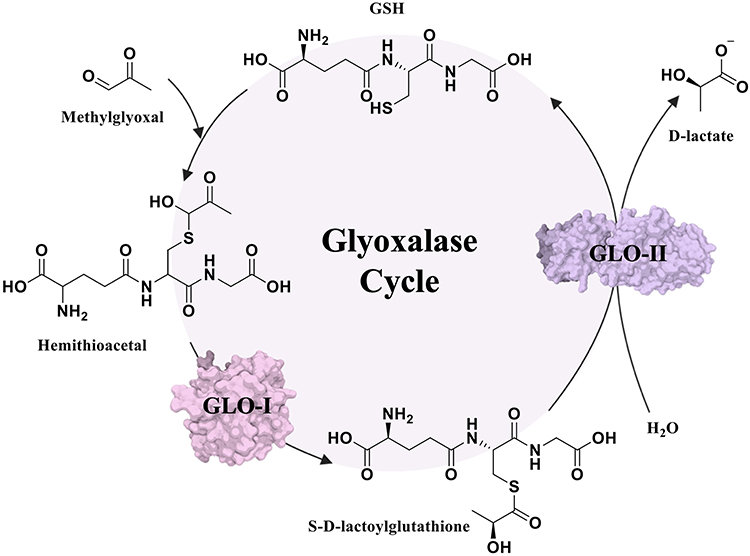 |
Figure 1 The glyoxalase system detoxification pathway. |
Consistent with this survival advantage, overexpression of GLO-I has been reported in various cancers, including breast,5 gastric,6 pancreatic,7 colon,8 prostate,9 and endometrial tumors.10 Such upregulation promotes the survival of cancer by mitigating MG-induced cytotoxicity. High levels of GLO-I have also been associated with multidrug resistance, metastasis, and poor clinical outcomes,11,12 highlighting its potential as a therapeutic target for anticancer drug development.12,13
Structurally, GLO-I is a homodimeric, zinc-dependent metalloenzyme consisting of two identical polypeptide chains of 183 amino acid residues each (Figure 2A and B).12,14 The dimer interface houses the catalytically active site and is characterized by three binding sites: a deep hydrophobic pocket, a central zinc (Zn2+) ion, and a positively charged entrance (Figure 2C).12,14 The entrance is delineated by Arg37, Arg122, Lys150, and Lys156 residues, while the Zn2+ ion is collectively stabilized by coordination with Gln33, Glu99, His126, and Glu172.12,14
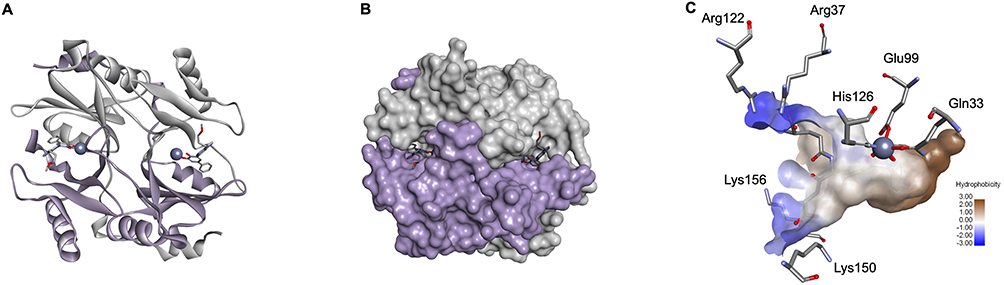 |
Figure 2 Structural Representation of Glyoxalase-I (PDB ID: 3VW9; resolution at 1.47 Å). (A) Homodimeric structures are shown in solid ribbon form, where monomer A is colored light purple and monomer B is grey. The zinc atoms are represented as a dark purple sphere. (B) Surface representation of the above-mentioned structure where co-crystallized ligands are presented in stick form. (C) Surface representation of the Glyoxalase-I Active Site (PDB ID: 1QIN; resolution at 2.00 Å, used for clearer surface visualization). Hydrophobic pocket in brown, positively charged mouth in blue, and the zinc atom in a dark purple sphere. |
Through the years, several strategies in drug discovery have been adopted to identify inhibitors of GLO-I. These compounds can be broadly divided into glutathione- and non-glutathione-based inhibitors.12 The earliest class consisted of competitive GSH analogs, of which PBBG (S-p-bromobenzylglutathione) exhibited significant potency,15 but had certain drawbacks like limited cellular permeability and rapid degradation by γ-glutamyl transferase (γ-GT).12,15 Structural modifications were subsequently introduced to rectify these limitations, resulting in more stable analogs with improved therapeutic potential, such as S-(N-aryl-N-hydroxycarbamoyl)glutathiones.16
In addition, natural flavonoids, including myricetin and luteolin,12,17 along with anthocyanidins like delphinidin, are some examples of non-glutathione-based inhibitors.18 Computer-Aided Drug Design (CADD) techniques have also been crucial in the identification and development of new GLO-I inhibitors. Al-Shar’i et al, for example, employed a structure-based pharmacophore modeling strategy and identified seven promising hits, of which the most active compound exhibited an IC50 of 0.34 μM.19
Despite these advances, conserved water molecules play an essential role in protein structure, stability, and enzyme catalysis.20 In ligand binding, they can mediate recognition by forming hydrogen-bond bridges between proteins and ligands, and can also influence binding thermodynamics when ordered water molecules are displaced and replaced by favorable ligand-protein interactions, thereby helping offset de/solvation penalties.20–22 Because of these dual roles (bridging vs displacement), the explicit treatment of crystallographic waters in structure-based workflows can influence docking scores and pose prediction, particularly when water molecules are modeled as rigid entities.
In this context, water molecules are also critical for the catalytic function of several zinc metalloenzymes.23 In GLO-I, Cameron et al reported that water contributes to catalysis by stabilizing key intermediates and facilitating proton transfer.24 Their X-ray crystallographic analysis indicated that a Zn2+-coordinated water molecule is involved in stabilizing both the transition state and the enediolate intermediate.24,25 Furthermore, two water molecules may occupy the coordination sphere of Zn2+ and can be displaced upon inhibitor binding, suggesting that they may be relevant not only for enzymatic activity but also for inhibitor recognition.24 Nonetheless, the functional significance of conserved water molecules within the GLO-I active site remains largely uninvestigated, as most of the previous structure-based studies on GLO-I removed water molecules during docking.19,26,27 Notably, Chiba et al designed 7-azaindole–substituted N-hydroxypyridones to engage an active site water network, supporting the idea that conserved waters can contribute to inhibitor recognition in GLO-I.28
Accordingly, this study aimed to identify potential GLO-I inhibitors and examine how conserved crystallographic water molecules influence pharmacophore modeling, docking pose quality, and activity prediction by comparing workflows performed with and without retained crystallographic waters. This was achieved by generating structure-based pharmacophore models with and without conserved active site water molecules and using them to virtually screen the OTAVA Lead-Like library (commercial lead-like compound library). The resulting hits were prioritized by molecular docking and binding energy calculations, and final compounds were selected based on multiple criteria, including visual inspection of binding poses/interactions and commercial availability, before being evaluated in vitro for GLO-I inhibitory activity. An overview of the study workflow is provided in Figure 3.
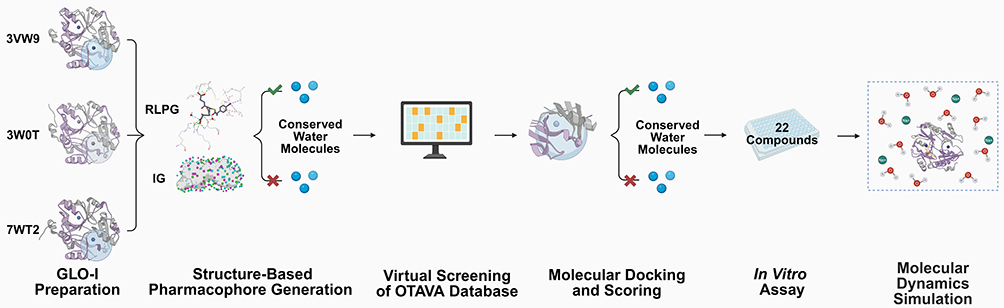 |
Figure 3 Overview of the structure-based workflow used in this study. Human GLO-I crystal structures (PDB IDs: 3VW9, 3W0T, and 7WT2) were prepared and used to generate structure-based pharmacophore models using receptor–ligand pharmacophore generation (RLPG) and interaction generation (IG) under two conditions (with and without conserved active site water molecules). The resulting models were applied to virtually screen the OTAVA library, followed by molecular docking and scoring to select 22 candidate compounds. These compounds were evaluated in vitro using a GLO-I inhibition assay. Molecular dynamics (MD) simulations were subsequently performed for the most active compound to assess binding-pose stability and the influence of conserved water molecules on binding. Created in BioRender. Isawi, (I) (2026) https://BioRender.com/olei0la. |
Materials and Methods
Computational Materials
2D chemical structures of compounds were sketched using ChemBioDraw Professional 16.0 (PerkinElmer Inc., MA). Preparation of the GLO-I crystal structures, pharmacophore generation, virtual screening, molecular docking, and binding energy calculations were conducted using Discovery Studio (DS) 2022 from BIOVIA Software Inc. (CA, USA). The commercial lead-like compound library was used for virtual screening to identify potential hits. Molecular images were generated using DS. Schematic illustrations and graphical representations were created with BioRender (Graphical Abstract and Figure 3) under a BioRender Publication License. Half-maximal inhibitory concentration (IC50) values and percent enzyme inhibition of the final hits were calculated using GraphPad Prism 8 (GraphPad Software Inc., CA). Molecular Dynamics simulations (MD) were performed using the Schrödinger Maestro software package (Schrödinger, New York, NY, USA).
