Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 8
Streptozotocin-induced type 1 diabetes in rodents as a model for studying mitochondrial mechanisms of diabetic β cell glucotoxicity
Received 5 February 2015
Accepted for publication 5 March 2015
Published 2 April 2015 Volume 2015:8 Pages 181—188
DOI https://doi.org/10.2147/DMSO.S82272
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ming-Hui Zou
Jinzi Wu, Liang-Jun Yan
Department of Pharmaceutical Sciences, UNT System College of Pharmacy, University of North Texas Health Science Center, Fort Worth, TX, USA
Abstract: Chronic hyperglycemia and the corresponding glucotoxicity are the main pathogenic mechanisms of diabetes and its complications. Streptozotocin (STZ)-induced diabetic animal models are useful platforms for the understanding of β cell glucotoxicity in diabetes. As diabetes induced by a single STZ injection is often referred to as type 1 diabetes that is caused by STZ’s partial destruction of pancreas, one question often being asked is whether the STZ type 1 diabetes animal model is a good model for studying the mitochondrial mechanisms of β cell glucotoxicity. In this mini review, we provide evidence garnered from the literature that the STZ type 1 diabetes is indeed a suitable model for studying mitochondrial mechanisms of diabetic β cell glucotoxicity. Evidence presented includes: 1) continued β cell derangement is due to chronic hyperglycemia after STZ is completely eliminated out of the body; 2) STZ diabetes can be reversed by insulin treatment, which indicates that β cell responds to treatment and shows ability to regenerate; and 3) STZ diabetes can be ameliorated or alleviated by administration of phytochemicals. In addition, mechanisms of STZ action and fundamental gaps in understanding mitochondrial mechanisms of β cell dysfunction are also discussed.
Keywords: diabetes, β cell, glucotoxicity, mitochondria, redox imbalance, streptozotocin
Introduction
Diabetes mellitus and its complications are chronic glucotoxicity diseases. The concept of β cell glucotoxicity (and other cells as well) implicates that persistent excessive glucose can exert adverse or toxic effect on β cell function after the establishment of diabetes induced by either genetic or environmental factors.1 Hence, diabetic glucotoxicity is believed to play a forceful role in driving secondary β cell failure in both type 1 and type 2 diabetes.2,3 Despite intensive and extensive studies on β cell glucotoxicity, detailed mitochondrial mechanisms still remain poorly understood. In this regard, experimental animal models of diabetes have been indispensable to research in diabetes and its complications.
Streptozotocin-induced animal models of diabetes
