Back to Journals » International Journal of Nanomedicine » Volume 16
Stem Cell Mimicking Nanoencapsulation for Targeting Arthritis
Authors Shin MJ, Park JY ![]() , Lee DH, Khang D
, Lee DH, Khang D ![]()
Received 14 August 2021
Accepted for publication 5 December 2021
Published 31 December 2021 Volume 2021:16 Pages 8485—8507
DOI https://doi.org/10.2147/IJN.S334298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Min Jun Shin,1,2,* Jun Young Park,1,2,* Dae Ho Lee,3,4 Dongwoo Khang1,2,5
1Department of Health Sciences and Technology, GAIHST, Gachon University, Incheon, 21999, South Korea; 2Lee Gil Ya Cancer and Diabetes Institute, Gachon University, Incheon, 21999, South Korea; 3Department of Internal Medicine, Gachon University Gil Medical Center, Incheon, 21999, South Korea; 4Department of Internal Medicine, Gachon University College of Medicine, Incheon, 21999, South Korea; 5Department of Physiology, School of Medicine, Gachon University, Incheon, 21999, South Korea
*These authors contributed equally to this work
Correspondence: Dongwoo Khang
Department of Physiology, School of Medicine, Gachon University, Incheon, 21999, South Korea
Tel +82 32 899 1525
Email [email protected]
Dae Ho Lee
Department of Internal Medicine, Gachon University College of Medicine, Incheon, 21565, South Korea
Tel +82 32 458 2733
Email [email protected]
Abstract: Mesenchymal stem cells (MSCs) are considered a promising regenerative therapy due to their ability to migrate toward damaged tissues. The homing ability of MSCs is unique compared with that of non-migrating cells and MSCs are considered promising therapeutic vectors for targeting major cells in many pathophysiological sites. MSCs have many advantages in the treatment of malignant diseases, particularly rheumatoid arthritis (RA). RA is a representative autoimmune disease that primarily affects joints, and secreted chemokines in the joints are well recognized by MSCs following their migration to the joints. Furthermore, MSCs can regulate the inflammatory process and repair damaged cells in the joints. However, the functionality and migration ability of MSCs injected in vivo still show insufficient. The targeting ability and migration efficiency of MSCs can be enhanced by genetic engineering or modification, eg, overexpressing chemokine receptors or migration-related genes, thus maximizing their therapeutic effect. However, there are concerns about genetic changes due to the increased probability of oncogenesis resulting from genome integration of the viral vector, and thus, clinical application is limited. Furthermore, it is suspected that administering MSCs can promote tumor growth and metastasis in xenograft and orthotopic models. For this reason, MSC mimicking nanoencapsulations are an alternative strategy that does not involve using MSCs or bioengineered MSCs. MSC mimicking nanoencapsulations consist of MSC membrane-coated nanoparticles, MSC-derived exosomes and artificial ectosomes, and MSC membrane-fused liposomes with natural or genetically engineered MSC membranes. MSC mimicking nanoencapsulations not only retain the targeting ability of MSCs but also have many advantages in terms of targeted drug delivery. Specifically, MSC mimicking nanoencapsulations are capable of encapsulating drugs with various components, including chemotherapeutic agents, nucleic acids, and proteins. Furthermore, there are fewer concerns over safety issues on MSC mimicking nanoencapsulations associated with mutagenesis even when using genetically engineered MSCs, because MSC mimicking nanoencapsulations use only the membrane fraction of MSCs. Genetic engineering is a promising route in clinical settings, where nano-encapsulated technology strategies are combined. In this review, the mechanism underlying MSC homing and the advantages of MSC mimicking nanoencapsulations are discussed. In addition, genetic engineering of MSCs and MSC mimicking nanoencapsulation is described as a promising strategy for the treatment of immune-related diseases.
Keywords: stem cell migration, stem cell mimicking nanoencapsulations, autoimmune disease targeting strategy, exosomes, ectosomes, liposomes
Introduction
Given the multi-lineage differentiation abilities of mesenchymal stem cells (MSCs) isolated from different tissues and organs, MSCs have been widely used in various medical fields, particularly regenerative medicine.1–3 The representative sources of MSCs are bone marrow, adipose, periodontal, muscle, and umbilical cord blood.4–10 Interestingly, slight differences have been reported in the characteristics of MSCs depending on the different sources, including their population in source tissues, immunosuppressive activities, proliferation, and resistance to cellular aging.11 Bone marrow-derived MSCs (BM-MSCs) are the most intensively studied and show clinically promising results for cartilage and bone regeneration.11 However, the isolation procedures for BM-MSCs are complicated because bone marrow contains a relatively small fraction of MSCs (0.001–0.01% of the cells in bone marrow).12 Furthermore, bone marrow aspiration to harvest MSCs in human bones is a painful procedure and the slower proliferation rate of BM-MSCs is a clinical limitation.13 In comparison with BM-MSCs, adipose-derived MSCs (AD-MSCs) are relatively easy to collect and can produce up to 500 times the cell population of BM-MSCs.14 AD-MSCs showed a greater ability to regenerate damaged cartilage and bone tissues with increased immunosuppressive ability.14,15 Umbilical cord blood-derived MSCs (UC-MSCs) proliferate faster than BM-MSCs and are resistant to significant cellular aging.11
MSCs have been investigated and gained worldwide attention as potential therapeutic candidates for incurable diseases such as arthritis, spinal cord injury, and cardiac disease.3,16–23 In particular, the inherent tropism of MSCs to inflammatory sites has been thoroughly studied.24 This inherent tropism, also known as homing ability, originates from the recognition of various chemokine sources in inflamed tissues, where profiled chemokines are continuously secreted and the MSCs migrate to the chemokines in a concentration-dependent manner.24 Rheumatoid arthritis (RA) is a representative inflammatory disease that primarily causes inflammation in the joints, and this long-term autoimmune disorder causes worsening pain and stiffness following rest. RA affects approximately 24.5 million people as of 2015, but only symptomatic treatments such as pain medications, steroids, and nonsteroidal anti-inflammatory drugs (NSAIDs), or slow-acting drugs that inhibit the rapid progression of RA, such as disease-modifying antirheumatic drugs (DMARDs) are currently available. However, RA drugs have adverse side effects, including hepatitis, osteoporosis, skeletal fracture, steroid-induced arthroplasty, Cushing’s syndrome, gastrointestinal (GI) intolerance, and bleeding.25–27 Thus, MSCs are rapidly emerging as the next generation of arthritis treatment because they not only recognize and migrate toward chemokines secreted in the inflamed joints but also regulate inflammatory progress and repair damaged cells.28
However, MSCs are associated with many challenges that need to be overcome before they can be used in clinical settings.29–31 One of the main challenges is the selective accumulation of systemically administered MSCs in the lungs and liver when they are administered intravenously, leading to insufficient concentrations of MSCs in the target tissues.32,33 In addition, most of the administered MSCs are typically initially captured by macrophages in the lungs, liver, and spleen.32–34 Importantly, the viability and migration ability of MSCs injected in vivo differed from results previously reported as favorable therapeutic effects and migration efficiency in vitro.35
To improve the delivery of MSCs, researchers have focused on chemokines, which are responsible for MSCs’ ability to move.36 The chemokine receptors are the key proteins on MSCs that recognize chemokines, and genetic engineering of MSCs to overexpress the chemokine receptor can improve the homing ability, thus enhancing their therapeutic efficacy.37 Genetic engineering is a convenient tool for modifying native or non-native genes, and several technologies for genetic engineering exist, including genome editing, gene knockdown, and replacement with various vectors.38,39 However, safety issues that prevent clinical use persist, for example, genome integration, off-target effects, and induction of immune response.40 In this regard, MSC mimicking nanoencapsulations can be an alternative strategy for maintaining the homing ability of MSCs and overcoming the current safety issues.41–43 Nanoencapsulation involves entrapping the core nanoparticles of solids or liquids within nanometer-sized capsules of secondary materials.44
MSC mimicking nanoencapsulation uses the MSC membrane fraction as the capsule and targeting molecules, that is chemokine receptors, with several types of nanoparticles, as the core.45,46 MSC mimicking nanoencapsulation consists of MSC membrane-coated nanoparticles, MSC-derived artificial ectosomes, and MSC membrane-fused liposomes. Nano drug delivery is an emerging field that has attracted significant interest due to its unique characteristics and paved the way for several unique applications that might solve many problems in medicine. In particular, the nanoscale size of nanoparticles (NPs) enhances cellular uptake and can optimize intracellular pathways due to their intrinsic physicochemical properties, and can therefore increase drug delivery to target tissues.47,48 However, the inherent targeting ability resulting from the physicochemical properties of NPs is not enough to target specific tissues or damaged tissues, and additional studies on additional ligands that can bind to surface receptors on target cells or tissues have been performed to improve the targeting ability of NPs.49 Likewise, nanoencapsulation with cell membranes with targeting molecules and encapsulation of the core NPs with cell membranes confer the targeting ability of the source cell to the NPs.50,51 Thus, MSC mimicking nanoencapsulation can mimic the superior targeting ability of MSCs and confer the advantages of each core NP. In addition, MSC mimicking nanoencapsulations have improved circulation time and camouflaging from phagocytes.52
This review discusses the mechanism of MSC migration to inflammatory sites, addresses the potential strategy for improving the tropism of MSCs using genetic engineering, and discusses the promising therapeutic agent, MSC mimicking nanoencapsulations.