Experimental Materials
The recombinant human GLO-I enzyme (rhGLO-I) was obtained from R&D Systems Corporation (USA) for in vitro assay. The final twenty-two compounds identified through virtual screening were purchased with high purity from OTAVA Chemicals (OTAVA LTD, Kyiv, Ukraine) via local vendors. All solvents and reagents used in the experiments, including mono- and di-phosphate buffers, deionized water, dimethyl sulfoxide (DMSO), glutathione, and methylglyoxal, were obtained from Acros (Thermo Fisher Scientific, New Jersey, USA) and Sigma-Aldrich Co. (USA) through local suppliers, ensuring analytical-grade quality. The biological activity of the compounds was analyzed in vitro using a double-beam UV-Vis spectrophotometer (Biotech Engineering Management Co. Ltd., UK) and a Synergy 2 UV microplate reader.
Computational Methods
Protein Selection and Preparation
Three GLO-I crystal structures with codes 3VW9 (resolution 1.47 Å), 3W0T (resolution 1.35 Å), and 7WT2 (resolution 2.00 Å) were retrieved from the Protein Data Bank (PDB).28–30 The structures were checked for missing loops, alternative conformations, or incomplete residues using the Clean Protein and Protein Report tools. The Prepare Protein protocol was used for protein preparation using default parameters, with the only change being to set the Keep Water option to True. This protocol standardizes atom names, fills missing atoms in residues, removes alternative conformations, and adjusts residue protonation states to pH 7.4.
To account for the role of water in GLO-I inhibition, water molecules within the active site were analyzed based on their proximity to the Zn2+ ion and their involvement in coordination or hydrogen bonding interactions. This evaluation was conducted using 2D interaction diagrams generated by the Receptor-Ligand Interactions tool to identify direct and water mediated contacts between the co-crystallized ligand and binding site residues. For each structure, crystallographic waters located in the binding site and prioritized based on (i) proximity to the catalytic Zn2+ ion and/or (ii) hydrogen-bonding interactions with the co-crystallized ligand were designated as conserved active site waters and retained in the “with water” workflows. Distances of the retained waters from Zn2+ are summarized in Table 1.
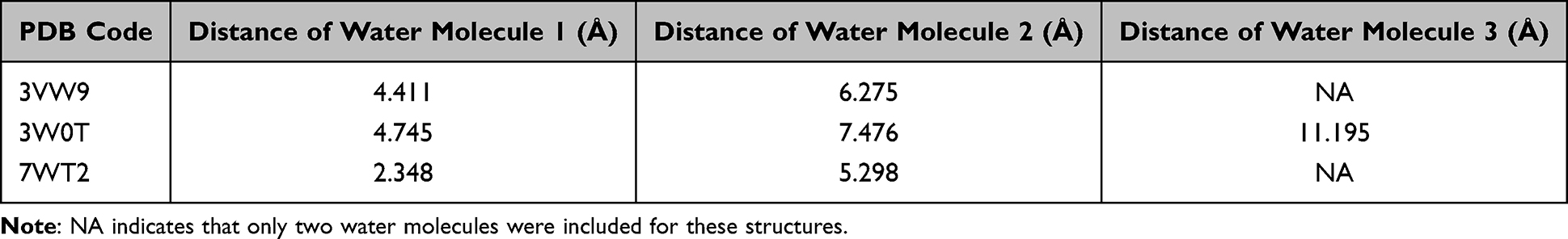 |
Table 1 Distances of Crystallographic Active Site Water Molecules from Zn2+ Ion in the Selected GLO-I Crystal Structures |
Structure-Based Pharmacophore Generation
For each crystal structure, active site pharmacophore models were generated under conditions that varied in the inclusion or exclusion of crystallographic water molecules. Two techniques were utilized: the Receptor-Ligand Pharmacophore Generation (RLPG) and the Interaction Generation (IG). The active site was defined from the co-crystallized ligand by creating a binding sphere with the Define and Edit Binding Site tool. The sphere radius was set to 12.5 Å to fit all key active site residues, including Arg37 and Arg122 located at the entrance of the cavity. Finally, a zinc-binding feature was subsequently incorporated using the Customize Pharmacophore Features tool, followed by the Add Feature from Dictionary option.
Validation of the pharmacophore models was performed using the Ligand Profiler protocol, which maps a validation set comprising known active GLO-I inhibitors and inactive/decoy compounds onto the generated pharmacophore models to compute fit scores. Subsequently, the Calculate ROC Curve protocol was utilized to assess predictive performance by ranking compounds according to fit score and plotting the true positive rate (TPR = TP/(TP+FN); sensitivity) against the false positive rate (FPR = FP/(FP+TN); 1–specificity) across thresholds, thereby indicating the ability of each pharmacophore model to discriminate between active and inactive/decoys compounds. Here, true positives (TP) were defined as known active inhibitors correctly retrieved above a given fit-score threshold, false positives (FP) as inactive/decoy compounds incorrectly retrieved above that threshold, true negatives (TN) as inactive/decoy compounds correctly not retrieved, and false negatives (FN) as known actives not retrieved. The overall discriminatory ability of each model was summarized by the area under the ROC curve (AUC), where AUC = 0.5 indicates random classification and AUC = 1.0 indicates perfect discrimination.
At the selected fit-score cutoff used to prioritize screening hits, sensitivity and specificity were recorded (Tables S1 and S2).
Receptor-Ligand Pharmacophore Generation
The RLPG protocol uses the interactions between the co-crystallized ligand and the protein to generate pharmacophore models. The parameter keep water molecules was set to True or False based on the desired condition (with or without conserved active site water molecules, respectively).
Interaction Generation
This protocol uses the defined active site to generate interaction features that complement the amino acid residues in the active site, represented as interaction maps. The generated features comprised H-bond donors, H-bond acceptors, and hydrophobic features. The Edit and Cluster Features tool was employed to identify the most significant features and to generate pharmacophore models for analysis. Exclusion spheres were incorporated according to the locations of active site residues to avoid steric clashes with the protein.
Virtual Screening
3D Library Preparation
The commercial lead-like compound library, consisting of ~90,000 compounds, was downloaded and prepared for virtual screening using the Build 3D Database protocol in DS. This protocol produced 3D conformations of each compound to facilitate accurate pharmacophore mapping. The conformation method was set to Best, and the other parameters were left at their default.
Pharmacophore-Based Virtual Screening
The prepared library underwent screening against the selected pharmacophore models utilizing the Search 3D Database, which aligns compounds to pharmacophore features and produces fit scores reflecting their compatibility. The search method was set to Best; other parameters were kept at default values. Virtual screening results were analyzed by filtering compounds based on their fit values. A threshold of ≥ 50% fit was applied; for instance, for a five-feature model, compounds with fit ≥ 2.5 were retained. This criterion ensured consistent compound selection across all pharmacophore models.
Molecular Docking
Protein solvation was performed using the Solvation protocol, which adds water molecules around the protein to simulate the aqueous conditions in biological systems. An explicit periodic boundary model was applied to add a cubic water box around the proteins, maintaining a minimum distance of 7 Å from the boundary. Counterions (Na+ and Cl−) were added according to the protein’s net charge to neutralize the system and maintain electrostatic balance. Energy minimization was then performed to relieve steric clashes and allow the protein structures to reach low-energy conformations for accurate docking.
