Back to Journals » Stem Cells and Cloning: Advances and Applications » Volume 8
Hedgehog signaling in cancer stem cells: a focus on hematological cancers
Authors Campbell V, Copland M
Received 3 October 2014
Accepted for publication 18 November 2014
Published 16 January 2015 Volume 2015:8 Pages 27—38
DOI https://doi.org/10.2147/SCCAA.S58613
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Bernard Binetruy
Victoria Campbell, Mhairi Copland
Paul O'Gorman Leukaemia Research Centre, Institute of Cancer Sciences, College of Medical, Veterninary and Life Sciences, University of Glasgow, Glasgow, UK
Abstract: The stem cell paradigm was first demonstrated in hematopoietic stem cells. Whilst classically it was cytokines and chemokines which were believed to control stem cell fate, more recently it has become apparent that the stem cell niche and highly conserved embryonic pathways play a key role in governing stem cell behavior. One of these pathways, the hedgehog signaling pathway, found in all organisms, is vitally important in embryogenesis, performing the function of patterning through early stages of development, and in adulthood, through the control of somatic stem cell numbers. In addition to these roles in health however, it has been found to be deregulated in a number of solid and hematological malignancies, components of the hedgehog pathway being associated with a poor prognosis. Further, these components represent viable therapeutic targets, with inhibition from a drug development perspective being readily achieved, making the hedgehog pathway an attractive potential therapeutic target. However, although the concept of cancer stem cells is well established, how these cells arise and the factors which influence their behavior are not yet fully understood. The role of the hedgehog signaling pathway and its potential as a therapeutic target in hematological malignancies is the focus of this review.
Keywords: hedgehog signaling pathway, stem cell, cancer stem cell, hematopoiesis, myeloid, lymphoid
Background
Stem cells
Adult somatic stem cells are defined as undifferentiated cells with three key properties: long life, multipotency, and the capacity to self-renew.1 However, recent evidence has shown that although their phenotype is tightly defined, these cells are functionally heterogeneous.2 The properties of stem cell self-renewal and survival are controlled by highly conserved embryonic signaling pathways, including the hedgehog (Hh), epithelial-mesenchymal transition, WNT, and Notch signaling pathways, the HOX transcription factors, and the BMI1/polycomb transcriptional regulators.3
Cancer stem cells
There is experimental evidence from a number of malignancies to suggest the presence of a small population of very primitive cells that share many of the properties of somatic stem cells. These cells have been termed cancer stem cells (CSCs).1
Whilst there is still much to learn in respect of the CSC, various experimental models have shown these cells to be quiescent, resistant to therapy, capable of self-renewal, and the initiation of tumors in secondary transplanted hosts.4 Malignancies with evidence of CSC origin include hematological cancers4,5 and solid tumors including: prostate,6 pancreas,7 lung,8 and certain neurological malignancies.9
Although the concept of the CSC is largely accepted, the model by which it is capable of generating a tumor, or leukemia, remains contentious.10 The stochastic model argues tumors are biologically homogeneous, with CSC behavior being determined by intrinsic or extrinsic factors; tumor heterogeneity arising because these factors are unpredictable.11 In contrast, the hierarchy model argues CSCs are highly organized in unidirectional cellular hierarchies (the pattern of normal tissue growth). In the hierarchical model, the CSCs are biologically unique.12 The fundamental difference between the models lies in which cell, or cells, are capable of behaving as a CSC with discrimination relying on accurate, well-defined experimental design.13
The CSC hypothesis is of considerable clinical importance, potentially explaining minimal residual disease, relapse and disease progression, and highlighting the need to target these cells in order to effect a cure.14 CSCs have been shown to be innately less sensitive to treatment, to continually develop genomic and epigenomic changes, and to uniquely interact with the stem cell niche.14,15 The influence each of these factors has on the CSCs treatment-resistant phenotype is however, unclear.
The pathways involved in self-renewal are of intense interest, with many, if not all being implicated in neoplastic proliferation when deregulated.15 It is the behavior of one of these pathways, the Hh signaling pathway, which is the focus of this review.
Hh signaling pathway
The Hh signaling pathway was initially discovered in 1980 by Nüsslein-Volhard and Weischaus whilst studying embryonic patterning in the Drosophila fruit fly, with absence of the Hh protein giving the Drosophila a characteristic “hairy” or “prickly” appearance.16,17 Subsequent work has shown the Hh pathway to be highly conserved across species and vitally important in embryogenesis, performing the function of patterning during the early stages of development through the expansion and contraction of stem cell numbers. In adult organisms, through its ability to affect stem cell behavior in responsive tissues, it is involved in aspects of tissue maintenance and regeneration – proliferation, apoptosis, chromatin modeling, and self-renewal, acting in concert with other stimuli and the stem cell niche.18
Canonical signaling