MSC Homing, Migratory Ability, and Genetic Engineering
The MSC migration mechanism can be exploited for diverse clinical applications.53 The MSC migration mechanism can be divided into five stages: rolling by selectin, activation of MSCs by chemokines, stopping cell rolling by integrin, transcellular migration, and migration to the damaged site (Figure 1).54,55 Chemokines are secreted naturally by various cells such as tumor cells, stromal cells, and inflammatory cells, maintaining high chemokine concentrations in target cells at the target tissue and inducing signal cascades.56–58 Likewise, MSCs express a variety of chemokine receptors, allowing them to migrate and be used as new targeting vectors.59–61 MSC migration accelerates depending on the concentration of chemokines, which are the most important factors in the stem cell homing mechanism.62,63 Chemokines consist of various cytokine subfamilies that are closely associated with the migration of immune cells. Chemokines are divided into four classes based on the locations of the two cysteine (C) residues: CC-chemokines, CXC-chemokine, C-chemokine, and CX3 Chemokine.64,65 Each chemokine binds to various MSC receptors and the binding induces a chemokine signaling cascade (Table 1).56,66
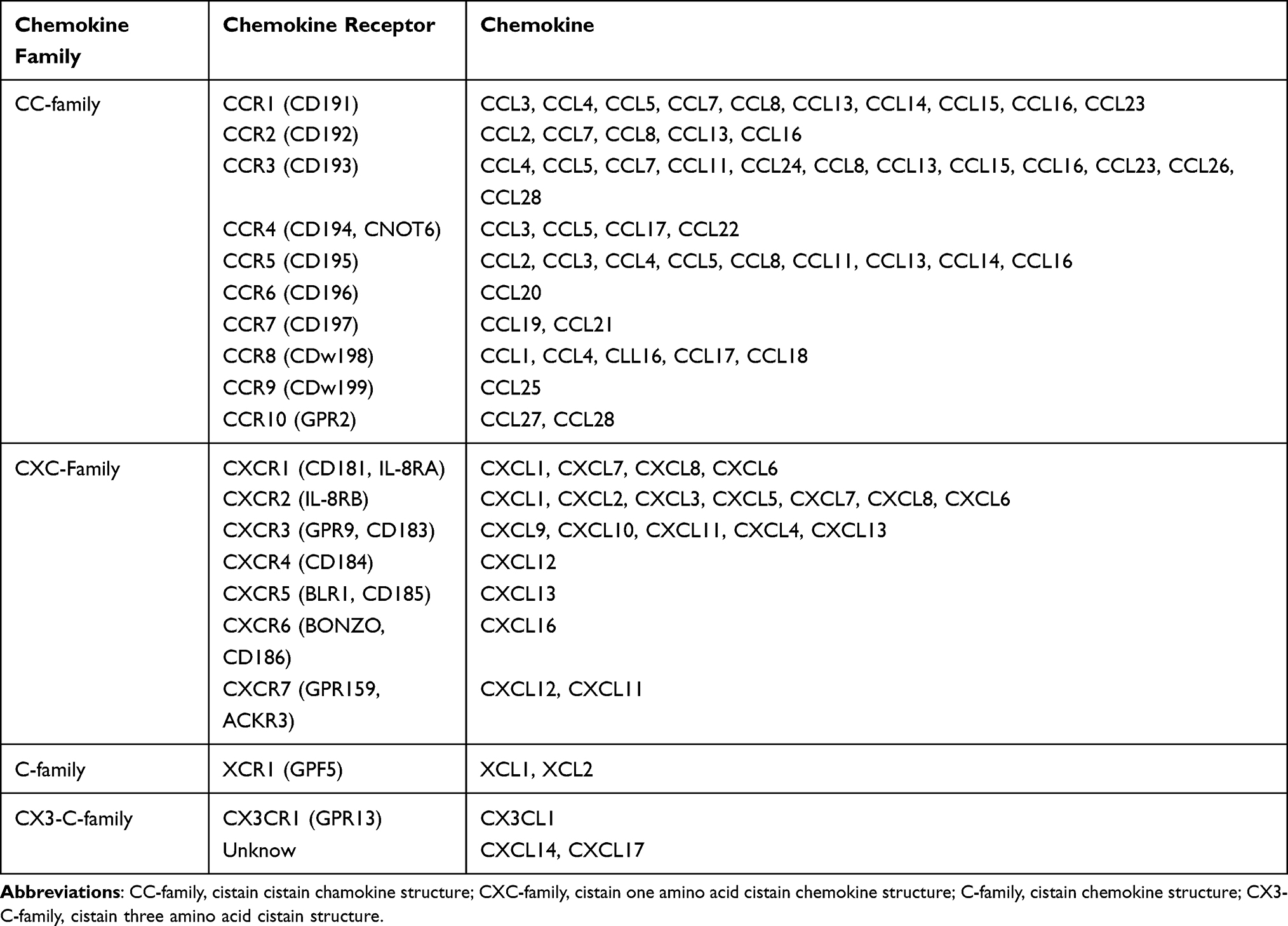 |
Table 1 Chemokine and Chemokine Receptors for Different Chemokine Families |
 |
Figure 1 Representation of stem cell homing mechanism. |
Mechanisms of MSC Homing
The mechanisms underlying MSC and leukocyte migration are similar in terms of their migratory dynamics.55 P-selectin glycoprotein ligand-1 (PSGL-1) and E-selectin ligand-1 (ESL-1) are major proteins involved in leukocyte migration that interact with P-selectin and E-selectin present in vascular endothelial cells. However, these promoters are not present in MSCs (Figure 2).53,67
 |
Figure 2 Differences in adhesion protein molecules between leukocytes and mesenchymal stem cells during rolling stages and rolling arrest stage of MSC. (A) The rolling stage of leukocytes starts with adhesion to endothelium with ESL-1 and PSGL-1 on leukocytes. (B) The rolling stage of MSC starts with the adhesion to endothelium with Galectin-1 and CD24 on MSC, and the rolling arrest stage was caused by chemokines that were encountered in the rolling stage and VLA-4 with a high affinity for VACM present in endothelial cells. Abbreviations: ESL-1, E-selectin ligand-1; PSGL-1, P-selectin glycoprotein ligand-1 VLA-4, very late antigen-4; VCAM, vascular cell adhesion molecule-1. |
The initial rolling is facilitated by selectins expressed on the surface of endothelial cells. Various glycoproteins on the surface of MSCs can bind to the selectins and continue the rolling process.68 However, the mechanism of binding of the glycoprotein on MSCs to the selectins is still unclear.69,70 P-selectins and E-selectins, major cell-cell adhesion molecules expressed by endothelial cells, adhere to migrated cells adjacent to endothelial cells and can trigger the rolling process.71 For leukocyte migration, P-selectin glycoprotein ligand-1 (PSGL-1) and E-selectin ligand-1 (ESL-1) expressed on the membranes of leukocytes interact with P-selectins and E-selectins on the endothelial cells, initiating the process.72,73 As already mentioned, MSCs express neither PSGL-1 nor ESL-1. Instead, they express galectin-1 and CD24 on their surfaces, and these bind to E-selectin or P-selectin (Figure 2).74–76
In the migratory activation step, MSC receptors are activated in response to inflammatory cytokines, including CXCL12, CXCL8, CXCL4, CCL2, and CCL7.77 The corresponding activation of chemokine receptors of MSCs in response to inflammatory cytokines results in an accumulation of MSCs.58,78 For example, inflamed tissues release inflammatory cytokines,79 and specifically, fibroblasts release CXCL12, which further induces the accumulation of MSCs through ligand–receptor interaction after exposure to hypoxia and cytokine-rich environments in the rat model of inflammation.79–82 Previous studies have reported that overexpressing CXCR4, which is a receptor to recognize CXCL12, in MSCs improves the homing ability of MSCs toward inflamed sites.83,84 In short, cytokines are significantly involved in the homing mechanism of MSCs.53
The rolling arrest stage is facilitated by integrin α4β1 (VLA-4) on MSC.85 VLA-4 is expressed by MSCs which are first activated by CXCL-12 and TNF-α chemokines, and activated VLA-4 binds to VCAM-1 expressed on endothelial cells to stop the rotational movement (Figure 2).86,87
Karp et al categorized the migration of MSCs as either “systemic homing” or “non-systemic homing.” Systemic homing refers to the process of migration through blood vessels and then across the vascular endothelium near the inflamed site.67,88 The process of migration after passing through the vessels or local injection is called non-systemic homing. In non-systemic migration, stem cells migrate through a chemokine concentration gradient (Figure 3).89 MSCs secrete matrix metalloproteinases (MMPs) during migration. The mechanism underlying MSC migration is currently undefined but MSC migration can be advanced by remodeling the matrix through the secretion of various enzymes.90–93 The migration of MSCs to the damaged area is induced by chemokines released from the injured site, such as IL-8, TNF-α, insulin-like growth factor (IGF-1), and platelet-derived growth factors (PDGF).94–96 MSCs migrate toward the damaged area following a chemokine concentration gradient.87
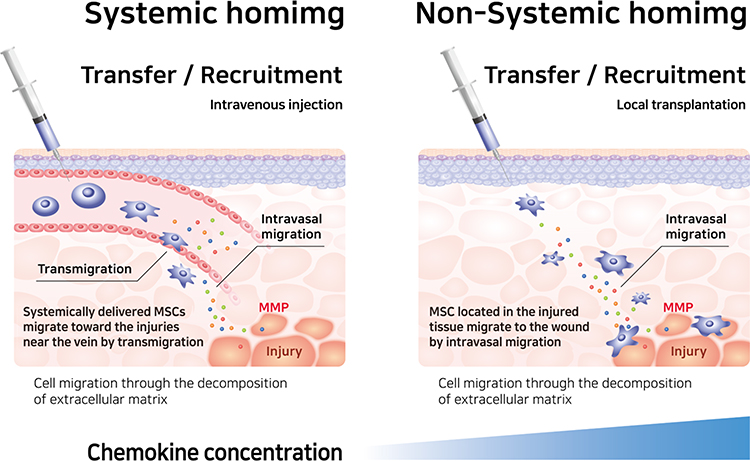 |
Figure 3 Differences between systemic and non-systemic homing mechanisms. Both systemic and non-systemic homing to the extracellular matrix and stem cells to their destination, MSCs secrete MMPs and remodel the extracellular matrix. Abbreviation: MMP, matrix metalloproteinase. |
Migratory Ability of MSCs to Arthritis