The minimization was performed in three consecutive stages to gradually reduce strain and avoid structural deformations that could lead to artificial distortions or biologically irrelevant conformations. This was achieved using the Minimization protocol with the CHARMM force field and the Smart Minimizer algorithm under default settings. In the first step, harmonic restraints were applied only to all heavy atoms (backbone and side chains). In the second step, restraints were applied only to the backbone; then, in the final step, no restraints were used, enabling the entire protein to relax into a low-energy conformation.
Ligand preparation was performed using the Prepare Ligands protocol to generate 3D coordinates of the selected hits. Ionization states were assigned with a pH-based method (pH 7.0–7.2), and canonical tautomers were enumerated to include all possible ligand isomers. The active site of the minimized protein was defined with the Define and Edit Binding Site tool, using a binding sphere of 12.5 Å. The influence of the conserved water molecules at the active site was assessed by docking under two scenarios: (1) with conserved active site waters retained and (2) with conserved active site waters removed.
Docking was performed using the CDocker protocol with default parameters. The resulting poses were scored using -CDocker energy (-CDE), -CDocker interaction energy, and inspection of ligand-protein interactions.
As a filtration step, the top-ranked poses, defined by -CDE ≥ 10 kcal·mol−1, were selected for further analysis. This value was selected based on the relatively good binding profiles shown by compounds at this threshold.
Total Binding Energy Calculation
Prior to binding energy calculations, top docking hits were minimized in situ using In Situ Ligand Minimization protocol, which minimizes ligands in the presence of the receptor to eliminate steric clashes that may arise during docking. The CHARMM force field is used in this protocol, and the parameters are set to default.
Binding energy calculations were then conducted for both conditions, with and without active site water molecules, using the Calculate Binding Energies protocol. This protocol estimates the free binding energy of minimized protein-ligand complexes with CHARMM-based models. Ligand conformational entropy was calculated, and Poisson Boltzmann with non-polar Surface Area (PBSA) was selected as the implicit solvent model; other parameters were left at default. Compounds were evaluated based on total binding energy (TBE) values, in which more negative values indicated stronger binding, followed by visual inspection of protein-ligand interactions.
Final Compound Selection
The final compounds for experimental evaluation were chosen based on a combination of factors, including chemical structure, enzyme-interaction profiles, docking scores, and binding energy values. After filtration, the final set of compounds was purchased from commercial lead-like compound library through local suppliers.
Molecular Dynamics Simulations
MD simulations were performed using Desmond (Schrödinger Maestro) to investigate the effect of retaining or removing conserved crystallographic active site water molecules on the dynamic behavior and binding stability of compound 19, the most active hit identified from docking into the human GLO-I crystal structure (PDB: 3W0T).
The MD systems were generated from the docking poses of compound 19 in 3W0T. As described in the Protein Selection and Preparation subsection, three conserved crystallographic water molecules in the 3W0T active site were identified based on their involvement in hydrogen-bonding interactions with the co-crystallized ligand and their proximity to the catalytic Zn2⁺ ion. In the with conserved active site water system, these three water molecules were retained during docking and subsequent MD setup, whereas in the without conserved active site water system, docking was performed after removal of these water molecules. Both the with conserved active site water and without conserved active site water protein–ligand complexes were then solvated under identical conditions in an explicit TIP3P water box, such that the intended difference between the two systems was limited to the presence or absence of the three conserved crystallographic active site water molecules.
The protein–ligand complexes were prepared using the Protein Preparation Wizard, with zero-order bonds assigned to the catalytic Zn2+ ion. Each system was solvated in an orthorhombic box of TIP3P water molecules with a 10 Å buffer region, ensuring comparable solvation conditions in both setups. The systems were neutralized with Na⁺ and Cl− ions, and 0.15 M NaCl was included. Ligand parameters for compound 19 were generated using the Force Field Builder tool to ensure compatibility with the OPLS4 forcefield. The MD protocol included gradual system relaxation using the default Desmond multistep relaxation protocol under NVT and NPT conditions, followed by a 1000 ns production run performed with the OPLS4 force field at 310 K and 1.01325 bar using a 2 fs time step. Trajectory analysis was carried out using the Simulation Interaction Diagram tool to evaluate protein backbone RMSD, ligand heavy-atom RMSD, and the persistence of protein–ligand interactions in both systems. To further assess the energetic impact of conserved water molecules on ligand binding, post-MD Molecular Mechanics Generalized Born Surface Area (MM-GBSA) calculations were performed using Prime in the Schrödinger Maestro suite. Binding free energy was evaluated from molecular mechanics energy contributions and solvation free energy terms, without inclusion of entropic contributions. The solvation term comprised polar Generalized Born and nonpolar surface area components. In the present study, the solvation/desolvation term was examined to compare differences in solvation free-energy contribution between the systems with and without conserved active site water molecules. Calculations were performed on the final 500 ns of each 1000 ns trajectory.
Experimental Methods
In vitro Enzyme Assay
The inhibitory activity of the selected compounds against rhGLO-I was evaluated according to the manufacturer’s protocol (R&D Systems, Minneapolis, MN, USA). The enzyme was reconstituted by dissolving it in sterile deionized water to a final concentration of 0.5 mg·mL−1 and stored at −80°C until use. On the day of the assay, the enzyme was thawed and used immediately.
Absorbance measurements were recorded at a wavelength of 240 nm for 200 s at 25°C using a double-beam UV-Vis spectrophotometer using a Synergy 2 UV microplate reader. Test compounds were dissolved in DMSO to prepare a 10 mM stock solution. Each compound was tested across different concentrations in triplicate, and inhibitory activity was evaluated by measuring the percent inhibition at 50 μM and by determining IC50 values using GraphPad Prism 8. IC50 values were calculated by plotting the logarithm of compound concentrations against the average percent inhibition. Myricetin, with a reported IC50 of 3.38 ± 0.41 μM,19 was used as a positive control.
For each well, absorbance was measured at 0 and 200 s. The mean absorbance of blank wells was subtracted from the corresponding mean absorbance values of enzyme and compound wells at both time points. Percent GLO-I inhibition was calculated as:
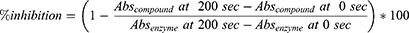
Abs: Absorbance
Results and Discussion
Protein Selection and Evaluation
In this study, the main goal was to employ a structure-based drug design (SBDD) approach to identify novel GLO-I inhibitors, with particular emphasis on evaluating the role of conserved active site water molecules. To capture active site variability relevant to inhibitor selection, three crystal structures were used, reducing dependence on a single receptor conformation and allowing pharmacophore generation and docking to capture multiple plausible interaction patterns.
Therefore, selecting appropriate crystal structures was critical. The selection criteria included the availability of a co-crystallized ligand, the presence of well-defined interactions within the active site, and a high crystallographic resolution (≤ 2.5 Å). In addition, the selected templates differed in metal-proximal hydration (Table 1), bound-ligand chemotypes, and structure resolution. Specifically, 3VW9 (1.47 Å) and 3W0T (1.35 Å) represent GLO-I complexes with N-hydroxypyridone derivative inhibitors, which coordinate Zn2+ through the two oxygen atoms of the hydroxypyridone moiety, whereas 7WT2 (2.00 Å) is bound to a TLSC702 derivative that coordinates Zn2+ via a carboxylate oxygen, which can promote distinct interaction patterns within the active site. Consistent with the value of multi-structure modeling, top ROC-performing models originated from different templates (eg, RLPG Model 2 from 3VW9 and IG Model 19 from 3W0T).
Across the three structures, the Zn2+-proximal hydration pattern differed: the closest retained water in 7WT2 was located 2.348 Å from Zn2+, whereas the closest retained waters in 3VW9 and 3W0T were at 4.411 Å and 4.745 Å, respectively (Table 1). This indicates a more tightly associated metal-proximal water site in 7WT2, which may influence whether ligands are stabilized by water-mediated bridges or instead displace these waters upon binding.
Figure 4A shows a 3D representation of the active site of 3VW9, where two water molecules are located near Zn2+and form H-bonds with the co-crystallized ligand. These interactions suggest that these water molecules may contribute to ligand stabilization within the binding site (Figure 4B).
