Back to Journals » Infection and Drug Resistance » Volume 11
Structural basis of development of multi-epitope vaccine against Middle East respiratory syndrome using in silico approach
Authors Srivastava S, Kamthania M ![]() , Singh S, Saxena AK, Sharma N
, Singh S, Saxena AK, Sharma N
Received 24 May 2018
Accepted for publication 1 August 2018
Published 21 November 2018 Volume 2018:11 Pages 2377—2391
DOI https://doi.org/10.2147/IDR.S175114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Sukrit Srivastava,1,2 Mohit Kamthania,1,3 Soni Singh,1 Ajay K Saxena,2 Nishi Sharma1
1Department of Biotechnology, Mangalayatan University, Aligarh, India; 2Molecular Medicine Lab, School of Life Sciences, Jawaharlal Nehru University, New Delhi, India; 3Department of Biotechnology, Faculty of Life Sciences, Institute of Applied Medicines and Research, Ghaziabad, Uttar Pradesh, India
Background: Middle East respiratory syndrome (MERS) is caused by MERS coronavirus (MERS-CoV). Thus far, MERS outbreaks have been reported from Saudi Arabia (2013 and 2014) and South Korea (2015). No specific vaccine has yet been reported against MERS.
Purpose: To address the urgent need for an MERS vaccine, in the present study, we have designed two multi-epitope vaccines (MEVs) against MERS utilizing several in silico methods and tools.
Methods: The design of both the multi-epitope vaccines (MEVs) are composed of cytotoxic T lymphocyte (CTL) and helper T lymphocyte (HTL) epitopes, screened form thirteen different proteins of MERS-CoV. Both the MEVs also carry potential B-cell linear epitope regions, B-cell discontinuous epitopes as well as interferon-γ-inducing epitopes. Human β-defensin-2 and β-defensin-3 were used as adjuvants to enhance the immune response of MEVs. To design the MEVs, short peptide molecular linkers were utilized to link screened most potential CTL epitopes, HTL epitopes and the adjuvants. Tertiary models for both the MEVs were generated, refined, and further studied for their molecular interaction with toll-like receptor 3. The cDNAs of both MEVs were generated and analyzed in silico for their expression in a mammalian host cell line (human).
Results: Screened CTL and HTL epitopes were found to have high propensity for stable molecular interaction with HLA alleles molecules. CTL epitopes were also found to have favorable molecular interaction within the cavity of transporter associated with antigen processing. The selected CTL and HTL epitopes jointly cover upto 94.0% of worldwide human population. Both the CTL and HTL MEVs molecular models have shown to have stable binding and complex formation propensity with toll-like receptor 3. The cDNA analysis of both the MEVs have shown high expression tendency in mammalian host cell line (human).
Conclusion: After multistage in silico analysis, both the MEVs are predicted to elicit humoral as well as cell mediated immune response. Epitopes of the designed MEVs are predicted to cover large human population worldwide. Hence both the designed MEVs could be tried in vivo as potential vaccine candidates against MERS.
Keywords: Middle East respiratory syndrome, MERS, Middle East respiratory syndrome coronavirus, MERS-CoV, human transporter associated with antigen processing, TAP, toll-like receptor 3, TLR-3, epitope, immunoinformatics, molecular docking, molecular dynamics simulation, MD simulation, multi-epitope vaccine
Introduction
Middle East respiratory syndrome (MERS) is a respiratory disease caused by Middle East respiratory syndrome coronavirus (MERS-CoV). MERS involves high fever, cough, difficulty in breathing, chills, chest pain, body aches, sore throat, diarrhea, nausea/vomiting, running nose, renal failure, and pneumonia. The first case of MERS in humans was reported in Saudi Arabia in 2012. After 2012, within a span of only 4 years, MERS-CoV infection was reported from 27 countries.1 Three MERS outbreaks have already occurred in Saudi Arabia (2013 and 2014) and South Korea (2015).2–4 The outbreak reported in South Korea also involved spread of MERS through hospital-to-hospital transit of patients and had the fatality rate as high as 40%. High attack rate and easy spread of MERS indicate an epidemic risk.5–7 To date, no specific vaccine is available for MERS. The steep increase in MERS cases and its high mortality rate demand an urgent need for specific and safe MERS vaccine. Thus far, only little information is known about the pathogenesis of MERS-CoV. Hence, an immunoinformatics approach to thoroughly study and screen immunogenic proteins from the available proteome sequence data of MERS-CoV is essential for vaccine design.
The proteome of MERS-CoV consists of several important vaccine candidate and drug target proteins. These proteins are involved in infection and pathogenesis of MERS-CoV to human host cells. Spike (S) glycoprotein, in particular, its receptor-binding domain, is involved in virus and host cell interaction.8 Envelope (E) protein plays an important role in host cell recognition.9 Nucleocapsid (N) protein is involved in RNA binding during ribonucleocapsid formation by MERS-CoV.10 Membrane (M) protein has interferon (IFN)-antagonizing properties, thus reducing IFN levels in infected patients.11 Open reading frame (ORF) proteins also play critical roles in viral infection and pathogenesis. A mutation study of ORFs (ORF3, ORF4a, ORF4b, and ORF5) indicated that ORFs have major implications in viral infection involving disruption of host cell processes, dysregulated IFN pathway activation, and abrupt inflammation.12 Proteins ORF1a (4P16) and ORF1ab (4WUR) are papain-like proteases (PL(pro)) involved in viral infection and are potential targets for the development of antiviral drugs.13 PL(pro) facilitate infection by their proteolytic, deubiquitinating, and deISGylating activities suppressing innate immune response from the host cell. Protein ORF1a (4RSP), a protease (3CLpro), facilitates proteolytic activity during viral infection and replication.14,15 Protein ORF1ab (5WWP) is a helicase protein of MERS-CoV, and it is one of the most conserved proteins among nidoviruses. Protein ORF8b is a highly conserved protein, and during infection, it induces various immune responses.16
In the present study, we propose two multi-epitope vaccine (MEV) designs for MERS. The proposed MEVs are composed of cytotoxic T lymphocyte (CTL) and helper T lymphocyte (HTL) epitopes. Both CTL and HTL MEVs contain overlapping regions of linear B-cell epitopes. Both MEVs also contain human β-defensin-2 (hBD-2) and human β-defensin-3 (hBD-3) as adjuvants to enhance the immunogenic response.17,18
Methodology
To design the MEVs, different in silico tools were used to screen potential CTL, HTL, and B-cell epitopes from 13 MERS-CoV proteins that are involved in virus–host interaction and viral proliferation. All three types of epitopes were studied for overlapping consensus regions. CTL and HTL epitopes showing partial or complete overlapping regions with all or any of the three kinds of epitopes and the epitopes with the highest number of human leukocyte antigen (HLA) allele binders were selected for detailed analysis. The selected CTL and HTL epitopes were further validated for their molecular interaction with their HLA allele binders. Furthermore, the selected CTL epitopes were validated for their molecular interaction with cavity of transporter associated with antigen processing (TAP) to analyze their smooth passage from the cytoplasm to the endoplasmic reticulum (ER) lumen.19 Three-dimensional (3D) structure model for both MEVs was generated, refined, validated, and analyzed for different physiochemical properties. Both CTL and HTL MEV models were further screened to have several B-cell discontinuous epitopes and IFN-γ-inducing epitopes. Because the immune system broadly recognizes pathogens partially due to the involvement of toll-like receptor 3 (TLR-3) and its signaling cascade, both CTL and HTL MEVs were further studied for their molecular interaction with TLR-3.20,21 Moreover, the cDNAs of both MEVs were analyzed and were predicted to be highly expressing in a mammalian host cell line (human) (Figure S1).
MERS-CoV proteins chosen for MEV preparation
For vaccine development, proteins critically involved in virus–host cell interaction, viral proliferation, and host cell disruption were chosen. Thirteen proteins of MERS-CoV proteome chosen for epitope screening included the following: S glycoprotein; E protein; N protein; M protein; ORFs (ORF3, ORF4a, ORF4b, and ORF5); ORF1a (4P16), a papain-like protease (PL(pro)); ORF1a (4RSP), a protease (3CLpro); ORF1ab (4WUR), a PL(pro); ORF1ab (5WWP); and ORF8b. For sequence-based epitope screening, full-length protein sequences of the abovementioned MERS-CoV proteins were retrieved from the NCBI database (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/protein). A total of 2,499 amino acid sequences belonging to different proteins of MERS-CoV of different strains and origins were retrieved. For structure-based epitope screening, available 3D structures of MERS-CoV proteins were retrieved from Protein Data Bank (PDB, http://www.rcsb.org/pdb/home/home.do). For the proteins with no structure available in PDB, homology modeling was performed using the Swiss-model, (http://swissmodel.expasy.org/) (Table S1).22
Screening of potential epitopes
T-cell epitope prediction
Screening of CTL epitope
To screen CTL epitopes, the Immune Epitope Database (IEDB) tool “Proteasomal cleavage/TAP transport/MHC class I combined predictor” (http://tools.iedb.org/processing/) was used.23–25 The total score generated by the tool is a combined score of proteasome, TAP (N-terminal interaction), major histocompatibility complex (MHC), and processing analysis scores. The score is generated using the combination of six methods, namely, Consensus, NN-align, SMM-align, Combinatorial Library, Sturniolo, and NetMHCIIpan. The IC50 (nM) value was also obtained using this IEDB tool. Epitopes with high, intermediate, and least affinity of binding to an HLA allele have IC50 values <50 nM, <500 nM, and <5,000 nM, respectively. Immunogenicity of the screened CTL epitopes was also obtained by the “MHC I Immunogenicity” tool of IEDB (http://tools.iedb.org/immunogenicity/) with all parameters set to default (first, second, and C-terminus amino acids).26 The tool predicts immunogenicity of a peptide MHC complex on the basis of physiochemical properties of amino acids and their position within the short peptide sequence.