RA is a chronic inflammatory autoimmune disease characterized by distinct painful stiff joints and movement disorders.97 RA affects approximately 1% of the world’s population.98 RA is primarily induced by macrophages, which are involved in the innate immune response and are also involved in adaptive immune responses, together with B cells and T cells.99 Inflammatory diseases are caused by high levels of inflammatory cytokines and a hypoxic low-pH environment in the joints.100,101 Fibroblast-like synoviocytes (FLSs) and accumulated macrophages and neutrophils in the synovium of inflamed joints also express various chemokines.102,103 Chemokines from inflammatory reactions can induce migration of white blood cells and stem cells, which are involved in angiogenesis around joints.101,104,105 More than 50 chemokines are present in the rheumatoid synovial membrane (Table 2). Of the chemokines in the synovium, CXCL12, MIP1-a, CXCL8, and PDGF are the main ones that attract MSCs.106 In the RA environment, CXCL12, a ligand for CXCR4 on MSCs, had 10.71 times higher levels of chemokines than in the normal synovial cell environment. MIP-1a, a chemokine that gathers inflammatory cells, is a ligand for CCR1, which is normally expressed on MSC.107,108 CXCL8 is a ligand for CXCR1 and CXCR2 on MSCs and induces the migration of neutrophils and macrophages, leading to ROS in synovial cells.59 PDGF is a regulatory peptide that is upregulated in the synovial tissue of RA patients.109 PDGF induces greater MSC migration than CXCL12.110 Importantly, stem cells not only have the homing ability to inflamed joints but also have potential as cell therapy with the anti-apoptotic, anti-catabolic, and anti-fibrotic effect of MSC.111 In preclinical trials, MSC treatment has been extensively investigated in collagen-induced arthritis (CIA), a common autoimmune animal model used to study RA. In the RA model, MSCs downregulated inflammatory cytokines such as IFN-γ, TNF-α, IL-4, IL-12, and IL1β, and antibodies against collagen, while anti-inflammatory cytokines, such as tumor necrosis factor-inducible gene 6 protein (TSG-6), prostaglandin E2 (PGE2), transforming growth factor-beta (TGF-β), IL-10, and IL-6, were upregulated.112–116
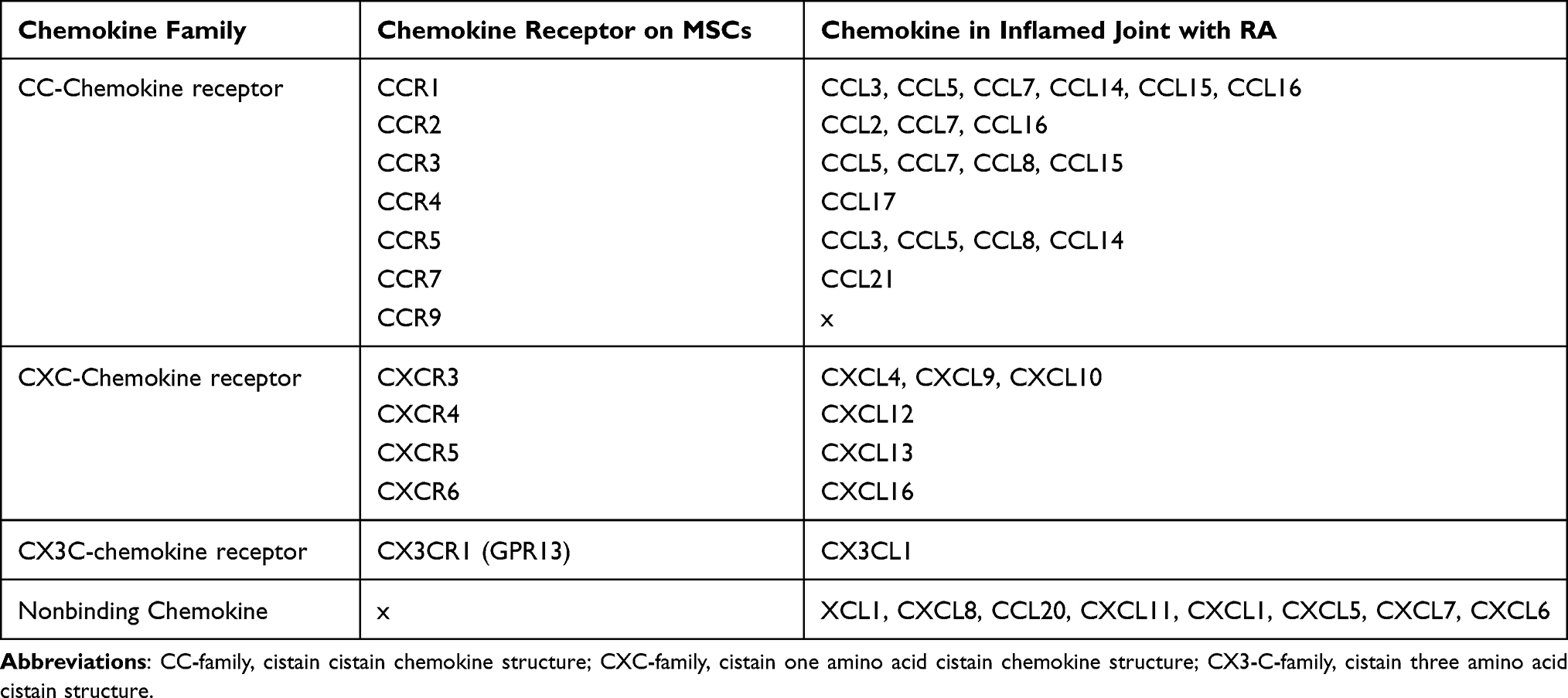 |
Table 2 Rheumatoid Arthritis (RA) Chemokines Present in the Pathological Environment and Chemokine Receptors Present in Mesenchymal Stem Cells |
Genetically Engineered MSCs Targeting Arthritis
Genetic engineering can improve the therapeutic potential of MSCs, including long-term survival, angiogenesis, differentiation into specific lineages, anti- and pro-inflammatory activity, and migratory properties (Figure 4).117,118 Although MSCs already have an intrinsic homing ability, the targeting ability of MSCs and their derivatives, such as membrane vesicles, which are utilized to produce MSC mimicking nanoencapsulation, can be enhanced.118 The therapeutic potential of MSCs can be magnified by reprogramming MSCs via upregulation or downregulation of their native genes, resulting in controlled production of the target protein, or by introducing foreign genes that enable MSCs to express native or non-native products, for example, non-native soluble tumor necrosis factor (TNF) receptor 2 can inhibit TNF-alpha signaling in RA therapies.28
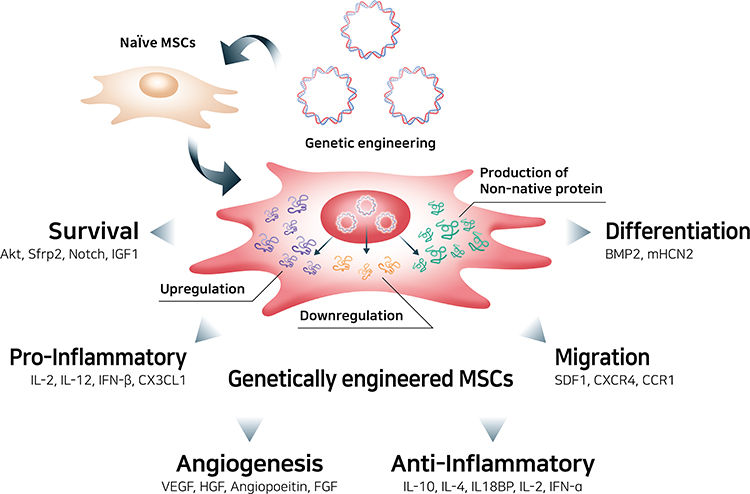 |
Figure 4 Genetic engineering of mesenchymal stem cells to enhance therapeutic efficacy. Abbreviations: Sfrp2, secreted frizzled-related protein 2; IGF1, insulin-like growth factor 1; IL-2, interleukin-2; IL-12, interleukin-12; IFN-β, interferon-beta; CX3CL1, C-X3-C motif chemokine ligand 1; VEGF, vascular endothelial growth factor; HGF, human growth factor; FGF, fibroblast growth factor; IL-10, interleukin-10; IL-4, interleukin-4; IL18BP, interleukin-18-binding protein; IFN-α, interferon-alpha; SDF1, stromal cell-derived factor 1; CXCR4, C-X-C motif chemokine receptor 4; CCR1, C-C motif chemokine receptor 1; BMP2, bone morphogenetic protein 2; mHCN2, mouse hyperpolarization-activated cyclic nucleotide-gated. |
MSCs can be genetically engineered using different techniques, including by introducing particular genes into the nucleus of MSCs or editing the genome of MSCs (Figure 5).119 Foreign genes can be transferred into MSCs using liposomes (chemical method), electroporation (physical method), or viral delivery (biological method). Cationic liposomes, also known as lipoplexes, can stably compact negatively charged nucleic acids, leading to the formation of nanomeric vesicular structure.120 Cationic liposomes are commonly produced with a combination of a cationic lipid such as DOTAP, DOTMA, DOGS, DOSPA, and neutral lipids, such as DOPE and cholesterol.121 These liposomes are stable enough to protect their bound nucleic acids from degradation and are competent to enter cells via endocytosis.120 Electroporation briefly creates holes in the cell membrane using an electric field of 10–20 kV/cm, and the holes are then rapidly closed by the cell’s membrane repair mechanism.122 Even though the electric shock induces irreversible cell damage and non-specific transport into the cytoplasm leads to cell death, electroporation ensures successful gene delivery regardless of the target cell or organism. Viral vectors, which are derived from adenovirus, adeno-associated virus (AAV), or lentivirus (LV), have been used to introduce specific genes into MSCs. Recombinant lentiviral vectors are the most widely used systems due to their high tropism to dividing and non-dividing cells, transduction efficiency, and stable expression of transgenes in MSCs, but the random genome integration of transgenes can be an obstacle in clinical applications.123 Adenovirus and AAV systems are appropriate alternative strategies because currently available strains do not have broad genome integration and a strong immune response, unlike LV, thus increasing success and safety in clinical trials.124 As a representative, the Oxford-AstraZeneca COVID-19 vaccine, which has been authorized in 71 countries as a vaccine for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which spread globally and led to the current pandemic, transfers the spike protein gene using an adenovirus-based viral vector.125 Furthermore, there are two AAV-based gene therapies: Luxturna for rare inherited retinal dystrophy and Zolgensma for spinal muscular atrophy.126
 |
Figure 5 Genetic engineering techniques used in the production of bioengineered mesenchymal stem cells. |
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 were recently used for genome editing and modification because of their simpler design and higher efficiency for genome editing, however, there are safety issues such as off-target effects that induce mutations at sites other than the intended target site.127 The foreign gene is then commonly transferred into non-integrating forms such as plasmid DNA and messenger RNA (mRNA).128
The gene expression machinery can also be manipulated at the cytoplasmic level through RNA interference (RNAi) technology, inhibition of gene expression, or translation using neutralizing targeted mRNA molecules with sequence-specific small RNA molecules such as small interfering RNA (siRNA) or microRNA (miRNA).129 These small RNAs can form enzyme complexes that degrade mRNA molecules and thus decrease their activity by inhibiting translation. Moreover, the pre-transcriptional silencing mechanism of RNAi can induce DNA methylation at genomic positions complementary to siRNA or miRNA with enzyme complexes.
CXC chemokine receptor 4 (CXCR4) is one of the most potent chemokine receptors that is genetically engineered to enhance the migratory properties of MSCs.130 CXCR4 is a chemokine receptor specific for stromal-derived factor-1 (SDF-1), also known as CXC motif chemokine 12 (CXCL12), which is produced by damaged tissues, such as the area of inflammatory bone destruction.131 Several studies on engineering MSCs to increase the expression of the CXCR4 gene have reported a higher density of the CXCR4 receptor on their outer cell membrane and effectively increased the migration of MSCs toward SDF-1.83,132,133 CXC chemokine receptor 7 (CXCR7) also had a high affinity for SDF-1, thus the SDF-1/CXCR7 signaling axis was used to engineer the MSCs.134 CXCR7-overexpressing MSCs in a cerebral ischemia-reperfusion rat hippocampus model promoted migration based on an SDF-1 gradient, cooperating with the SDF-1/CXCR4 signaling axis (Figure 6).37
 |
Figure 6 Engineered mesenchymal stem cells with enhanced migratory abilities. Abbreviations: CXCR4, C-X-C motif chemokine receptor 4; CXCR7, C-X-C motif chemokine receptor 7; SDF1, stromal cell-derived factor 1; CXCR1, C-X-C motif chemokine receptor 1; IL-8, interleukin-8; Aqp1, aquaporin 1; FAK, focal adhesion kinase. |
CXC chemokine receptor 1 (CXCR1) enhances MSC migratory properties.59 CXCR1 is a receptor for IL-8, which is the primary cytokine involved in the recruitment of neutrophils to the site of damage or infection.135 In particular, the IL-8/CXCR1 axis is a key factor for the migration of MSCs toward human glioma cell lines, such as U-87 MG, LN18, U138, and U251, and CXCR1-overexpressing MSCs showed a superior capacity to migrate toward glioma cells and tumors in mice bearing intracranial human gliomas.136
The migratory properties of MSCs were also controlled via aquaporin-1 (Aqp1), which is a water channel molecule that transports water across the cell membrane and regulates endothelial cell migration.137 Aqp1-overexpressing MSCs showed enhanced migration to fracture gap of a rat fracture model with upregulated focal adhesion kinase (FAK) and β-catenin, which are important regulators of cell migration.138
Nur77, also known as nerve growth factor IB or NR4A1, and nuclear receptor-related 1 (Nurr1), can play a role in improving the migratory capabilities of MSCs.139,140 The migrating MSCs expressed higher levels of Nur77 and Nurr1 than the non-migrating MSCs, and overexpression of these two nuclear receptors functioning as transcription factors enhanced the migration of MSCs toward SDF-1. The migration of cells is closely related to the cell cycle, and normally, cells in the late S or G2/M phase do not migrate.141 The overexpression of Nur77 and Nurr1 increased the proportion of MSCs in the G0/G1-phase similar to the results of migrating MSCs had more cells in the G1-phase.
MSC Mimicking Nanoencapsulation Targeting Arthritis
MSC mimicking nanoencapsulations are nanoparticles combined with MSC membrane vesicles and these NPs have the greatest advantages as drug delivery systems due to the sustained homing ability of MSCs as well as the advantages of NPs. Particles sized 10–150 nm have great advantages in drug delivery systems because they can pass more freely through the cell membrane by the interaction with biomolecules, such as clathrin and caveolin, to facilitate uptake across the cell membrane compared with micron-sized materials.142,143 Various materials have been used to formulate NPs, including silica, polymers, metals, and lipids.144,145 NPs have an inherent ability, called “passive targeting,” to accumulate at specific sites based on their physicochemical properties such as size, surface charge, surface hydrophilicity, and geometry.146–148 However, physicochemical properties are not enough to target specific tissues or damaged tissues, and thus “active targeting” is a clinically approved strategy involving the addition of ligands that can bind to surface receptors on target cells or tissues.149,150 MSC mimicking nanoencapsulation uses natural or genetically engineered MSC membranes to coat synthetic NPs, producing artificial ectosomes and fusing them with liposomes to increase their targeting ability (Figure 7).151 Especially, MSCs have been studied for targeting inflammation and regenerative drugs, and the mechanism and efficacy of migration toward inflamed tissues have been actively investigated.152 MSC mimicking nanoencapsulation can mimic the well-known migration ability of MSCs and can be equally utilized without safety issues from the direct application of using MSCs. Furthermore, cell membrane encapsulations have a wide range of functions, including prolonged blood circulation time and increased active targeting efficacy from the source cells.153,154 MSC mimicking encapsulations enter recipient cells using multiple pathways.155 MSC mimicking encapsulations can fuse directly with the plasma membrane and can also be taken up through phagocytosis, micropinocytosis, and endocytosis mediated by caveolin or clathrin.156 MSC mimicking encapsulations can be internalized in a highly cell type-specific manner that depends on the recognition of membrane surface molecules by the cell or tissue.157 For example, endothelial colony-forming cell (ECFC)-derived exosomes were shown CXCR4/SDF-1α interaction and enhanced delivery toward the ischemic kidney, and Tspan8-alpha4 complex on lymph node stroma derived extracellular vesicles induced selective uptake by endothelial cells or pancreatic cells with CD54, serving as a major ligand.158,159 Therefore, different source cells may contain protein signals that serve as ligands for other cells, and these receptor–ligand interactions maximized targeted delivery of NPs.160 This natural mechanism inspired the application of MSC membranes to confer active targeting to NPs.
 |
Figure 7 Mesenchymal stem cell mimicking nanoencapsulation. |
MSC Membrane-Coated Nanoparticles Targeting Arthritis
Cell membrane-coated NPs (CMCNPs) are biomimetic strategies developed to mimic the properties of cell membranes derived from natural cells such as erythrocytes, white blood cells, cancer cells, stem cells, platelets, or bacterial cells with an NP core.161 Core NPs made of polymer, silica, and metal have been evaluated in attempts to overcome the limitations of conventional drug delivery systems but there are also issues of toxicity and reduced biocompatibility associated with the surface properties of NPs.162,163 Therefore, only a small number of NPs have been approved for medical application by the FDA.164 Coating with cell membrane can enhance the biocompatibility of NPs by improving immune evasion, enhancing circulation time, reducing RES clearance, preventing serum protein adsorption by mimicking cell glycocalyx, which are chemical determinants of “self” at the surfaces of cells.151,165 Furthermore, the migratory properties of MSCs can also be transferred to NPs by coating them with the cell membrane.45 Coating NPs with MSC membranes not only enhances biocompatibility but also maximizes the therapeutic effect of NPs by mimicking the targeting ability of MSCs.166 Cell membrane-coated NPs are prepared in three steps: extraction of cell membrane vesicles from the source cells, synthesis of the core NPs, and fusion of the membrane vesicles and core NPs to produce cell membrane-coated NPs (Figure 8).167 Cell membrane vesicles, including extracellular vesicles (EVs), can be harvested through cell lysis, mechanical disruption, and centrifugation to isolate, purify the cell membrane vesicles, and remove intracellular components.168 All the processes must be conducted under cold conditions, with protease inhibitors to minimize the denaturation of integral membrane proteins. Cell lysis, which is classically performed using mechanical lysis, including homogenization, sonication, or extrusion followed by differential velocity centrifugation, is necessary to remove intracellular components. Cytochalasin B (CB), a drug that affects cytoskeleton–membrane interactions, induces secretion of membrane vesicles from source cells and has been used to extract the cell membrane.169 The membrane functions of the source cells are preserved in CB-induced vesicles, forming biologically active surface receptors and ion pumps.170 Furthermore, CB-induced vesicles can encapsulate drugs and NPs successfully, and the vesicles can be harvested by centrifugation without a purification step to remove nuclei and cytoplasm.171 Clinically translatable membrane vesicles require scalable production of high volumes of homogeneous vesicles within a short period. Although mechanical methods (eg, shear stress, ultrasonication, or extrusion) are utilized, CB-induced vesicles have shown potential for generating membrane encapsulation for nano-vectors.168 The advantages of CB-induced vesicles versus other methods are compared in Table 3.
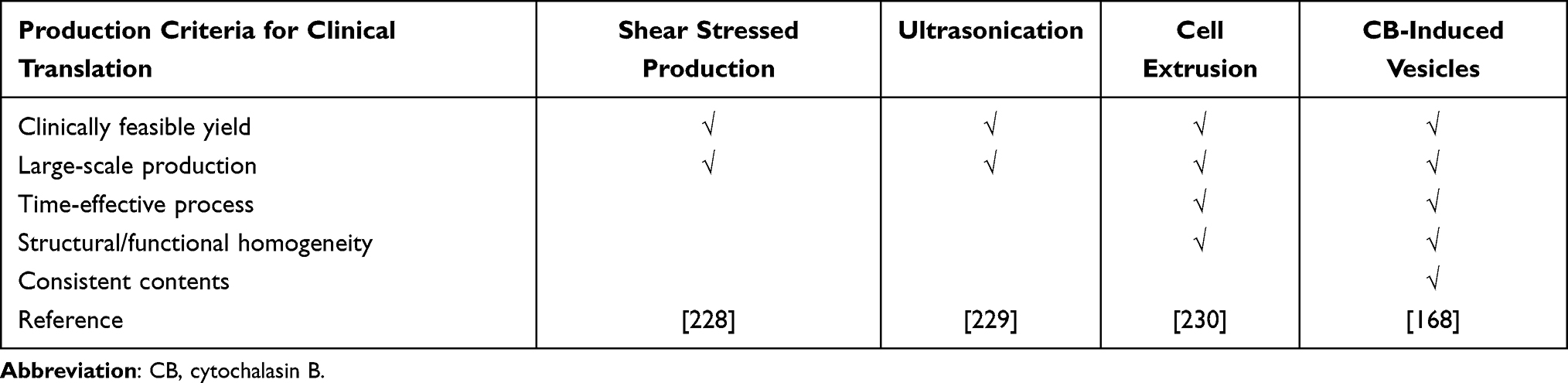 |
Table 3 Comparison of Membrane Vesicle Production Methods |
 |
Figure 8 MSC membrane-coated nanoparticles. Abbreviations: EVs, extracellular vesicles; NPs, nanoparticles. |
After extracting cell membrane vesicles, synthesized core NPs are coated with cell membranes, including surface proteins.172 Polymer NPs and inorganic NPs are adopted as materials for the core NPs of CMCNPs, and generally, polylactic-co-glycolic acid (PLGA), polylactic acid (PLA), chitosan, and gelatin are used. PLGA has been approved by FDA is the most common polymer of NPs.173 Biodegradable polymer NPs have gained considerable attention in nanomedicine due to their biocompatibility, nontoxic properties, and the ability to modify their surface as a drug carrier.174 Inorganic NPs are composed of gold, iron, copper, and silicon, which have hydrophilic, biocompatible, and highly stable properties compared with organic materials.175 Furthermore, some photosensitive inorganic NPs have the potential for use in photothermal therapy (PTT) and photodynamic therapy (PDT).176 The fusion of cell membrane vesicles and core NPs is primarily achieved via extrusion or sonication.165 Cell membrane coating of NPs using mechanical extrusion is based on a different-sized porous membrane where core NPs and vesicles are forced to generate vesicle-particle fusion.177 Ultrasonic waves are applied to induce the fusion of vesicles and NPs. However, ultrasonic frequencies need to be optimized to improve fusion efficiency and minimize drug loss and protein degradation.178
CMCNPs have extensively employed to target and treat cancer using the membranes obtained from red blood cell (RBC), platelet and cancer cell.165 In addition, membrane from MSC also utilized to target tumor and ischemia with various types of core NPs, such as MSC membrane coated PLGA NPs targeting liver tumors, MSC membrane coated gelatin nanogels targeting HeLa cell, MSC membrane coated silica NPs targeting HeLa cell, MSC membrane coated PLGA NPs targeting hindlimb ischemia, and MSC membrane coated iron oxide NPs for targeting the ischemic brain.179–183 However, there are few studies on CMCNPs using stem cells for the treatment of arthritis. Increased targeting ability to arthritis was introduced using MSC-derived EVs and NPs.184,185 MSC membrane-coated NPs are proming strategy for clearing raised concerns from direct use of MSC (with or without NPs) in terms of toxicity, reduced biocompatibility, and poor targeting ability of NPs for the treatment of arthritis.
MSC Derived Exosomes and Artificial Ectosomes Targeting Arthritis
Exosomes are natural NPs that range in size from 40 nm to 120 nm and are derived from the multivesicular body (MVB), which is an endosome defined by intraluminal vesicles (ILVs) that bud inward into the endosomal lumen, fuse with the cell surface, and are then released as exosomes.186 Because of their ability to express receptors on their surfaces, MSC-derived exosomes are also considered potential candidates for targeting.187 Exosomes are commonly referred to as intracellular communication molecules that transfer various compounds through physiological mechanisms such as immune response, neural communication, and antigen presentation in diseases such as cancer, cardiovascular disease, diabetes, and inflammation.188
However, there are several limitations to the application of exosomes as targeted therapeutic carriers. First, the limited reproducibility of exosomes is a major challenge. In this field, the standardized techniques for isolation and purification of exosomes are lacking, and conventional methods containing multi-step ultracentrifugation often lead to contamination of other types of EVs. Furthermore, exosomes extracted from cell cultures can vary and display inconsistent properties even when the same type of donor cells were used.189 Second, precise characterization studies of exosomes are needed. Unknown properties of exosomes can hinder therapeutic efficiencies, for example, when using exosomes as cancer therapeutics, the use of cancer cell-derived exosomes should be avoided because cancer cell-derived exosomes may contain oncogenic factors that may contribute to cancer progression.190 Finally, cost-effective methods for the large-scale production of exosomes are needed for clinical application. The yield of exosomes is much lower than EVs. Depending on the exosome secretion capacity of donor cells, the yield of exosomes is restricted, and large-scale cell culture technology for the production of exosomes is high difficulty and costly and isolation of exosomes is the time-consuming and low-efficient method.156
Ectosome is an EV generated by outward budding from the plasma membrane followed by pinching off and release to the extracellular parts. Recently, artificially produced ectosome utilized as an alternative to exosomes in targeted therapeutics due to stable productivity regardless of cell type compared with conventional exosome. Artificial ectosomes, containing modified cargo and targeting molecules have recently been introduced for specific purposes (Figure 9).191,192 Artificial ectosomes are typically prepared by breaking bigger cells or cell membrane fractions into smaller ectosomes, similar size to natural exosomes, containing modified cargo such as RNA molecules, which control specific genes, and chemical drugs such as anticancer drugs.193 Naturally secreted exosomes in conditioned media from modified source cells can be harvested by differential ultracentrifugation, density gradients, precipitation, filtration, and size exclusion chromatography for exosome separation.194 Even though there are several commercial kits for isolating exosomes simply and easily, challenges in compliant scalable production on a large scale, including purity, homogeneity, and reproducibility, have made it difficult to use naturally secreted exosomes in clinical settings.195 Therefore, artificially produced ectosomes are appropriate for use in clinical applications, with novel production methods that can meet clinical production criteria. Production of artificially produced ectosomes begins by breaking the cell membrane fraction of cultured cells and then using them to produce cell membrane vesicles to form ectosomes. As mentioned above, cell membrane vesicles are extracted from source cells in several ways, and cell membrane vesicles are extracted through polycarbonate membrane filters to reduce the mean size to a size similar to that of natural exosomes.196 Furthermore, specific microfluidic devices mounted on microblades (fabricated in silicon nitride) enable direct slicing of living cells as they flow through the hydrophilic microchannels of the device.197 The sliced cell fraction reassembles and forms ectosomes. There are several strategies for loading exogenous therapeutic cargos such as drugs, DNA, RNA, lipids, metabolites, and proteins, into exosomes or artificial ectosomes in vitro: electroporation, incubation for passive loading of cargo or active loading with membrane permeabilizer, freeze and thaw cycles, sonication, and extrusion.198 In addition, protein or RNA molecules can be loaded by co-expressing them in source cells via bio-engineering, and proteins designed to interact with the protein inside the cell membrane can be loaded actively into exosomes or artificial ectosomes.157 Targeting molecules at the surface of exosomes or artificial ectosomes can also be engineered in a manner similar to the genetic engineering of MSCs.
 |
Figure 9 Mesenchymal stem cell-derived exosomes and artificial ectosomes. (A) Wound healing effect of MSC-derived exosomes and artificial ectosomes,231 (B) treatment of organ injuries by MSC-derived exosomes and artificial ectosomes,42,232–234 (C) anti-cancer activity of MSC-derived exosomes and artificial ectosomes.200,202,235 |
Most of the exosomes derived from MSCs for drug delivery have employed miRNAs or siRNAs, inhibiting translation of specific mRNA, with anticancer activity, for example, miR-146b, miR-122, and miR-379, which are used for cancer targeting by membrane surface molecules on MSC-derived exosomes.199–201 Drugs such as doxorubicin, paclitaxel, and curcumin were also loaded into MSC-derived exosomes to target cancer.202–204 However, artificial ectosomes derived from MSCs as arthritis therapeutics remains largely unexplored area, while EVs, mixtures of natural ectosomes and exosomes, derived from MSCs have studied in the treatment of arthritis.184 Artificial ectosomes with intrinsic tropism from MSCs plus additional targeting ability with engineering increase the chances of ectosomes reaching target tissues with ligand–receptor interactions before being taken up by macrophages.205 Eventually, this will decrease off-target binding and side effects, leading to lower therapeutic dosages while maintaining therapeutic efficacy.206,207
MSC Membrane-Fused Liposomes Targeting Arthritis
Liposomes are spherical vesicles that are artificially synthesized through the hydration of dry phospholipids.208 The clinically available liposome is a lipid bilayer surrounding a hollow core with a diameter of 50–150 nm. Therapeutic molecules, such as anticancer drugs (doxorubicin and daunorubicin citrate) or nucleic acids, can be loaded into this hollow core for delivery.209 Due to their amphipathic nature, liposomes can load both hydrophilic (polar) molecules in an aqueous interior and hydrophobic (nonpolar) molecules in the lipid membrane. They are well-established biomedical applications and are the most common nanostructures used in advanced drug delivery.210 Furthermore, liposomes have several advantages, including versatile structure, biocompatibility, low toxicity, non-immunogenicity, biodegradability, and synergy with drugs: targeted drug delivery, reduction of the toxic effect of drugs, protection against drug degradation, and enhanced circulation half-life.211 Moreover, surfaces can be modified by either coating them with a functionalized polymer or PEG chains to improve targeted delivery and increase their circulation time in biological systems.212 Liposomes have been investigated for use in a wide variety of therapeutic applications, including cancer diagnostics and therapy, vaccines, brain-targeted drug delivery, and anti-microbial therapy. A new approach was recently proposed for providing targeting features to liposomes by fusing them with cell membrane vesicles, generating molecules called membrane-fused liposomes (Figure 10).213 Cell membrane vesicles retain the surface membrane molecules from source cells, which are responsible for efficient tissue targeting and cellular uptake by target cells.214 However, the immunogenicity of cell membrane vesicles leads to their rapid clearance by macrophages in the body and their low drug loading efficiencies present challenges for their use as drug delivery systems.156 However, membrane-fused liposomes have advantages of stability, long half-life in circulation, and low immunogenicity due to the liposome, and the targeting feature of cell membrane vesicles is completely transferred to the liposome.215 Furthermore, the encapsulation efficiencies of doxorubicin were similar when liposomes and membrane-fused liposomes were used, indicating that the relatively high drug encapsulation capacity of liposomes was maintained during the fusion process.216 Combining membrane-fused liposomes with macrophage-derived membrane vesicles showed differential targeting and cytotoxicity against normal and cancerous cells.217 Although only a few studies have been conducted, these results corroborate that membrane-fused liposomes are a potentially promising future drug delivery system with increased targeting ability. MSCs show intrinsic tropism toward arthritis, and further engineering and modification to enhance their targeting ability make them attractive candidates for the development of drug delivery systems. Fusing MSC exosomes with liposomes, taking advantage of both membrane vesicles and liposomes, is a promising technique for future drug delivery systems.
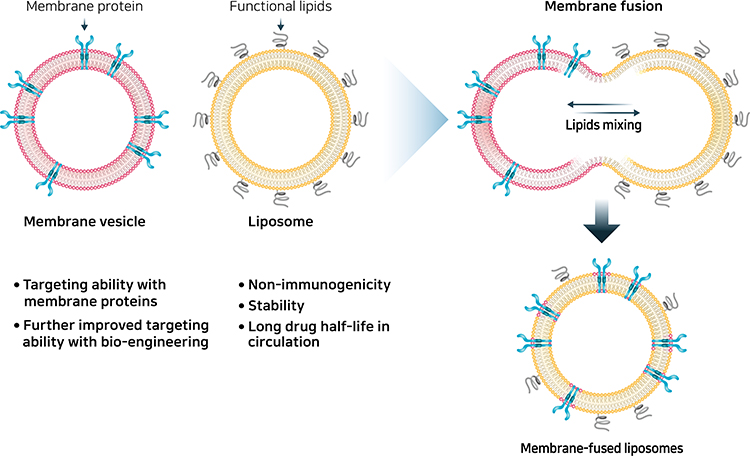 |
Figure 10 Mesenchymal stem cell membrane-fused liposomes. |
Discussion and Conclusion
MSCs have great potential as targeted therapies due to their greater ability to home to targeted pathophysiological sites. The intrinsic ability to home to wounds or to the tumor microenvironment secreting inflammatory mediators make MSCs and their derivatives targeting strategies for cancer and inflammatory disease.218,219 Contrary to the well-known homing mechanisms of various blood cells, it is still not clear how homing occurs in MSCs. So far, the mechanism of MSC tethering, which connects long, thin cell membrane cylinders called tethers to the adherent area for migration, has not been clarified. Recent studies have shown that galectin-1, VCAM-1, and ICAM are associated with MSC tethering,53,220 but more research is needed to accurately elucidate the tethering mechanism of MSCs. MSC chemotaxis is well defined and there is strong evidence relating it to the homing ability of MSCs.53 Chemotaxis involves recognizing chemokines through chemokine receptors on MSCs and migrating to chemokines in a gradient-dependent manner.221 RA, a representative inflammatory disease, is associated with well-profiled chemokines such as CXCR1, CXCR4, and CXCR7, which are recognized by chemokine receptors on MSCs. In addition, damaged joints in RA continuously secrete cytokines until they are treated, giving MSCs an advantage as future therapeutic agents for RA.222 However, there are several obstacles to utilizing MSCs as RA therapeutics. In clinical settings, the functional capability of MSCs is significantly affected by the health status of the donor patient.223 MSC yield is significantly reduced in patients undergoing steroid-based treatment and the quality of MSCs is dependent on the donor’s age and environment.35 In addition, when MSCs are used clinically, cryopreservation and defrosting are necessary, but these procedures shorten the life span of MSCs.224 Therefore, NPs mimicking MSCs are an alternative strategy for overcoming the limitations of MSCs. Additionally, further engineering and modification of MSCs can enhance the therapeutic effect by changing the targeting molecules and loaded drugs. In particular, upregulation of receptors associated with chemotaxis through genetic engineering can confer the additional ability of MSCs to home to specific sites, while the increase in engraftment maximizes the therapeutic effect of MSCs.36,225
Furthermore, there are several methods that can be used to exploit the targeting ability of MSCs as drug delivery systems. MSCs mimicking nanoencapsulation, which consists of MSC membrane-coated NPs, MSC-derived artificial ectosomes, and MSC membrane-fused liposomes, can mimic the targeting ability of MSCs while retaining the advantages of NPs. MSC-membrane-coated NPs are synthesized using inorganic or polymer NPs and membranes from MSCs to coat inner nanosized structures. Because they mimic the biological characteristics of MSC membranes, MSC-membrane-coated NPs can not only escape from immune surveillance but also effectively improve targeting ability, with combined functions of the unique properties of core NPs and MSC membranes.226 Exosomes are also an appropriate candidate for use in MSC membranes, utilizing these targeting abilities. However, natural exosomes lack reproducibility and stable productivity, thus artificial ectosomes with targeting ability produced via synthetic routes can increase the local concentration of ectosomes at the targeted site, thereby reducing toxicity and side effects and maximizing therapeutic efficacy.156 MSC membrane-fused liposomes, a novel system, can also transfer the targeting molecules on the surface of MSCs to liposomes; thus, the advantages of liposomes are retained, but with targeting ability. With advancements in nanotechnology of drug delivery systems, the research in cell-mimicking nanoencapsulation will be very useful. Efficient drug delivery systems fundamentally improve the quality of life of patients with a low dose of medication, low side effects, and subsequent treatment of diseases.227 However, research on cell-mimicking nanoencapsulation is at an early stage, and several problems need to be addressed. To predict the nanotoxicity of artificially synthesized MSC mimicking nanoencapsulations, interactions between lipids and drugs, drug release mechanisms near the targeted site, in vivo compatibility, and immunological physiological studies must be conducted before clinical application.
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2019M3A9H1103690), by the Gachon University Gil Medical Center (FRD2021-03), and by the Gachon University research fund of 2020 (GGU-202008430004).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chapel A, Bertho JM, Bensidhoum M, et al. Mesenchymal stem cells home to injured tissues when co-infused with hematopoietic cells to treat a radiation-induced multi-organ failure syndrome. J Gene Med. 2003;5(12):1028–1038. doi:10.1002/jgm.452
2. Park JS, Suryaprakash S, Lao YH, Leong KW. Engineering mesenchymal stem cells for regenerative medicine and drug delivery. Methods. 2015;84:3–16. doi:10.1016/j.ymeth.2015.03.002
3. Ringe J, Burmester GR, Sittinger M. Regenerative medicine in rheumatic disease-progress in tissue engineering. Nat Rev Rheumatol. 2012;8(8):493–498. doi:10.1038/nrrheum.2012.98
4. Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6(2):230–247. doi:10.1097/00007890-196803000-00009
5. Zuk PA, Zhu M, Ashjian P, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13(12):4279–4295. doi:10.1091/mbc.e02-02-0105
6. Crisan M, Yap S, Casteilla L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–313. doi:10.1016/j.stem.2008.07.003
7. Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A. 2000;97(25):13625–13630. doi:10.1073/pnas.240309797
8. Young HE, Steele TA, Bray RA, et al. Human reserve pluripotent mesenchymal stem cells are present in the connective tissues of skeletal muscle and dermis derived from fetal, adult, and geriatric donors. Anat Rec. 2001;264(1):51–62. doi:10.1002/ar.1128
9. Campagnoli C, Roberts IA, Kumar S, Bennett PR, Bellantuono I, Fisk NM. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood. 2001;98(8):2396–2402. doi:10.1182/blood.V98.8.2396
10. Wang HS, Hung SC, Peng ST, et al. Mesenchymal stem cells in the Wharton’s jelly of the human umbilical cord. Stem Cells. 2004;22(7):1330–1337. doi:10.1634/stemcells.2004-0013
11. Heo JS, Choi Y, Kim HS, Kim HO. Comparison of molecular profiles of human mesenchymal stem cells derived from bone marrow, umbilical cord blood, placenta and adipose tissue. Int J Mol Med. 2016;37(1):115–125. doi:10.3892/ijmm.2015.2413
12. Drela K, Stanaszek L, Snioch K, et al. Bone marrow-derived from the human femoral shaft as a new source of mesenchymal stem/stromal cells: an alternative cell material for banking and clinical transplantation. Stem Cell Res Ther. 2020;11(1):262. doi:10.1186/s13287-020-01697-5
13. Li J, Wong WH, Chan S, et al. Factors affecting mesenchymal stromal cells yield from bone marrow aspiration. Chin J Cancer Res. 2011;23(1):43–48. doi:10.1007/s11670-011-0043-1
14. Melief SM, Zwaginga JJ, Fibbe WE, Roelofs H. Adipose tissue-derived multipotent stromal cells have a higher immunomodulatory capacity than their bone marrow-derived counterparts. Stem Cells Transl Med. 2013;2(6):455–463. doi:10.5966/sctm.2012-0184
15. Trivanovic D, Jaukovic A, Popovic B, et al. Mesenchymal stem cells of different origin: comparative evaluation of proliferative capacity, telomere length and pluripotency marker expression. Life Sci. 2015;141:61–73. doi:10.1016/j.lfs.2015.09.019
16. Lefevre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15(12):1414–1420. doi:10.1038/nm.2050
17. Cyranoski D. Japan’s approval of stem-cell treatment for spinal-cord injury concerns scientists. Nature. 2019;565(7741):544–545. doi:10.1038/d41586-019-00178-x
18. Cofano F, Boido M, Monticelli M, et al. Mesenchymal stem cells for spinal cord injury: current options, limitations, and future of cell therapy. Int J Mol Sci. 2019;20(11):2698. doi:10.3390/ijms20112698
19. Liau LL, Looi QH, Chia WC, Subramaniam T, Ng MH, Law JX. Treatment of spinal cord injury with mesenchymal stem cells. Cell Biosci. 2020;10:112. doi:10.1186/s13578-020-00475-3
20. Williams AR, Hare JM, Dimmeler S, Losordo D. Mesenchymal stem cells: biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ Res. 2011;109(8):923–940. doi:10.1161/CIRCRESAHA.111.243147
21. Karantalis V, Hare JM. Use of mesenchymal stem cells for therapy of cardiac disease. Circ Res. 2015;116(8):1413–1430. doi:10.1161/CIRCRESAHA.116.303614
22. Bernstein HS, Srivastava D. Stem cell therapy for cardiac disease. Pediatr Res. 2012;71(4 Pt 2):491–499. doi:10.1038/pr.2011.61
23. Guo Y, Yu Y, Hu S, Chen Y, Shen Z. The therapeutic potential of mesenchymal stem cells for cardiovascular diseases. Cell Death Dis. 2020;11(5):349. doi:10.1038/s41419-020-2542-9
24. Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15(10):730–738. doi:10.1038/gt.2008.39
25. Vos T, Allen C, Arora M, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–1602.
26. Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;(2):CD008794. doi:10.1002/14651858.CD008794.pub2
27. Majithia V, Geraci SA. Rheumatoid arthritis: diagnosis and management. Am J Med. 2007;120(11):936–939. doi:10.1016/j.amjmed.2007.04.005
28. Park N, Rim YA, Jung H, et al. Etanercept-synthesising mesenchymal stem cells efficiently ameliorate collagen-induced arthritis. Sci Rep. 2017;7:39593. doi:10.1038/srep39593
29. Herberts CA, Kwa MS, Hermsen HP. Risk factors in the development of stem cell therapy. J Transl Med. 2011;9:29. doi:10.1186/1479-5876-9-29
30. Rodriguez-Fuentes DE, Fernandez-Garza LE, Samia-Meza JA, Barrera-Barrera SA, Caplan AI, Barrera-Saldana HA. Mesenchymal stem cells current clinical applications: a systematic review. Arch Med Res. 2021;52(1):93–101. doi:10.1016/j.arcmed.2020.08.006
31. Kabat M, Bobkov I, Kumar S, Grumet M. Trends in mesenchymal stem cell clinical trials 2004–2018: is efficacy optimal in a narrow dose range? Stem Cells Transl Med. 2020;9(1):17–27. doi:10.1002/sctm.19-0202
32. Leibacher J, Henschler R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res Ther. 2016;7:7. doi:10.1186/s13287-015-0271-2
33. Zheng B, von See MP, Yu E, et al. Quantitative magnetic particle imaging monitors the transplantation, biodistribution, and clearance of stem cells in vivo. Theranostics. 2016;6(3):291–301. doi:10.7150/thno.13728
34. Gholamrezanezhad A, Mirpour S, Bagheri M, et al. In vivo tracking of 111In-oxine labeled mesenchymal stem cells following infusion in patients with advanced cirrhosis. Nucl Med Biol. 2011;38(7):961–967. doi:10.1016/j.nucmedbio.2011.03.008
35. Pittenger MF, Discher DE, Peault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med. 2019;4:22. doi:10.1038/s41536-019-0083-6
36. Marquez-Curtis LA, Janowska-Wieczorek A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res Int. 2013;2013:561098. doi:10.1155/2013/561098
37. Liu L, Chen JX, Zhang XW, et al. Chemokine receptor 7 overexpression promotes mesenchymal stem cell migration and proliferation via secreting Chemokine ligand 12. Sci Rep. 2018;8(1):204. doi:10.1038/s41598-017-18509-1
38. Rittiner JE, Moncalvo M, Chiba-Falek O, Kantor B. Gene-editing technologies paired with viral vectors for translational research into neurodegenerative diseases. Front Mol Neurosci. 2020;13:148. doi:10.3389/fnmol.2020.00148
39. Srifa W, Kosaric N, Amorin A, et al. Cas9-AAV6-engineered human mesenchymal stromal cells improved cutaneous wound healing in diabetic mice. Nat Commun. 2020;11(1):2470. doi:10.1038/s41467-020-16065-3
40. van Haasteren J, Li J, Scheideler OJ, Murthy N, Schaffer DV. The delivery challenge: fulfilling the promise of therapeutic genome editing. Nat Biotechnol. 2020;38(7):845–855. doi:10.1038/s41587-020-0565-5
41. Gowen A, Shahjin F, Chand S, Odegaard KE, Yelamanchili SV. Mesenchymal stem cell-derived extracellular vesicles: challenges in clinical applications. Front Cell Dev Biol. 2020;8:149. doi:10.3389/fcell.2020.00149
42. Lou G, Chen Z, Zheng M, Liu Y. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases. Exp Mol Med. 2017;49(6):e346. doi:10.1038/emm.2017.63
43. Phinney DG, Di Giuseppe M, Njah J, et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun. 2015;6:8472. doi:10.1038/ncomms9472
44. Villemin E, Ong YC, Thomas CM, Gasser G. Polymer encapsulation of ruthenium complexes for biological and medicinal applications. Nat Rev Chem. 2019;3(4):261–282. doi:10.1038/s41570-019-0088-0
45. Su YQ, Zhang TY, Huang T, Gao JQ. Current advances and challenges of mesenchymal stem cells-based drug delivery system and their improvements. Int J Pharma. 2021;600:120477.
46. Kwon S, Kim SH, Khang D, Lee JY. Potential therapeutic usage of nanomedicine for glaucoma treatment. Int J Nanomed. 2020;15:5745–5765. doi:10.2147/IJN.S254792
47. Sanna V, Sechi M. Therapeutic potential of targeted nanoparticles and perspective on nanotherapies. ACS Med Chem Lett. 2020;11(6):1069–1073. doi:10.1021/acsmedchemlett.0c00075
48. Arias-Alpizar G, Kong L, Vlieg RC, et al. Light-triggered switching of liposome surface charge directs delivery of membrane impermeable payloads in vivo. Nat Commun. 2020;11(1):3638. doi:10.1038/s41467-020-17360-9
49. Tietjen GT, Bracaglia LG, Saltzman WM, Pober JS. Focus on fundamentals: achieving effective nanoparticle targeting. Trends Mol Med. 2018;24(7):598–606. doi:10.1016/j.molmed.2018.05.003
50. Murphy DE, de Jong OG, Brouwer M, et al. Extracellular vesicle-based therapeutics: natural versus engineered targeting and trafficking. Exp Mol Med. 2019;51(3):1–12. doi:10.1038/s12276-019-0223-5
51. Bliss CM, Parsons AJ, Nachbagauer R, et al. Targeting antigen to the surface of evs improves the in vivo immunogenicity of human and non-human adenoviral vaccines in mice. Mol Ther Methods Clin Dev. 2020;16:108–125. doi:10.1016/j.omtm.2019.12.003
52. Wu M, Le W, Mei T, et al. Cell membrane camouflaged nanoparticles: a new biomimetic platform for cancer photothermal therapy. Int J Nanomed. 2019;14:4431–4448. doi:10.2147/IJN.S200284
53. Ullah M, Liu DD, Thakor AS. Mesenchymal stromal cell homing: mechanisms and strategies for improvement. iScience. 2019;15:421–438. doi:10.1016/j.isci.2019.05.004
54. Nitzsche F, Muller C, Lukomska B, Jolkkonen J, Deten A, Boltze J. Concise review: MSC adhesion cascade-insights into homing and transendothelial migration. Stem Cells. 2017;35(6):1446–1460. doi:10.1002/stem.2614
55. Sackstein R. The lymphocyte homing receptors: gatekeepers of the multistep paradigm. Curr Opin Hematol. 2005;12(6):444–450. doi:10.1097/01.moh.0000177827.78280.79
56. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572. doi:10.1038/nri.2017.49
57. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7(5):a016303. doi:10.1101/cshperspect.a016303
58. Kyurkchiev D, Bochev I, Ivanova-Todorova E, et al. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells. 2014;6(5):552–570. doi:10.4252/wjsc.v6.i5.552
59. Ringe J, Strassburg S, Neumann K, et al. Towards in situ tissue repair: human mesenchymal stem cells express chemokine receptors CXCR1, CXCR2 and CCR2, and migrate upon stimulation with CXCL8 but not CCL2. J Cell Biochem. 2007;101(1):135–146. doi:10.1002/jcb.21172
60. Sordi V, Malosio ML, Marchesi F, et al. Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood. 2005;106(2):419–427. doi:10.1182/blood-2004-09-3507
61. Kwon S, Yoo KH, Sym SJ, Khang D. Mesenchymal stem cell therapy assisted by nanotechnology: a possible combinational treatment for brain tumor and central nerve regeneration. Int J Nanomed. 2019;14:5925–5942. doi:10.2147/IJN.S217923
62. Hocking AM. The role of chemokines in mesenchymal stem cell homing to wounds. Adv Wound Care. 2015;4(11):623–630. doi:10.1089/wound.2014.0579
63. Kang SK, Shin IS, Ko MS, Jo JY, Ra JC. Journey of mesenchymal stem cells for homing: strategies to enhance efficacy and safety of stem cell therapy. Stem Cells Int. 2012;2012:342968. doi:10.1155/2012/342968
64. Miller MC, Mayo KH. Chemokines from a structural perspective. Int J Mol Sci. 2017;18(10):2088. doi:10.3390/ijms18102088
65. Gustavsson M. New insights into the structure and function of chemokine receptor: chemokine complexes from an experimental perspective. J Leukoc Biol. 2020;107(6):1115–1122. doi:10.1002/JLB.2MR1219-288R
66. Shachar I, Karin N. The dual roles of inflammatory cytokines and chemokines in the regulation of autoimmune diseases and their clinical implications. J Leukoc Biol. 2013;93(1):51–61. doi:10.1189/jlb.0612293
67. De Becker A, Riet IV. Homing and migration of mesenchymal stromal cells: how to improve the efficacy of cell therapy? World J Stem Cells. 2016;8(3):73–87. doi:10.4252/wjsc.v8.i3.73
68. Tedder TF, Steeber DA, Chen A, Engel P. The selectins: vascular adhesion molecules. FASEB J. 1995;9(10):866–873. doi:10.1096/fasebj.9.10.7542213
69. Lo CY, Weil BR, Palka BA, Momeni A, Canty JM, Neelamegham S. Cell surface glycoengineering improves selectin-mediated adhesion of mesenchymal stem cells (MSCs) and cardiosphere-derived cells (CDCs): pilot validation in porcine ischemia-reperfusion model. Biomaterials. 2016;74:19–30. doi:10.1016/j.biomaterials.2015.09.026
70. Burdick MM, Henson KA, Delgadillo LF, et al. Expression of E-selectin ligands on circulating tumor cells: cross-regulation with cancer stem cell regulatory pathways? Front Oncol. 2012;2:103. doi:10.3389/fonc.2012.00103
71. Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423(6936):190–193. doi:10.1038/nature01605
72. Barthel SR, Gavino JD, Descheny L, Dimitroff CJ. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin Ther Targets. 2007;11(11):1473–1491. doi:10.1517/14728222.11.11.1473
73. Kim DK, Nishida H, An SY, Shetty AK, Bartosh TJ, Prockop DJ. Chromatographically isolated CD63+CD81+ extracellular vesicles from mesenchymal stromal cells rescue cognitive impairments after TBI. Proc Natl Acad Sci U S A. 2016;113(1):170–175. doi:10.1073/pnas.1522297113
74. Lo CY, Antonopoulos A, Dell A, Haslam SM, Lee T, Neelamegham S. The use of surface immobilization of P-selectin glycoprotein ligand-1 on mesenchymal stem cells to facilitate selectin mediated cell tethering and rolling. Biomaterials. 2013;34(33):8213–8222. doi:10.1016/j.biomaterials.2013.07.033
75. Liao W, Pham V, Liu L, et al. Mesenchymal stem cells engineered to express selectin ligands and IL-10 exert enhanced therapeutic efficacy in murine experimental autoimmune encephalomyelitis. Biomaterials. 2016;77:87–97. doi:10.1016/j.biomaterials.2015.11.005
76. Mardpour S, Hamidieh AA, Taleahmad S, Sharifzad F, Taghikhani A, Baharvand H. Interaction between mesenchymal stromal cell-derived extracellular vesicles and immune cells by distinct protein content. J Cell Physiol. 2019;234(6):8249–8258. doi:10.1002/jcp.27669
77. Marcuzzi E, Angioni R, Molon B, Cali B. Chemokines and chemokine receptors: orchestrating tumor metastasization. Int J Mol Sci. 2018;20(1):96. doi:10.3390/ijms20010096
78. Ma S, Xie N, Li W, Yuan B, Shi Y, Wang Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014;21(2):216–225. doi:10.1038/cdd.2013.158
79. Hernandez-Rodriguez J, Segarra M, Vilardell C, et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology. 2004;43(3):294–301. doi:10.1093/rheumatology/keh058
80. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis. 2018;7(1):10. doi:10.1038/s41389-017-0011-9
81. Liu T, Han C, Wang S, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12(1):86. doi:10.1186/s13045-019-0770-1
82. Zheng XB, He XW, Zhang LJ, et al. Bone marrow-derived CXCR4-overexpressing MSCs display increased homing to intestine and ameliorate colitis-associated tumorigenesis in mice. Gastroenterol Rep. 2019;7(2):127–138. doi:10.1093/gastro/goy017
83. Chen W, Li M, Cheng H, et al. Overexpression of the mesenchymal stem cell Cxcr4 gene in irradiated mice increases the homing capacity of these cells. Cell Biochem Biophys. 2013;67(3):1181–1191. doi:10.1007/s12013-013-9632-6
84. Yang JX, Zhang N, Wang HW, Gao P, Yang QP, Wen QP. CXCR4 receptor overexpression in mesenchymal stem cells facilitates treatment of acute lung injury in rats. J Biol Chem. 2015;290(4):1994–2006. doi:10.1074/jbc.M114.605063
85. Alon R, Kassner PD, Carr MW, Finger EB, Hemler ME, Springer TA. The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J Cell Biol. 1995;128(6):1243–1253. doi:10.1083/jcb.128.6.1243
86. Acharyya S, Oskarsson T, