Back to Archived Journals » Journal of Neurorestoratology » Volume 5
Stem cell-transplantation therapy for adrenoleukodystrophy: current perspectives
Authors Miller W
Received 7 October 2016
Accepted for publication 1 December 2016
Published 9 January 2017 Volume 2017:5 Pages 5—19
DOI https://doi.org/10.2147/JN.S99304
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Hongyun Huang
Weston Miller
Department of Pediatrics, Division of Blood and Marrow Transplantation, University of Minnesota, Minneapolis, MN, USA
Abstract: Adrenoleukodystrophy (ALD) is a rare, X-linked peroxisomal disorder of impaired very long-chain fatty-acid metabolism. It results from various mutations in the ABCD1 gene (Xq28). All males with the biochemical defect of ALD are at risk of developing cerebral white-matter disease (cALD) during their lifetime. Thirty-five percent of ALD patients develop cALD in boyhood, a life-threatening phenotype characterized by rapidly expanding, neuroinflammatory demyelination and irreversible clinical neurologic decline. The ABCD1 genotype does not predict susceptibility to or protection from the childhood cALD phenotype; therefore, clinicians must remain ever vigilant for its development when monitoring ALD patients. Currently, allogeneic hematopoietic cell transplantation (HCT) is the standard of care for boyhood cALD. While HCT provides dramatic functional survival benefit in boys with early, presymptomatic cALD, outcomes are less favorable and less predictable for those with more advanced disease. Furthermore, little is known about how successful HCT in childhood might impact the onset of central nervous system disease in adulthood. Finally, investigations of experimental gene-therapy strategies are ongoing. This review explores current perspectives of stem cell transplantation in cALD.
Keywords: adrenoleukodystrophy, cerebral adrenoleukodystrophy, stem cell transplantation, bone marrow transplantation, umbilical cord-blood transplantation, hematopoietic cell transplantation
Introduction
Adrenoleukodystrophy (ALD) is an X-linked disorder affecting approximately one in 21,000 males.1 Over 700 unique mutations in the disease-causing ABCD1 gene have been reported to date.2 Affected males may manifest disease in the adrenal glands, testes, and/or central nervous system (CNS).3 The most feared disease phenotype is the cerebral variant (cALD) with onset in childhood, for which allogeneic hematopoietic cell transplantation (HCT) is presently considered the standard of care.4 This review discusses current considerations for HCT and cellular therapies in cALD.
Adrenoleukodystrophy: pathophysiology, diagnosis, and phenotypes
The common biochemical defect in ALD is that of insufficient very long-chain fatty acid (VLCFA) catabolism resulting from abnormal ALD-protein (ALDP) function. ALDP, the protein product of ABCD1 at Xq28, is a membrane-bound transporter necessary for shuttling cytosolic VLCFA into the peroxisome for β-oxidation. Without functioning ALDP, supranormal VLCFA concentrates in tissue and plasma in a pathognomonic pattern. Also, while their mechanistic linkage to end-organ disease is not entirely clear, elevated VLCFAs are thought to play a central role in ALD’s pathogenesis.5–7
The diagnosis of ALD, therefore, can be made with the highly sensitive and specific plasma VLCFA assay. When an abnormal pattern is detected with an identifiable ABCD1 mutation in a presymptomatic patient (or in the setting of classic clinical disease in others), males are determined to have the biochemical defect of ALD. Disease may manifest at any age. Most ALD males will develop primary adrenal insufficiency (AI) in childhood or adolescence, and most surviving males will develop spinal cord disease (adrenomyeloneuropathy [AMN]) during adulthood. Approximately half of ALD males will manifest cALD, and boyhood is the highest-risk period for onset.8 Importantly, the ABCD1 genotype does not predict cALD phenotype, including age of onset, as is apparent in large kinships demonstrating variable disease manifestations. This finding underscores the likely role of modifier genes, epigenetics, or environmental factors in the onset of CNS disease.9
During boyhood, 35% of ALD patients will develop cALD. On average in this phenotype, cerebral disease becomes radiographically evident at age 5 years and clinically evident at age 7 years. In 85% of childhood cALD cases, the lesion initiates in the splenium of the corpus callosum, followed by contiguous demyelination into the parieto-occipital lobes. Less commonly, the initial lesion is seen in the anterior/genu of the corpus callosum, followed by contiguous demyelination into the frontal lobes.10–12 Usually, cALD lesions are contrast-enhancing on magnetic resonance imaging (MRI), a finding that strongly predicts disease progression.13 Clinical progression among boys with cALD tends to be predictable: behavioral or academic decline, visual/auditory dysfunction, cognitive decline, gait/motor abnormalities, bulbar dysfunction, and fatal complete cerebral neurologic devastation. The clinical sequence generally reflects progressive demyelination in key functional pathways.8
Allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy
Rationale and mechanism of action
Since the early 1980s, HCT has been recognized to ameliorate various life-threatening, neuropathic lysosomal storage diseases, including those characterized by cerebral white-matter loss.14,15 Mechanistically, it was known that host-cell endocytosis of soluble lysosomal enzymes secreted by donor cells could restore catabolism of pathogenic substrate in the diseased, enzyme-deficient host tissues.16 In contrast, when HCT was first considered for cALD, it was already recognized that a membrane-bound protein product (ALDP) – not a soluble enzyme – was implicated in the pathogenesis. However, investigators hypothesized that by providing metabolically competent, donor-derived cells of hematopoietic lineage, the natural history of progressive white-matter loss in cALD could be favorably altered.
Most current evidence points to several likely pathologic mechanisms in childhood cALD. First, VLCFA accumulation over time may simply destabilize myelin structure and directly mediate its subsequent loss. With time, supranormal VLCFA-mediated myelin destabilization may expose antigen that triggers the abnormal, mononuclear cellular infiltrate observed on cALD histopathology. Intersecting with these is the effect of blood–brain barrier (BBB) disruption, as trauma has been observed to precede inflammatory cALD and active disease is nearly universally characterized by intravenous (IV) contrast enhancement.17,18 It is noteworthy that a common observation following successful HCT for boys with cALD is resolution of MRI contrast enhancement. This suggests reestablishment of BBB integrity, perhaps in part by intense immunomodulation, to be a key therapeutic event of HCT. Additionally, oxidative stress and damage likely contribute to cALD pathology.19–21 Rockenbach et al analyzed plasma samples in four cALD boys before and after successful HCT, and demonstrated favorable changes in lipid and protein oxidative damage markers, thus suggesting HCT’s ability to decrease oxidative stress pathways in cALD.22
To date, the field has lacked animal models that recapitulate the dramatic inflammatory demyelination seen in boyhood cALD, thus impeding full elucidation of both the pathophysiology and transplant’s mechanistic effect. Yamada et al analyzed HCT’s metabolic impact in the murine ABCD1-knockout model. Although the ALDP-deficient mouse is not observed to develop inflammatory cerebral demyelination, the model does manifest elevated VLCFA patterns in tissue and plasma. The authors described significant reductions in VLCFA ratios in spleens and lungs of transplanted mice following total-body irradiation (TBI) conditioning. Interestingly, VLCFA reductions in the CNS were only observed in animals exhibiting donor-cell presence in the brain and spinal cord. Finally, the investigators observed ex vivo reductions in VLCFA when ALD cells were cocultured with wild-type microglia, even though ALDP was not detected in diseased cells at culture termination. They concluded that donor cells of hematopoietic lineage are able to reduce tissue VLCFA burden by providing metabolically competent β-oxidation.23
Regardless of its mechanism, allogeneic HCT is currently considered standard treatment for cALD, based on overwhelming evidence that in select patients it can prolong functional survival. However, given the lack of evidence that HCT can prevent destined cALD, as well as the significant risk profile of allogeneic transplantation, current recommendations are that this therapy be considered only for ALD patients with active cerebral disease (cALD).