Streptozotocin (STZ) is a widely used chemical for the induction of experimental diabetes in rodents.4,5 Since the initial report of its diabetogenic properties in 1963,6 STZ has been used alone or in combination with other chemicals or with dietary manipulations for induction of either type 1 or type 2 diabetes.7,8 Type 1 diabetes can be induced in rodents by a single STZ injection,9,10 while type 2 diabetes can be induced by at least three approaches, which include STZ injection after administration of nicotinamide,11,12 high fat diet (HFD) feeding followed by a low-dose STZ injection,13 and STZ injection during the neonatal period.14,15 All these STZ-involved diabetic animal models have been very useful in elucidating the mechanisms of diabetic pathogenesis and in screening artificial chemicals, natural products, and pharmacological agents that are potentially capable of lowering blood glucose levels.16,17
Mechanisms of STZ action
STZ is an antibiotic produced by the bacterium Streptomyces achromogens and possesses a broad spectrum of antibacterial properties.18 It contains a glucose molecule (in deoxy form) that is linked to a highly reactive methylnitrosourea moiety (Figure 1) that is thought to exert STZ’s cytotoxic effects, while the glucose moiety directs the chemical to the pancreatic β cells.19 STZ recognizes the GLUT2 receptor that is abundant on β cell plasma membranes.5 Therefore, pancreatic β cell is a specific target of STZ. As GLUT2 also exists in liver and kidney to a less extent,20 high doses of STZ could also impair the functions of liver and kidney.20 Upon ingestion, STZ is rapidly metabolized in the liver and quickly eliminated by renal excretion;21 therefore, STZ has really a short life (with a half-life of 15 minutes in the serum after IV injection22) and its acute toxicity to the liver and the kidney can be neglected after persistent hyperglycemia is obtained.23 After STZ is eliminated out of the body, any further functional impairment of the liver and the kidney may be attributed to the effects of diabetic hyperglycemia. This is the basis for studying the mechanisms of STZ diabetic complications in these organs23 as well as other organs such as the brain, the heart, and the muscles.24
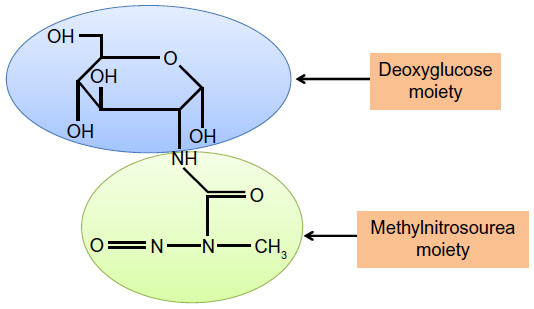 | Figure 1 Chemical structure of streptozotocin. |
Potential mechanisms of diabetic β cell glucotoxicity
When there is a status of persistent hyperglycemia as opposed to episodic hyperglycemia, cellular metabolic system is under constant pressure due to glucose overloading.25 This glucose overloading can activate many metabolic or signaling pathways that not only attempt to dispose excessive glucose but also generate more reactive oxygen species, leading to oxidative stress and β cell failure.1 As shown in Figure 2, these hyperglycemia-stimulated pathways include an increased NADH/NAD+ ratio linked to pseudohypoxia and reductive stress,25 the hexosamine pathway responsible for protein O-GlcNAc modifications,26 protein kinase C activation,27 activation of the polyol pathway resulting in accumulation of sorbitol and fructose,28 formation of methylglyoxal and advanced glycation products,29 enediol formation,30 and endoplasmic reticulum stress.31 The establishment that all these pathways culminate in reactive oxygen species production,32 together with the evidence of a low-level antioxidant capacity in β cells,1 is thought to be responsible for secondary diabetic β cell failure.3 Nonetheless, the mitochondrial mechanisms of β cell glucotoxicity in diabetes still remain poorly understood.