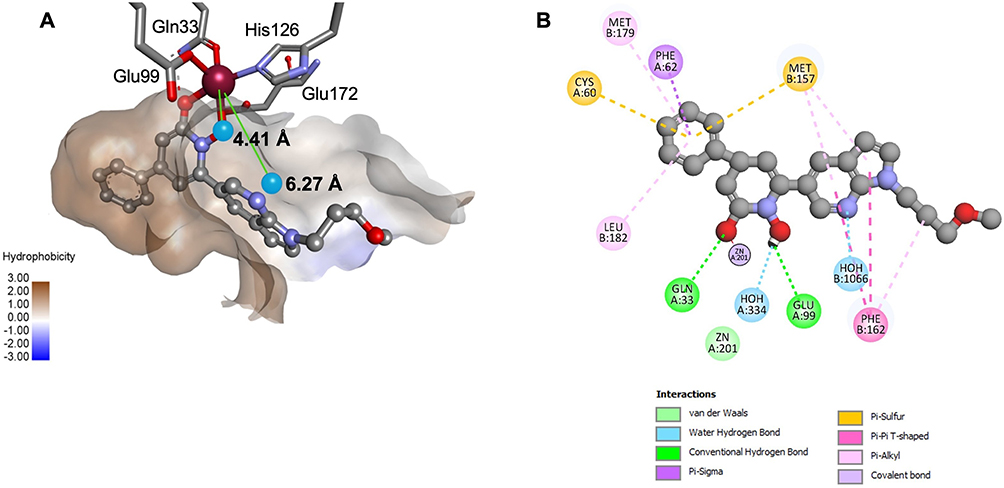 |
Figure 4 Water molecules and their interactions within the GLO-I active site (PDB ID: 3VW9, 1.47 Å). (A) 3D representation showing water molecules and their distances from the Zn2+ (in Å). (B) 2D interaction diagram highlighting H-bonds between water molecules and the co-crystallized ligand. |
Structure-Based Pharmacophore Modeling
Receptor-Ligand Pharmacophore Models and ROC Performance
The RLPG protocol was applied under two conditions: with or without crystallographic active site water molecules. For each protein–ligand complex, ten pharmacophore models were generated. Each model was subsequently modified to incorporate a zinc binding feature (ZBF) to account for the critical role of the Zn2+ in ligand coordination and binding stability.
Pharmacophore models were selected based on a combination of criteria, including feature diversity, representation of key binding-site interactions, and the area under the ROC curve (AUC) value. Based on these parameters, a total of nine pharmacophore models were shortlisted, with AUC values ranging from 0.856 to 0.951 (Table S1 and Figure S1). The highest AUC (0.951) was achieved by pharmacophore model 2 derived from PDB: 3VW9 with conserved active site water molecules included (Figure 5).
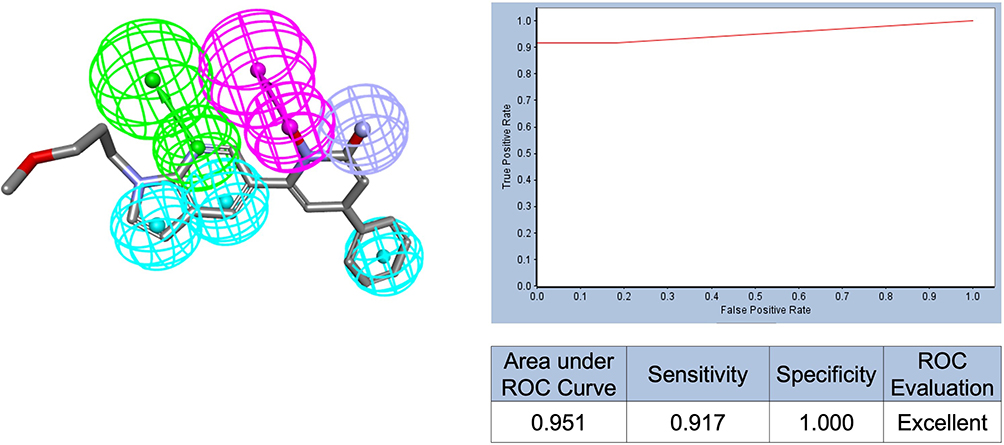 |
Figure 5 Best-performing pharmacophore model generated by RLPG (Model 2), aligned with the co-crystallized ligand, along with its corresponding ROC curve. Feature colors: HBA (green), HBD (magenta), ZBF (light purple), HA (blue). Exclusion spheres are omitted for clarity. |
Interaction Generation Pharmacophore Models and ROC Performance
The IG protocol was used as an additional approach to ensure comprehensive coverage of key ligand–protein interaction features. Following the generation of features using Ludi interaction maps, the Edit and Cluster Features tool was used to construct final models, emphasizing features corresponding to residues critical for binding. All models were subsequently modified to incorporate customized ZBFs, consistent with the RLPG workflow.
As with RLPG, two conditions were tested, with and without crystallographic active site water molecules. Model selection followed the same criteria, focusing on feature relevance and ROC AUC performance. Ten pharmacophore models were retained, with AUC values from 0.818 to 0.955 (Table S2 and Figure S2). The best-performing model (Model 19), derived from the PDB: 3W0T structure with conserved active site water molecules included, achieved the highest AUC score (0.955) (Figure 6).
 |
Figure 6 Best-performing pharmacophore model generated by IG (Model 19), with its ROC curve. Feature colors: HBA (green), HBD (magenta), ZBF (light purple), HA (blue). Exclusion spheres are omitted for clarity. |
Collectively, the best RLPG model with conserved active site water molecules (Model 2; AUC = 0.951) and the best IG model with conserved active site water molecules (Model 19; AUC = 0.955) demonstrated a strong ability to rank known actives ahead of inactive/decoy compounds, indicating robust discrimination beyond random behavior. Importantly, AUC was interpreted alongside the TPR/FPR trade-off; at low FPR (high specificity), these models maintained elevated TPR (high sensitivity), supporting their suitability for practical virtual screening where only a small top-ranked fraction of compounds is prioritized for docking and purchase. For example, RLPG Model 2 achieved sensitivity = 0.917 and specificity = 1.000 at the selected cutoff (Table S1).
Virtual Screening Results
All selected pharmacophore models, generated under both sets of conditions, with and without conserved active site water molecules, were used to screen the commercial lead-like compound library using the Search 3D Database protocol. The library offers compounds with drug-like properties, complying with Lipinski’s Rule of Five (molecular weight < 500 Da, LogP < 5, hydrogen bond donors < 5, hydrogen bond acceptors < 10), and also having ≤ 10 rotatable bonds. Compliance was validated using the Filter by Lipinski and Veber Rules protocol; hence, no additional physicochemical filtration was necessary.
A fit value threshold of ≥ 50% was applied to filter the screening hits. In cases where multiple pharmacophore models existed under the same condition, only compounds common to all models were retained. These filtered compounds were subsequently selected for molecular docking studies.
Molecular Docking and Total Binding Energy Calculation
Docking of the screened hits was carried out using the CDocker under two parallel conditions: with and without conserved active site water molecules. Interestingly, whether or not water molecules were present had a noticeable impact on the docking outcomes, including -CDE scores, ligand binding poses, and interaction profiles. In most instances, docking with conserved active site water molecules improved the docking scores, likely due to favorable water-mediated H-bonds. However, in some cases, the absence of water improved performance, particularly when the presence of water disrupted key interactions or introduced unfavorable donor-donor contacts. For further analysis, only compounds with a -CDE value greater than or equal to 10 kcal·mol−1 were selected for TBE calculations.
TBE served as an additional scoring metric to estimate the overall binding affinity of each compound. Generally, across different structures, the inclusion of water molecules resulted in more favorable (ie, more negative) TBE values, indicative of water-mediated hydrogen bonding contributing to a more stable protein-ligand complex.
For instance, RLPG-derived hits for the 7WT2 structure gave the most favorable TBE when water was included: −160.0 kcal·mol−1, compared to −89.96 kcal·mol−1 without water. By contrast, water had little effect on RLPG-derived hits against the 3VW9 structure, while omission of water improved binding affinity for IG-derived hits: 134.74 versus −36.64 kcal·mol−1. These findings indicate that conserved water molecules play a context-dependent role in stabilizing ligand binding.
Final Hit Selection
Selection of the compounds for in vitro testing was guided by an integrated assessment approach, including -CDE scores, TBE values, key protein-ligand interactions (especially those involving Zn2+), and chemical diversity. Initial filtering considered docking performance and binding potential to Zn2+. From there, representative compounds were selected from each scenario based on their structural uniqueness and their relevance as potential GLO-I inhibitors.
Notably, most of the selected compounds possessed a carboxylate group, which either directly coordinated to the catalytic Zn2+ ion or formed favorable electrostatic interactions with the positively charged entrance of the binding pocket. Final selections from both docking conditions, with and without conserved active site water molecules, were chosen to ensure broad modeling coverage. A higher preference was given to compounds which performed well under both conditions. Some compounds were excluded based on vendor-reported quality control issues, thus giving a total of 22 compounds. These were then purchased and assayed for GLO-I inhibitory activity using an in-house developed in vitro assay31 (Table 2).