Screening of HTL epitopes
To screen HTL epitopes, the IEDB tool “MHC-II Binding Predictions” (http://tools.iedb.org/mhcii/) was used. The percentile rank for each peptide is generated by the combination of three methods (Combinatorial Library, SMM-align, and Sturniolo) by comparing the score of peptide against the scores of other random five million 15-mer peptides from the SwissProt database.27–30 The rank for the Consensus method was generated by the median percentile rank of the three methods.
Population coverage by CTL and HTL epitopes
The “Population Coverage” tool of IEDB (http://tools.iedb.org/population/) was used to analyze the world human population coverage by the shortlisted 28 CTL and 28 HTL epitopes.31 Use of multi-epitopes involving both CTL and HTL epitopes would have the higher probability of larger human population coverage worldwide.
B-cell epitope prediction
Sequence-based B-cell epitope prediction
Protein sequence-based linear B-cell epitopes were predicted by six different prediction methods available at “B Cell Epitope Prediction Tools” tool of IEDB server (http://tools.iedb.org/bcell/); these tools are based on the propensity scale method and physicochemical properties of the antigenic sequence. These methods include “BepiPred Linear Epitope Prediction” (propensity scale method such as hidden Markov model), “Chou & Fasman Beta-Turn Prediction”, “Emini Surface Accessibility Prediction”, “Karplus & Schulz Flexibility Prediction”, “Kolaskar & Tongaonkar Antigenicity”, and “Parker Hydrophilicity Prediction”.32–37
Structure-based B-cell epitope prediction
Two structure-based B-cell epitope prediction methods, namely, DiscoTope 2.0 (Structure-based Antibody Prediction tool; http://tools.iedb.org/discotope/) and Ellipro (Antibody Epitope Prediction tool; http://tools.iedb.org/ellipro/), available in IEDB, were used for linear and discontinuous B-cell epitope prediction.38,39 The ElliPro method is based on the location of the residue in the protein’s 3D structure. The residues lying outside of the ellipsoid covering 90% of the inner core residues of the protein score highest protrusion index (PI) of 0.9 and so on. Discontinuous epitopes predicted by ElliPro are clustered on the basis of distance R in Å between two residues’ centers of mass lying outside the largest possible ellipsoid. The larger the value of R, the larger will be the number of discontinuous epitopes clustered. DiscoTope 2.0 is based on the number of contacts of the residues’ Cα carbon atom with other Cα carbon atoms in the 3D structure within the 10 Å distance and the propensity of a residue to be a part of an epitope.
Characterization of potential epitopes
Epitope conservation analysis
Shortlisted CTL, HTL, and B-cell epitopes were subjected to conservancy analysis by the “Epitope Conservancy Analysis” tool (http://tools.iedb.org/conservancy/) of IEDB among all their respective source protein sequences of MERS-CoV retrieved from the NCBI protein database.40
Epitope toxicity prediction
Toxicity assessment of CTL and HTL epitopes and B-cell epitopes was performed by ToxinPred (http://crdd.osdd.net/raghava/toxinpred/multi_submit.php) analysis. This analysis allows to identify highly toxic or nontoxic peptides. The analysis was performed by the “support vector machine (SVM) (SwissProt)-based” method with all the parameters set to default.41
Overlapping residue analysis
Multiple Sequence Alignment (MSA) analysis using the Clustal Omega tool (https://www.ebi.ac.uk/Tools/msa/clustalo/) of European Bioinformatics Institute was performed for all the shortlisted CTL, HTL, and B-cell epitopes from 13 MERS-CoV proteins.42 MSA by Clustal Omega virtually aligns any number of protein sequences and delivers accurate alignments.
Epitope selected for molecular interaction study with HLA allele and TAP
CTL and HTL epitopes were shortlisted for further in silico analysis on the basis of their overlapping sequence regions among all three types of epitopes (CTL, HTL, and B-cell) or complete overlap among any two types of epitopes or the highest number of HLA allele binders.
Molecular interaction analysis of the selected epitopes with HLA alleles
Tertiary structure modeling of HLA alleles and selected T-cell epitopes
Template-based homology modeling for HLA class I and II allele binders of the shortlisted epitopes was performed by the Swiss-model.22 Protein sequences of HLA class I and II alleles were retrieved from Immuno Polymorphism Database (IPD-IMGT/HLA) (https://www.ebi.ac.uk/ipd/imgt/hla/allele.html).69 Template with high sequence identity was chosen for modeling, and the models thus generated were validated for quality by the Qualitative Model Energy ANalysis (QMEAN) analysis. The QMEAN value is a composite (both global and local [ie, per residue] structure) quality estimate of the generated model.43 Models with an acceptable QMEAN value with a cutoff of −4.0 were chosen for further studies (Table S2).
“Natural Peptides Module for Beginners” module of the PEPstrMOD tool (http://osddlinux.osdd.net/raghava/pepstrmod/nat_ss.php) was used to generate tertiary structures of the selected epitopes.44 Prediction was carried out with a simulation time window of 100 ps, and the peptide environment was set to vacuum.
Molecular docking and molecular dynamics (MD) simulation study of the selected epitopes with HLA alleles
AutoDock Tool 4.2 and AutoDock Vina were used for molecular docking study of the selected epitopes and their respective HLA allele binders.45,46 Further, the docked complexes were subjected to MD simulation study by Gromacs 5.1.4 using Optimized Potentials for Liquid Simulations – all atom (OPLS-AA) force field.47,48
Molecular interaction analysis (docking) of the selected CTL epitopes with TAP
Molecular docking study of the shortlisted CTL epitopes with TAP receptor was performed by AutoDock Vina.45,46 For more accurate prediction, instead of homology modeling, the cryo-EM structure of TAP (PDB ID: 5u1d) was used.49 For docking, the antigen from the TAP cavity of the original structure was removed.
Design, characterization, and interaction analysis of MEVs with TLR-3
Design of MEVs
From the screened and shortlisted CTL and HTL epitopes, two MEVs were designed using EAAAK and GGGGS as short peptide rigid and flexible linkers, respectively (Figure 1A and B). To enhance the immune response, hBD-2 (PDB ID: 1FD3, sequence: GIGDPVTCLKSGAICHPVFCPRRYKQIGTCGLPGTKCCKKP) and hBD-3 (PDB ID: 1KJ6, sequence: GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK) were used as adjuvants for both MEVs at N and C terminals, respectively.17,18,50–53 Upon lung infection, the expression of hBD-2 and hBD-3 was found to be increased. β-Defensins are involved in chemotactic activity for memory T cells, monocytes, and immature dendritic cells as well as in degranulation of mast cells. Thus, hBDs enhance innate and adaptive immunity and therefore were chosen here as adjuvants for the design of MEVs.