Initial experiences
The first report of HCT for cALD was in a 12-year-old boy transplanted in 1982 for a several-year history of clinically symptomatic disease. He was noted on pretransplant head computed tomography (CT) to have a symmetric, contrast-enhancing lesion in the corpus callosum and white matter of the temporal, parietal, and occipital lobes. He was conditioned with a myeloablative (MA) regimen consisting of busulfan (BU) and cyclophosphamide (CY) and grafted with marrow from a human leukocyte antigen (HLA)-matched unaffected brother. Despite donor hematopoietic engraftment, normalization of plasma VLCFA ratios, and resolution of contrast enhancement on head CT, the boy experienced rapid neurologic decline in the immediate posttransplant period. He died of adenovirus infection 5 months after HCT.24
In 1990, Aubourg et al reported the first favorable outcome following HCT for cALD. An 8-year-old boy was diagnosed with cALD following clinical presentation with primary AI. Serial MRI studies showed progressive white-matter lesions in the internal capsules, pallidum, caudate nuclei, and pons, though contrast-enhancement status was not specified. The boy underwent an MA BU–CY regimen and was grafted with HLA-matched, unaffected-sibling marrow. He demonstrated rapid donor hematopoietic recovery. Longitudinal radiographic and clinical examinations demonstrated resolution of his mild pretransplant disease: he showed normalization of the plasma VLCFA profile at 18 months. Although MRI normalization following transplantation for cALD is now recognized as anomalous, the investigators concluded at the time that HCT could reverse early cerebral demyelination and clinical disease in cALD. They speculated this effect was mediated by metabolically competent cells of donor hematopoietic origin (microglial or other) that penetrated the CNS.25
In 1994, Loes et al used the newly described, 34-point ALD-specific MRI radiographic severity scale (Loes demerit scale) to describe short-term, posttransplant radiographic outcomes of seven boys with cALD.26,27 Details of the HCT regimens were not provided. When compared to nontransplanted cALD controls at an average posttransplant follow-up of 19 months, the investigators concluded that HCT stabilized (but in contrast to Aubourg et al, did not resolve) existing white-matter lesions. In 1997, Swedish investigators reported outcomes following HCT for three boys with cALD. All received HLA-matched unrelated donor marrow following MA BU–CY conditioning regimens with antithymocyte globulin (ATG). The three boys demonstrated full donor hematopoietic engraftment. Despite this, inferior outcomes were observed for two patients who had more advanced or rapidly progressing cerebral disease at the time of HCT. In contrast, a favorable outcome followed the transplant for the boy with very early, presymptomatic (but radiographically evident) cALD. Interestingly, donor-specific cells were detected in the CNS at autopsy in one patient dying from progressive cALD. The authors advised HCT in the window of early cALD, when radiographically evident, but clinically silent disease is present.28
Nowaczyk et al provided evidence that transplant efficacy for cALD depended upon successful donor hematopoietic engraftment, and not only the intense immunomodulation provided by the preparative and posttransplant maintenance regimen. They reported a boy with autologous hematopoietic recovery following MA conditioning (etoposide, CY, high-dose TBI) and an HLA-mismatched, T-cell-depleted marrow graft. In the months following the transplant, the child demonstrated clinical and radiographic disease progression; the status of MR contrast enhancement was not specified.29
The first reported use of umbilical cord blood (UCB) as the allograft source in HCT for cALD came from Kapelushnik et al in 1998. The recipient was an 11-year-old boy diagnosed with ALD after presenting with primary AI. On screening MRI, he was found to have enhancing, radiographically minimal, presymptomatic cALD. Because no HLA-matched donors were available and because a follow-up MRI showed extension of his white-matter disease, he underwent an HLA-matched UCB transplantation (UCBT) following ATG, CY, and high-dose TBI (with partial sparing of the head). Complete donor hematopoietic engraftment and clinical and radiographic stability (enhancement status not specified) were observed at 13 months after the transplant. The authors concluded that the growing number of publicly banked UCB units – a rapidly available graft source – could particularly benefit boys with cALD, as the accumulating international experience exposed the need for urgent HCT once a suitable cALD transplant candidate was identified.30 In the same vein of expeditious allografting, Lin et al were the first to describe HLA-haploidentical (half-matched) paternal donor transplantation for cALD. Their patient was a 14-year-old with symptomatically advanced cerebral disease. Although he experienced donor engraftment and biochemical improvement, he had a poor neurologic outcome.31
Predicting outcomes after transplantation
With growing HCT experience in cALD, investigators turned their attention to identifying which cALD boys were most likely to benefit from the risky intervention. The initial robust description of long-term outcomes after HCT for cALD was provided by Shapiro et al, who studied 18 boys transplanted at multiple international centers. Deaths (n=6, 33%) resulted from transplant-related complications (n=2) or cALD progression (n=4). In the latter subgroup, two patients had advanced radiographic disease prior to HCT (Loes MRI scores 14 and 16.5), while two had rapid radiographic and clinical progression in the peritransplant period. All received MA BU–CY regimens; two were grafted from HLA-matched unrelated donors, while ten underwent HLA-matched sibling transplantation (one from a known carrier sister; however, the boy was found to have normal levels of ALDP expression in leukocytes following donor hematopoietic recovery). Among 12 engrafted survivors followed for a minimum of 5 years post-HCT, the median pretransplant baseline Loes MRI score was 4 and that at 1–2 years post-HCT was 7.5. At the furthest reported follow-up (range 5–10 years), the mean Loes MRI score remained 7.5. A mean 55% decrease in plasma VLCFA concentration (C26:0) was seen from baseline to furthest follow-up. All patients entered HCT with primary AI; none recovered function. Neurologically, engrafted survivors demonstrated aggregately stable favorable sensory and motor function at furthest follow-up. Similarly, neuropsychological profiles remained relatively stable for the cohort, and eight engrafted survivors (75%) continued to function normally in school. Although the study was insufficiently powered to determine clear predictors of treatment outcome, the authors noted superior results in patients with lower pretransplant Loes MRI severity scores.32
Wilken et al followed suit, and described a cohort of 14 boys transplanted for cALD. Two patients died of transplant-related complications, and the remaining 12 were thoroughly studied up to 5.5 years following HCT. Ten boys received marrow grafts, while two underwent peripheral blood stem cell HCT. Ten donors were unrelated, and two were related (including one carrier sister). All patients received MA BU–CY conditioning; ten received either ATG or antilymphocyte globulin. Eleven patients had stable, near-total donor hematopoietic engraftment. Six patients demonstrated clinical deterioration posttransplant (defined as any worsening on an ALD clinical severity scale), with two dying of progressive cALD. The other half (n=6) exhibited clinically stable disease. The authors observed that in some instances, clinical worsening may have been related to medications or transplant complications. No patients exhibited reversal of existing signs or symptoms of cALD following HCT. Based on their cohort and existing evidence at the time, the investigators recommended considering HCT for boys with cALD only if the following three criteria were met: 1) brain MRI showed characteristic white-matter disease; 2) two or more of the following were present: worsening neurologic symptoms, worsening neuropsychological function, increasing Loes score over time, contrast-enhancing disease, or elevated choline:N-acetylaspartate ratio on MR spectroscopy (MRS); and 3) the patient did not demonstrate “advanced” disease, defined as Loes MRI severity score >10 and either performance IQ (PIQ) <75 or severe neurological symptoms.33 The same group assessed whether pretransplant MRS characteristics might predict the clinical course of their cohort following HCT. Interestingly, they found rising choline-containing compounds to correlate with pretransplant disease progression and high pretransplant N-acetylaspartate levels within abnormal-appearing white matter to correlate with better clinical outcome.34
In 2004, Peters et al reported what remains to date the largest cALD HCT cohort. Of 126 boys transplanted between 1982 and 1999 at 43 international centers, 94 (from 32 centers, and several described in previous reports) had sufficient data for analysis. All boys had characteristic white-matter disease on brain MRI at the time of HCT, and their Loes MRI severity scores were captured. Patients were assigned neurologic deficit scores at baseline and follow-up according to the cumulative number of abnormalities in the following domains: vision, hearing, speech, gait, fine-motor skills, activities of daily living, or other. The median age at HCT was 9 years (range 4.9–18.6 years). Most boys were grafted with related (45%) or unrelated (43%) marrow; some received UCB (12%). The majority (68%) received HLA-matched transplants by antigen-level criteria at HLA-A, -B, and -C. Half of the patients were conditioned for HCT with MA BU–CY regimens, while the other half received MA TBI + chemotherapy for conditioning.