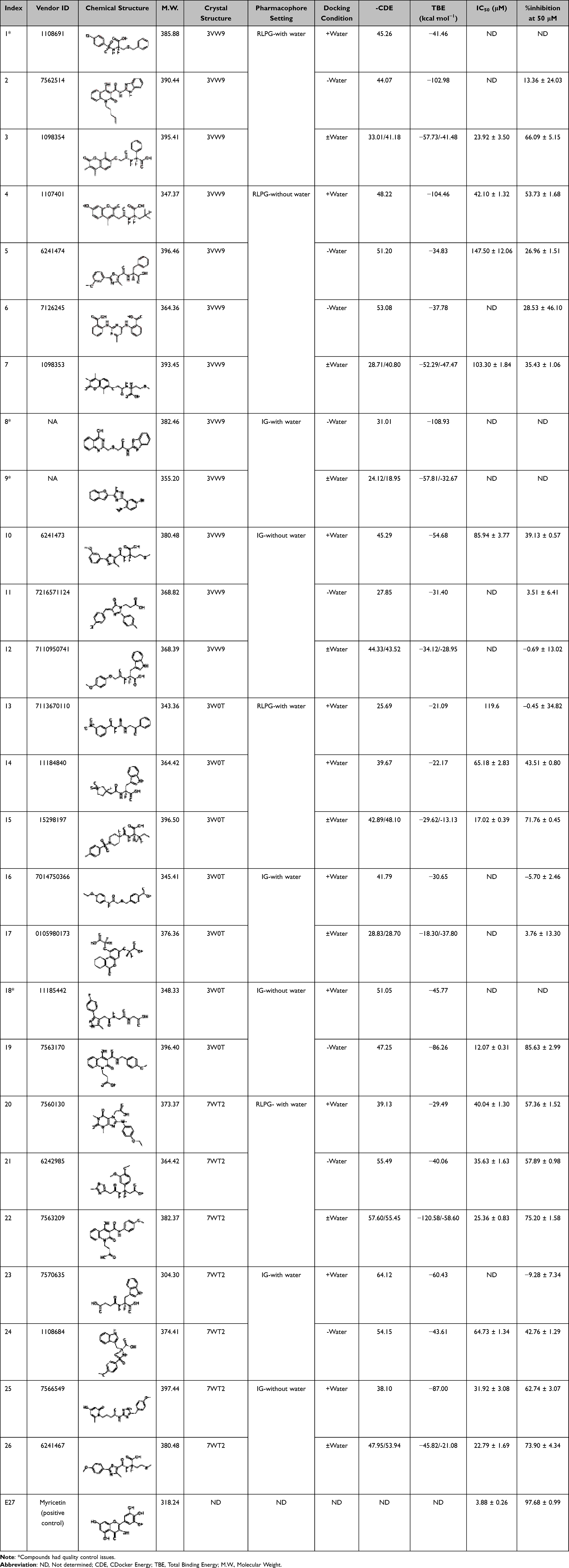 |
Table 2 Final Set of Selected Compounds with Their Chemical Structures, Docking and Binding Energy Scores Derived from PDB 3VW9, 3W0T, and 7WT2 Under Conditions with and without Conserved Active-Site Water Molecules, Together with Their Biological Screening Results |
Experimental Analysis
In vitro Biological Evaluation
Among the 22 compounds tested, five exhibited promising inhibitory activity, with IC50 values below 30 μM and percent inhibition ranging from 66% to 85% at 50 μM (Table 2).
Although the identified hits were less potent than the positive control myricetin (IC50 = 3.88 ± 0.26 μM), the active compounds showed reproducible low-micromolar inhibition (IC50 = 12.07–25.36 μM) and are therefore best viewed as lead-like starting points rather than optimized inhibitors. This is consistent with an early hit-identification workflow using a lead-like library, particularly as these compounds represent new scaffolds distinct from benchmark flavonoids. The computational workflow (pharmacophore validation and docking) supports specific binding and provides a structure-guided basis for hit-to-lead optimization to improve potency toward the control benchmark.
Analysis of the Most Active Compound
Compound 19 (Vendor ID 7563170) emerged as the most potent compound with an IC50 value of 12.07 μM and an 85.63% inhibition at 50 μM. It was obtained from an IG-derived pharmacophore model based on the 3W0T crystal structure under without water conditions, suggesting that the presence of water may not be necessary for strong inhibitory activity in this case and that direct interactions with the enzyme may be sufficient. Docking results correlated with the experimental findings, yielding a -CDE of 47.25 and a TBE of −86.26 kcal·mol−1. Visual inspection showed that compound 19 engages with all three key binding regions of the GLO-I active site (Figure 7A), forming several strong interactions. The p-methoxyphenyl group is embedded within the hydrophobic pocket, forming multiple hydrophobic contacts, including π-π T-shaped interactions with Phe62. The 4-hydroxy group of the 1,2-dihydroquinoline-3-carboxamide moiety coordinates and electrostatically interacts with Zn2+, likely facilitated by tautomerization-induced ionization. Furthermore, the terminal carboxylate engages the positively charged entrance of the active site via electrostatic interaction with Arg37 and an H-bond with Asn103, collectively contributing to complex stabilization (Figure 7B).
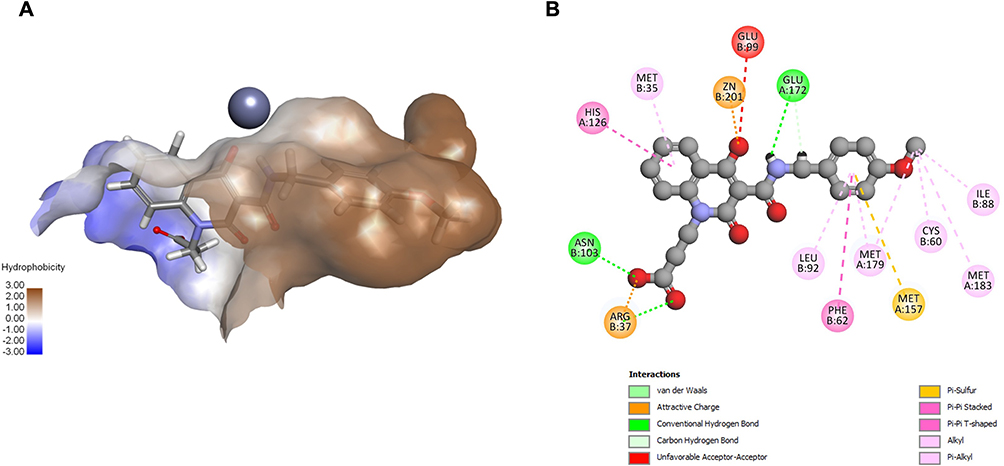 |
Figure 7 Visual representation of the most potent compound 19 within the GLO-I active site of PDB ID: 3W0T without conserved active site water molecules. (A) 3D representation of the docking pose. (B) 2D interaction diagram. |
Effect of Water on Activity Prediction
The primary objective of our structure-based approach was to evaluate the impact of conserved active site water molecules on activity prediction by comparing docking outcomes obtained with and without conserved active site waters. To illustrate the strengths and limitations of each docking setup, we highlight two active compounds (15 and 26) and one inactive compound (12) that exhibited clear pose differences between water-inclusion and no water docking.
Although water-inclusive docking often produced higher scores, detailed analysis of binding poses, and interaction profiles revealed that no-water docking generally aligned better with activity trends.