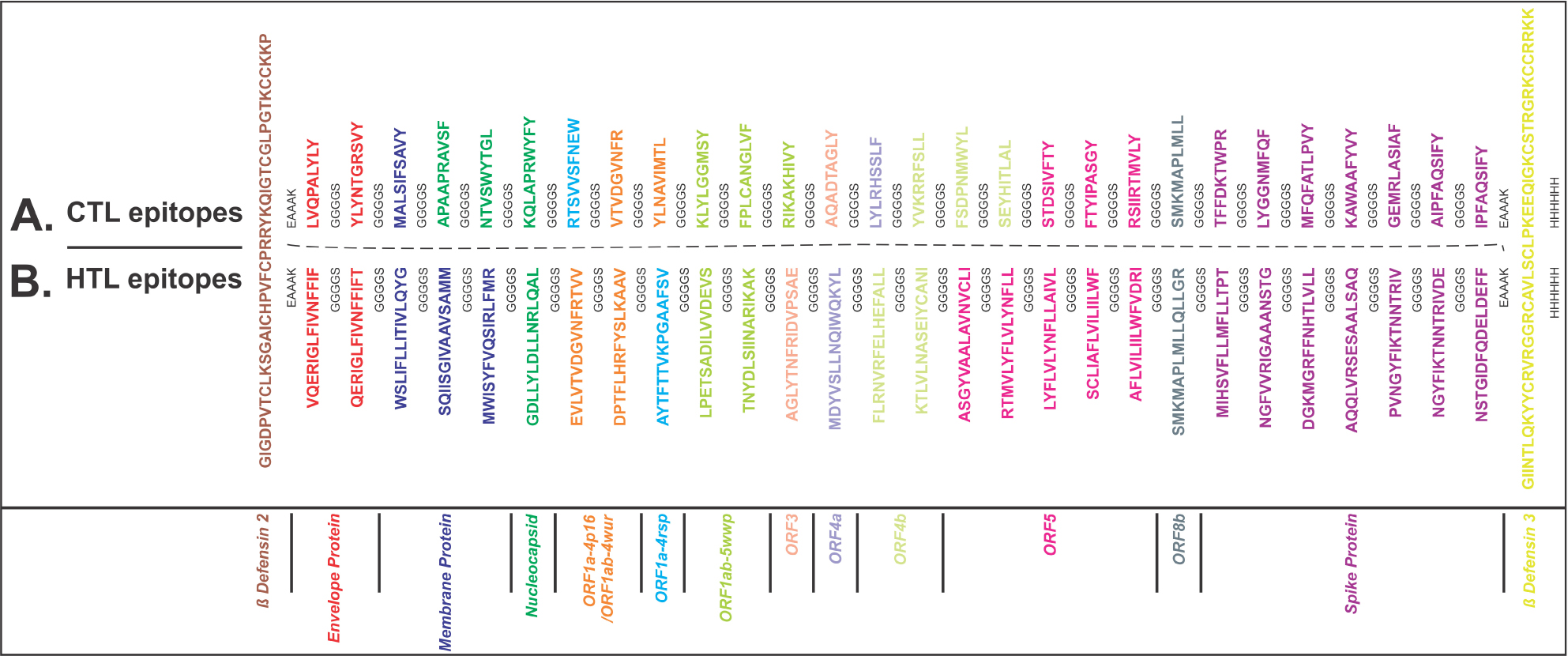 | Figure 1 Multi-epitope vaccine design. Notes: (A) CTL and (B) HTL epitopes were linked by the short peptide linker GGGGS. Human β-defensin-2 and β-defensin-3 were used as adjuvant at N and C terminals, respectively, linked by the short peptide linker EAAAK. Epitopes from different proteins are colored in different colors. C-terminal 6xHis is designated as His tag. Abbreviations: CTL, cytotoxic T lymphocyte; HTL, helper T lymphocyte. |
IFN-γ-inducing epitope prediction
IFN-γ epitopes with potential to induce the release of IFN-γ from CD4+ T cells from both MEVs were predicted by “IFNepitope” server (http://crdd.osdd.net/raghava/ifnepitope/scan.php) using the “Motif and SVM hybrid” (MERCI: Motif-EmeRging and with Classes-Identification, and SVM) approach. The tool generates overlapping sequences from the query protein/antigen and uses it for IFN-γ-inducing epitope prediction. The prediction is based on a dataset of IFN-γ-inducing and IFN-γ-noninducing MHC class II binders.54,55
Allergenicity and antigenicity prediction of MEVs
For both MEVs, AlgPred was used for allergenicity prediction (http://crdd.osdd.net/raghava/algpred/submission.html).56 The AlgPred prediction of allergens is based on similarity of the known epitope with any region of the submitted protein. VaxiJen (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used to analyze the probability of antigenicity of both MEVs.57 The VaxiJen analysis applies an alignment-free approach that is solely based on the physicochemical properties of the sequences of the submitted proteins.
Physicochemical analysis of MEVs
To analyze the physiochemical properties of CTL and HTL MEVs, ProtParam (https://web.expasy.org/protparam/) was used.58 ProtParam analysis performs empirical investigation for a given protein amino acid sequence.
3D structure modeling and refinement of MEVs
Both the CTL and HTL MEVs were further subjected to 3D structure modeling by RaptorX structure prediction server (http://raptorx.uchicago.edu/StructurePrediction/predict/).59 The quality of the generated model is indicated by its P-value. The P-value is the probability of a predicted model being worse than the best. It indicates relative quality in terms of modeling error by combining global distance test (GDT) and un-normalized GDT, that is, error at each residue. The smaller the P-value, the higher is the quality of the predicted model.
The generated MEV models were further refined by ModRefiner and GalaxyRefin.60,70 GalaxyRefine performs repeated structure perturbation along with subsequent structural relaxation by dynamics simulation to refine a protein structure. To avoid breaks in model structures, GalaxyRefine uses the triaxial loop closure method.
In silico validation of refined MEV models
The refined 3D models of both MEVs were then subjected to RAMPAGE analysis for generating a Ramachandran plot (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php).61 The Ramachandran plot shows the residues that form energetically allowed and disallowed dihedral angles psi (ψ) and phi (ϕ), which are calculated on the basis of van der Waal radius of the amino acid side chain.
Discontinuous B-cell epitope prediction from MEVs
Both the designed CTL and HTL MEVs were subjected to discontinuous B-cell epitope prediction to analyze structure-based humoral immunogenic potential of both MEVs using the ElliPro tool.39
Molecular docking and MD simulation study of MEVs and the immunological receptor TLR-3
To study the molecular interaction, the refined models of CTL and HTL MEVs were docked with TLR-3 by PatchDock server (http://bioinfo3d.cs.tau.ac.il/PatchDock/).62–64 The 3D structure of human TLR-3 ectodomain (ECD) was retrieved from PDB databank (PDB ID: 2A0Z). To analyze the stable complex formation, both complexes were subjected to MD simulation by Gromacs 5.1.4 using OPLS-AA.47,48
Analysis of cDNAs of both MEVs for cloning and expression
Optimized cDNAs for both MEVs were generated by the Codon Usage Wrangler Tool with the option of a mammalian host cell line (human) as the expression system (http://www.mrc-lmb.cam.ac.uk/ms/methods/codon.html). Further, the GenScript Rare Codon Analysis Tool (https://www.genscript.com/tools/rare-codon-analysis) was used to analyze the cDNAs of both MEVs. The tool provides GC content, codon adaptation index (CAI), and tandem rare codon frequency for cDNA.65–67
Results and discussion
Screening of potential epitopes
T-cell epitope prediction
Screening of CTL epitope
On the basis of “total score” and acceptably low IC50 (nM) value of epitope-HLA class I allele pairs, 75 CD8+ CTL epitopes were chosen. Later, 28 epitopes were shortlisted with high scoring and larger number of HLA class I allele binders for further studies. Immunogenicity of the screened CTL epitopes was also determined. The higher the immunogenicity score, the greater is the immunogenicity of the epitope (Tables S3 and S4).
Screening of HTL epitopes
Screening of HTL epitopes was performed on the basis of “percentile rank”. Small percentile rank shows the high affinity of the peptide for its respective HLA allele. Initially, from the 13 MERS-CoV proteins, 70 CD4+ T-cell epitopes with the highest percentile rank were screened, and 28 epitopes with high percentile rank and the highest number of HLA class II allele binders were then shortlisted (Tables S5 and S6). Several CTL and HTL epitopes predicted in the present study show overlapping regions with the epitopes detected to induce T-cell responses in previous studies done using peripheral blood mononuclear cells from infected patients.68 Hence, epitopes screened and reported in the present study could be predicted to originate from the highly immunogenic stretch of MERS-CoV protein sequences.
Population coverage by CTL and HTL epitopes
In this study, most geographical regions of the world, and in particular, the countries of Middle East, South Asia, East Asia, and Northeast Asia, were included. This study indicates that the combined use of all shortlisted CTL and HTL epitopes would have an average worldwide population coverage of 94.0%, with an SD of 20.19 (Table S7).
B-cell epitope prediction
Sequence-based B-cell epitope prediction
Initially, a total of 144 B-cell epitopes were screened from 13 MERS-CoV proteins, with epitope length of four or more than four amino acids by the BepiPred Linear Epitope Prediction method. The epitopes predicted by five other methods based on different physicochemical properties showed a significant consensus overlap of amino acid sequences with that of BepiPred Linear Epitope Prediction. From the 144 B-cell epitopes, 12 with the length of 4–19 amino acids were shortlisted (Table S8 and Figure 2).