At a median follow-up of 3.1 years, the estimated 5-year survival following related-donor transplant (64%) was superior to that following unrelated-donor transplant (53%, P=0.07). The main cause of death was progressive cALD. Survival depended significantly upon several ALD-related, pretransplant characteristics: number of neurologic deficits (increasing number, inferior survival), ALD disability rating scale score (greater disability, inferior survival), and Loes MRI severity score (score ≥9, inferior survival). A dramatic difference in 5-year survival (92%) was noted between boys with favorable baseline disease characteristics (one or less neurologic deficit and Loes MRI score <9) and all others (45%, P<0.01). Neurologic function was analyzed for patients with data at 1+ year(s) posttransplant. Of 32 boys with zero baseline neurologic deficits, 56% remained stable, 13% acquired one new deficit, and 28% demonstrated two or more new deficits. Of 28 patients with one neurologic deficit pretransplant, 47% had stable or improved function, while 32% had two or more new deficits after HCT. Of patients with two baseline deficits, the majority (67%) worsened posttransplant. A baseline PIQ <80 predicted inferior posttransplant disability-rating scores and worsening posttransplant PIQ; however, it did not correlate with Loes score or number of neurologic deficits following HCT. With this large, multicenter experience, the authors urged the importance of timely HCT in early cALD and extreme caution in transplanting in advanced cerebral disease.35
Beam et al next reported 12 boys with cALD, some of whom were not included in Peters et al’s cohort. Varying baseline disease severity was noted in their cohort; all patients underwent unrelated UCBT following a uniform MA regimen of BU–CY with ATG. No boys had an available HLA-matched related or unrelated marrow donor. One patient died from adrenal crisis during pretransplant conditioning, two died from progressive cALD (baseline Loes MRI scores of 13 and 23), and one died from hepatic veno-occlusive disease, a chemotherapy-related complication. One patient had graft failure requiring second transplantation. Among engrafted survivors followed for 2.3–6.3 years, long-term cognitive and motor function showed a striking dependence on pretransplant Loes score (≤10 with superior outcomes). Due to insufficient patient numbers, the authors were not able to determine the precise impact of other baseline cALD features, such as full and subscale IQ, on functional outcome. They found no definitive correlation between pretransplant neuroelectrophysiologic characteristics and various neurologic outcomes. The investigators concluded that UCB grafting was an effective alternative to marrow transplantation, but that similar trends in survival and functional outcome existed as previously described: HCT in early, presymptomatic cALD was strongly advisable. They also stated the likely benefit of inclusion of ALD in newborn-screening programs.36
Recognizing that progressive visual dysfunction was particularly common following transplants for cALD, Gess et al aimed to determine pretransplant predictors of this outcome. They retrospectively reviewed 14 boys undergoing HCT for whom complete ophthalmologic and radiographic data were available. Perhaps unsurprisingly, they found greater visual dysfunction after HCT in boys with baseline Loes MRI scores >11, PIQ <76, and parieto-occipital disease. They found a trend toward greater visual dysfunction in patients whose cALD diagnosis followed clinical manifestations (as opposed to screening for a family history of ALD). They uncovered no correlations among major transplant-related characteristics and inferior posttransplant visual function. The authors concluded that the same baseline characteristics previously shown to predict poor survival and overall neurologic function overlapped as predictors of vision failure after HCT. They urged clinical caution in transplanting this higher-risk group.37
In 2011, Miller et al analyzed a previously unreported cohort of 60 consecutive cALD boys undergoing HCT at their center between 2000 and 2009. Given numerous previous reports identifying pretransplant Loes MRI severity score as a predictor of both survival and function, the investigators retrospectively parsed boys into two strata: standard risk (Loes MRI score <10, n=30) and higher risk (Loes MRI score ≥10, n=30). Gross neurologic function at baseline and most recent evaluation were scored using the ALD Neurologic Function Scale (NFS), a demerit clinical rating system that has been used by several ALD investigators (0, no clinically evident dysfunction; 25, maximum score characterized by neurologic devastation; Table 1). Conditioning strategies changed over time, and included MA CY/TBI-based, MA BU–CY-based, or a reduced-intensity conditioning (RIC) regimen (alemtuzumab, clofarabine, melphalan, and low-dose TBI) that was used exclusively in some higher-risk boys. Beginning in 2005, all patients received peritransplant N-acetylcysteine (NAC) for antioxidant support.
 | Table 1 cALD Neurologic Function Scale Notes: The cALD Neurologic Function Scale is a demerit clinical scoring system used by various investigators to describe disease severity. Any non-zero score denotes clinically evident disease. Maximal disease within a domain scores the total of all grades within that domain (eg, a patient with “loss of communication” scores 5). Bolded items indicate “major functional disability” end points used by some investigators. Abbreviation: cALD, cerebral adrenoleukodystrophy. |
Forty-seven (78%) patients survived. Of 13 deaths, five (38%) were from progressive cALD and eight (62%) were due to transplant-related complications. A significant difference in 5-year estimated survival was seen between standard-risk versus higher-risk patients (89% versus 60%, P=0.03). On multivariable analysis, this risk stratification continued to strongly predict survival (relative risk of death with Loes MRI score ≥10 9.2, 95% confidence interval 1.7–49.4; P<0.01). Of eight standard-risk patients receiving HLA-matched related marrow grafts, 100% survived. For higher-risk patients, NAC therapy dramatically increased 1-year survival (84% versus 45%, P=0.02). Most standard-risk patients at baseline scored 0 on the NFS (67%), while most higher-risk patients had a pretransplant NFS score of 1 (27%) or 2+ (63%). The authors evaluated the posttransplant progression of clinical disease on the ALD NFS, and analyzed factors that may have affected it. Higher baseline Loes MRI score (≥10), NFS score (≥1), and lower baseline PIQ (<80) and full-scale IQ (<80) correlated with statistically significant, dramatic worsening on the functional scale (median change of 7 at 2 years). In contrast, for patients who demonstrated favorably low pretransplant disease burden (either Loes <10, NFS =0, PIQ ≥80, or full-scale IQ ≥80), the aggregate median change on the NFS was 0 at a median follow-up of approximately 3 years. The authors concluded by highlighting the continuing striking disparity in functional survival outcomes after HCT between boys with early (standard-risk) and advanced/symptomatic (higher-risk) cALD. They stressed the importance of early ALD diagnosis and rapid intervention if active radiographic cerebral disease developed.38
Evidence for the efficacy of transplantation
Soon after the initial transplant experiences, expert ALD clinicians recognized the capacity for HCT to arrest cerebral disease and provide favorable functional survival. However, formal assessments between HCT and natural history with head-to-head trials were considered ethically problematic, due to the high likelihood of untreated cerebral disease progressing to neurologic devastation and death. Still, a few reports have retrospectively aimed to compare transplanted versus untreated cohorts. In the largest of these, Mahmood et al reviewed their large natural history cohort of 283 boys with cALD and identified a subgroup of 30 patients who at initial diagnosis were matched at key variables to the “favorable” HCT cohort in Peters et al’s report (n=19). Therefore, all boys in this natural history subgroup demonstrated Loes MRI scores <9 and one or less neurologic deficit at initial cALD diagnosis. Demographic and disease-related characteristics were similar to the transplanted comparators, as there was no difference at diagnosis in age or Loes MRI severity score between the two groups. Estimated 5-year survival for the untreated group was 54% (compared to 95% with HCT, P<0.01), and estimated 10-year survival was 42% (compared to 95% with HCT). When the entire untreated cohort (n=283) was considered, estimated 5-year survival was 66% and 10-year survival 43%. The authors concluded that this evidence strongly supported using HCT in early cALD, and urged clinicians to seize this “narrow window” for transplants should it present itself.39 Similarly, in their early multicenter HCT series, Shapiro et al described a small comparator group comprising 13 nontransplanted boys with cALD. Most were untreated due to the lack of acceptable donors, and not because of severe baseline disease. In total, seven (54%) died from progressive cALD and four (31%) entered a vegetative state at furthest follow-up.32
Transplant limitations and unknowns
An ongoing challenge for cALD transplants is whether treatment regimens might be optimized to improve outcomes, especially for boys with advanced cerebral disease at presentation. Given the observation of increased oxidative stress pathways in ALD, Tolar et al reasoned that intense antioxidant therapy in the peritransplant period could improve survival in boys with advanced cerebral disease. They reported three consecutive boys (baseline Loes MRI score ≥14.5 and PIQ <70) administered NAC (70 mg/kg, four times daily, both IV and orally) before and after HCT with an MA BU–CY and ATG regimen. One patient received an HLA-matched sibling-marrow graft, and the other two underwent two-unit, HLA-mismatched UCBT. All patients had complete donor hematopoietic engraftment and radiographic evidence of disease stabilization at 6 months following HCT. At follow-up of 7, 8, and 11 months, all patients survived, a significant finding compared to untreated comparators.40