Classically, the Hh signaling pathway is believed to be ligand-dependent. Three Hh ligands (Sonic [SHH], Indian [IHH], and Desert [DHH]) have been identified in vertebrates, affecting stem cell behavior in a time- and concentration-dependent manner.19 SHH is widely expressed, particularly during embryogenesis, with SHH deficiency being embryonically lethal.17 IHH is produced in hematopoietic cells, bone, and cartilage,20 whilst DHH is found in the peripheral nervous system and testes.21 Hh ligands are initially synthesized as an inactive 45 kDa precursor, undergoing post-translational modifications to form a 19 kDa amino-terminal active signaling molecule.22 This cholesterol and palmitoyl modification, catalyzed by Hh acyltransferase,23 not only enhances ligand activity but also modifies its diffusion capacity.24 The Hh ligands bind to the 12 trans-membrane receptor protein Patched 1 (PTCH1), causing its internalization and removing its repression of the 7-span trans-membrane protein Smoothened (SMO), allowing pathway activity.25 In vertebrates, activity of the Hh pathway appears intrinsically related to primary immotile cilia; in the absence of ligand, PTCH1 is located within the primary cilia. Following ligand binding, and the internalization of PTCH1, SMO is able to concentrate in the primary cilia where it interacts with the GLI transcription factors shifting the balance toward pathway activation.25 Whilst the intricacies of this interaction remain poorly understood, studies suggest these receptors do not physically interact, rather PTCH1 is thought to regulate SMO through an intermediary, with studies suggesting that oxysterols, including vitamin D3, are involved.26 SMO subsequently causes accumulation of the full length active form of the zinc transcription factors GLI-2 and GLI-3 in the nucleus, and potentiates the activity of other positive regulators of the pathway including serine threonine kinase 36 (STK36) and kinesin family member 7 (KIF7), resulting in transcription of key downstream targets such as GLI-1 and PTCH1, regulators of chromatin formation, cell cycle activity, cell mobility, and apoptosis, eg, bone morphogenetic protein 4, forkhead box protein M1, and WNT2a,27 Figure 1A. Of the transcription factors, GLI-1 functions as a positive regulator, GLI-3 a transcriptional repressor, and GLI-2 both a positive and negative transcriptional regulator determined by post-transcriptional and post-translational modifications.28 It is the balance between these transcription factors which determines pathway activity.29
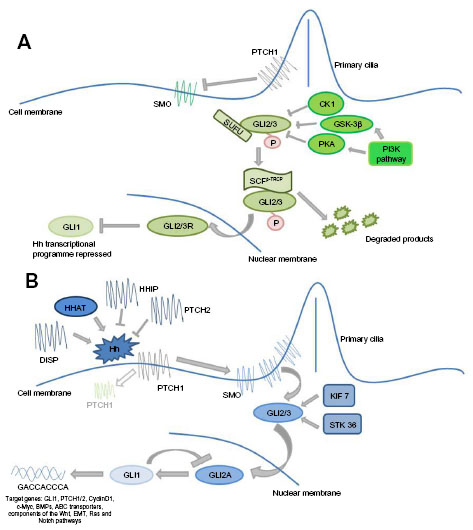 | Figure 1 Canonical Hedgehog (Hh) signaling. |
Alternatively, the Hh ligands can bind to a number of membrane-associated glycoproteins: Patched 2 (PTCH2), Hh-interacting protein (HHIP), and Dispatched (DISP). PTCH2, although structurally similar to PTCH1, has distinct functional properties and notably different tissue distribution, PTCH1 being broadly expressed whilst PTCH2 is primarily restricted to the skin and testes.30,31 HHIP is an endogenous Hh ligand inhibitor, with a binding affinity comparable to PTCH1, preventing pathway activation.32 DISP, a 12 trans-membrane receptor protein, is not involved in Hh ligand synthesis or processing but rather facilitates ligand movement, thereby modulating canonical pathway activity.33
In the inactive state the transcription factors GLI-2 and GLI-3 are retained in the cytoplasm in a protein complex associated with the inhibitory molecule, suppressor of fused (SUFU)34 and non-specifically phosphorylated by casein kinase (CKI), glycogen synthase 3β (GSK3β), and protein kinase A (PKA). This complex subsequently undergoes E3 ubiquitin-mediated proteolysis by the Skp1-Cullin1-F-box protein (SCFβ-TRCP) to the truncated repressor form which, on translocating to the nucleus, strongly inhibits the Hh pathway,35 Figure 1B. It is the complex interplay between the active and inactive state of the pathway and the positive and negative feedback loops that maintain the careful balance of Hh signaling in normal tissue.
Non-canonical signaling
The notion of non-canonical signaling has arisen following observations that the pathway response does not always appear to follow the classical canonical signaling paradigm, although the evidence is not conclusive. Three scenarios of non-canonical Hh signaling have been described, Figure 2: direct interaction with components of other pathways, atypical interaction of components, and activity independent of GLI-mediated transcription, defined as Type I (PTCH-dependent) and Type II (SMO-dependent).36 In reality, it is likely the canonical and non-canonical pathways act in parallel.
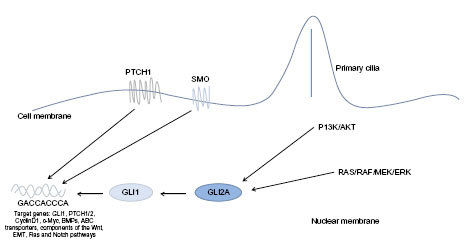 | Figure 2 Non-canonical Hh signaling. |
Downstream targets
There are numerous downstream targets of the Hh pathway: BCL-2, the ATP-binding cassette transporter family members; the multi-drug resistance protein-1 and components of the epithelial-mesenchymal transition, WNT, and Notch signaling pathways, Figure 3. There is already evidence to show up-regulation and involvement of many of these pathways in chemo-resistance in human malignancies.37,38 Additionally, studies have highlighted several of these downstream targets to be over-expressed in both acute myeloid leukemia (AML) and chronic myeloid leukemia (CML), with expression linked to chemo-resistance and poorer survival.39
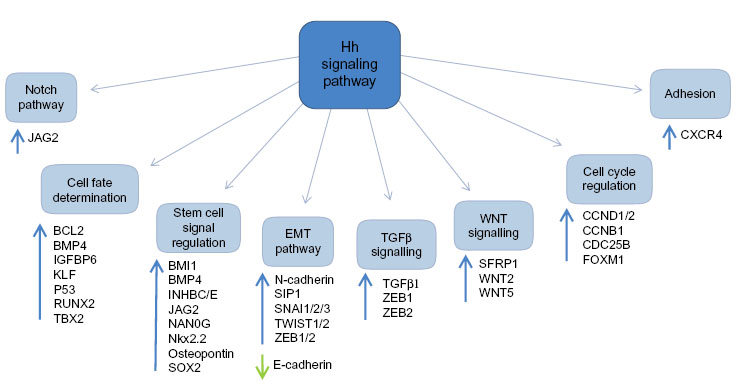 | Figure 3 Targets of the Hh signaling pathway. |
Hh signaling in hematopoiesis
In vertebrates, hematopoiesis is broadly divided into two major phases, primitive (embryonic) and definitive.40 The Hh signaling pathway has a complex role in both embryonic and adult hematopoiesis. This role appears to be dependent on developmental stage, cell lineage, and whether the hematopoietic system is under regenerative pressure.27,41 Whilst evidence has shown it to be vital for early hematopoietic development,20 there remains controversy over its role in normal hematopoiesis in adult organisms,41–43 although some of this may be explained by experimental method. Interestingly, recent early phase clinical trials looking at SMO inhibition have shown little or no hematopoietic toxicity potentially indicating Hh signaling may be dispensable in certain situations.44 Importantly, abrogation of canonical Hh signaling by knockout of SMO does not adversely affect steady-state normal hematopoiesis.42,45
Hh signaling in malignancy
Crucially, abnormal Hh signaling has been associated with diverse human malignancies including basal cell carcinoma,46 medulloblastoma,47 pancreatic,48 and lung cancer.49 Interestingly, data suggest different mechanisms of action in the various tumor environments. Constitutive pathway activation through loss-of-function mutations,46 epigenetic modifications,50 or reduced expression of the negative regulators PTCH, HHIP, and SUFU51 or gain-of-function mutations and epigenetic changes52 in the positive regulator SMO have been observed in a number of solid malignancies.53 To date, no mutations have been identified in hematological malignancies; however, epigenetic modifications have been observed in a cohort of pediatric AML patients, correlating with disease status.54 Ligand-dependent canonical pathway activation involves autocrine or paracrine Hh signaling.55 Autocrine Hh signaling has been identified in multiple myeloma (MM),56 prostate,57 and lung cancer.58 Paracrine Hh signaling has been observed in lymphoma,59 colon, and pancreatic cancer.60
Hh signaling in myeloid malignancies