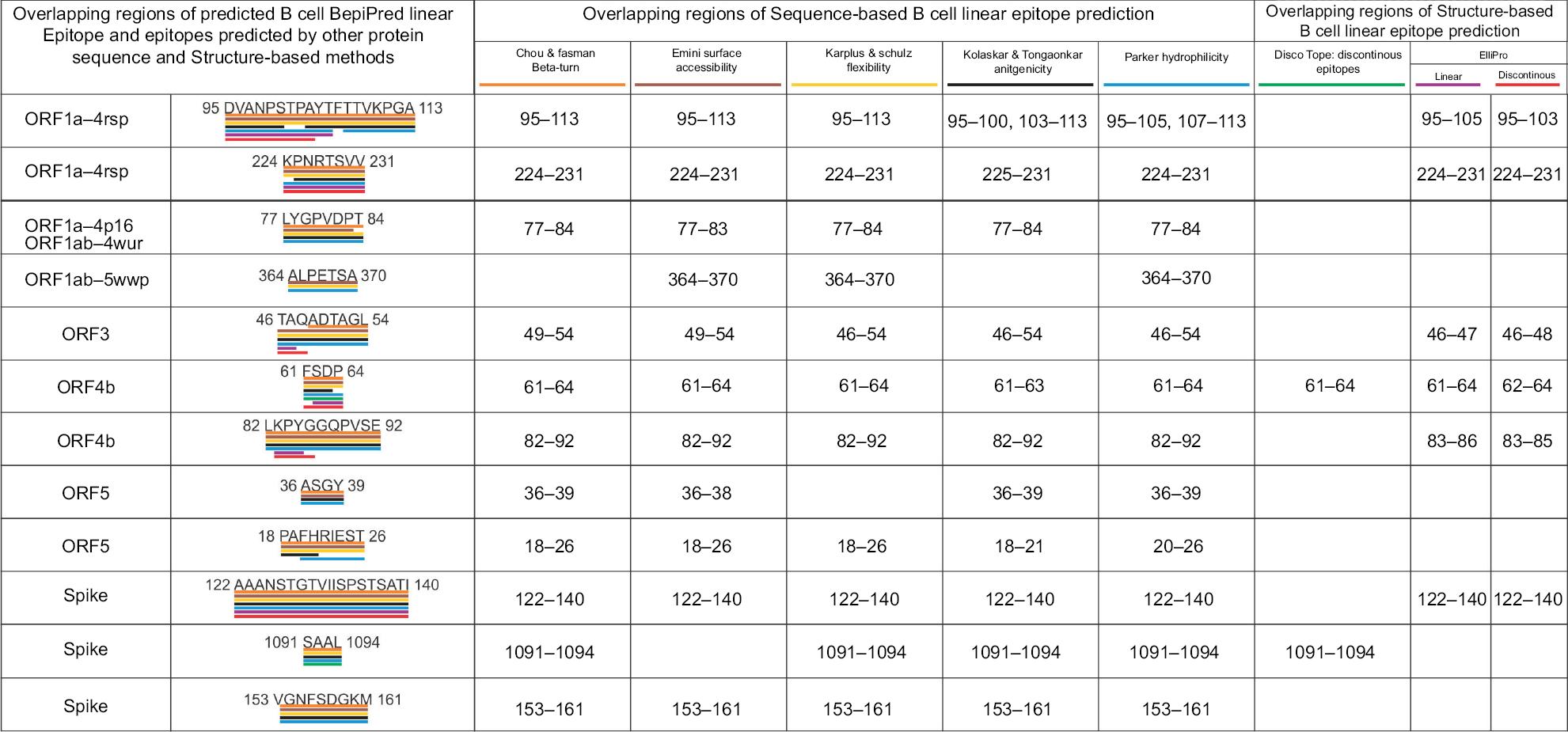 | Figure 2 Overlapping regions of the BepiPred linear B-cell epitopes and the epitopes predicted by other methods. Notes: This analysis shows a strong consensus between the BepiPred linear B-cell epitopes and the epitopes predicted by other sequence-based and structure-based epitope prediction methods. Different colors are used to highlight overlapping regions of epitopes predicted by the different methods. |
Structure-based B-cell epitope prediction
Discontinuous and linear epitopes predicted by DiscoTope 2.0 and Ellipro methods showed significant consensus overlap of amino acid sequence with the linear epitopes predicted by the BepiPred linear epitope method (Table S8 and Figure 2). This result confirms that the shortlisted BepiPred linear epitopes are highly immunogenic B-cell epitopes.
Characterization of potential epitopes
Epitope conservation analysis
The conservation analysis of shortlisted 28 CTL, 28 HTL, and 12 B-cell epitopes shows that amino acid sequence conservancy of CTL epitopes varied from 72.7% to 100%, that of HTL epitopes varied from (50% for two epitopes) 68.18% to 100%, and that of B-cell epitopes varied from 85.71% to 99.26% (Tables S3, S5, and S8). This result indicates high conservancy nature of the shortlisted CTL, HTL, and B-cell epitopes.
Epitope toxicity prediction
All the shortlisted CTL, HTL, and B-cell epitopes analyzed by the ToxinPred tool were predicted to be nontoxic (Tables S3, S5, and S8). The analysis was based on the ToxinPred main dataset consisting of 1,805 toxic peptides for its prediction.
Overlapping residue analysis
MSA analysis of all the screened CTL, HTL, and B-cell epitopes from the 13 MERS-CoV protein candidates revealed that several epitopes of CTL, HTL, and B-cell have overlapping amino acid sequence regions. Epitopes with two or more than two residues in the overlapping region are shown in Figure 3.
 | Figure 3 Overlapping CTL, HTL, and B-cell epitopes. Notes: Overlapping CTL (red), HTL (blue), and B-cell epitopes (green) were sorted out by MSA using Clustal Omega at EBI server. The ringed clusters of epitopes involve all three types of epitopes, epitopes with full sequence overlap, and the epitopes with highest number of HLA allele binders. Abbreviations: CTL, cytotoxic T lymphocyte; EBI, European Bioinformatics Institute; HLA, human leukocyte antigen; HTL, helper T lymphocyte; MSA, multiple sequence alignment. |
Epitope selected for molecular interaction study with HLA allele and TAP
Overlapping CTL and HTL epitopes clustering with all three (CTL, HTL, and B-cell) types of epitopes or with complete epitope sequence overlap or with the highest number of HLA allele binders were identified and selected for their molecular interaction analysis with HLA alleles and TAP (Figure 3). These include CTL epitopes:47 AQADTAGLY,31,55 FTYIPASGY,25,39 STDSIVFTY,33,58 SMKMAPLMLL,14,67 VTVDGVNFR,22,51 and LVQPALYLY;59 and HTL epitopes:52 AGLYTNFRIDVPSAE,36,66 ASGYVAALAVNVCLI,11,50 EVLVTVDGVNFRTVV,25,58 SMKMAPLMLLQLLGR,72,117 and FLRNVRFELHEFALL.131
Molecular interaction analysis of the selected epitopes with HLA allele
Molecular docking and MD simulation study of the selected epitopes with HLA alleles