For example, compound 15, the second most active compound, (IC50 =17.02 μM; 71.76% inhibition at 50 μM) was identified from an RLPG-derived model based on the 3W0T structure and exhibited a favorable binding profile under both conditions. Interestingly, while water-inclusive docking yielded superior numerical scores, the without water configuration generated biologically more relevant poses. As shown in Figure 8A, the water-inclusive docking pose positioned the carboxylate in direct coordination with Zn2+, yielding a stronger interaction than the metal-acceptor interaction in the without water condition. However, it formed only limited interactions with the other binding regions, including minimal hydrophobic interactions and no interactions with the positive entrance residues. In contrast, the without water pose (Figure 8B) exhibited a more optimal binding pose, which allowed compound 15 to improve hydrophobic packing, while the carboxylate group engaged with the positively charged entrance residues, specifically, an electrostatic interaction with Arg37 and an H-bond with His126, features associated with potent GLO-I inhibition.
 |
Figure 8 Comparison of compound 15 docking poses and interactions within the active site of PDB ID: 3W0T. (A) With conserved active site water molecules. (B) Without conserved active site water molecules. |
A similar trend occurred for compound 26 (IC50 = 22.8 μM; 73.9% inhibition at 50 μM). As shown in Figure 9A, the inclusion of water revealed that the compound exhibited a poor fit, failing to align properly with the hydrophobic pocket and losing key interactions in that area. This phenomenon likely resulted from steric hindrance imposed by the presence of water molecules, which prevented the ligand from fully accessing the hydrophobic pocket and forming essential interactions. In contrast, without water (Figure 9B), the ligand fits optimally within the pocket, establishing multiple stabilizing interactions, including an additional H-bond with Arg37 at the entrance.
 |
Figure 9 Comparison of compound 26 docking poses and interactions within the GLO-I active site of PDB ID: 7WT2. (A) With conserved active site water molecules. (B) Without conserved active site water molecules. |
Even among inactive compounds, differences between water-inclusive and water-excluded docking were evident. For example, compound 12 showed higher binding energies with conserved active site water and a more favorable and realistic pose: the indole occupied the hydrophobic pocket, and the carboxylate established metal-acceptor and ionic interactions with Zn2+, plus H-bonds with Glu99, Gln33, and Arg37 (Figure 10A). In contrast, without conserved active site water (Figure 10B), the pose was less optimal, in which the amide and ether oxygen atoms coordinated with Zn2+ via weaker metal-acceptor interactions, and the indole ring failed to fully occupy the hydrophobic pocket. Collectively, this explains its weak activity in vitro, which was better predicted in the without water setup.
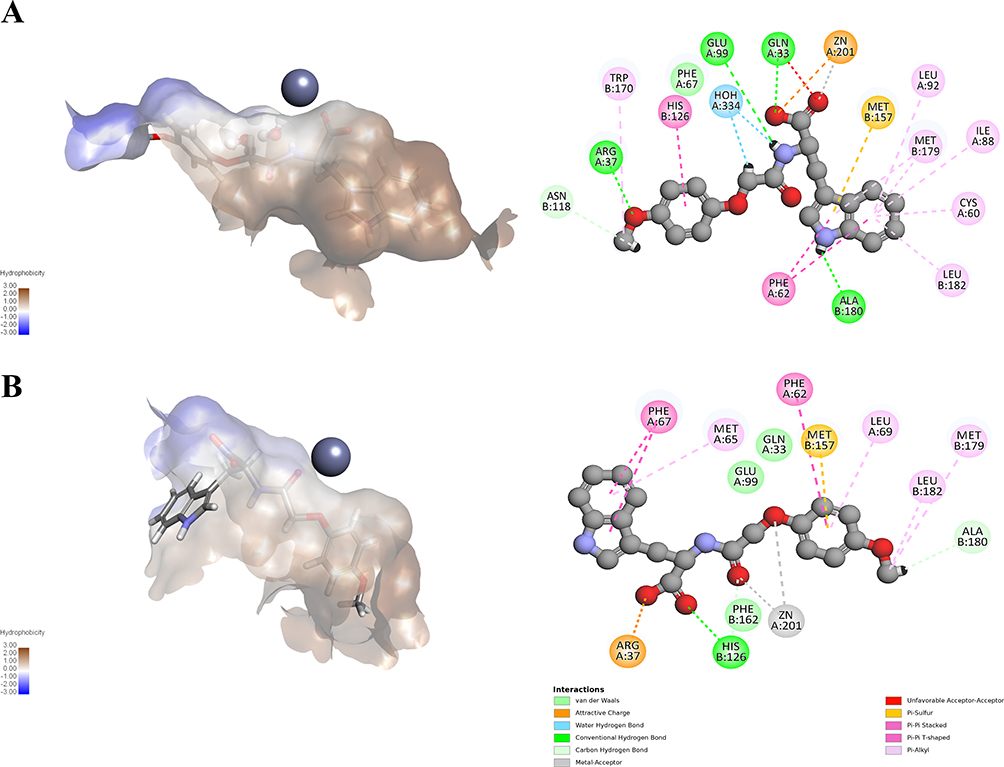 |
Figure 10 Comparison of compound 12 docking poses and interactions within the GLO-I active site of PDB ID: 3VW9. (A) With conserved active site water molecules. (B) Without conserved active site water molecules. |
Overall, these examples highlight the novelty of our workflow: although water inclusion can improve numerical scores via water-mediated contacts, the without water setup more consistently produced chemically and biologically plausible poses that engaged the hydrophobic pocket and positively charged entrance in a manner that better tracked experimental activity.
Docking and Binding Energy Correlation with Experimental Activity
To provide a quantitative comparison between the two docking setups, we assessed the association between docking metrics and experimental activity using Spearman rank correlation across compounds with available data. For the with conserved active site water condition, both -CDE and TBE showed weak, non-significant associations with inhibitory activity (-CDE: ρ = 0.143, n = 15, p = 0.612; TBE: ρ = −0.318, n = 15, p = 0.248). In contrast, for the without conserved active site water condition, -CDE showed a moderate positive association with % inhibition at 50 μM (ρ = 0.547, n = 14, p = 0.043), whereas TBE showed a weak, non-significant negative association (ρ = −0.235, n = 14, p = 0.418). A Spearman rank correlation analysis between docking metrics and % inhibition at 50 μM is provided in Table 3. These results indicate that the without conserved active site water docking workflow showed better agreement with experimental activity than the with conserved active site water workflow.
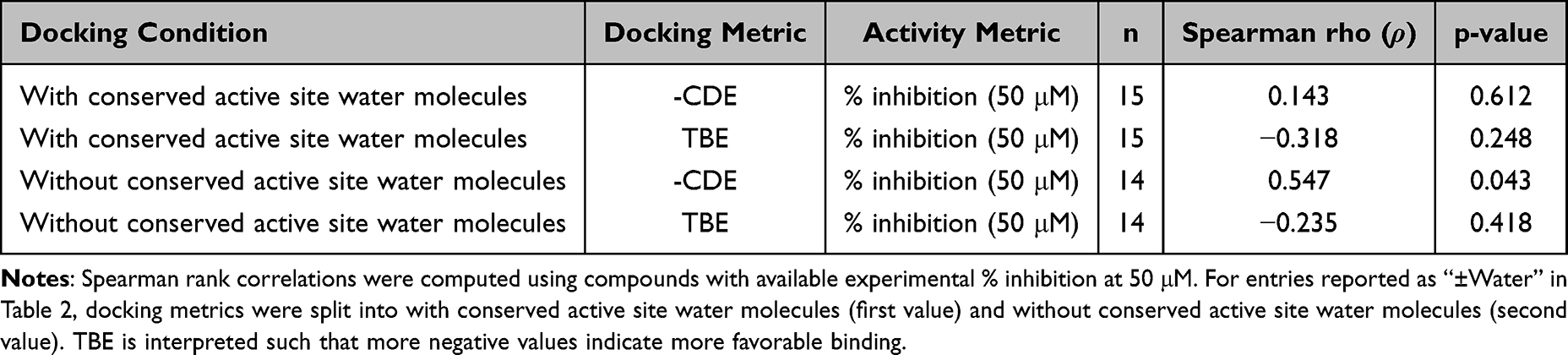 |
Table 3 Correlation Between Docking Metrics and Experimental Activity |
Molecular Dynamics Stability and Hydration Analysis
MD simulations were performed to examine the dynamic behavior of compound 19 in the GLO-I active site in two systems derived from docking into 3W0T: a with conserved active site water system, in which the three conserved crystallographic active site water molecules were retained, and a without conserved active site water system, in which these three water molecules were removed prior to docking and subsequent MD setup. Both systems were subsequently solvated under identical explicit-solvent conditions; therefore, the comparison reflects the effect of the conserved active site crystallographic water molecules rather than the absence of bulk solvent.
Protein backbone and ligand RMSD values were monitored over 1000 ns to assess complex stability. As shown in Figure 11A and B, the protein backbone remained relatively stable in both systems, with RMSD values of 2.992 ± 0.485 Å for the with conserved active site water system and 2.763 ± 0.281 Å for the without conserved active site water system. In contrast, ligand RMSD analysis revealed clearer differences between the two conditions. The ligand displayed greater deviation in the with conserved active site water system, with an average RMSD of 1.857 ± 1.228 Å, compared with 1.114 ± 0.232 Å in the without conserved active site water system. Notably, the with conserved active site water system showed a marked increase in ligand fluctuation near ~850 ns, indicating late binding-mode rearrangement, whereas the without conserved active site water system maintained a more stable ligand pose throughout the simulation.