In myeloid malignancies, Hh signaling has been found to be vital in the maintenance and expansion of the CSC or “leukemic stem cell” (LSC), either as a survival and proliferation signal or through direction of the LSC fate.27,61–64
CML is a clonal myeloproliferative disease driven by the Philadelphia chromosome which encodes the constitutively active BCR-ABL tyrosine kinase. For CML to develop, this mutation must originate in a multi-potent hematopoietic stem cell (HSC), producing the leukemia-initiating cell or LSC.65 Acquisition of BCR-ABL by an HSC results in a number of functional changes, including increased proliferation, differentiation block, inhibition of apoptosis, and altered cell adhesion and stromal interactions, producing the clinical phenotype of CML.66
The development of tyrosine kinase inhibitors (TKIs) has revolutionized the treatment of CML, with the majority of patients now achieving long-term survival with a good quality of life.67 TKI therapy, however, is not without side effects, nor is it effective in all patients; TKI resistance and disease persistence despite TKI therapy remain ongoing issues. Persistent disease is believed to be due to lack of dependence on BCR-ABL signaling for survival and TKI resistance in CML stem cells.68
Hh signaling appears to be intimately involved in the persistence and expansion of these CML stem cells.35,43,61 Two different SMO-deficient murine models have shown supporting results – reduced incidence of leukemia in primary and secondary transplant recipients and a prolonged latency in primary transplantation.43,61 Additionally, these groups demonstrated improved survival, with an accompanying reduction in the LSC population following treatment with the SMO antagonist cyclopamine; combination therapy with a TKI and cyclopamine resulting in the largest reduction in LSCs in vitro and in vivo. These findings are supported by studies using clinical grade SMO inhibitors alone, and combined with TKIs, showing an improved survival, with a reduced incidence of leukemia in secondary transplant recipients and a marked reduction in measures of self-renewal.69 Supporting work in NOD scid gamma mice, looking at molecular targets, has shown targeting the Hh pathway with dasatinib and GDC-0449 resulted in reduced expression of GLI-1, GLI-2, BCL-2, and Cyclin D2, and increased expression of p21, pATM, pChk2, and γH2AX.70 Interestingly, low PTCH1 expression has been found to be an independent predictor of imatinib failure and reduced overall survival.71
AML is an extremely heterogeneous clonal disorder. Whilst there is clear evidence to support the CSC theory in AML,4,72 recent work has suggested the LSC population is phenotypically variable, and may not be confined to a single clonal subpopulation. Further, whether this LSC arises following progenitor cell acquisition of abnormal self-renewal potential or from an HSC remains unclear.4,5,72
There is increasing evidence showing the Hh pathway is deregulated in AML. Leukemic cell lines and primary AML cells express components of the Hh pathway, SHH and GLI-1.73 GLI-1 expression correlating with cytogenetic risk, inferior event-free survival and a reduced overall survival, with GLI-1 conferring drug resistance through UGT1A-dependent glucuronidation.74 Further, high GLI-1 expression predicts poor remission status and reduced overall survival in secondary AML.75 GLI-2 has been shown to be a negative prognostic indicator in a number of microarrays. Additionally, aberrant Hh signaling has been linked to drug resistance with inhibition of the pathway restoring chemosensitivity in AML.36,62 Further, pathway inhibition with PF-04449913 sensitized AML cell lines and primary cells to the standard chemotherapy agent cytarabine; additional inhibition of SMO modulated cell cycle and self-renewal signaling.76 In pediatric AML the hypo- and hypermethylation of pathway promoters were highly associated with AML diagnosis and relapse.54 The complex interplay between the intrinsic and extrinsic signals governing LSC behavior may, however, mean targeting a single pathway, such as the Hh pathway, is not sufficient to eradicate these CSCs. For example, in an MLL-AF9-mediated murine model of myeloid leukemia Hh signaling was completely dispensable for the development of acute leukemia.42,45
The role of the Hh pathway in myelodysplastic syndrome (MDS) is not so well understood. However, there is increasing evidence to show it is deregulated. Analysis of primary MDS samples found overexpression of SHH, DHH, PTCH1, and SMO.77 Further, there was a correlation between ligand expression and the progenitor/stem cell marker c-KIT.77
Interestingly, work has shown the Hh pathway to act on the microenvironmental stromal cells, thereby regulating HSC behavior,64 with the Hh ligands acting in an autocrine and/or paracrine manner.59,62,78 Moreover, HHIP was highly expressed in primary stromal cells, but not in primary peripheral blood, bone marrow, or cord blood CD34+ selected cells, with stromal HHIP expression suppressing leukemic cell proliferation.79 Further, studies have demonstrated HHIP expression in AML and MDS-derived bone marrow stromal cells to be markedly reduced, with these cells supporting the proliferation of leukemic cells; pretreatment with azacitidine increased stromal cell HHIP expression, with a parallel reduction in leukemic cell proliferation in co-culture.79
Myeloproliferative neoplasms is a term encompassing several BCR-ABL-negative neoplasms – myelofibrosis (MF), polycythemia vera, and essential thrombocythemia – characterized by stem cell-derived clonal myeloproliferation and the Janus kinase 2 (JAK2V617F)80 or Calreticulin mutation.81 There are limited data available on the Hh pathway in these malignancies. GLI-1 and PTCH1 are increased 20–100-fold in primary myeloproliferative neoplasms samples, with pathway activity demonstrated in a murine bone marrow transplant model of MF using a GLI-luciferase reporter system. Combining the SMO antagonist LDE225 with INC424, a JAK1/2 dual kinase inhibitor, in this murine model saw a reduction in mutant allele burden and an improved clinical phenotype.82 Another murine model of MF (Gata1low) has shown alterations in the Hh pathway at the gene level in the bone marrow and spleen, components of the Hh pathway coordinating with TGFβ, p53, and mTOR-related genes to produce the biological phenotype of MF.83 However, a recent Phase II clinical trial using IPI-926 in MF saw the majority of patients taken off therapy due to side effects or no response.84 How the Hh inhibitors work in combination with other novel drugs remains to be fully ascertained.
Hh signaling in lymphoid disorders
Hh signaling is vital for normal B- and T-cell development; with Hh ligands, produced by either the bone marrow or lymphoid organ microenvironment, determining lymphoid cell behavior.63
B-cell disorders
B-cell acute lymphoblastic leukemia (B-ALL) is thought to develop from the malignant transformation of immature hematopoietic progenitor cells. Unlike AML, in which the CSC hypothesis was first demonstrated and is widely accepted, the B-ALL LSC has yet to be isolated or characterized,85 blasts from all stages of maturation being able to reconstitute and establish a leukemic phenotype in vivo.86 Interestingly however, up-regulation of components of the Hh pathway has been observed in precursor B-ALL,87 SMO inhibition reducing in vitro and in vivo measures of self-renewal.88
B-cell chronic lymphocytic leukemia (B-CLL) is a clonal disorder of mature, differentiated lymphocytes. Although not a CSC disease, both components of the Hh pathway (GLI-1, GLI-2, and SUFU) and key downstream targets (BCL-2, BCL-XL) are increased, and importantly correlate with clinical outcome.89 Expression of BCL-2 is increased in the presence of active Hh signaling and down-regulated upon inhibition of the pathway.59 Further, cyclopamine increased apoptosis of B-CLL cells in vitro, in stromal co-culture conditions and in combination with fludarabine.59,89 Gene expression of components of the Hh pathway is extremely variable in CLL patients, with expression correlating with response to SMO inhibition. Sixty percent of treated patient samples responded to at least one SMO antagonist; likelihood of response correlating with elevated GLI-1 and PTCH1 expression and the presence of trisomy 12 (a poor prognostic indicator). This work also showed evidence of autocrine DHH signaling, potentially enabling non-canonical signaling in B-CLL cells.90 In contrast, another study found PTCH1, SMO, and GLI-1 to be reduced in primary CLL samples, although there was considerable heterogeneity – about 25% showing high transcript levels compared to normal B-lymphocytes. GANT61, a direct GLI inhibitor, induced apoptosis in a time- and concentration-dependent manner in association with STAT3 phosphorylation.91 A further study found SMO and GLI-1 to be significantly down-regulated in B-CLL cells compared to normal B cells.92 Moreover, whilst cyclopamine and knockdown of SMO had only minor specific effects on B-CLL cell survival in vitro, GANT61 caused significant apoptosis in B-CLL cells but not normal B cells.92 Combined treatment with GANT61 and fludarabine causing increased apoptosis compared to either agent alone. Interestingly however, whilst CLL viability improved in the presence of the stromal cell lines HS-5 and M210-B4, known to produce Hh ligands,59,92 the same effect was not achieved with soluble SHH indicating other factors are involved. So, whilst these results support previous reports of Hh pathway activity in CLL, they would support redundancy of canonical Hh signaling in vitro.
The term lymphoma encompasses a broad spectrum of diseases from the relatively indolent to the aggressive and rapidly fatal; each has a complex and heterogeneous pathogenesis. Broadly there are two categories, Hodgkin’s lymphoma (HL) and non-Hodgkin’s lymphoma (NHL). Components of the Hh pathway and key downstream targets (BCL-2 and BCL-XL) are expressed in a variety of NHL cell lines and primary tissue,59,93 with expression of the downstream targets being influenced by the Hh pathway.
Whilst an Eμ-myc murine model of Burkitt’s lymphoma, an extremely aggressive form of NHL, demonstrated the importance of stromal co-culture for lymphoma cell survival and expansion, stroma could be replaced with soluble SHH or IHH.59 Moreover, Burkitt’s cells underwent apoptosis in the absence of Hh signaling both in vitro and in vivo.
In diffuse large B-cell lymphoma (DLBCL), an aggressive form of NHL, response to Hh signaling is dependent on subtype, predominantly inducing cell cycle arrest in germinal center B-cell type, and apoptosis in the activated B-cell type.93 In addition to responding to the Hh ligands, these DLBCL cells were also shown to synthesize and secrete Hh ligands, supporting a role for autocrine signaling.94 Pharmacological and silencing techniques have shown expression of the AKT genes to be modulated by the Hh pathway in DLBCL at the transcriptional level,95 with another study showing that the Hh pathway activates the NF-kβ pathway.96
In another form of aggressive NHL, mantle cell lymphoma, a therapy-resistant murine model showed up-regulation of the GLI transcription factors at the gene level,97 confirming previous work showing the GLI transcription factors to be over-expressed in mantle cell lymphoma, both in cell lines and primary lymphoma cells, compared to normal B cells.98 Further, targeting the GLI transcription factors with antisense oligonucleotides down-regulated BCL-2 and Cyclin D1 resulting in decreased proliferation and increased susceptibility to chemotherapy.98
Whilst these represent high grade forms of NHL, the Hh pathway has also been found to be deregulated and amenable to therapeutic intervention in low-grade lymphomas. In Waldenstrom’s macroglobulinemia, a characteristically indolent form of lymphoma secreting immunoglobulin M (IgM), GLI-2 has been found to influence IgM levels. Further, GANT61 resulted in a significant reduction in IgM secretion across a number of Waldenstrom’s macroglobulinemia cell lines potentially through the interleukin-6 receptor; this effect was not seen with cyclopamine.99
In classical HL, whilst the Hh ligands and GLI transcription factors GLI-1 and -2 were expressed at a relatively low level, GLI-3 was highly expressed in all cell lines. Furthermore, immunohistochemistry for GLI-3 showed strong, uniform nuclear expression in virtually all Hodgkin/Reed-Stenberg cells whilst expression was variable in nodular lymphocyte predominant HL and NHL.100 Interestingly GLI-3 in thymic stromal cells has been shown to regulate T-cell selection and thymocyte differentiation;101 whether it is responsible for a similar role in HL remains to be determined.
MM is a CSC disorder, arising from the proliferation of a clonal population of plasma cells, and associated with the production of monoclonal immunoglobulin. Despite a variety of therapeutic approaches MM is incurable with a relapsing natural history; it is the MM CSC which is believed to be resistant to standard therapies, including lenalidomide, bortezomib, dexamethasone, and cyclophosphamide.
In MM, Hh pathway activity has been implicated in the maintenance and differentiation of the CSC.56,78 Pathway inhibition, with cyclopamine, or neutralization, using the monoclonal antibody 5E1, resulted in contraction of the CSC compartment, whereas exogenous Hh ligand caused expansion.78 In addition, using LDE225 to target canonical signaling, and Forskolin a GLI-1 modulating compound, thereby bypassing PTCH1 and SMO and targeting non-canonical signaling, investigators have shown both mechanisms are amenable to therapeutic intervention in vitro in cell lines and patient samples.56 Supporting evidence, using LDE225, confirmed differentiation was significantly induced and de-differentiation blocked in both cell lines and primary cells in vitro.102 Moreover, there was a marked discrepancy in Hh pathway component expression, activity, and cyclopamine sensitivity between the stem cell and differentiated cell compartments.78 Interestingly, Hh pathway gene expression was significantly altered in vivo suggesting the cell microenvironment markedly affects expression of Hh pathway components.78
T-cell disorders
T-cell disorders, characterized by the abnormal proliferation of T-cell precursors, are generally aggressive, and far rarer than B-cell disorders. Interestingly, in comparison to B-ALL and B-cell lymphomas there are significant similarities between T-ALL and T-cell lymphomas; many arguing these entities are variations of the same disease, the two conditions often being treated in the same way.103
The Hh pathway has been shown to be important in T-cell development;104 it has also been implicated in the etiology of T-cell malignancies. In ALK+ anaplastic large cell lymphoma cell lines and primary tissue, SHH was amplified. In addition, GLI-1 expression at both the gene and protein level was increased; both SHH and GLI1 expression being influenced by the PI3K/AKT pathway although the mechanism remains unclear.105 Supporting the importance of these components in cell survival, inhibition of SHH signaling with cyclopamine or silencing GLI-1 with ribonucleic acid interference resulted in cell cycle arrest and apoptosis.105
Epigenetic modifications of the Hh pathway
Epigenetic modifications are stable, heritable alterations in gene expression caused without a change in coding DNA but rather DNA methylation and histone modification.106 The bromodomain and extra-terminal domain (BET) protein family, responsible for reading differentially acetylated histones and thereby communicating changes in transcription potential, comprises four distinct genes, BRD2, -3,and -4 which are ubiquitously expressed and BRDT which is restricted to the testes. Importantly, there is increasing evidence to support the epigenetic modification of components of the Hh pathway. Work has predominantly focused on neural tissue and medulloblastoma, finding GLI-1 and GLI-2, but not GLI-3, to be acetylated, with a careful regulatory balance controlling GLI acetylation, and thereby transcriptional activity.107 Transcriptional activity occurs through histone deacetylation, whilst the binding of RENKCTD11, an endogenous histone deacetylase inhibitor, to SCF(B-TrCP)3-like E3 ubiquitin ligase complex, promotes GLI acetylation inhibiting transcription.108 Recent work has shown BET proteins to further modulate GLI transcription and thereby pathway activity.109 Targeting BRD4 with JQ1, a small-molecule bromodomain inhibitor, preventing BRD4 acetyl-lysine binding resulted in a global down-regulation of GLI associated genes in both patient and genetically engineered mouse model-derived Hh-driven tumors.109 It remains to be seen whether this will translate into clinical practice. Further, how epigenetic modification of other components of the pathway will affect Hh antagonism in clinical practice and whether these too will be amenable to manipulation remains to be investigated.
Targeting the Hh pathway in hematological malignancies
There is increasing evidence to support the role of the Hh signaling pathway in hematological malignancies, with components representing viable therapeutic targets, making it a very attractive potential treatment strategy. Further, from a drug development perspective, inhibition of SMO and GLI-1 can be readily achieved, Table 1.
 | Table 1 Hedgehog inhibitors |
The rationale for Hh inhibition in hematological malignancies is to target the LSC population, eradicating these therapy resistant cells, potentially affecting a cure. However, whilst there is evidence that the Hh pathway is important for LSC maintenance and expansion in CML and MM, its role in other diseases such as AML, MF, CLL, and lymphoma appears more complex, representing a delicate balance between the diseased cells and the stromal microenvironment. Whether Hh antagonism will be therapeutically useful in hematologic malignancies not only depends on its ability to target the diseased cells but also on the anticipated level of toxicity, both hematologic and systemic. To date, there appears to be little hematologic toxicity reported, particularly in solid tumor studies.130
Whilst experimental data suggest Hh antagonism may address the persistence of CSC and the protective effect of the tumor microenvironment in both myeloid and lymphoid malignancies, clinical trials are only in the early stages, Table 2. Interestingly, it is only the SMO antagonists which are currently being trialed clinically. Excitingly however, several of these clinical grade SMO inhibitors are now in Phase II trial; with a number of Phase III trials in solid tumors and the licensing of vismodegib for use in basal cell carcinoma in 2012 hinting these early results could translate into clinical practice.
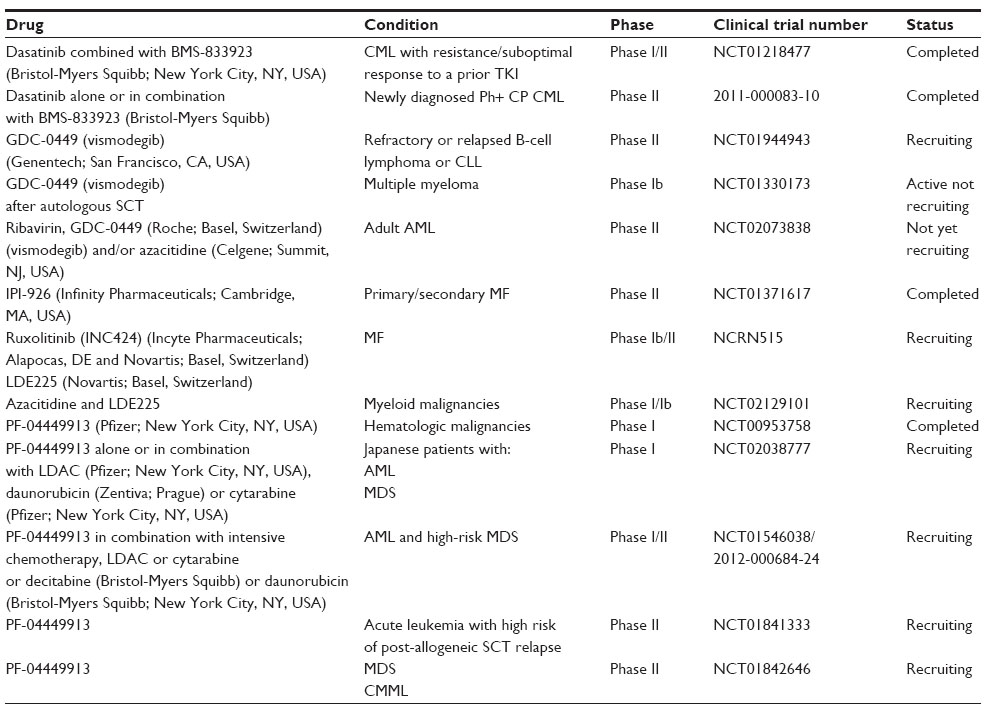 | Table 2 Clinical trials involving hedgehog inhibitors |
In hematological malignancies, results of a Phase Ia study using PF-04449913 in refractory, resistant, or intolerant myeloid malignancies were presented in 2011.131 Thirty-two patients were enrolled, with early indications of efficacy seen across all diseases.131 Notably, one patient with AML achieved a complete remission albeit with incomplete blood recovery and five had a >50% reduction in bone marrow blast counts. Additionally, one patient with MF achieved a >50% reduction in extramedullary disease and five attained stable disease. Results of the current Phase II trials are eagerly awaited.
Conclusion
Deregulated expression of components of the Hh signaling pathway is found throughout the huge range of hematological malignancies; the pathway appearing to either determine CSC behavior directly or indirectly via the stromal microenvironment.
As our knowledge and understanding of the Hh and other conserved embryonic pathways in hematologic malignancies increases, our ability to manipulate and potentially use these as therapeutic targets can only increase. Further, the sheer number of compounds coming onto the market, targeting both canonical and non-canonical mechanisms and the array of clinical trials, makes this an exciting time looking to tailor therapy and minimize treatment-related side effects.
Disclosure
M Copland has received research funding from Bristol-Myers Squibb and Novartis, honoraria from/attended Advisory Boards for Bristol-Myers Squibb, Pfizer, Ariad and Novartis, Bristol-Myers Squibb and Pfizer and travel funding from Bristol-Myers Squibb and Novartis. V Campbell is funded by the Wellcome Trust via the STMTI programme. M Copland is funded by Leukaemia and Lymphoma Research (Grant No: 11017). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in this manuscript apart from those discussed.
References
Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12(2):133–143. | |
Copley MR, Beer PA, Eaves CJ. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell. 2012;10(6):690–697. | |
Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 2008;453(7193):306–313. | |
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. | |
Goardon N, Marchi E, Atzberger A, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138–152. | |
Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. | |
Buglino JA, Resh MD. Palmitoylation of hedgehog proteins. Vitam Horm. 2012;88:229–252. | |
O’Flaherty JD, Barr M, Fennell D, et al. The cancer stem-cell hypothesis: its emerging role in lung cancer biology and its relevance for future therapy. J Thorac Oncol. 2012;7(12):1880–1890. | |
Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. | |
Valent P, Bonnet D, De Maria R, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12(11):767–775. | |
Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15(9):494–501. | |
Dick JE. Stem cell concepts renew cancer research. Blood. 2008; 112(13):4793–4807. | |
Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17(3):313–319. | |
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. | |
Alison MR, Lim SM, Nicholson LJ. Cancer stem cells: problems for therapy? J Pathol. 2011;223(2):147–161. | |
Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. | |
Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–3087. | |
Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9(7):873–886. | |
Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet. 2006;7(11):841–850. | |
Cridland SO, Keys JR, Papathanasiou P, Perkins AC. Indian hedgehog supports definitive erythropoiesis. Blood Cells Mol Dis. 2009;43(2):149–155. | |
Bitgood MJ, Shen L, McMahon AP. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol. 1996;6(3):298–304. | |
Lee JJ, Ekker SC, von Kessler DP, et al. Autoproteolysis in hedgehog protein biogenesis. Science. 1994;266(5190):1528–1537. | |
Buglino JA, Resh MD. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J Biol Chem. 2008;283(32):22076–22088. | |
Porter JA, von Kessler DP, Ekker SC, et al. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature. 1995;374(6520):363–366. | |
Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–376. | |
Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418(6900):892–897. | |
Bhardwaj G, Murdoch B, Wu D, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol. 2001;2(2):172–180. | |
Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126(17):3915–3924. | |
Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007;17(9):438–447. | |
Carpenter D, Stone DM, Brush J, et al. Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc Natl Acad Sci U S A. 1998;95(23):13630–13634. | |
Rahnama F, Toftgard R, Zaphiropoulos PG. Distinct roles of PTCH2 splice variants in Hedgehog signalling. Biochem J. 2004;378(Pt 2):325–334. | |
Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397(6720):617–621. | |
Kawakami T, Kawcak T, Li YJ, et al. Mouse dispatched mutants fail to distribute hedgehog proteins and are defective in hedgehog signaling. Development. 2002;129(24):5753–5765. | |
Chen MH, Wilson CW, Li YJ, et al. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009;23(16):1910–1928. | |
Irvine DA, Copland M. Targeting hedgehog in hematologic malignancy. Blood. 2012;119(10):2196–2204. | |
Jenkins D. Hedgehog signalling: emerging evidence for non-canonical pathways. Cellular Signal. 2009;21(7):1023–1034. | |
Reed JC. Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer. Adv Pharmacol. 1997;41:501–532. | |
Ling V. Charles F. Kettering Prize. P-glycoprotein and resistance to anticancer drugs. Cancer. 1992;69(10):2603–2609. | |
Kurinna S, Konopleva M, Palla SL, et al. Bcl2 phosphorylation and active PKC alpha are associated with poor survival in AML. Leukemia. 2006;20(7):1316–1319. | |
Keller G, Lacaud G, Robertson S. Development of the hematopoietic system in the mouse. Exp Hematol. 1999;27(5):777–787. | |
Trowbridge JJ, Scott MP, Bhatia M. Hedgehog modulates cell cycle regulators in stem cells to control hematopoietic regeneration. Proc Natl Acad Sci U S A. 2006;103(38):14134–14139. | |
Gao J, Graves S, Koch U, et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell. 2009;4(6):548–558. | |
Dierks C, Beigi R, Guo GR, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14(3):238–249. | |
Cohen DJ. Targeting the hedgehog pathway: role in cancer and clinical implications of its inhibition. Hematol Oncol Clin North Am. 2012; 26(3):565–588. | |
Hofmann I, Stover EH, Cullen DE, et al. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell. 2009;4(6):559–567. | |
Xie J, Murone M, Luoh SM, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–92. | |
Berman DM, Karhadkar SS, Hallahan AR, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297(5586):1559–1561. | |
Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003; 425(6960):851–856. | |
Park KS, Martelotto LG, Peifer M, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011;17(11):1504–1508. | |
Martin ST, Sato N, Dhara S, et al. Aberrant methylation of the Human Hedgehog interacting protein (HHIP) gene in pancreatic neoplasms. Cancer Biol Ther. 2005;4(7):728–733. | |
Slade I, Murray A, Hanks S, et al. Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma. Fam Cancer. 2011;10(2):337–342. | |
Shahi MH, Lorente A, Castresana JS. Hedgehog signalling in medulloblastoma, glioblastoma and neuroblastoma. Oncol Rep. 2008;19(3):681–688. | |
Car D, Sabol M, Musani V, Ozretic P, Levanat S. Epigenetic regulation of the Hedgehog-Gli signaling pathway in cancer. Period Biol. 2010; 112(4):419–423. | |
Ochs M, Farrar J, Considine M, et al. Genome wide promoter methylation patterns predict AML subtype outcomes and identify novel pathways characterizing diagnostic and relapsed disease in children. Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta. GA, USA. | |
Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30(6):303–312. | |
Blotta S, Jakubikova J, Calimeri T, et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood. 2012;120(25):5002–5013. | |
Karhadkar SS, Bova GS, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–712. | |
Watkins DN, Berman DM, Burkholder SG, et al. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422(6929):313–317. | |
Dierks C, Grbic J, Zirlik K, et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;13(8):944–951. | |
Theunissen JW, de Sauvage FJ. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009;69(15):6007–6010. | |
Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–779. | |
Kobune M, Takimoto R, Murase K, et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009;100(5):948–955. | |
Kobune M, Kato J, Kawano Y, et al. Adenoviral vector-mediated transfer of the Indian hedgehog gene modulates lymphomyelopoiesis in vivo. Stem Cells. 2008;26(2):534–542. | |
Kobune M, Ito Y, Kawano Y, et al. Indian hedgehog gene transfer augments hematopoietic support of human stromal cells including NOD/SCID-beta2m-/- repopulating cells. Blood. 2004;104(4):1002–1009. | |
Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96(10):3343–3356. | |
Hochhaus A, Hughes T. Clinical resistance to imatinib: mechanisms and implications. Hematol Oncol Clin North Am. 2004;18(3):641–656. | |
Hochhaus A, O’Brien SG, Guilhot F, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23(6):1054–1061. | |
Hamilton A, Helgason GV, Schemionek M, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–1510. | |
Irvine DA, Zhang B, Allan EK, et al. Combination of the hedgehog pathway inhibitor LDE225 and nilotinib eliminates chronic myeloid leukemia stem and progenitor cells. Presented at: 51st ASH Annual Meeting and Exposition; December 5–8, 2009; New Orleans, LA. | |
Okabe S, Tauchi T, Tanaka Y, Katagiri S, Ohyashiki K. Effects of the hedgehog inhibitor GDC-0449, alone or in combination with dasatinib, on BCR-ABL-positive leukemia Cells. Stem Cells Dev. 2012;21(16):2939–2948. | |
Alonso-Dominguez JM, Grinfeld J, Alikian M, et al. PTCH1 expression at diagnosis reliably predicts treatment failure in imatinib-treated chronic myeloid leukaemia patients. Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, GA. | |
Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. | |
Bai LY, Chiu CF, Lin CW, et al. Differential expression of Sonic hedgehog and Gli1 in hematological malignancies. Leukemia. 2008;22(1):226–228. | |
Zahreddine HA, Culjkovic-Kraljacic B, Assouline S, et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature. 2014;511(7507):90–93. | |
Campbell V, Tholouli E, Quigley MT, et al. Evidence that activated hedgehog signaling predicts for poor clinical outcome in acute myeloid leukemia. Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, GA. | |
Fukushima N, Minami Y, Hayakawa F, et al. Treatment with hedgehog inhibitor, PF-04449913, attenuates leukemia-initiation potential in acute myeloid leukemia cells. Presented at: 55th ASH Annual Meeting and Exposition; December 7–10, 2013; New Orleans, LA. | |
Xavier JM, Duarte ASS, Pericole FV, et al. Hedgehog pathway is deregulated in myelodysplastic syndrome progenitor bone marrow cells. Presented at: 55th ASH Annual Meeting and Exposition; December 7–10, 2013; New Orleans, LA. | |
Peacock CD, Wang Q, Gesell GS, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104(10):4048–4053. | |
Kobune M, Iyama S, Kikuchi S, et al. Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms. Blood Cancer J. 2012;2:e87. | |
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | |
Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. | |
Bhagwat N, Keller MD, Rampal RK, et al. Improved efficacy of combination of JAK2 and hedgehog inhibitors in myelofibrosis. Presented at: 55th ASH Annual Meeting and Exposition; December 7–10, 2013; New Orleans, LA. | |
Zingariello M, Martelli F, Ciaffoni F, et al. Characterization of the TGF-beta1 signaling abnormalities in the Gata1low mouse model of myelofibrosis. Blood. 2013;121(17):3345–3363. | |
Sasaki KG, Gotlib JR, Mesa RA, et al. A phase 2 study of IPI-926, an oral hedgehog inhibitor, in patients with myelofibrosis. J Clin Oncol. 2014;32:5s (suppl; abstr 7111). | |
Bernt KM, Armstrong SA. Leukemia stem cells and human acute lymphoblastic leukemia. Semin Hematol. 2009;46(1):33–38. | |
le Viseur C, Hotfilder M, Bomken S, et al. In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell. 2008;14(1):47–58. | |
Lang F, Badura S, Ruthardt M, Rieger MA, Ottmann OG. Modulation of leukemic stem cell self-renewal and cell fate decisions by inhibition of hedgehog signalling in human acute lymphoblastic leukemia (ALL). Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, GA. | |
Lin TL, Wang QH, Brown P, et al. Self-renewal of acute lymphocytic leukemia cells is limited by the Hedgehog pathway inhibitors cyclopamine and IPI-926. PloS One. 2010;5(12):e15262. | |
Hegde GV, Peterson KJ, Emanuel K, et al. Hedgehog-induced survival of B-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: a potential new therapeutic target. Mol Cancer Res. 2008;6(12):1928–1936. | |
Decker S, Zirlik K, Djebatchie L, et al. Trisomy 12 and elevated GLI1 and PTCH1 transcript levels are biomarkers for Hedgehog-inhibitor responsiveness in CLL. Blood. 2012;119(4):997–1007. | |
Wang C, Lu K, Wang X. GLI1 Inhibitor GANT61 Deregulates stat3 Phosphorylation In Chronic Lymphocytic Leukemia Cells. Blood. 2013;122(21). | |
Desch P, Asslaber D, Kern D, et al. Inhibition of GLI, but not Smoothened, induces apoptosis in chronic lymphocytic leukemia cells. Oncogene. 2010;29(35):4885–4895. | |
Patole SP, Patole AS, Rhen DS, et al. Patterned carbon nanotube growth using an electron beam sensitive direct writable catalyst. Nanotechnology. 2009;20(31):315302. | |
Singh RR, Kim JE, Davuluri Y, et al. Hedgehog signaling pathway is activated in diffuse large B-cell lymphoma and contributes to tumor cell survival and proliferation. Leukemia. 2010;24(5):1025–1036. | |
Agarwal NK, Qu CJ, Kunkalla K, Liu YD, Vega F. GLI1 directly regulates the transcription of AKT genes in diffuse large B-cell lymphoma. Blood. 2012;120(21). Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, GA, USA. | |
Qu CJ, Liu YD, Kunkalla K, Agarwal NK, Vega F. Smoothened (SMO) activates NF-Kb pathway through activation of PKC beta/CARMA1 and TRAF6 stabilization in diffuse large B-cell lymphoma. Blood. 2012;120(21). Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, GA, USA. | |
Hegde GV, Nordgren TM, Munger CM, et al. Novel therapy for therapy-resistant mantle cell lymphoma: multipronged approach with targeting of hedgehog signaling. Int J Cancer. 2012;131(12):2951–2960. | |
Hegde GV, Munger CM, Emanuel K, et al. Targeting of sonic hedgehog-GLI signaling: a potential strategy to improve therapy for mantle cell lymphoma. Mol Cancer Ther. 2008;7(6):1450–1460. | |
Amarsaikhan N, Dennison JR, Boi SK, Neil MS, Elsawa SF. GLI transcription factors modulate IgM secretion in waldenstrom macroglobulinemia. Blood. 2013;122(21). | |
Greaves WO, Kim JE, Singh RR, et al. Glioma-associated oncogene homologue 3, a hedgehog transcription factor, is highly expressed in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Hum Pathol. 2011;42(11):1643–1652. | |
Hager-Theodorides AL, Dessens JT, Outram SV, Crompton T. The transcription factor Gli3 regulates differentiation of fetal CD4- CD8- double-negative thymocytes. Blood. 2005;106(4):1296–1304. | |
Yang SB, RD, Loh YS, et al. Blockade of the hedgehog signaling pathway by the novel agent NVP-LDE225 induces differentiation, prevents de-differentiation and inhibits proliferation of multiple myeloma stem cells in vitro. Blood. 2012;120(120). Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, GA, USA. | |
Hoelzer D, Gokbuget N. T-cell lymphoblastic lymphoma and T-cell acute lymphoblastic leukemia: a separate entity? Clin Lymphoma Myeloma. 2009;9 Suppl 3:S214–S221. | |
El Andaloussi A, Graves S, Meng F, et al. Hedgehog signaling controls thymocyte progenitor homeostasis and differentiation in the thymus. Nat Immunol. 2006;7(4):418–426. | |
Singh RR, Cho-Vega JH, Davuluri Y, et al. Sonic hedgehog signaling pathway is activated in ALK-positive anaplastic large cell lymphoma. Cancer Res. 2009;69(6):2550–2558. | |
Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612. | |
Canettieri G, Di Marcotullio L, Coni S, Greco A, Gulino A. Turning off the switch in medulloblastoma: the inhibitory acetylation of an oncogene. Cell Cycle. 2010;9(11):2047–2048. | |
Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12(2):132–142. | |
Tang Y, Gholamin S, Schubert S, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20(7):732–740. | |
Kim J, Lee JJ, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010;107(30):13432–13437. | |
Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280(5369):1603–1607. | |
Firestone AJ, Weinger JS, Maldonado M, et al. Small-molecule inhibitors of the AAA plus ATPase motor cytoplasmic dynein. Nature. 2012;484(7392):125–129. | |
Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16(21):2743–2748. | |
Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104(20):8455–8460. | |
Hyman JM, Firestone AJ, Heine VM, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci U S A. 2009;106(33):14132–14137. | |
Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388–399. | |
Lee J, Wu X, Pasca di Magliano M, et al. A small-molecule antagonist of the hedgehog signaling pathway. Chembiochem. 2007;8(16):1916–1919. | |
Manetti F, Faure H, Roudaut H, et al. Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as smoothened antagonists. Mol Pharmacol. 2010; 78(4):658–665. | |
Rohner A, Spilker ME, Lam JL, et al. Effective targeting of hedgehog signaling in a medulloblastoma model with PF-5274857, a potent and selective smoothened antagonist that penetrates the blood-brain barrier. Mol Cancer Ther. 2012;11(1):57–65. | |
Stanton BZ, Peng LF. Small-molecule modulators of the Sonic Hedgehog signaling pathway. Mol Biosyst. 2010;6(1):44–54. | |
Petrova E, Rios-Esteves J, Ouerfelli O, Glickman JF, Resh MD. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nat Chem Biol. 2013;9(4):247–249. | |
Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A. 2002; 99(22):14071–14076. | |
Wang Y, Arvanites AC, Davidow L, et al. Selective identification of hedgehog pathway antagonists by direct analysis of smoothened ciliary translocation. ACS Chem Biol. 2012;7(6):1040–1048. | |
Incardona JP, Gaffield W, Lange Y, et al. Cyclopamine inhibition of sonic hedgehog signal transduction is not mediated through effects on cholesterol transport. Developmental Biology. 2000;224(2):440–452. | |
Gendreau SB, Hawkins D, Ho C, et al. Preclinical characterization of BMS-833923 (XL139), a hedgehog (HH) pathway inhibitor in early clinical development. Mol Cancer Ther. 2009;8:B192. | |
Wong H, Alicke B, West KA, et al. Pharmacokinetic-pharmacodynamic analysis of vismodegib in preclinical models of mutational and ligand-dependent Hedgehog pathway activation. Clin Cancer Res. 2011;17(14):4682–4692. | |
Peluso MO, Campbell VT, Harari JA, et al. Impact of the smoothened inhibitor, IPI-926, on smoothened ciliary localization and hedgehog pathway activity. PloS One. 2014;9(3):e90534. | |
Pan SF, Wu X, Jiang JQ, et al. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med Chem Lett. 2010;1(3):130–134. | |
Munchhof MJ, Li Q, Shavnya A, et al. Discovery of PF-04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med Chem Lett. 2012;3(2):106–111. | |
Ohashi T, Oguro Y, Tanaka T, et al. Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: Modification of the core skeleton for improved solubility. Bioorg Med Chem. 2012;20(18):5507–5517. | |
Jamieson C, Cortes JE, Oehler V, et al. Phase 1 dose-escalation study of PF-04449913, an oral hedgehog (Hh) inhibitor, in patients with select hematologic malignancies. Blood. 2011;118(21):195–196. Presented at: 53rd ASH Annual Meeting and Exposition; December 10-13, 2011; San Diego, CA, USA. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.