Molecular docking study of all the selected epitopes with their respective HLA allele binders revealed a significantly favorable molecular interaction. Docking complexes thus formed have significantly negative binding energy, and several amino acid residues of epitopes and HLA alleles were involved in hydrogen bond formation (Figure 4). To analyze the stability of binding, docking complexes were further subjected to MD simulation study with an analysis time window of 0.1 ns at the reasonably invariable temperature (~300 K) and pressure (~1 bar). The results of MD simulation for all the epitope-HLA allele complexes showed a very convincing reasonably invariant root mean square deviation (RMSD) value between ~0.1 and 0.2 nm, thus indicating stable complex formation (Figure 5). Moreover, the reasonably invariant radius of gyration (Rg) of the complexes (Figure S2) and RMS fluctuation (RMSF) for all the atoms in the complexes (Figure S3) indicate that the epitope-HLA complexes remain very stable in their folded form. B-factor of the epitope and HLA allele complexes is shown in the rainbow color presentation in Figure S4. Most of the regions of the complexes are stable (blue) with a very small region being acceptably fluctuating (yellow and orange).
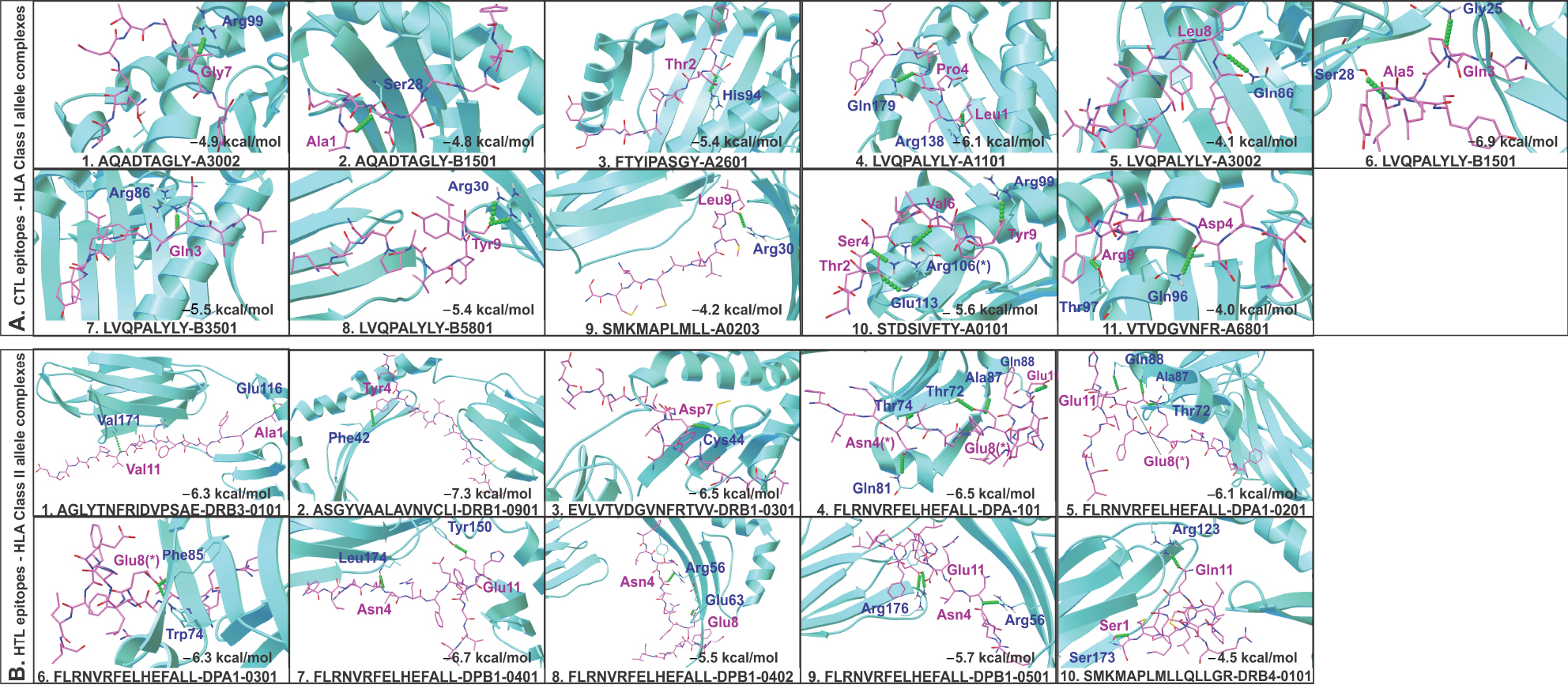 | Figure 4 Molecular docking study of CTL and HTL epitopes with respective HLA allele binders. Notes: (A) CTL and (B) HTL epitopes docked with their respective HLA allele binders. The docking study shows acceptable negative binding energy and hydrogen bonds (green balls) formation between epitopes (magenta sticks) and HLA alleles (cyan cartoons). (*) Residues forming hydrogen bonds with more than one residue. Abbreviations: CTL, cytotoxic T lymphocyte; HLA, human leukocyte antigen; HTL, helper T lymphocyte. |
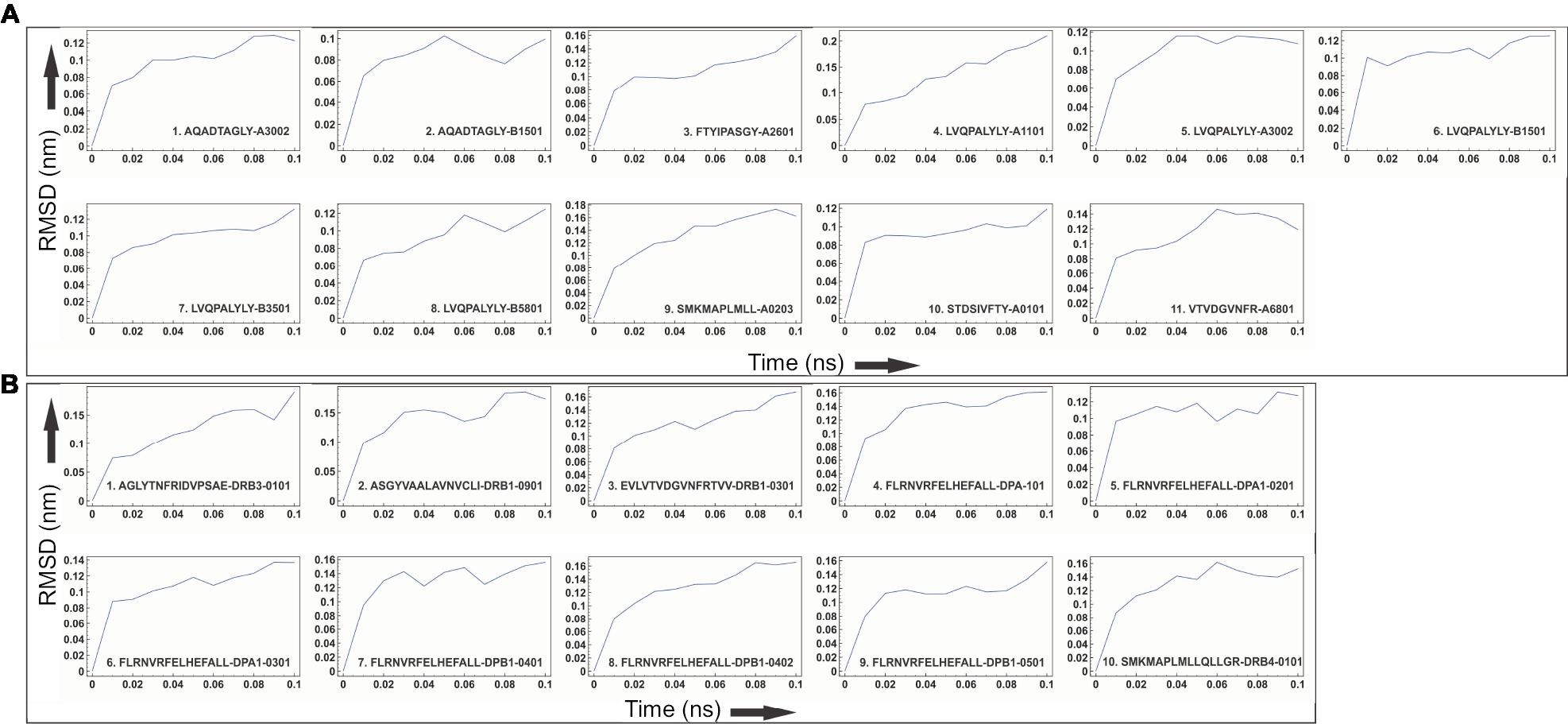 | Figure 5 Molecular dynamics simulation study of the epitope and HLA allele complexes. Notes: Molecular dynamics simulation study of (A) CTL and (B) HTL epitope complexes with their respective HLA allele binders. The reasonably invariable RMSD value indicates a stable complex formation. Abbreviations: CTL, cytotoxic T lymphocyte; HLA, human leukocyte antigen; HTL, helper T lymphocyte; RMSD, root mean square deviation. |
Molecular interaction analysis (docking) of the selected CTL epitopes with TAP
Molecular docking results show a favorable molecular interaction between the selected CTL epitopes and the TAP cavity. The result reveals several molecular interaction sites for the selected epitopes within the TAP cavity. Two sites of interaction—one close to the cytoplasm and another close to the ER lumen—are shown in Figure 6. All the interactions showed a significantly negative binding energy with one or more than one hydrogen bond formation at both sites of interaction. From this study, we may predict a smooth passage for selected CTL epitopes from the cytoplasm to the ER lumen throughout the entire passage of the TAP cavity (Figure 6).
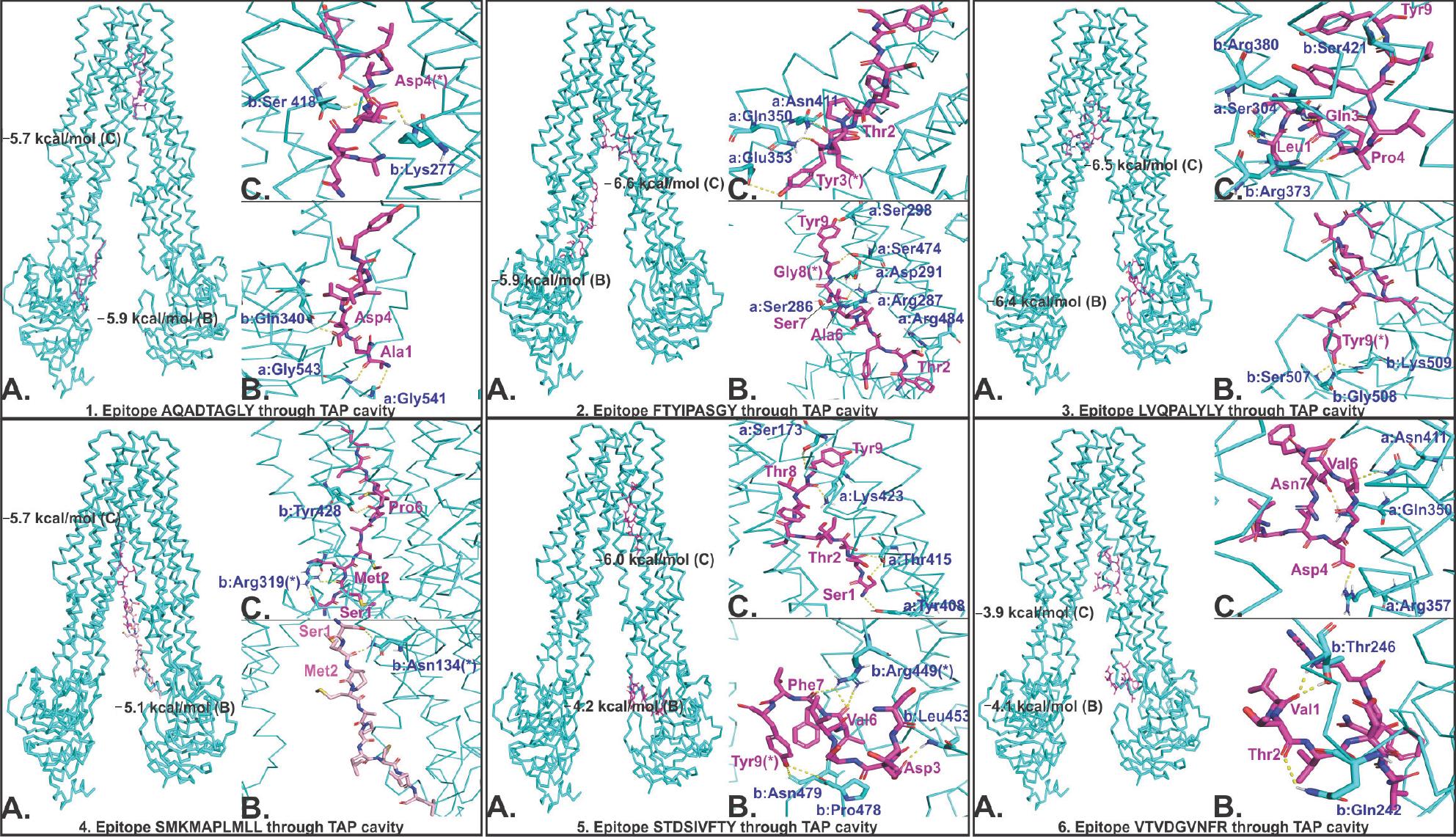 | Figure 6 Molecular docking study of CTL epitopes with the TAP cavity. Notes: TAP is shown by cyan ribbon, and epitopes are shown by magenta. In every panel of the epitope–TAP complex, (A) shows the binding of epitope at two different sites of the TAP cavity, (B) and (C) show the hydrogen bond formation between epitopes and TAP cavity residues at two respective sites of the TAP cavity. (*) Residues forming hydrogen bonds with more than one residue. (a and b) Chain A and B of TAP. Binding energy is shown in kcal/mol. Hydrogen bonds are shown by yellow dots. Abbreviations: CTL, cytotoxic T lymphocyte; TAP, transporter associated with antigen processing. |
Characterization and interaction analysis of designed MEVs with TLR-3
IFN-γ-inducing epitope prediction
IFN-γ is involved in both adaptive and innate immune responses. It stimulates macrophages and natural killer cells. IFN-γ-inducing 15-mer peptide epitopes were predicted from both CTL and HTL MEVs by IFNepitope server. A total of 486 and 640 IFN-γ epitopes were predicted from CTL and HTL MEVs, respectively. Out of these, 35 CTL MEVs and 18 HTL MEVs, IFN-γ-inducing POSITIVE epitopes, with score of one or more than one were chosen (Table S9 and Figure 7C and G).
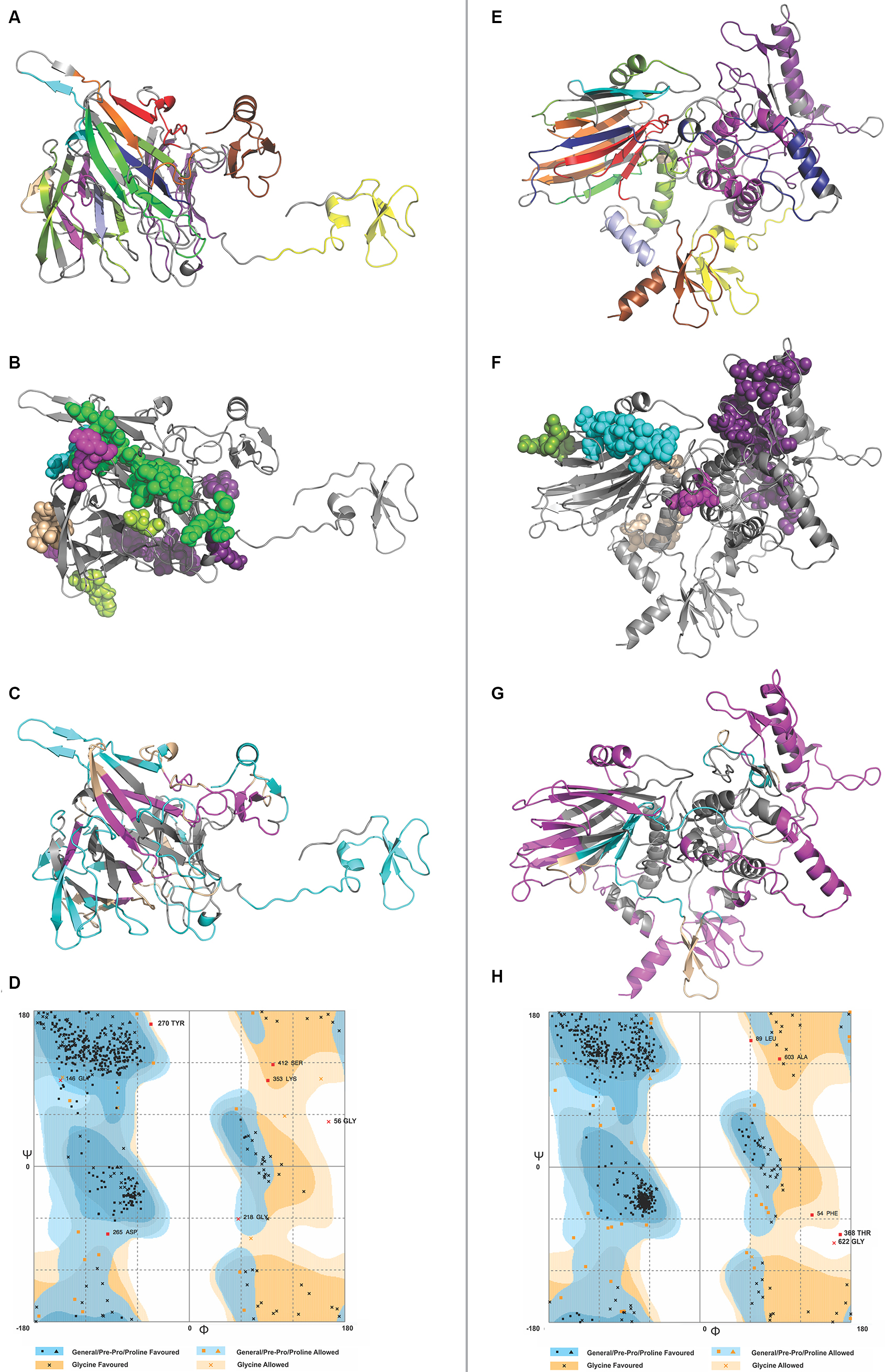 | Figure 7 3D structure modeling of CTL and HTL MEVs. Notes: (A and E) 3D structure of CTL and HTL MEVs models, respectively, showing epitopes from different proteins in different colors (epitope colors are the same as those shown in Figure 1). (B and F) Overlapping regions belonging to B-cell linear epitopes from both CTL and HTL MEVs, respectively, are shown by spheres. From (C) CTL and (G) HTL MEVs, IFN-γ-inducing epitopes are shown in cyan, while discontinuous B-cell epitopes are shown in magenta. The regions shown in wheat color are common to both IFN-γ and discontinuous B-cell epitopes. (D and H) RAMPAGE analysis of refined CTL and HTL MEV models, respectively. Abbreviations: CTL, cytotoxic T lymphocyte; HTL, helper T lymphocyte; IFN, interferon; MEV, multi-epitope vaccine. |
Allergenicity and antigenicity prediction of MEVs
The AlgPred analysis of CTL and HTL MEVs revealed that both MEVs are nonallergens with the score of −0.95415008 and −0.8647714, respectively, with sensitivity and specificity of 88.87% and 81.86%, respectively, while the default threshold value was −0.4. The VaxiJen analysis indicated both MEVs to be probable ANTIGENS with prediction score of 0.5302 and 0.5097 for CTL MEVs and HTL MEVs, respectively, while the default threshold value was 0.4 for viral proteins. Hence, both MEVs are predicted to be nonallergic and potentially antigenic in nature.
Physicochemical analysis of MEVs
ProtParam analysis showed that CTL MEVs have 500 amino acids, 50.4 kDa molecular weight, and 9.72 theoretical pI. The half-life in mammalian reticulocytes, yeast, and Escherichia coli was 30 hours, 20 hours, and 10 hours, respectively; aliphatic index was 61.68, and grand average of hydropathicity (GRAVY) was −0.020, indicating globular and hydrophilic nature of CTL MEVs; the instability index was 50.70, indicating that CTL MEVs are theoretically close to stable in nature. ProtParam study showed HTL MEV has 657 amino acids, 67.583 kDa molecular weight, and 9.29 theoretical pI. The half-life in mammalian reticulocytes, yeast, and E. coli was 30 hours, 20 hours, and 10 hours, respectively; aliphatic index was 94.22, and grand average of hydropathicity (GRAVY) was 0.337, both indicating globular and hydrophilic nature of HTL MEV; the instability index was 37.66, indicating theoretically stable nature of HTL MEV.
3D structure modeling and refinement of MEVs
The model generated by RaptorX for CTL MEV has 4% helix, 37% β-sheet, and 58% coil; the model is 26% exposed, 41% medium, and 31% buried. It has three domains: first domain (1–46 aa, template-1fd3:A); second domain (47–444 aa, template-4dou:A, 4zch:A); and third domain (445–500 aa, template-1kj6:A) (Figure 7A). The model generated by RaptorX for HTL MEV has 35% helix, 20% β-sheet, and 44% coil; the model is 26% exposed, 35% medium, and 37% buried. It has four domains: first domain (1–46 aa, template-1fd3:A); second domain (47–546 aa, templates-4ufcA, 2eabA, and 2rdyA); third domain (547–601 aa, template-1tue:B); and fourth domain (602–657 aa, template-1kj6:A) (Figure 7E). The P-values of the best template of CTL and HTL MEV models are 3.98e-05 and 5.41e-04, respectively. Good quality alpha proteins and good quality beta proteins should have P-value of less than 10-3 and 10-4 respectively. Hence, both the models of CTL and HTL MEVs generated by RaptorX are of good quality. Because the CTL and HTL epitopes used for the design of both MEVs show overlapping with B-cell linear epitopes (Figure 3), both the CTL and HTL MEV models also carry overlapping regions of B-cell linear epitopes (Figure 7B and F).
The structural accuracy of initial model including joining of the gaps was performed by ModRefiner refinment. GalaxyRefine was used to further refine the 3D models of CTL and HTL MEVs. For both MEVs, refined model 1 was chosen on the basis of best scorings. For CTL MEV refinement, the score of model 1 was as follows: Rama favored was 94.2%, GDT-HA was 0.9500, RMSD was 0.433, MolProbity was 2.211, Clash score was 18.3, and Poor rotamers was 1.2. For HTL MEV refinement, the score of model 1 was as follows: Rama favored was 93.4%, GDT-HA was 0.9502, RMSD was 0.412, MolProbity was 2.262, Clash score was 21.9, and Poor rotamers was 0.6. Here, MolProbity is the log-weighted combination of clash score, percentage Ramachandran not favored, and percentage bad side-chain rotamers. After refinement, all the mentioned parameters improved significantly as compared to those of the initial model (Table S10).
In silico validation of refined MEV models
The RAMPAGE analysis showed that the refined CTL MEV model has 94% residues in the favored region, 3.8% residues in the allowed region, and only 1.4% residues in the outlier region, while the refined HTL MEV model has 94% of residues in the favored region, 5.2% residues in the allowed region, and only 0.8% residues in the outlier region (Figure 7D and H).
Discontinuous B-cell epitope prediction from MEVs
Discontinuous B-cell epitope prediction from both MEVs was performed by the ElliPro tool. The screening revealed CTL MEV to have six discontinuous epitopes and HTL MEV to have five discontinuous epitopes. The PI score for CTL MEV epitopes ranged from 0.549 to 0.906 and that for HTL MEV ranged from 0.56 to 0.729 (Table S11 and Figure 7C and G). The higher the score, the greater is the potential of the B-cell discontinuous epitope.
Molecular docking and MD simulation study of MEVs and the immunological receptor TLR-3
The refined models of CTL and HTL MEVs were further analyzed for their interaction with the ECD of human TLR-3 by molecular docking using the PatchDock tool. Docking conformation chosen for the CTL and HTL MEVs showed scores of 18,096 and 23,690, respectively, which were highest among all docked complexes. The highest score indicates the best geometric shape complementarity fitting of ligand and receptor predicted by the tool. The docking complex shows a fitting confirmation of both the MEVs within the ECD of TLR-3 (Figure 8A and E). Further, the MD simulation analysis of the docked CTL-TLR-3 and HTL-TLR-3 complexes showed a very convincing reasonably stable RMSD value between ~0.4 and 0.5 nm for a given time window of 6 ns at the reasonably invariable temperature (~300 K) and pressure (~1 bar). These results indicate a stable complex formation for both MEVs with TLR-3 (Figure 8B and F). The reasonably invariant Rg of MEV-TLR-3 complexes (Figure 8C and G) and RMSF for all the atoms in the complexes (Figure 8D and H) indicate that both the MEV-TLR-3 complexes are reasonably stable and are properly folded. The B-factor of CTL and HTL MEV complexes with the TLR-3 receptor is shown by rainbow color presentation in Figure 8A and E. Most of the regions of both complexes are stable (blue) with a very small region being acceptably fluctuating (yellow and orange).
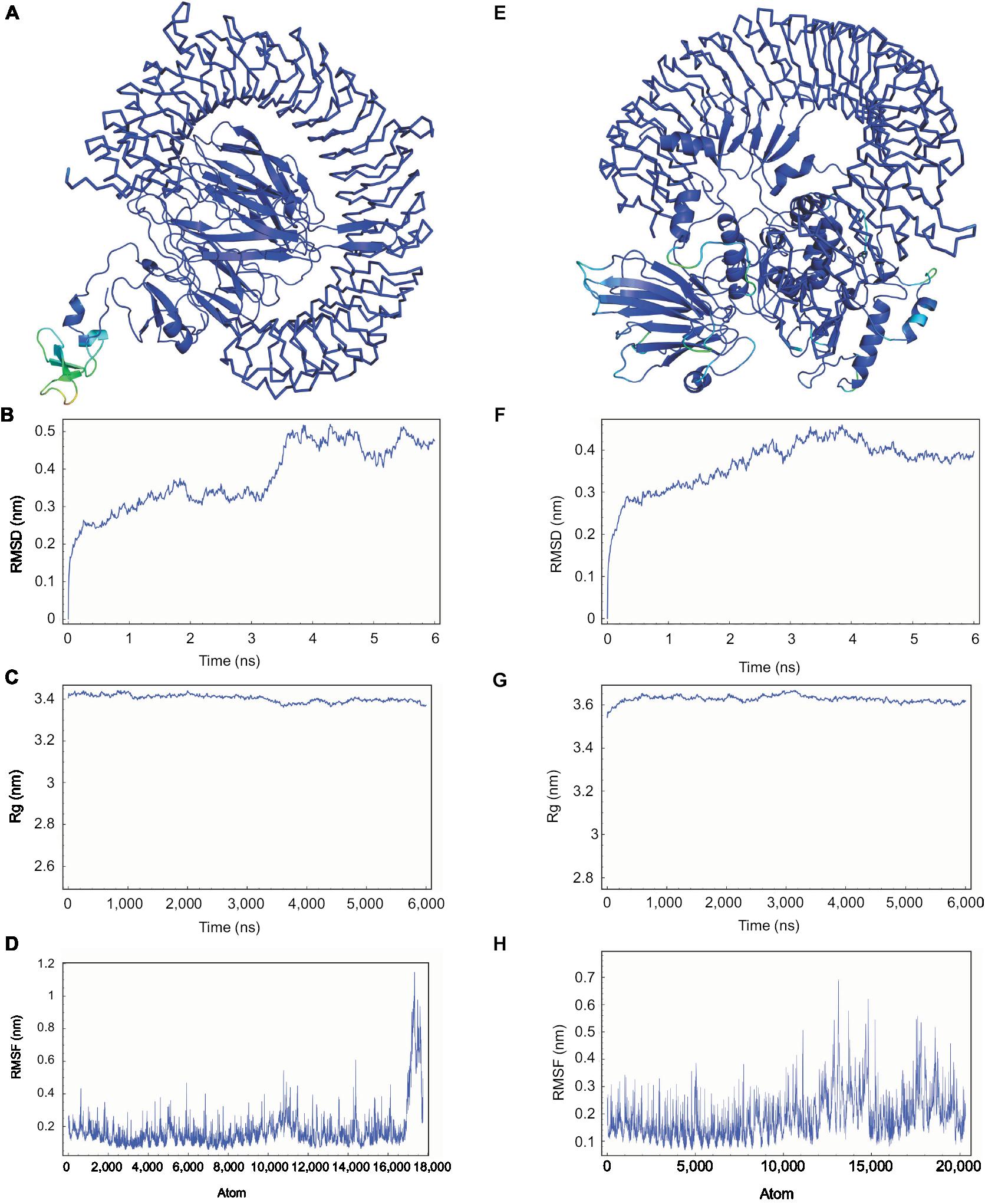 | Figure 8 Molecular docking and MD simulation study of CTL and HTL MEVs with TLR-3. Notes: (A) CTL and (E) HTL MEVs (cartoon) docked complex with TLR-3 (ribbon), respectively. B-factor of the complexes is shown by rainbow (VIBGYOR) presentation; the regions in blue are very stable, and the region in red is unstable. In the above complexes, most of the regions are in blue and a very small region is in yellow and orange, indicating that both complexes are very stable. (B and F) MD simulation study of CTL and HTL MEV complexes with TLR-3, respectively. (C and G) Rg across the time window of 6,000 ps for CTL and HTL MEV complexes with TLR-3, respectively. (D and H) RMSF for all the atoms of the CTL and HTL MEV complex with TLR-3, respectively. Abbreviations: CTL, cytotoxic T lymphocyte; HTL, helper T lymphocyte; MD, molecular dynamics; MEV, multi-epitope vaccine; Rg, radius of gyration; RMSD, root mean square deviation; RMSF, root mean square fluctuation; TLR-3, toll-like receptor 3. |
Analysis of cDNAs of both MEVs for cloning and expression
Optimized cDNAs for both CTL and HTL MEVs were generated by the Codon Usage Wrangler Tool with a mammalian host cell line (human) as the choice of the expression system. The GenScript Rare Codon Analysis Tool showed that the GC content of optimized CTL MEV cDNA was 69.74% and CAI score was 1.0 with 0% tandem rare codons. Likewise, the GC content of optimized HTL MEV cDNA was 66.63%, and the CAI score was 1.0 with 0% tandem rare codons. Ideally, the GC content should be 30%–70%, the CAI score indicating the possibility of cDNA expression in the chosen expression system should be between 0.8 and 1.0, and tandem rare codon frequency indicating the percentage of low-frequency codons present in cDNA for the chosen expression system should be <30%. Tandem rare codons may hinder the expression of cDNA or even interrupt the translational machinery. Hence, the optimized cDNAs of both MEVs are predicted to be highly expressing in the mammalian host cell line (human).
Conclusion
In this study, we propose the design of two MEVs against MERS-CoV that consisted of CTL and HTL epitopes. The selected CTL and HTL epitopes were validated by in silico methods for their molecular interaction with HLA alleles and TAP (for CTL epitopes). The population coverage by the designed MEVs is as high as 94% of the world population. Both MEVs were found to contain IFN-γ epitopes and B-cell discontinuous epitopes. Moreover, they also showed stable interaction with the immunoreceptor TLR-3. On the basis of the design and in silico validation of both the CTL and HTL MEVs, their joint administration is predicted to induce humoral and cell-mediated immune response. Codon-based cDNAs of both MEVs are predicted to have high expression in the mammalian host cell line (human); hence, they could be cloned and expressed at the laboratory level for in vivo trials on humanized HLA-expressing mice for further study.