Besides the provision of antioxidant therapy in the peritransplant period, it stands to reason that RIC regimens may spare toxicity to the vulnerable cALD CNS and promote more favorable functional survival following HCT. To this end, Awaya et al reported outcomes after an RIC regimen followed by HLA-matched UCBT for a boy with advanced baseline disease (Loes MRI score 11.5, cortical blindness, and VIQ 75). He was conditioned with melphalan, fludarabine, and low-dose TBI (400 cGy). He gradually achieved full donor hematopoietic chimerism and exhibited resolution of MRI contrast enhancement. His standardized disability-scale scores improved with time, and he remained radiographically and clinically stable 3 years out. The authors cautiously speculated that should full donor hematopoietic chimerism follow less intense conditioning regimens, then more favorable outcomes might be achievable after HCT for advanced cALD.41
In 2012, Niizuma et al described two brothers also successfully grafted following RIC UCBT. Both received regimens with fludarabine, melphalan, and brain-sparing low-dose TBI. Both showed resolution of gadolinium enhancement posttransplant. Interestingly, the proband’s baseline Loes MRI score was 14.5, and he had obvious visual, auditory, and motor deficits that were progressing up to the time of UCBT. Twenty months following HLA-matched UCBT, he was neurologically and radiographically stable. His younger brother, with a Loes MRI score of 1 at transplant and clinically presymptomatic, received a 4/6 HLA-matched UCBT. At 13 months, he had stable radiographic and clinical disease. The authors concluded that RIC regimens followed by UCB grafting may lead to favorable engrafted, functional survival following disease arrest in boys with advanced and early cALD alike.42
Resnick et al were the first to explore and report outcomes following RIC HCT for cALD. Three children were described; although full details were not provided, all could be inferred to have favorable baseline disease characteristics. All three underwent HLA-matched, non-carrier-sibling marrow transplantation after fludarabine, BU, and ATG conditioning. Interestingly, one patient was an 8.5-year-old girl with a known family history of ALD, predominant frontal lobe white-matter lesions on brain MRI, declining academic performance, and worsening behavior dysfunction. Further investigation revealed a pathogenic frameshift mutation in the maternal ABCD1 allele and absence of the paternal allele due to a distal Xq deletion spanning the Xq28 locus. At a follow-up of 2–3 years, all three patients had complete donor hematopoietic engraftment, uneventful peritransplant courses, and clinically stable disease. The authors concluded that further exploration of RIC regimens for cALD was warranted.43
However, a significant problem with less intense HCT conditioning regimens remains a higher risk of failed donor engraftment. It is possible that similar attempts at RIC HCT for cALD that resulted in failed engraftment have simply not been reported. At a recent annual joint meeting of the American Society of Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplantation Research, Fernandes et al briefly described engraftment outcomes of eight boys with cALD who underwent T-cell-replete, related HLA-haploidentical transplantation following a very low-intensity preparative regimen. This strategy utilizes posttransplantation CY to eliminate alloreactive T cells and minimize the historically high rates of GVHD and rejection observed in T-cell-replete HLA-haploidentical transplantation. Based on accumulating reports in the transplant literature of the success of this RIC + haploidentical posttransplant CY platform for patients with hematologic malignancies, the team hypothesized this approach could merge two desirable characteristics in transplanting cALD: expeditious allografting and minimization of neurotoxicity. In all, eight boys received the fludarabine, CY, low-dose TBI (200 cGy) ± ATG regimen followed by haploidentical posttransplant CY. Some patients were undergoing subsequent transplantation for previous graft failures. Three patients failed to engraft after the regimen, and the remaining five were all still less than 1 year out from HCT. An updated description of engrafted, functional survival for this cohort has yet to be reported.44
One hypothesis of a transplant’s therapeutic effect in cALD is by replacement of metabolically incompetent host microglia with wild-type, donor-derived ones. Therefore, it is perhaps important to consider the ability of an HCT regimen to achieve donor engraftment within the CNS, as well as within the marrow/peripheral blood compartments. Currently, real-time longitudinal monitoring of this phenomenon is not technically feasible in the clinic. However, preclinical experiments in murine HCT models (both in wild-type mice and those with neuropathic lysosomal storage disease) suggest that certain conditioning strategies, eg, MA BU-based regimens, may provide superior long-term donor CNS microglial presence. Intriguingly, mice undergoing conditioning with the CNS-sparing agent treosulfan showed long-term full donor engraftment within marrow and peripheral blood, but substantially low-level donor microglial presence in the CNS and inferior neurologic outcomes. How the clinician should balance such preclinical data with concerns for neurotoxicity that highly penetrating CNS agents may induce in the cALD brain is not presently clear, and highlights the need for better animal models of inflammatory cALD.45,46
Still another challenge with regard to transplants is predicting within the higher-risk cALD population the extent of neurologic decline and the degree of residual function following disease stabilization in patients who might undergo HCT. In their 2011 analysis of 30 boys transplanted for advanced cALD (baseline Loes MRI score ≥10), Miller et al observed aggregately poor neurologic outcomes for these advanced patients; however, they also noted wide interpatient variability in posttransplant function. Among 18 evaluable boys at a median follow-up of 2 years, the median change in ALD clinical NFS score from baseline was 7.5, with an interquartile range of 4–19 and a range of 0–23.47 Aiming for better prognostic predictors in this population, they subsequently retrospectively assessed the correlation of a simple baseline brain-MRI gadolinium-intensity scoring system with posttransplant neurologic outcomes in higher-risk patients (Loes MRI score ≥10). Among 25 evaluable boys in that analysis, greater pre-HCT gadolinium-intensity scores correlated with worsening posttransplant neurologic function (mean change on NFS scale of 10.4 versus 4.3 for patients with lower baseline gadolinium intensity, P=0.05). The investigators also observed a strong correlation between pretransplant gadolinium intensity and CSF chitotriosidase activity, a cALD biomarker they had previously described.48 They concluded that higher-risk cALD patients manifesting less intense gadolinium enhancement pretransplant might be expected to have more favorable neurologic function following HCT.49
While short- and medium-range functional outcomes after HCT for childhood cALD are relatively well described, the medical literature lacks similar catalogs of transplanted patients with very long-term follow-up. In addition to Shapiro et al’s32 early description of outcomes at 5–10 years following transplant, Gassas et al reported six consecutive boys who underwent HCT for cALD with a median follow-up of approximately 7 years (maximum 12 years). Two patients died of cALD progression. The remaining four engrafted survivors were all described to be “completely ambulatory and independent in terms of daily living activities”. One was noted to be in the workforce following school graduation, and the other three were still in school.50
A Minnesota group attempted to overcome follow-up attrition and describe very late functional outcomes in their cALD HCT cohort by remotely gathering data from surviving patients and their caretakers. Among 16 participants (n=8 with pre-HCT Loes MRI score <10, n=8 with pre-HCT Loes MRI score ≥10) at a median age of 15 years (maximum 36 years), it was observed that long-term protection from any deficits and major functional disabilities on the ALD clinical severity NFS scale depended highly upon baseline pretransplant Loes MRI severity score. An encouraging observation was of continued favorable long-term gross neurologic functioning in standard-risk cALD patients (Figure 1; Miller, unpublished data, 2016).
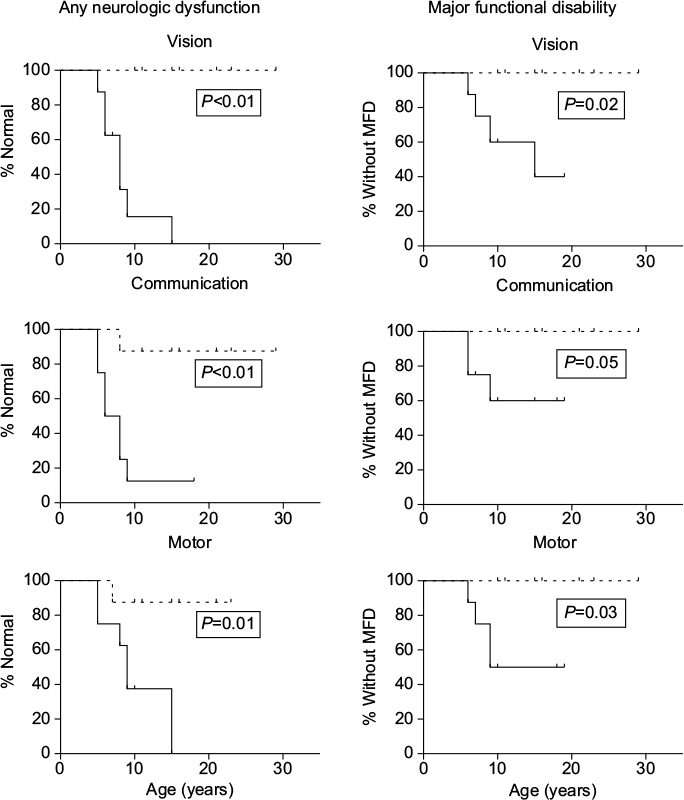 | Figure 1 Proportions of patients without clinical abnormalities (left column) or major functional disability (right column) on the ALD NFS for evaluable long-term survivors in the Minnesota cohort. Notes: The clinical domains of vision, communication, and motor function are shown. Dashed lines: standard-risk cALD patients (pretransplant Loes MRI score <10, n=8). Solid lines: higher-risk cALD patients (pretransplant Loes MRI score 10+, n=8). Patients were censored at age of most recent assessment (vertical marks). Abbreviations: ALD, adrenoleukodystrophy; cALD, cerebral adrenoleukodystrophy; NFS, Neurologic Function Scale; MRI, magnetic resonance imaging; MFD, major functional disability. |
Little is currently known regarding the ability of successful HCT to prevent or lessen the severity of future spinal cord disease (AMN) in patients with ALD. To address this, van Geel et al reported five adult patients, all of whom underwent HCT in boyhood for cALD. Three (60%) were found to have clinical myelopathy. The authors concluded that HCT may not prevent evolution to AMN in adulthood.51 Larger analyses are clearly needed to address this important question more definitively; a multicenter international study is currently under consideration.
Despite its ability to arrest active cerebral disease, published data suggest that successful HCT neither reverses existing nor protects against future AI. Petryk et al retrospectively scrutinized their cALD HCT cohort for this outcome. In all, 20 patients were evaluable. Of 16 with pretransplant AI, none recovered function; furthermore, three of the four patients without AI at baseline manifested adrenal failure within 1–2 years posttransplant.52
Who, when, and how to transplant
The collective HCT experience for childhood cALD champions the crucial roles of early ALD diagnosis and vigilance for conversion to cerebral disease. One possible route to diagnosis follows screening for males who demonstrate “idiopathic” primary AI. Several reports catalog a relatively high incidence of ALD as causative of primary hypoadrenalism in patients who do not have true Addison’s disease (ACTH receptor antibody-negative primary AI).53,54 Polgreen et al retrospectively reviewed their cohort, and identified boys whose initial and sole clinical manifestation of ALD was adrenal crisis and who already had radiographic cerebral disease upon diagnosis of ALD. They then studied the impact of delay from AI presentation to ALD diagnosis on long-term clinical outcomes. Ten such boys were grouped into the “early” ALD diagnosis category (mean lag between AI and ALD/cALD diagnosis of 2 months, range 0–12 months), while seven boys were assigned to the “late” ALD diagnosis category (mean lag between AI and ALD/cALD diagnosis of 3.9 years, range 1.4–10 years). AI was diagnosed at the same average age, 6 years, for both groups. The investigators found that a lag in diagnosis of ALD/cALD after initial diagnosis of AI led to significantly higher cerebral disease burden at the time of transplant, and urged that all males with unexplained primary AI be biochemically screened for ALD by VLCFA assay.55
ALD clinical experts have long recognized the likely utility of broad-based population screening for the purpose of presymptomatic ALD detection.56 Currently, a small but growing number of US states have added ALD to their newborn-screening programs on the basis of available, affordable, and highly sensitive and specific diagnostic assays. Experts urge that cases identified through such programs be referred to highly experienced centers for longitudinal surveillance of disease onset (AI and cALD). They caution against the risk of “overtreatment”, such as HCT performed for patients without clear evidence of cerebral disease, and underscore the potential for high-quality patient registries that might improve understanding about ALD and phenotypic manifestations. Finally, they highlight the challenges in identifying other at-risk relatives within kinships.57 In short, ALD patients identified by public newborn-screening programs should be referred to centers with extensive experience in the disease for counseling, surveillance, and management. Furthermore, extensive genetic counseling with a focus on identifying at-risk first-degree and extended relatives should be provided upon identification of probands, no matter the route to diagnosis.
A vexing challenge for ALD clinicians remains predicting which young patients known to have the biochemical defect of the disease will ultimately develop cerebral disease (cALD) in boyhood or beyond. As no ABCD1 genotype–phenotype correlations have been uncovered in this regard, the current recommended practice is frequent surveillance with conventional brain MRI in all ALD boys (every 6 months during the highest-risk period between ages 2 years and adolescence; yearly monitoring thereafter) until hyperintense white-matter disease becomes evident on T2-weighted imaging. Few published reports of efforts to predict MRI conversion currently exist. Oz et al studied whether neurochemical profiles of cerebral white matter using high-field MRS might portend impending lesions on conventional MRI. Three distinct groups, all with relatively small numbers, underwent longitudinal MRS: 1) cALD patients (with early white-matter lesions evident on conventional MRI), 2) patients with the known biochemical defect of ALD, but without cerebral disease evident on conventional MRI, and 3) healthy controls. Although few patients in the series demonstrated white-matter lesion conversion on conventional MRI during the period of monitoring (ALD → cALD), the authors noted the possible emergence of an MRS fingerprint of impending lesions.58 Larger studies to corroborate these findings have not been reported.
An additional challenge is posed by newly diagnosed boys with ALD who on first MRI show radiographically evident low-level cerebral disease. In these patients, the managing clinician may consider the possibility of inactive cerebral disease. To address this, Loes et al retrospectively analyzed historical cALD patients in a Kennedy Krieger Institute cohort for radiographic and demographic patterns that might predict MRI progression of white-matter disease. In total, 140 cALD patients were evaluable with two or more MRI studies over time and without intervention; the average time between first and last follow-up MRI was 3.5 years. When considering the variables of age, neuroanatomic regions involved, and status of contrast enhancement, they found that young age, high baseline Loes MRI severity score, and contrast-enhancing parieto-occipital and/or frontal white-matter disease strongly predicted rapid MRI progression.13 Similarly, Musolino et al studied whether parameters of cerebral perfusion, as measured with dynamic susceptibility-contrast MR perfusion, might predict disease progression on follow-up conventional MRI. In untreated boys with cALD, hypoperfusion in the zone centrifugal to the expanding rim of enhancement correlated with eventual enhancement in the area on follow-up imaging. Interestingly, one transplanted boy demonstrated normalization of previous hypoperfusion in this zone following engraftment. On his follow-up conventional MRI studies, the region remained radiographically unaffected.59
In published studies of large cALD HCT cohorts, survival and gross neurological functional outcomes for standard-risk patients (Loes MRI score <10 before HCT) have been favorable. These data may tempt the ALD clinician to “wait watchfully” when presented with boys with early cALD. However, it is important to consider the impact such an approach may have on longer-term neuropsychological functioning of cALD patients as well. Within a Minnesota cohort, 33 standard-risk patients had sufficient baseline and posttransplant (2+ years) neuropsychology data for rigorous analysis. It was noted that very low pretransplant cerebral burden correlated with clearly superior longer-term cognitive functioning when compared to boys who were still “standard risk”, but demonstrated higher pre-HCT Loes MRI severity scores (Miller, unpublished data, 2016). These potential consequences of delay to transplantation should be considered when clinicians and family members are deciding if and when to proceed to HCT. In general, strong consideration to urgent transplantation should be given to boys with presymptomatic, early cALD who demonstrate either contrast enhancement on a single MRI or expanding white-matter lesions on sequential scans, regardless of contrast uptake status. For boys with higher-risk cALD (Loes MRI score >10 or clinically evident cerebral dysfunction), it is recommended that expert transplant centers carefully evaluate the particular case and that the family receive extensive counseling on the spectrum of potential outcomes before HCT is considered.
Graft selection for HCT in cALD should aim to minimize the complications of graft failure and graft-versus-host disease (GVHD), as several of the reports cited in this review have noted these to be accompanied by clinical cALD progression following transplant. When available, current evidence supports prioritization for HLA-matched sibling donors. However, a conundrum arises if the only HLA-matched sibling is an ALD-carrier sister. While outcomes following transplant for lysosomal storage disease are known to be inferior when donors who produce subnormal enzymes are used, there are currently insufficient reports to support or refute carrier-donor grafting in cALD.60 The author’s practice has been to avoid using carrier donors when acceptable alternative grafts are available. In select circumstances where future transplant of an ALD patient is possible, but not urgently indicated, private banking of noncarrier HLA-matched, sibling UCB may be advisable; furthermore, strategic family planning by preimplantation genetic selection with in vitro fertilization may be considered, as has been previously described for ALD.61
When appropriate HLA-matched sibling allografts are not available, several alternative donor sources may be considered. Growing experience with UCBT in cALD and similar inherited neuropathic metabolic storage disorders suggests this graft source to be an effective one. Furthermore, UCB has the advantage of being rapidly available for immediate HCT. However, limited cell dosing, as well as escalating risks of graft failure with increasing HLA disparity between the UCB unit and recipient, may preclude its use in certain circumstances. In general, HLA-matched (allele-level, HLA-A, -B, -C, and –DRB1) unrelated donor-marrow grafts are an acceptable option in transplanting cALD. Immunomediated complications, particularly GVHD, become common in the setting of HLA-mismatched unrelated donor transplantation that uses conventional GVHD-prevention strategies. An additional obstacle is the delay to donation that is commonly encountered in anonymous, volunteer registry-mediated unrelated-donor HCT; in certain clinical scenarios, such delays may not be in the best interest of the cALD boy. Related HLA-haploidentical donor HCT strategies are attractive in cALD for the nearly universal and immediate access to such familial, noncarrier donors; however, this approach should be considered experimental until longer-term outcomes are reported.31,44
Numerous HCT conditioning strategies have been described in cALD transplantation, but no single regimen has emerged as “superior”. Most boys have received MA BU–CY-based conditioning. Though disease stabilization and long-term survival have been described in advanced cALD patients transplanted with less intense regimens, the lure of this RIC approach must be weighed against the risk of graft rejection with autologous hematopoietic recovery. Trends in pediatric transplantation for nonmalignant disease have included efforts to reduce alkylator-type chemotherapy exposure, and Bartelink et al recently documented exceptionally favorable engrafted survival and low toxicity rates in children, including many with nonmalignant indications for HCT, using a reduced intensity but still MA regimen comprising BU (targeted exposure), fludarabine, and ATG.62 This author has recently used this same regimen in eleven consecutive boys with cALD who received various allografts. In total, ten boys are alive and fully donor-engrafted, though one of these required second transplantation for graft failure. One boy died of complications of graft failure (Miller, unpublished data, 2016).
Careful consideration to adrenal function should be given before beginning HCT conditioning. For boys without obvious evidence of hypoadrenalism, it is the author’s practice to perform dynamic screening with low-dose cosyntropin to exclude subclinical AI fully. In patients with known or suspected AI, empiric stress dosing with exogenous adrenocortical hormones in the peritransplant period should be considered, and expert pediatric endocrinologic consultation should be sought.
Allogeneic hematopoietic cell transplantation for adult cerebral adrenoleukodystrophy
In contrast to HCT for childhood-onset disease, a dearth of published experience can be found for that in adult-onset cALD. In 2005, Hitomi et al reported a 2-year follow-up on a 20-year-old man transplanted for contrast-enhancing MRI-apparent lesions in the left corticospinal tracts. He had right hemiparesis and gait disturbance, and his pretransplant Expanded Disability Status Scale (EDSS) score was 4.5. At 2 months posttransplant, lesional gadolinium uptake on MRI had decreased. At 2-year follow-up, his brain MRI remained stable, his EDSS score was 2, and he exhibited normal motor function and ambulation, though abnormalities persisted on running.63 Fitzpatrick et al later reported HCT for a 36-year-old man with clinical disease onset similar to that seen in boyhood cALD. He presented with declining cognitive, visual, and auditory function. MRI showed symmetric, enhancing white-matter disease in the parieto-occipital lobes, and ALD was diagnosed. A baseline Loes severity score was not reported. He underwent an HLA-matched, non-carrier-sibling marrow transplant after MA BU–CY conditioning, and despite presumed donor hematopoietic engraftment, developed irreversible cortical blindness 1 month out. He died from GVHD at 3 months following HCT.64 For adult ALD patients with contrast-enhancing or expanding cerebral white-matter lesions, consideration to urgent HCT should be given. However, it is recommended that counseling, treatment planning, and transplantation (if indicated) be undertaken at experienced ALD centers.
Autologous, genetically modified hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy
The risks of treatment-related complications and death following allogeneic HCT are substantial, even in the setting of HLA-matched sibling and/or RIC regimens. After laying the preclinical groundwork for efficient ex vivo lentiviral vector-mediated transduction of human hematopoietic stem cells with wild type ABCD1, a French group described the initial human experience with this gene-therapy approach.65–68 Two boys with ALD, both aged 7 years, lacked HLA-matched donors or UCB units for allogeneic HCT. Both demonstrated progressive cerebral disease on MRI. Both had identifiable ABCD1 mutations, and neither expressed ALDP on baseline leukocyte immunocytochemistry. Autologous peripheral blood mononuclear cell collection was performed following mobilization with granulocyte colony-stimulating factor, and the product was purified for CD34+ cells with immunomagnetic selection. Ex vivo CD34+ transduction occurred under cytokine stimulation with a replication-defective lentiviral vector, and the genetically modified product was infused into recipients following an MA BU–CY regimen for eradication of uncorrected hematopoietic stem and progenitor cells.
At 2 years from treatment, both boys demonstrated persistent expression of ALDP in 10%–15% of lymphocytes and monocytes. No dominant hematopoietic clones were identified on longitudinal monitoring, suggesting no concerning insertional genotoxicity. Reductions from baseline (~40%) in plasma VLCFA were seen in both patients at near 2 years out. Radiographically, one patient showed resolution of contrast enhancement at 12 months following treatment, and the Loes score changed from 2.25 at baseline to 6.75 at last evaluation. The other patient, although experiencing waxing and waning contrast enhancement and contiguous extension of white-matter disease, technically maintained a stable Loes score over time (7 at baseline and last follow-up). Clinically, both patients developed mild cognitive or functional deficits following treatment, though these stabilized with longitudinal assessments. The investigators concluded that this initial human experience approximated outcomes seen in standard allogeneic HCT, even as the majority of circulating monocytes (the cell type presumed to mediate mechanistic effects in transplants) remained of original, genetically uncorrected lineage.
A larger, multicenter international trial followed upon the initial experience of the French group, and studied ex vivo autologous HSC ABCD1 correction in 17 boys with early cALD. In April 2016, the interim results were presented at the annual American Academy of Neurology meeting. All boys, ages ranging 4–13 years and with a median baseline Loes MRI severity score of 2, had been reinfused with lentiviral vector ABCD1 corrected autologous HSC following an MA BU–CY regimen. Follow-up ranged from 6 months to 2 years. All patients survived and none had yet developed a major functional disability on the ALD NFS. Sixteen patients were noted to demonstrate stable ALD NFS scores (defined as change of fewer than 3 points from baseline and an absolute score ≤4 at last follow-up). Sixteen patients demonstrated initial extinction of gadolinium enhancement, five developed recurrent enhancement on subsequent scans, and two once again resolved enhancement at later MRI. The overall safety of the procedure appeared highly favorable. Similarly to Cartier et al’s report, no concerning clonal dominance among hematopoietic lines had been observed.69 Detailed reports of longer-term outcomes of this gene-therapy cohort are anxiously awaited.
In vivo gene-correction strategies for ameliorating ALD are also being explored, as such an approach avoids delay required for ex vivo autologous HSC correction, as well as MA cytotoxic conditioning. Gong et al recently reported effects in the ABCD1-knockout mouse following in vivo gene therapy with the adenoassociated virus vector serotype 9. When administered either intracerebroventricularly or IV, the vector effectively transduced various neural cell types in the brain and spinal cord, the gene product was found to localize to peroxisomes, and CNS VLCFA levels were markedly lower compared to untreated controls. As IV delivery appeared to provide greater correction of clinically relevant CNS cell types, as well as somatic tissue, including the adrenal cortex, the authors held optimism that this strategy could soon be translated to clinical trials for ALD and AMN.70
Other (nonhematopoietic) stem cell therapies for adrenoleukodystrophy
To date, there has been no published experience using enriched, other (nonhematopoietic) stem cell therapies for patients with ALD. However, as various other stem therapies emerge in the clinical armamentarium, it is important to consider their potential utilities in this disease. Accumulating reports document the therapeutic potential of mesenchymal stem cells, which possess capacity for both modulating pathologic immune responses and facilitating tissue regeneration.71–84 Mesenchymal stem cells modified for improved homing and survivability can provide even superior clinical efficacy in neuroinflammatory and neurodegenerative disorders.85,86 Both glial progenitor (or less committed neural stem cell) and endothelial progenitor-cell therapies are theoretically attractive in cALD, which is characterized by rapid demyelination following BBB disruption.87–92 In practice, among other potential barriers to clinical implementation of such stem cell therapies, histoincompatibility between random donors and patients may limit the long-term in vivo survival of therapeutic stem cells and their progeny.93 While autologous, induced pluripotent stem cell manufacturing followed by genetic correction is one means to overcome the allogeneic hurdle, another potential solve in the post-HCT cALD population is the use of HCT donor-derived, enriched stem cell populations (from UCB units, for example).
Conclusion
In conclusion, allogeneic HCT remains standard therapy for cALD, particularly when onset is in childhood. Since the first HCT for cALD performed over 30 years ago, outcomes have improved and a clearer understanding of HCT’s limitations in the disease has emerged, despite the ongoing lack of animal models approximating cALD that could better inform investigators. Boys with early, presymptomatic cALD and sufficiently HLA-matched allografts face favorable estimates of long-term functional survival following HCT. A persisting challenge remains if and how HCT can be optimized to improve outcomes for boys who already manifest advanced cALD upon coming to medical attention. With increased disease awareness and the advent of newborn screening, earlier ALD diagnosis is expected to translate to better aggregate outcomes, including for those destined to develop cALD. In all males with the known biochemical defect of ALD, clinicians should remain vigilant for the emergence of early, presymptomatic cerebral disease.