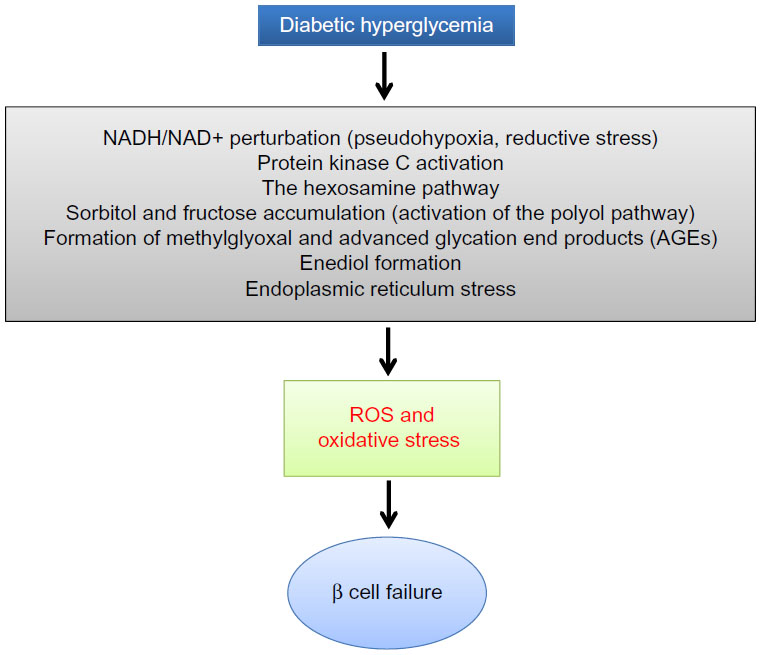 | Figure 2 Mechanisms by which diabetic hyperglycemia can impose glucotoxicity on β cells. |
Glucose combustion, mitochondrial ATP production, and β cell insulin secretion
It is well established that glucose-stimulated insulin secretion is tightly linked to glucose-driven ATP production by β cell mitochondria.33,34 As β cell has a very low level of lactate dehydrogenase,35 most of the pyruvate generated by the glycolytic pathway is transported into mitochondria to generate the reducing compounds NADH and FADH2, along with a complete combustion of pyruvate to CO2.36 This process is achieved not only by formation of acetyl-CoA that feeds into the Krebs cycle but also by formation of oxaloacetate that replenishes the intermediates in the Krebs cycle.37 When blood glucose gets higher after a meal, ATP production in β cell mitochondria also gets higher (Figure 3). This leads to an elevated level of ATP/ADP ratio in the cytoplasm and a consequent closure of the kATP channels on the cell membrane. The closure of the kATP channels then depolarizes the membranes and renders the opening of the calcium channel, resulting in calcium influx that triggers insulin granule exocytosis and insulin release38–40 (Figure 3). In diabetes, however, this episodic process of glucose-stimulated insulin secretion is believed to be impaired.41,42
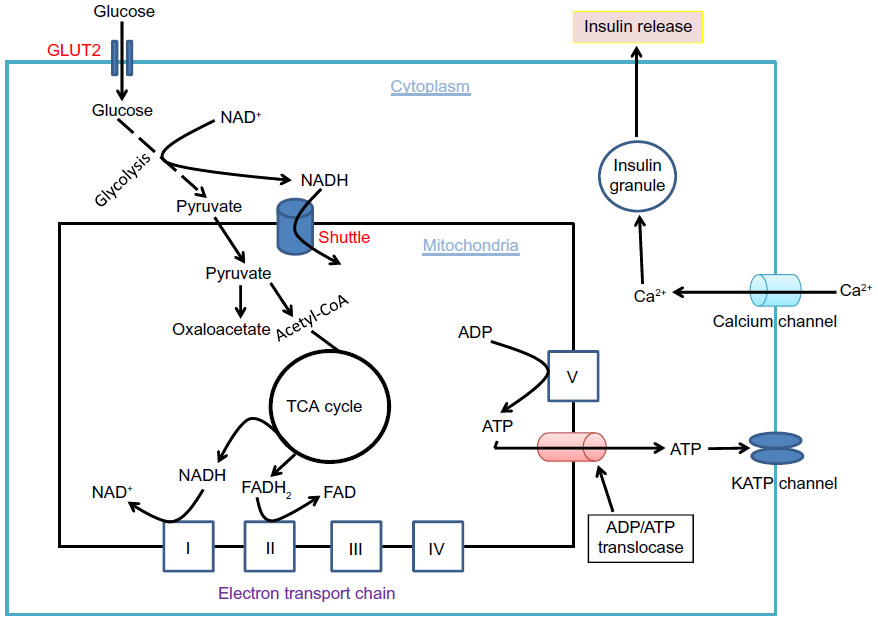 | Figure 3 Glucose combustion is tightly coupled to insulin secretion in pancreatic β cells. |
Redox imbalance and mitochondrial deregulation in β cell dysfunction
Although pancreatic β cell function operates by a supply driven mechanism by which glucose or nutrient metabolism is coupled to insulin secretion,43 persistent stimulation of β cell by a high level of glucose could eventually lead to β cell exhaustion and cell death.3,44 This glucotoxicity has been thought to originate from redox imbalance between NADH and NAD+.45 On one hand, NADH can be overproduced by hyperglycemia and the activation of polyol pathway that makes NADH from NADPH;25 on the other hand, NAD+ can be depleted by the activation of enzymes such as poly ADP-ribosylase, sirtuins, and CD38 that use NAD+ as their substrate.46 Therefore, exceedingly accumulation of NADH and potential depletion of NAD+ indicate a severe problem in NADH/NAD+ recycling under diabetic conditions.46 As mitochondria is the major cellular site for maintaining redox balance by oxidizing NADH via complex I and FADH2 via complex II, mitochondrial election transport chain plays a critical role in β cell function and dysfunction.36 Nonetheless, the role of each component of the electron transport chain, in particular, that of complex I (Figure 4), in diabetic β cell glucotoxicity remains largely unknown. Additionally, the roles of those mitochondrial dehydrogenases in making NADH from NAD+ (Figure 4) are also unknown. From this perspective, STZ diabetic animal models should provide a broad platform for exploring the mitochondrial mechanism of diabetic glucotoxicity, implicating each component of the electron transport chain.
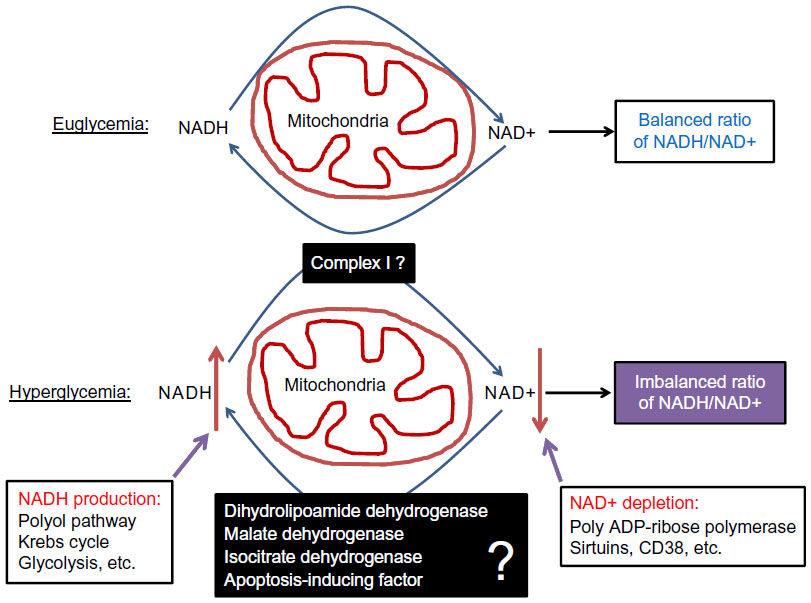 | Figure 4 Role of redox imbalance between NADH and NAD+ in β cell dysfunction. |
STZ-induced type 1diabetes as an animal model for further exploring the mitochondrial mechanisms of diabetic β cell glucotoxicity
Our laboratory recently embarked on studies of mitochondrial mechanisms of β cell glucotoxicity using animal models of diabetes induced by a single intraperitoneal STZ injection. While there is no doubt that STZ-induced type 2 diabetes are good models for studying mitochondrial glucotoxicity in β cells, one question often being asked is that whether the STZ type 1 diabetes model is suitable for studying β cell diabetic glucotoxicity as STZ partially destructs pancreas and reduces β cell mass.47 After reviewing the literature, we firmly believe that the single STZ injection diabetic animal model is appropriate for studying the mitochondrial mechanisms of β cell glucotoxicity. Specifically, our belief is based on the following experimental evidence reported in the literature.
- STZ is rapidly eliminated after ingestion. For intraperitoneal injection, STZ can be eliminated within 48 hours of ingestion21 and its DNA methylating effect quickly diminishes as no further increase in DNA methylation can be detected after 24 hours of STZ exposure.48,49 These pieces of evidence indicate that STZ acute toxicity is very short-lived after ingestion. Yet, β cell function continues to deteriorate in the absence of detectable STZ.50 It is now known that it is the hyperglycemic state established by acute STZ toxicity that drives further β cell derangement.51,52 Therefore, after induction of diabetes, β cell dysfunction is maintained by persistent hyperglycemia, and such β cell can be explored to provide insights into the mitochondrial mechanisms of glucotoxicity.
- STZ diabetes can be reversed by insulin treatment. Grossman et al have reported that glycemic control in STZ diabetic mice by insulin can promote β cell regeneration in the diabetic pancreas,53 demonstrating again that further β cell dysfunction, in the absence of STZ, is due to diabetic hyperglycemia. Once this hyperglycemic status is reversed to euglycemia by insulin supplement, β cell function improves via β cell regeneration.53 In fact, while animals show insulin deficiency after STZ ingestion, they do not require insulin treatment for survival.54 Therefore, many diabetic animals can live beyond 24 weeks without any interventions after a single STZ injection,24 indicating that the dosage of STZ given as a single injection only partially destructs islets and β cells54,55 and that diabetes created as such is due to β cell glucotoxicity rather than STZ acute toxicity. The results of Grossman’s study agree well with those of other studies that β cell function indeed responds to treatment and may have limited regeneration after partial pancreatic destruction by STZ.52,56,57 On the other hand, the use of high STZ doses (eg, 100 mg/kg for rats) could lead to nearly a complete destruction of β cells, which is often associated with a quick demise of the animals.54 However, the use of a very high STZ dose is never the case for studies of drug-screening and of glucotoxicity in diabetes and diabetic complications.
- β cell dysfunction in STZ diabetes can be alleviated by plant natural products or phytochemicals. In fact, this is the basis for screening diabetic drugs or compounds using STZ diabetic animal models. In addition to DNA alkylation thought to be involved in STZ β cell toxicity,4,5 oxidative damage by reactive oxygen or reactive nitrogen species has also been implicated in β cell destruction by STZ.4,5 Therefore, numerous studies have demonstrated that plant extracts or phytochemicals having antioxidant properties can ameliorate β cell dysfunction in STZ diabetes. For example, grape seed proanthocyanidins,58 curcumin,59 resveratrol,60 and pycnogenol61 all have been reported to improve β cell function in STZ diabetic animals. The underlying mechanisms of these phytochemicals are likely due to their abilities to attenuate hyperglycemic glucotoxicity by decreasing blood glucose levels and/or facilitating glucose combustion, leading to an eventual rebalanced redox state between NADH and NAD+,62–64 which is conducive to proliferation of surviving β cell or regeneration of β cell from other type of cells such as acinar and ductal cells.10,20,56 It is unlikely that hyperglycemia-impaired β cell would be a direct target for repairing by these phytochemicals or other glucose lowering agents.