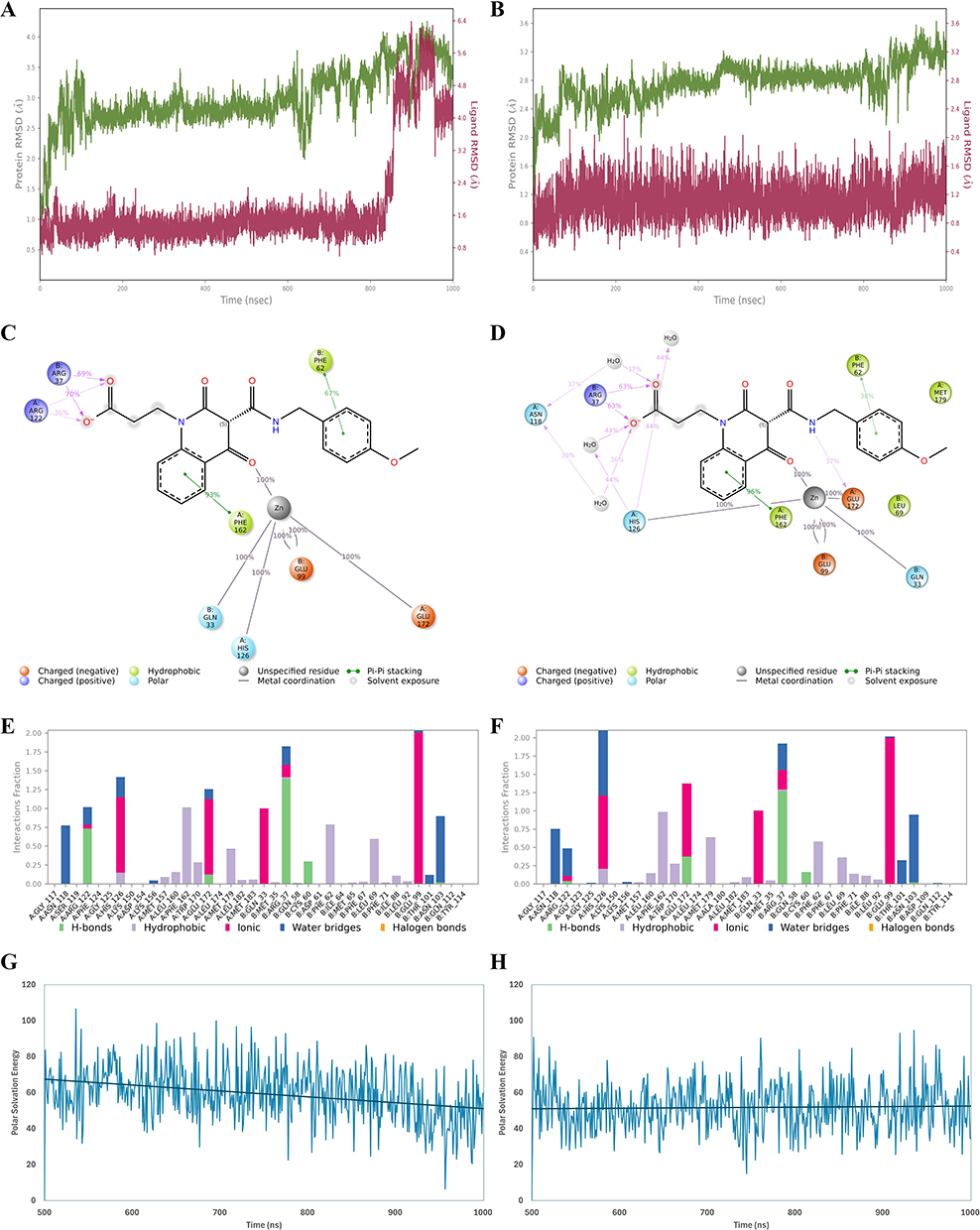 |
Figure 11 Molecular dynamics (MD) stability, interaction persistence, and MM/GBSA polar solvation contribution for the GLO-I/compound 19 complex over 1000 ns with conserved active site water molecules (left) and without conserved active site water molecules (right). (A and B) Time evolution of protein backbone RMSD (green) and ligand heavy-atom RMSD (magenta) during the 1000 ns simulations for the with and without conserved active site water molecule systems, respectively. (C and D) Schematic representative 2D interaction diagrams highlighting the persistent protein–ligand contacts in the with and without conserved active site water molecule systems, respectively; only interactions present for >30% of the simulation time are shown. (E and F) Protein–ligand interaction fractions across the trajectory for the with and without conserved active site water molecule systems, respectively; categorized into four types: Hydrogen Bonds, Hydrophobic, Ionic and Water Bridges. (G and H) MM/GBSA polar solvation energy contribution (ΔG_solv, GB) calculated over the last 500 ns (500–1000 ns) for the with and without conserved active site water molecule systems, respectively. |
Interaction analysis indicated that both systems retained coordination with the Zn2+ ion, together with several protein–ligand contacts during the simulation (Figure 11C–F). However, notable differences in interaction profiles were observed. In the without conserved active site water system, compound 19 formed more sustained direct contacts with important active site residues, including Glu172 and His126, along with more frequent water-bridge interactions (Figure 11D and F). Importantly, these solvent-mediated contacts arose from bulk solvent water molecules entering the binding site during MD rather than from the three crystallographic water molecules removed prior to docking. In contrast, in the with conserved active site water system, the interaction pattern differed, with reduced direct contacts involving Glu172 and His126, fewer water-bridge interactions, and higher occupancy of Arg122 interactions at the pocket entrance (Figure 11C and E). These differences are consistent with the lower ligand RMSD and greater pose stability observed for the without conserved active site water molecules.
To further evaluate energetic effects, the MM-GBSA polar solvation term, used here as an indicator of the solvation/desolvation contribution, was analyzed. The with conserved active site water system exhibited a higher average MM-GBSA polar solvation value (59.425 ± 15.130 kcal/mol) than the without conserved active site water system (51.911 ± 12.966 kcal/mol) (Figure 11G and H), suggesting a less favorable solvation contribution to binding in the with conserved active site water system.
Overall, these findings suggest that, for GLO-I, removal of the conserved crystallographic active site water molecules prior to docking yielded a more stable ligand-binding model for compound 19, and the subsequent MD simulations supported the greater stability of this binding model.
Overall Assessment
Across all analyzed compounds, docking without conserved crystallographic active site water molecules consistently produced binding poses and interaction profiles that aligned more closely with experimental inhibitory activity, as it allowed the compounds to adopt their favorable binding orientations without any structural or functional constraints produced by these water molecules.
In contrast, docking performed with active site water molecules often led to inconsistent predictions, failing to accurately reflect the actual binding modes of ligands, and sometimes generating unrealistic interaction profiles. This is likely due to artificial stabilization introduced by fixed water molecules.
These findings can be explained by the fact that, under physiological conditions, water molecules present in the active site are frequently displaced when a ligand binds.32 As a result, including them in docking protocols can be unnecessary—or even misleading—by exaggerating specific interactions and inflating binding scores that do not truly reflect ligand behavior.
As a result, docking without conserved active site water molecules appears to offer more reliable predictions of biological activity and is better suited for virtual screening and lead optimization in the discovery of GLO-I inhibitors.
Future work will focus on hit-to-lead optimization of the most active scaffolds; subsequently, optimized analogs will be evaluated in cell-based assays to assess cellular activity and further validate these leads.
Conclusion
An SBDD strategy was employed to identify potential GLO-I inhibitors while assessing the influence of conserved active site water molecules on ligand recognition and binding. Three high-resolution crystal structures of human GLO-I were utilized to ensure a comprehensive assessment of the enzyme’s active site.
Structure-based pharmacophore models were generated under two conditions, with and without active site water molecules, and subsequently used for virtual screening of the commercial lead-like compound library. The resulting hits were further refined through molecular docking under both water-inclusive and no-water conditions. Based on docking scores, binding energies, interaction profiles, and chemical diversity, 22 compounds were selected for in vitro biological evaluation.
Of these, five compounds demonstrated notable inhibitory activity, with compound 19 identified as the most potent, showing an IC50 of 12.07 μM and 85.6% inhibition at 50 μM. Comparative analysis revealed that the inclusion of water molecules during docking significantly affected ligand orientation and interaction patterns, often producing artificially high docking scores and distorted poses. MD simulations of compound 19 further supported the docking results, showing that excluding conserved water molecules yielded a more favorable interaction profile and a lower solvation/desolvation penalty than the water-retained system.
Taken together, the results suggest that, for GLO-I, omission of conserved crystallographic active-site water molecules during docking setup offers a more reliable strategy for predicting ligand binding, with MD simulations further supporting the stability of the resulting binding mode under explicit-solvent conditions. This insight offers valuable guidance for future virtual screening and lead optimization, highlighting the importance of context-dependent treatment of water molecules in SBDD.
Acknowledgments
The authors used ChatGPT (OpenAI) for language editing of the manuscript; all scientific content and interpretations were verified by the authors.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by the Deanship of Scientific Research at Jordan University of Science and Technology, grant number 192/2024.
Disclosure
The authors report there are no conflicts of interest in this work.
References
1. Cancer Tomorrow. Available from: https://gco.iarc.who.int/tomorrow/en/dataviz/trends?multiple_populations=1.
2. Li C, Zhang G, Zhao L, Ma Z, Chen H. Metabolic reprogramming in cancer cells: glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer. World J Surg Oncol. 2016;14(1). doi:10.1186/S12957-016-0769-9
3. Phillips SA, Thornalley PJ. The formation of methylglyoxal from triose phosphates: investigation using a specific assay for methylglyoxal. Eur J Biochem. 1993;212(1):101–23. doi:10.1111/J.1432-1033.1993.TB17638.X
4. Thornalley PJ. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J. 1990;269(1):1–11. doi:10.1042/bj2690001
5. Rulli A, Carli L, Romani R, et al. Expression of glyoxalase I and II in normal and breast cancer tissues. Breast Cancer Res Treat. 2001;66(1):67–72. doi:10.1023/A:1010632919129
6. Cheng WL, Tsai MM, Tsai CY, et al. Glyoxalase-I is a novel prognosis factor associated with gastric cancer progression. PLoS One. 2012;7(3):e34352. doi:10.1371/journal.pone.0034352
7. Wang Y, Kuramitsu Y, Ueno T, et al. Glyoxalase I (GLO1) is up-regulated in pancreatic cancerous tissues compared with related non-cancerous tissues. Anticancer Res. 2012;32(8):3219–3222.
8. Chen Y, Fang L, Li G, et al. Synergistic inhibition of colon cancer growth by the combination of methylglyoxal and silencing of glyoxalase I mediated by the STAT1 pathway. Oncotarget. 2017;8(33):54838–54857. doi:10.18632/oncotarget.18601
9. Burdelski C, Shihada R, Hinsch A, et al. High-level Glyoxalase 1 (GLO1) expression is linked to poor prognosis in prostate cancer. Prostate. 2017;77(15):1528–1538. doi:10.1002/pros.23431
10. Jiang Y, Chen X, Wei Y, Feng Y, Zheng W, Zhang Z. Metformin sensitizes endometrial cancer cells to progestin by targeting TET1 to downregulate glyoxalase I expression. Biomed Pharmacother. 2019;113. doi:10.1016/j.biopha.2019.108712
11. Tiku NAB. Oxidative Stress and Glyoxalase Pathway in Cancer. In: Handbook of Oxidative Stress in Cancer: Mechanistic Aspects. Vol. 1. Springer Nature;2022:119–136. doi:10.1007/978-981-15-9411-3_12
12. Al-Balas QA, Hassan MA, Al-Shar’i NA, Al Jabal GA, Almaaytah AM. Recent advances in Glyoxalase-I inhibition. Mini-Rev Med Chem. 2019;19(4):281–291. doi:10.2174/1389557518666181009141231
13. Kim JY, Jung JH, Lee SJ, Han SS, Hong SH. Glyoxalase 1 as a therapeutic target in cancer and cancer stem cells. Mol Cells. 2022;45(12):869–876. doi:10.14348/molcells.2022.0109
14. Ridderström M, Cameron AD, Jones TA, Mannervik B. Involvement of an active-site Zn2+ Ligand in the catalytic mechanism of human Glyoxalase I. J Biol Chem. 1998;273(34):21623–21628. doi:10.1074/jbc.273.34.21623
15. Vince R, Wadd WB, Daluge S. Studies on the inhibition of glyoxalase I by S-Substituted glutathiones. J Med Chem. 1971;14(5):402–404. doi:10.1021/JM00287A006
16. Hamilton DS, Creighton DJ. Inhibition of glyoxalase I by the enediol mimic S-(N-hydroxy-N-methylcarbamoyl)glutathione. The possible basis of a tumor-selective anticancer strategy. J Biol Chem. 1992;267(35):24933–24936.
17. Takasawa R, Takahashi S, Saeki K, Sunaga S, Yoshimori A, Ichi TS. Structure-activity relationship of human GLO I inhibitory natural flavonoids and their growth inhibitory effects. Bioorg Med Chem. 2008;16(7):3969–3975. doi:10.1016/J.BMC.2008.01.031
18. Takasawa R, Saeki K, Tao A, et al. Delphinidin, a dietary anthocyanidin in berry fruits, inhibits human glyoxalase i. Bioorg Med Chem. 2010;18(19):7029–7033. doi:10.1016/J.BMC.2010.08.012
19. Al-Shar’i NA, Al-Balas QA, Al-Waqfi RA, Hassan MA, Alkhalifa AE, Ayoub NM. Discovery of a nanomolar inhibitor of the human glyoxalase-I enzyme using structure-based poly-pharmacophore modelling and molecular docking. J Comput Aided Mol Des. 2019;33(9):799–815. doi:10.1007/s10822-019-00226-8
20. de Beer S, Vermeulen N, Oostenbrink C. The role of water molecules in computational drug design. Curr Top Med Chem. 2010;10(1):55–66. doi:10.2174/156802610790232288
21. Kumar A, Zhang KYJ. Investigation on the effect of key water molecules on docking performance in CSARdock exercise. J Chem Inf Model. 2013;53(8):1880–1892. doi:10.1021/ci400052w
22. Poornima CS, Dean PM. Hydration in drug design. 3. Conserved water molecules at the ligand-binding sites of homologous proteins. J Comput Aided Mol Des. 1995;9(6):521–531. doi:10.1007/BF00124323
23. McCall KA, chin HC, Fierke CA. Function and mechanism of zinc metalloenzymes. J Nutr. 2000;130(5):1437S–1446S. doi:10.1093/jn/130.5.1437S
24. Cameron AD, Ridderström M, Olin B, Kavarana MJ, Creighton DJ, Mannervik B. Reaction mechanism of glyoxalase I explored by an x-ray crystallographic analysis of the human enzyme in complex with a transition state analogue. Biochemistry. 1999;38(41):13480–13490. doi:10.1021/bi990696c
25. Sellin S, Rosevear PR, Mannervik B, Mildvan AS. Nuclear relaxation studies of the role of the essential metal in glyoxalase I. J Biol Chem. 1982;257(17):10023–10029.
26. Al-Balas Q, Hassan M, Al-Oudat B, Alzoubi H, Mhaidat N, Almaaytah A. Generation of the first structure-based pharmacophore model containing a selective “zinc binding group” feature to identify potential glyoxalase-1 inhibitors. Molecules. 2012;17(12):13740–13758. doi:10.3390/molecules171213740
27. Al-Sha’er MA, Al-Balas QA, Hassan MA. Combined high throughput screening with QSAR analysis unravelling potential glyoxalase-i inhibitors. Curr Comput Aided Drug Des. 2021;16(6):814–832. doi:10.2174/1573409916666200117100326
28. Chiba T, Ohwada J, Sakamoto H, et al. Design and evaluation of azaindole-substituted N-hydroxypyridones as glyoxalase i inhibitors. Bioorg Med Chem Lett. 2012;22(24):7486–7489. doi:10.1016/j.bmcl.2012.10.045
29. Usami M, Ando K, Shibuya A, Takasawa R, Yokoyama H. Crystal structures of human glyoxalase I and its complex with TLSC702 reveal inhibitor binding mode and substrate preference. FEBS Lett. 2022;596(11):1458–1467. doi:10.1002/1873-3468.14344
30. Cameron AD. Crystal structure of human glyoxalase I_evidence for gene duplication and 3D domain swapping. EMBO J. 1997;16(12):3386–3395. doi:10.1093/emboj/16.12.3386
31. Al-Balas Q, Hassan M, Alshari N, et al. Novel glyoxalase-I inhibitors possessing a “zinc-binding feature” as potential anticancer agents. Drug Des Devel Ther. 2016;10:2623–2629. doi:10.2147/DDDT.S110997
32. Xiao W, Ren J, Hao J, Wang H, Li Y, Lin L. Predicting conserved water molecules in binding sites of proteins using machine learning methods and combining features. Comput Math Meth Med. 2022;2022:1–11. doi:10.1155/2022/5104464
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.