Acknowledgments
The authors acknowledge the Advance Instrumentation Research Facility (AIRF) at Jawaharlal Nehru University, New Delhi, for providing advance computational facility to conduct experiments. The current research was also supported by DST-PURSE grant to Jawaharlal Nehru University.
Author contributions
S Srivastava and MK designed the protocol and performed the experiments. S Srivastava, MK, S Singh, AKS, and NS contributed toward data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
WHO MERS-CoV Global Summary and Risk Assessment. 2016. Available from: www.who.int/emergencies/mers-cov/mers-summary-2016.pdf. Accessed May 20, 2018. | ||
Assiri A, McGeer A, Perl TM, et al; KSA MERS-CoV Investigation Team. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med Overseas Ed. 2013;369(5):407–416. | ||
Oboho IK, Tomczyk SM, Al-Asmari AM, et al. 2014 MERS-CoV outbreak in Jeddah – a link to health care facilities. N Engl J Med. 2015;372(9):846–854. | ||
Park JW, Lee KJ, Lee KH, et al. Hospital Outbreaks of Middle East Respiratory Syndrome, Daejeon, South Korea, 2015. Emerg Infect Dis. 2017;23(6):898–905. | ||
Kim KM, Ki M, Cho SI, et al. Epidemiologic features of the first MERS outbreak in Korea: focus on Pyeongtaek St. Mary’s Hospital. Epidemiol Health. 2015;37:e2015041. | ||
Ki M. 2015 MERS outbreak in Korea: hospital-to-hospital transmission. Epidemiol Health. 2015;37:e2015033. | ||
World Health Organization. Emergencies Preparedness, Reponse: Coronavirus Infections [cited October 25, 2015]. Available from: http://www.who.int/csr/don/25-october-2015-mers-korea/en/. Accessed May 20, 2018. | ||
Wang L, Shi W, Joyce MG, et al. Evaluation of candidate vaccine approaches for MERS-CoV. Nat Commun. 2015;6:7712. | ||
Xie Q, He X, Yang F, et al. Analysis of the genome sequence and prediction of B-cell epitopes of the envelope protein of Middle East respiratory syndrome-coronavirus. IEEE/ACM Trans Comput Biol Bioinform. 2018;15(4):1344–1350. | ||
Wang YS, Chang CK, Hou MH. Crystallographic analysis of the N-terminal domain of Middle East respiratory syndrome coronavirus nucleocapsid protein. Acta Crystallogr F Struct Biol Commun. 2015;71(Pt 8):977–980. | ||
Lui PY, Wong LY, Fung CL, et al. Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg Microbes Infect. 2016;5(4):e39. | ||
Menachery VD, Mitchell HD, Cockrell AS, et al. MERS-CoV Accessory ORFs Play Key Role for Infection and Pathogenesis. MBio. 2017;8(4):e00665-17. | ||
Lei J, Mesters JR, Drosten C, Anemüller S, Ma Q, Hilgenfeld R. Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antiviral Res. 2014;109:72–82. | ||
Tomar S, Johnston ML, St John SE, et al. Ligand-induced Dimerization of Middle East Respiratory Syndrome (MERS) Coronavirus nsp5 Protease (3CLpro): IMPLICATIONS FOR nsp5 REGULATION AND THE DEVELOPMENT OF ANTIVIRALS. J Biol Chem. 2015;290(32):19403–19422. | ||
Lei J, Hilgenfeld R. Structural and mutational analysis of the interaction between the Middle-East respiratory syndrome coronavirus (MERS-CoV) papain-like protease and human ubiquitin. Virol Sin. 2016;31(4):288–299. | ||
Lu R, Zou L, Wang Y, et al. Sequencing and Phylogenetic Analyses of Structural and Accessory Proteins of Middle East Respiratory Syndrome Coronavirus from the First Imported Case in China, 2015. Bing Du Xue Bao. 2015;31(4):333–340. Chinese. | ||
Wilson SS, Wiens ME, Smith JG. Antiviral mechanisms of human defensins. J Mol Biol. 2013;425(24):4965–4980. | ||
Duits LA, Nibbering PH, van Strijen E, et al. Rhinovirus increases human β-defensin-2 and-3 mRNA expression in cultured bronchial epithelial cells. Pathogens and Disease. 2003;38(1):59–64. | ||
Antoniou AN, Powis SJ, Elliott T. Assembly and export of MHC class I peptide ligands. Curr Opin Immunol. 2003;15(1):75–81. | ||
Delneste Y, Beauvillain C, Jeannin P. Innate immunity: structure and function of TLRs. Med Sci (Paris). 2007;23(1):67–73. | ||
Totura AL, Whitmore A, Agnihothram S, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. MBio. 2015;6(3):e00638-15. | ||
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. | ||
Tenzer S, Peters B, Bulik S, et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci. 2005;62(9):1025–1037. | ||
Peters B, Bulik S, Tampe R, Van Endert PM, Holzhütter HG. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J Immunol. 2003;171(4):1741–1749. | ||
Hoof I, Peters B, Sidney J, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1–13. | ||
Calis JJ, Maybeno M, Greenbaum JA, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 2013;9(10):e1003266. | ||
Wang P, Sidney J, Kim Y, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics. 2010;11(1):568. | ||
Sidney J, Assarsson E, Moore C, et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008;4(1):2. | ||
Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8(1):238. | ||
Sturniolo T, Bono E, Ding J, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17(6):555–561. | ||
Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics. 2006;7:153. | ||
Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. | ||
Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. | ||
Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55(3):836–839. | ||
Karplus PA, Schulz GE. Prediction of chain flexibility in proteins. Naturwissenschaften. 1985;72(4):212–213. | ||
Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1–2):172–174. | ||
Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25(19):5425–5432. | ||
Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol. 2012;8(12):e1002829. | ||
Ponomarenko J, Bui HH, Li W, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9:514. | ||
Bui HH, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007;8:361. | ||
Gupta S, Kapoor P, Chaudhary K, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8(9):e73957. | ||
Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. | ||
Benkert P, Tosatto SC, Schomburg D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins. 2008;71(1):261–277. | ||
Singh S, Singh H, Tuknait A, et al. PEPstrMOD: structure prediction of peptides containing natural, non-natural and modified residues. Biol Direct. 2015;10:73. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Abraham MJ, Murtola T, Schulz R, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2(2):19–25. | ||
Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. | ||
Oldham ML, Grigorieff N, Chen J. Structure of the transporter associated with antigen processing trapped by herpes simplex virus. Elife. 2016;5:e21829. | ||
Hu W, Li F, Yang X, et al. A flexible peptide linker enhances the immunoreactivity of two copies HBsAg preS1 (21-47) fusion protein. J Biotechnol. 2004;107(1):83–90. | ||
Hoover DM, Rajashankar KR, Blumenthal R, et al. The structure of human beta-defensin-2 shows evidence of higher order oligomerization. J Biol Chem. 2000;275(42):32911–32918. | ||
Hajighahramani N, Nezafat N, Eslami M, Negahdaripour M, Rahmatabadi SS, Ghasemi Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect Genet Evol. 2017;48:83–94. | ||
Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357–1369. | ||
Nagpal G, Gupta S, Chaudhary K, Dhanda SK, Prakash S, Raghava GP. VaccineDA: Prediction, design and genome-wide screening of oligodeoxynucleotide-based vaccine adjuvants. Sci Rep. 2015;5:12478. | ||
Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30. | ||
Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34(Web Server issue):W202–W209. | ||
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8:4. | ||
Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The Proteomics Protocols Handbook. Chicago, IL: Humana Press; 2005:571–607. | ||
Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7(8):1511–1522. | ||
Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40(Web Server issue):W294–W297. Available from: http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE. Accessed October 17, 2018. | ||
Lovell SC, Davis IW, Arendall WB 3rd, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50(3):437–450. | ||
Bell JK, Botos I, Hall PR, et al. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci U S A. 2005;102(31):10976–10980. | ||
Duhovny D, Nussinov R, Wolfson HJ. Efficient unbound docking of rigid molecules. In: Guigó R, Gusfield D, editors. Proceedings of the 2’nd Workshop on Algorithms in Bioinformatics (WABI). Vol. 2452. Rome: Lecture Notes in Computer Science; 2002:185–200. | ||
Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(Web Server issue):W363–W367. | ||
Nezafat N, Eslami M, Negahdaripour M, Rahbar MR, Ghasemi Y. Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol Biosyst. 2017;13(4):699–713. | ||
Morla S, Makhija A, Kumar S. Synonymous codon usage pattern in glycoprotein gene of rabies virus. Gene. 2016;584(1):1–6. | ||
Wu X, Wu S, Li D, et al. Computational identification of rare codons of Escherichia coli based on codon pairs preference. BMC Bioinformatics. 2010;11(1):61. | ||
Zhao J, Alshukairi AN, Baharoon SA, et al. Recovery from the Middle East respiratory syndrome is associated with antibody and T-cell responses. Sci Immunol. 2017;2(14):eaan5393. | ||
Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. The IPD and IMGT/HLA database: allele variant databases. Nucleic acids research. 2014:43(D1); D423–D431. | ||
Xu D and Zhang Y. Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-step Atomic-level Energy Minimization. Biophys J. 2011;101(10):2525–34. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.