Summary and conclusion
In summary, as the initial STZ toxicity to β cells is short-lived21 and further impairment of the surviving β cell’s function is due to hyperglycemic toxicity,24,51,65 the mitochondrial mechanisms of β cell glucotoxicity in these surviving β cells can thus be elucidated (Figure 5). Additionally, as STZ-destructed cells undergo necrosis and are eliminated by macrophages,66,67 no intact mitochondria may be isolated from these STZ-destructed cells; thus, mitochondria isolated from STZ-exposed pancreas should be from those surviving or hyperglycemia-impaired β cells (Figure 5). Based on these findings, we believe that animal models of diabetes induced by single STZ injection can serve as a platform for studying the mitochondrial mechanisms of β cell glucotoxicity. Of course, no mitochondria isolation and functional analysis should be performed right after STZ injection in order to avoid any acute toxic effects of STZ on mitochondria. Rather, mitochondria should be isolated when overt diabetes develops after STZ is completely eliminated out of the body. The point here is that it should be distinguished between STZ acute toxicity and diabetic glucotoxicity when needed for given STZ diabetes settings.
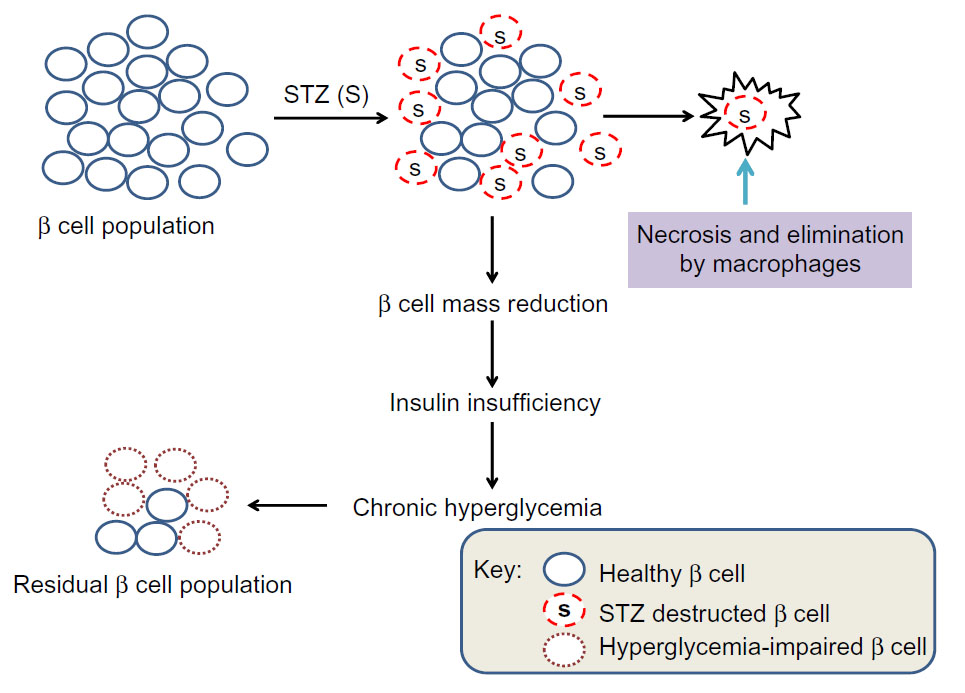 | Figure 5 Scheme showing partial destruction of β cell population by STZ and reduction in β cell mass that induces insulin insufficiency and chronic hyperglycemia. |
In conclusion, STZ-induced type 1 diabetes in rodents is a well-established and well-accepted practice for the studies of pathogenesis of diabetes and its complications. However, it should be pointed out that this STZ model has both advantages and disadvantages over other STZ-involved experimental models, including STZ-neonatal and STZ-HFD. On one hand, when compared with the STZ-neonatal and STZ-HFD models that can also be used to study the mitochondrial mechanisms of β cell glucotoxicity, the single STZ injection model is much less expensive and much less time-consuming. For example, in the STZ-HFD model, HFD feeding usually takes a few weeks or a few months followed by low-dose STZ injections.13,16,68 Similarly, the STZ-neonatal model also takes a longer time for diabetes to develop in addition to caring and manipulating neonatal animals.15,69 On the other hand, the disadvantage of the single STZ injection model is that it does not create the insulin resistance pathophysiology70 that can be observed in the STZ-neonatal and STZ-HFD models. Additionally, no standard protocols exist for STZ preparation and injections, and the diabetic state can be highly variable due to factors such as age of animals, sex, body weight, species, and strains.71 Moreover, it is by no means that the STZ type 1 diabetic animal model would be equivalent to the human disease state. Nonetheless, the single STZ injection model should continue to provide a cost-effective, time-saving, convenient platform for the study of the pathophysiological mitochondrial mechanisms of β cell derangement induced by diabetic glucotoxicity.
Acknowledgment
This publication was supported in part by National Institute of Neurological Disorders and Stroke, Grant R01NS079792.
Disclosure
The authors declare that there is no conflict of interest.
References
Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29(3):351–366. | |
Korsgren O, Jansson L, Sandler S, Andersson A. Hyperglycemia-induced B cell toxicity. The fate of pancreatic islets transplanted into diabetic mice is dependent on their genetic background. J Clin Invest. 1990;86(6):2161–2168. | |
Poitout V, Robertson RP. Minireview: secondary beta-cell failure in type 2 diabetes – a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143(2):339–342. | |
Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50(6):537–546. | |
Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. 2008;51(2):216–226. | |
Rakieten N, Rakieten ML, Nadkarni MR. Studies on the diabetogenic action of streptozotocin (NSC-37917). Cancer Chemother Rep. 1963; 29:91–98. | |
Gajdosik A, Gajdosikova A, Stefek M, Navarova J, Hozova R. Streptozotocin-induced experimental diabetes in male Wistar rats. Gen Physiol Biophys. 1999;18:54–62. | |
Ghasemi A, Khalifi S, Jedi S. Streptozotocin-nicotinamide-induced rat model of type 2 diabetes (review). Acta Physiol Hung. 2014;101(4):408–420. | |
Junod A, Lambert AE, Stauffacher W, Renold AE. Diabetogenic action of streptozotocin: relationship of dose to metabolic response. J Clin Invest. 1969;48(11):2129–2139. | |
Yin D, Tao J, Lee DD, et al. Recovery of islet beta-cell function in streptozotocin-induced diabetic mice: an indirect role for the spleen. Diabetes. 2006;55(12):3256–3263. | |
Wu MS, Liang JT, Lin YD, Wu ET, Tseng YZ, Chang KC. Aminoguanidine prevents the impairment of cardiac pumping mechanics in rats with streptozotocin and nicotinamide-induced type 2 diabetes. Br J Pharmacol. 2008;154(4):758–764. | |
Szkudelski T. Streptozotocin-nicotinamide-induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med (Maywood). 2012;237(5):481–490. | |
Skovso S. Modeling type 2 diabetes in rats using high fat diet and streptozotocin. J Diabetes Investig. 2014;5(4):349–358. | |
Patil MA, Suryanarayana P, Putcha UK, Srinivas M, Reddy GB. Evaluation of neonatal streptozotocin induced diabetic rat model for the development of cataract. Oxid Med Cell Longev. 2014;2014:463264. | |
Portha B, Blondel O, Serradas P, et al. The rat models of non-insulin dependent diabetes induced by neonatal streptozotocin. Diabete Metab. 1989;15(2):61–75. | |
Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005; 52(4):313–320. | |
Kumar S, Vasudeva N, Sharma S. GC-MS analysis and screening of antidiabetic, antioxidant and hypolipidemic potential of Cinnamomum tamala oil in streptozotocin induced diabetes mellitus in rats. Cardiovasc Diabetol. 2012;11:95. | |
Vavra JJ, Deboer C, Dietz A, Hanka LJ, Sokolski WT. Streptozotocin, a new antibacterial antibiotic. Antibiot Annu. 1959;7:230–235. | |
Johansson EB, Tjalve H. Studies on the tissue-disposition and fate of [14C]streptozotocin with special reference to the pancreatic islets. Acta Endocrinol. 1978;89(2):339–351. | |
Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiol Rev. 2005;85(4):1255–1270. | |
Karunanyake EH, Hearse DJ, Mellows G. The synthesis of C14Streptozotocin and its distribution and excretion in the rat. Biochem J. 1974;142:673–683. | |
Eleazu CO, Eleazu KC, Chukwuma S, Essien UN. Review of the mechanism of cell death resulting from streptozotocin challenge in experimental animals, its practical use and potential risk to humans. J Diabetes Metab Disord. 2013;12(1):60. | |
Tesch GH, Allen TJ. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology. 2007;12(3):261–266. | |
Wei M, Ong L, Smith MT, et al. The streptozotocin-diabetic rat as a model of the chronic complications of human diabetes. Heart Lung Circ. 2003;12(1):44–50. | |
Yan LJ. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res. 2014;2014:137919. | |
Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010;36(6 pt 1):423–435. | |
Naruse K, Rask-Madsen C, Takahara N, et al. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55(3):691–698. | |
Williamson JR, Chang K, Frangos M, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42(6):801–813. | |
Queisser MA, Yao D, Geisler S, et al. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010;59(3):670–678. | |
Wolff SP, Dean RT. Glucose autoxidation and protein modification. The potential role of ‘autoxidative glycosylation’ in diabetes. Biochem J. 1987;245(1):243–250. | |
Leibowitz G, Kaiser N, Cerasi E. beta-Cell failure in type 2 diabetes. J Diabetes Investig. 2011;2(2):82–91. | |
Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279(41):42351–42354. | |
Maechler P, Wollheim CB. Role of mitochondria in metabolism-secretion coupling of insulin release in the pancreatic beta-cell. Biofactors. 1998;8(3–4):255–262. | |
Malmgren S, Nicholls DG, Taneera J, et al. Tight coupling between glucose and mitochondrial metabolism in clonal beta-cells is required for robust insulin secretion. J Biol Chem. 2009;284(47):32395–32404. | |
Sekine N, Cirulli V, Regazzi R, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J Biol Chem. 1994;269(7):4895–4902. | |
Wiederkehr A, Wollheim CB. Minireview: implication of mitochondria in insulin secretion and action. Endocrinology. 2006;147(6):2643–2649. | |
Schuit F, De Vos A, Farfari S, et al. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem. 1997;272(30):18572–18579. | |
Maechler P. Mitochondrial function and insulin secretion. Mol Cell Endocrinol. 2013;379(1–2):12–18. | |
Maechler P, Carobbio S, Rubi B. In beta-cells, mitochondria integrate and generate metabolic signals controlling insulin secretion. Int J Biochem Cell Biol. 2006;38(5–6):696–709. | |
Maechler P. Mitochondrial signal transduction in pancreatic beta-cells. Best Pract Res Clin Endocrinol Metab. 2012;26(6):739–752. | |
Zou CY, Gong Y, Liang J. Metabolic signaling of insulin secretion by pancreatic beta-cell and its derangement in type 2 diabetes. Eur Rev Med Pharmacol Sci. 2014;18(15):2215–2227. | |
Satin LS, Butler PC, Ha J, Sherman AS. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Mol Aspects Med. In press 2015. | |
Wiederkehr A, Wollheim CB. Mitochondrial signals drive insulin secretion in the pancreatic beta-cell. Mol Cell Endocrinol. 2012;353(1–2):128–137. | |
Bensellam M, Laybutt DR, Jonas JC. The molecular mechanisms of pancreatic beta-cell glucotoxicity: recent findings and future research directions. Mol Cell Endocrinol. 2012;364(1–2):1–27. | |
Teodoro JS, Rolo AP, Palmeira CM. The NAD ratio redox paradox: why does too much reductive power cause oxidative stress? Toxicol Mech Methods. 2013;23(5):297–302. | |
Luo X, Li R, Yan L-J. Roles of pyruvate, NADH, and mitochondrial complex I in redox balance and imbalance in β cell function and dysfunction. J Diabetes Res. 2015. | |
Gomes A, Vedasiromoni JR, Das M, Sharma RM, Ganguly DK. Anti-hyperglycemic effect of black tea (Camellia sinensis) in rat. J Ethnopharmacol. 1995;45(3):223–226. | |
Mossman BT, Ireland CM, Filipak M, LeDoux S, Wilson GL. Comparative interactions of streptozotocin and chlorozotocin with DNA of an insulin-secreting cell line (RINr). Diabetologia. 1986;29(3):186–191. | |
Pettepher CC, LeDoux SP, Bohr VA, Wilson GL. Repair of alkali-labile sites within the mitochondrial DNA of RINr 38 cells after exposure to the nitrosourea streptozotocin. J Biol Chem. 1991;266(5):3113–3117. | |
Rerup CC. Drugs producing diabetes through damage of the insulin secreting cells. Pharmacol Rev. 1970;22(4):485–518. | |
Kullin M, Li Z, Hansen JB, Bjork E, Sandler S, Karlsson FA. K(ATP) channel openers protect rat islets against the toxic effect of streptozotocin. Diabetes. 2000;49(7):1131–1136. | |
Matsuda M, Kawasaki F, Mikami Y, et al. Rescue of beta-cell exhaustion by diazoxide after the development of diabetes mellitus in rats with streptozotocin-induced diabetes. Eur J Pharmacol. 2002;453(1):141–148. | |
Grossman EJ, Lee DD, Tao J, et al. Glycemic control promotes pancreatic beta-cell regeneration in streptozotocin-induced diabetic mice. PLoS One. 2010;5(1):e8749. | |
Rodrigues B, Poucheret P, Battell ML, Mcneill JH. Streptozotocin-induced diabetes: Induction, mechanism(s), and dose dependency. In: McNeill JH, editor. Experimental Models of Diabetes. Boca Raton: CRC Press; 1999:3–18. | |
Kozuka C, Sunagawa S, Ueda R, et al. Gamma-oryzanol protects pancreatic beta-cells against endoplasmic reticulum stress in male mice. Endocrinology. 2015;156(4):1242–1250. | |
Marchand KC, Arany EJ, Hill DJ. Effects of atorvastatin on the regeneration of pancreatic {beta}-cells after streptozotocin treatment in the neonatal rodent. Am J Physiol Endocrinol Metab. 2010;299(1):E92–E100. | |
Hamming NA, Reynolds WA. DNA synthesis in pancreatic islet and acinar cells in rats with streptozotocin-induced diabetes. Horm Metab Res. 1977;9(2):114–116. | |
Ding Y, Zhang Z, Dai X, et al. Grape seed proanthocyanidins ameliorate pancreatic beta-cell dysfunction and death in low-dose streptozotocin- and high-carbohydrate/high-fat diet-induced diabetic rats partially by regulating endoplasmic reticulum stress. Nutr Metab. 2013;10:51. | |
Chanpoo M, Petchpiboonthai H, Panyarachun B, Anupunpisit V. Effect of curcumin in the amelioration of pancreatic islets in streptozotocin-induced diabetic mice. J Med Assoc Thai. 2010;93(Suppl 6):S152–S159. | |
Ku CR, Lee HJ, Kim SK, Lee EY, Lee MK, Lee EJ. Resveratrol prevents streptozotocin-induced diabetes by inhibiting the apoptosis of pancreatic beta-cell and the cleavage of poly (ADP-ribose) polymerase. Endocr J. 2012;59(2):103–109. | |
Parveen K, Ishrat T, Malik S, Kausar MA, Siddiqui WA. Modulatory effects of Pycnogenol in a rat model of insulin-dependent diabetes mellitus: biochemical, histological, and immunohistochemical evidences. Protoplasma. 2013;250(1):347–360. | |
Kannadhasan R, Ashwini Anjana AJ. Phytochemical and antioxidant activity of polyherbal hydroethanolic extract in streptozotocin induced β cell destruction in rats. Int J Pharm Sci Drug Res. 2012;4(1):25–30. | |
Erejuwa OO, Sulaiman SA, Wahab MS, Sirajudeen KN, Salleh MS, Gurtu S. Antioxidant protection of Malaysian tualang honey in pancreas of normal and streptozotocin-induced diabetic rats. Ann Endocrinol. 2010;71(4):291–296. | |
Niture NT, Ansari AA, Naik SR. Anti-hyperglycemic activity of rutin in streptozotocin-induced diabetic rats: an effect mediated through cytokines, antioxidants and lipid biomarkers. Indian J Exp Biol. 2014;52(7):720–727. | |
Mellado-Gil JM, Aguilar-Diosdado M. High glucose potentiates cytokine- and streptozotocin-induced apoptosis of rat islet cells: effect on apoptosis-related genes. J Endocrinol. 2004;183(1):155–162. | |
Fujimoto H, Hirase T, Miyazaki Y, et al. IL-27 inhibits hyperglycemia and pancreatic islet inflammation induced by streptozotocin in mice. Am J Pathol. 2011;179(5):2327–2336. | |
O’Brien BA, Harmon BV, Cameron DP, Allan DJ. Beta-cell apoptosis is responsible for the development of IDDM in the multiple low-dose streptozotocin model. J Pathol. 1996;178(2):176–181. | |
Zhang M, Lv XY, Li J, Xu ZG, Chen L. The characterization of high-fat diet and multiple low-dose streptozotocin induced type 2 diabetes rat model. Exp Diabetes Res. 2008;2008:704045. | |
Takada J, Machado MA, Peres SB, et al. Neonatal streptozotocin-induced diabetes mellitus: a model of insulin resistance associated with loss of adipose mass. Metabolism. 2007;56(7):977–984. | |
Larsen MO. Beta-cell function and mass in type 2 diabetes. Dan Med Bull. 2009;56(3):153–164. | |
Deeds MC, Anderson JM, Armstrong AS, et al. Single dose streptozotocin-induced diabetes: considerations for study design in islet transplantation models. Lab Anim. 2011;45(3):131–140. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.