Disclosure
The author reports no conflicts of interest in this work.
References
Bezman L, Moser A, Raymond G, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49(4):512–517. | ||
Academic Medical Center, Amsterdam. Adrenoleukodystrophy database. 2016. Available from: http://www.x-ald.nl. Accessed September 30, 2016. | ||
Moser H. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997;120(Pt 8):1485–1508. | ||
Engelen M, Kemp S, de Visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7:51. | ||
Wanders R, Vreken P, Ferdinandusse S, et al. Peroxisomal fatty acid α- and β-oxidation in humans: enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem Soc Trans. 2001;29(Pt 2):250–267. | ||
Igarashi M, Schaumburg HH, Powers J, Kishmoto Y, Kolodny E, Suzuki K. Fatty acid abnormality in adrenoleukodystrophy. J Neurochem. 1976;26(4):851–860. | ||
Moser AB, Kreiter N, Bezman L, et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol. 1999;45(1):100–110. | ||
Berger J, Gärtner J. X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta. 2006;1763(12):1721–1732. | ||
Berger J, Molzer B, Faé I, Bernheimer H. X-linked adrenoleukodystrophy (ALD): a novel mutation of the ALD gene in 6 members of a family presenting with 5 different phenotypes. Biochem Biophys Res Commun. 1994;205(3):1638–1643. | ||
Moser H, Dubey P, Fatemi A. Progress in X-linked adrenoleukodystrophy. Curr Opin Neurol. 2004;17(3):263–269. | ||
Mahmood A, Dubey P, Moser H, Moser A. X-linked adrenoleukodystrophy: therapeutic approaches to distinct phenotypes. Pediatr Transplant. 2005;9 Suppl 7:55–62. | ||
Kim J, Kim H. Childhood X-linked adrenoleukodystrophy: clinical-pathologic overview and MR imaging manifestations at initial evaluation and follow-up. Radiographics. 2005;25(3):619–631. | ||
Loes DJ, Fatemi A, Melhem ER, et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology. 2003;61(3):369–374. | ||
Hobbs JR, Hugh-Jones K, Barrett AJ, et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981;2(8249):709–712. | ||
Pridjian G, Humbert J, Willis J, Shapira E. Presymptomatic late-infantile metachromatic leukodystrophy treated with bone marrow transplantation. J Pediatr. 1994;125(5):755–758. | ||
Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968;162(853):570–572. | ||
Berger J, Forss-Petter S, Eichler FS. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie. 2014;98:135–142. | ||
Ferrer I, Aubourg P, Pujol A. General aspects and neuropathology of X-linked adrenoleukodystrophy. Brain Pathol. 2010;20(4):817–830. | ||
Vargas CR, Wajner M, Sirtori LR, et al. Evidence that oxidative stress is increased in patients with X-linked adrenoleukodystrophy. Biochim Biophys Acta. 2004;1688(1):26–32. | ||
Deon M, Sitta A, Barschak AG, et al. Induction of lipid peroxidation and decrease of antioxidant defenses in symptomatic and asymptomatic patients with X-linked adrenoleukodystrophy. Int J Dev Neurosci. 2007;25(7):441–444. | ||
Powers JM, Pei Z, Heinzer AK, et al. Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol. 2005;64(12):1067–1079. | ||
Rockenbach FJ, Deon M, Marchese DP, et al. The effect of bone marrow transplantation on oxidative stress in X-linked adrenoleukodystrophy. Mol Genet Metab. 2012;106(2):231–236. | ||
Yamada T, Ohyagi Y, Shinnoh N, et al. Therapeutic effects of normal cells on ABCD1 deficient cells in vitro and hematopoietic cell transplantation in the X-ALD mouse model. J Neurol Sci. 2004;218(1–2):91–97. | ||
Moser HW, Tutschka PJ, Brown FR, et al. Bone marrow transplant in adrenoleukodystrophy. Neurology. 1984;34(11):1410–1417. | ||
Aubourg P, Blanche S, Jambaqué I, et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med. 1990;322(26):1860–1866. | ||
Loes D, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol. 1994;15(9):1761–1766. | ||
Loes DJ, Stillman AE, Hite S, et al. Childhood cerebral form of adrenoleukodystrophy: short-term effect of bone marrow transplantation on brain MR observations. AJNR Am J Neuroradiol. 1994;15(9):1767–1771. | ||
Malm G, Ringdén O, Anvret M, et al. Treatment of adrenoleukodystrophy with bone marrow transplantation. Acta Paediatr. 1997;86(5):484–492. | ||
Nowaczyk MJ, Saunders EF, Tein I, Blaser SI, Clarke JT. Immunoablation does not delay the neurologic progression of X-linked adrenoleukodystrophy. J Pediatr. 1997;131(3):453–455. | ||
Kapelushnik J, Varadi G, Nagler A. Matched unrelated human umbilical cord blood transplantation for X-linked adrenoleukodystrophy. J Pediatr Hematol Oncol. 1998;20(3):257-259. | ||
Lin HC, Lin KH, Wang PJ. Transplantation for adrenoleukodystrophy with HLA-A and B nonidentical paternal marrow: report of one case. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1998;39(4):260–263. | ||
Shapiro E, Krivit W, Lockman L, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356(9231):713–718. | ||
Baumann M, Korenke GC, Weddige-Diedrichs A, et al. Haematopoietic stem cell transplantation in 12 patients with cerebral X-linked adrenoleukodystrophy. Eur J Pediatr. 2003;162(1):6–14. | ||
Wilken B, Dechent P, Brockmann K, et al. Quantitative proton magnetic resonance spectroscopy of children with adrenoleukodystrophy before and after hematopoietic stem cell transplantation. Neuropediatrics. 2003;34(5):237–246. | ||
Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104(3):881–888. | ||
Beam D, Poe M, Provenzale J, et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol Blood Marrow Transplant. 2007;13(6):665–674. | ||
Gess A, Christiansen SP, Pond D, Peters C. Predictive factors for vision loss after hematopoietic cell transplant for X-linked adrenoleukodystrophy. J AAPOS. 2008;12(3):273–276. | ||
Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118(7):1971–1978. | ||
Mahmood A, Raymond G, Dubey P, Peters C, Moser H. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol. 2007;6(8):687–692. | ||
Tolar J, Orchard PJ, Bjoraker KJ, Ziegler RS, Shapiro EG, Charnas L. N-acetyl-L-cysteine improves outcome of advanced cerebral adrenoleukodystrophy. Bone Marrow Transplant. 2007;39(4):211–215. | ||
Awaya T, Kato T, Niwa A, et al. Successful cord blood transplantation using a reduced-intensity conditioning regimen for advanced childhood-onset cerebral adrenoleukodystrophy. Pediatr Transplant. 2011;15(6):E116–E120. | ||
Niizuma H, Uematsu M, Sakamoto O, et al. Successful cord blood transplantation with reduced-intensity conditioning for childhood cerebral X-linked adrenoleukodystrophy at advanced and early stages. Pediatr Transplant. 2012;16(2):E63–E70. | ||
Resnick I, Hai AA, Shapira M, et al. Treatment of X-linked childhood cerebral adrenoleukodystrophy by the use of an allogeneic stem cell transplantation with reduced intensity conditioning regimen. Clin Transplant. 2005;19(6):840–847. | ||
Fernandes J, Bonfim C, Kerbauy F, et al. T-cell-replete haploidentical stem cell transplantation with post-transplant cyclophosphamide for patients with X-linked adrenoleukodystrophy: an immediate choice for an urgent situation. Biol Blood Marrow Transplant. 2014;20(2 Suppl):S168–S169. | ||
Capotondo A, Milazzo R, Politi LS, et al. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc Natl Acad Sci U S A. 2012;109(37):15018–15023. | ||
Wilkinson FL, Sergijenko A, Langford-Smith KJ, Malinowska M, Wynn RF, Bigger BW. Busulfan conditioning enhances engraftment of hematopoietic donor-derived cells in the brain compared with irradiation. Mol Ther. 2013;21(4):868–876. | ||
Miller WP, Rothman SM, Nascene D, et al. Outcomes following allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118(7):1971–1978. | ||
Orchard PJ, Lund T, Miller W, et al. Chitotriosidase as a biomarker of cerebral adrenoleukodystrophy. J Neuroinflammation. 2011;8:144. | ||
Miller WP, Mantovani LF, Muzic J, et al. Intensity of MRI gadolinium enhancement in cerebral adrenoleukodystrophy: a biomarker for inflammation and predictor of outcome following transplantation in higher risk patients. AJNR Am J Neuroradiol. 2016;37(2):367–372. | ||
Gassas A, Raiman J, White L, Schechter T, Clarke J, Doyle J. Long-term adaptive functioning outcomes of children with inherited metabolic and genetic diseases treated with hematopoietic stem cell transplantation in a single large pediatric center: parents’ perspective. J Pediatr Hematol Oncol. 2011;33(3):216–220. | ||
van Geel BM, Poll-The BT, Verrips A, Boelens JJ, Kemp S, Engelen M. Hematopoietic cell transplantation does not prevent myelopathy in X-linked adrenoleukodystrophy: a retrospective study. J Inherit Metab Dis. 2015;38(2):359–361. | ||
Petryk A, Polgreen LE, Chahla S, Miller W, Orchard PJ. No evidence for the reversal of adrenal failure after hematopoietic cell transplantation in X-linked adrenoleukodystrophy. Bone Marrow Transplant. 2012;47(10):1377–1378. | ||
Horn MA, Erichsen MM, Wolff AS, et al. Screening for X-linked adrenoleukodystrophy among adult men with Addison’s disease. Clin Endocrinol (Oxf). 2013;79(3):316–320. | ||
Laureti S, Casucci G, Santeusanio F, Angeletti G, Aubourg P, Brunetti P. X-linked adrenoleukodystrophy is a frequent cause of idiopathic Addison’s disease in young adult male patients. J Clin Endocrinol Metab. 1996;81(2):470–474. | ||
Polgreen LE, Chahla S, Miller W, et al. Early diagnosis of cerebral X-linked adrenoleukodystrophy in boys with Addison’s disease improves survival and neurological outcomes. Eur J Pediatr. 2011;170(8):1049–1054. | ||
Raymond G, Jones R, Moser A. Newborn screening for adrenoleukodystrophy: implications for therapy. Mol Diagn Ther. 2007;11(6):381–384. | ||
Kemper AR, Brosco J, Comeau AM, et al. Newborn screening for X-linked adrenoleukodystrophy: evidence summary and advisory committee recommendation. Genet Med. Epub 2016 Jun 23. | ||
Oz G, Tkáč I, Charnas LR, et al. Assessment of adrenoleukodystrophy lesions by high field MRS in non-sedated pediatric patients. Neurology. 2005;64(3):434–441. | ||
Musolino PL, Rapalino O, Caruso P, Caviness VS, Eichler FS. Hypoperfusion predicts lesion progression in cerebral X-linked adrenoleukodystrophy. Brain. 2012;135(Pt 9):2676–2683. | ||
Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125(13):2164–2172. | ||
Kahraman S, Beyazyurek C, Yesilipek MA, et al. Successful haematopoietic stem cell transplantation in 44 children from healthy siblings conceived after preimplantation HLA matching. Reprod Biomed Online. 2014;29(3):340–351. | ||
Bartelink IH, van Reij EM, Gerhardt CE, et al. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: maintaining efficacy with less toxicity. Biol Blood Marrow Transplant. 2014;20(3):345–353. | ||
Hitomi T, Mezaki T, Tomimoto H, et al. Long-term effect of bone marrow transplantation in adult-onset adrenoleukodystrophy. Eur J Neurol. 2005;12(10):807–810. | ||
Fitzpatrick AS, Loughrey CM, Johnston P, et al. Haematopoietic stem-cell transplant for adult cerebral adrenoleukodystrophy. Eur J Neurol. 2008;15(3):e21–e22. | ||
Benhamida S, Pflumio F, Dubart-Kupperschmitt A, et al. Transduced CD34+ cells from adrenoleukodystrophy patients with HIV-derived vector mediate long-term engraftment of NOD/SCID mice. Mol Ther. 2003;7(3):317–324. | ||
Doerflinger N, Miclea JM, Lopez J, et al. Retroviral transfer and long-term expression of the adrenoleukodystrophy gene in human CD34+ cells. Hum Gene Ther. 1998;9(7):1025–1036. | ||
Cartier N, Hacein-Bey-Abina S, Bartholomae C, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326(5954):818–823. | ||
Cartier N, Lopez J, Moullier P, et al. Retroviral-mediated gene transfer corrects very-long-chain fatty acid metabolism in adrenoleukodystrophy fibroblasts. Proc Natl Acad Sci U S A. 1995;92(5):1674–1678. | ||
Eichler F, Duncan C, Thrasher A, et al. Interim results from a phase 2/3 study of the efficacy and safety of ex vivo gene therapy with lentiviral vector (Lenti-D) for childhood cerebral adrenoleukodystrophy. Neurology. 2016;86(16 Suppl):PL02.002. | ||
Gong Y, Mu D, Prabhakar S, et al. Adenoassociated virus serotype 9-mediated gene therapy for X-linked adrenoleukodystrophy. Mol Ther. 2015;23(5):824–834. | ||
Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8(9):726–736. | ||
Amado LC, Saliaris AP, Schuleri KH, et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci U S A. 2005;102(32):11474–11479. | ||
Kan I, Melamed E, Offen D. Autotransplantation of bone marrow-derived stem cells as a therapy for neurodegenerative diseases. Handb Exp Pharmacol. 2007;(180):219–242. | ||
Kopen GC, Prockop DJ, Phinney DG. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc Natl Acad Sci U S A. 1999;96(19):10711–10716. | ||
Mansilla E, Marin GH, Sturla F, et al. Human mesenchymal stem cells are tolerized by mice and improve skin and spinal cord injuries. Transplant Proc. 2005;37(1):292–294. | ||
Miller RH, Bai L. Cellular approaches for stimulating CNS remyelination. Regen Med. 2007;2(5):817–829. | ||
Müller I, Kustermann-Kuhn B, Holzwarth C, et al. In vitro analysis of multipotent mesenchymal stromal cells as potential cellular therapeutics in neurometabolic diseases in pediatric patients. Exp Hematol. 2006;34(10):1413–1419. | ||
Prockop DJ, Azizi SA, Colter D, Digirolamo C, Kopen G, Phinney DG. Potential use of stem cells from bone marrow to repair the extracellular matrix and the central nervous system. Biochem Soc Trans. 2000;28(4):341–345. | ||
Chen X, Armstrong MA, Li G. Mesenchymal stem cells in immunoregulation. Immunol Cell Biol. 2006;84(5):413–421. | ||
Crop M, Baan C, Weimar W, Hoogduijn M. Potential of mesenchymal stem cells as immune therapy in solid-organ transplantation. Transpl Int. 2009;22(4):365–376. | ||
Gordon D, Pavlovska G, Glover CP, Uney JB, Wraith D, Scolding NJ. Human mesenchymal stem cells abrogate experimental allergic encephalomyelitis after intraperitoneal injection, and with sparse CNS infiltration. Neurosci Lett. 2008;448(1):71–73. | ||
Iyer SS, Rojas M. Anti-inflammatory effects of mesenchymal stem cells: novel concept for future therapies. Expert Opin Biol Ther. 2008;8(5):569–581. | ||
Nasef A, Ashammakhi N, Fouillard L. Immunomodulatory effect of mesenchymal stromal cells: possible mechanisms. Regen Med. 2008;3(4):531–546. | ||
Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110(10):3499–3506. | ||
Nowakowski A, Walczak P, Lukomska B, Janowski M. Genetic engineering of mesenchymal stem cells to induce their migration and survival. Stem Cells Int. 2016;2016:4956063. | ||
Park JS, Suryaprakash S, Lao YH, Leong KW. Engineering mesenchymal stem cells for regenerative medicine and drug delivery. Methods. 2015;84:3–16. | ||
Windrem MS, Schanz SJ, Guo M, et al. Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell. 2008;2(6):553–565. | ||
Duncan ID. Oligodendrocytes and stem cell transplantation: their potential in the treatment of leukoencephalopathies. J Inherit Metab Dis. 2005;28(3):357–368. | ||
Ricca A, Rufo N, Ungari S, et al. Combined gene/cell therapies provide long-term and pervasive rescue of multiple pathological symptoms in a murine model of globoid cell leukodystrophy. Hum Mol Genet. 2015;24(12):3372–3389. | ||
Gupta N, Henry RG, Strober J, et al. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med. 2012;4(155):155ra137. | ||
Osorio MJ, Goldman SA. Glial progenitor cell-based treatment of the childhood leukodystrophies. Exp Neurol. 2016;283(Pt B):476–488. | ||
Boyer-Di Ponio J, El-Ayoubi F, Glacial F, et al. Instruction of circulating endothelial progenitors in vitro towards specialized blood-brain barrier and arterial phenotypes. PLoS One. 2014;9(1):e84179. | ||
Goldman SA. Stem and progenitor cell-based therapy of the central nervous system: hopes, hype, and wishful thinking. Cell Stem Cell. 2016;18(2):174–188. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.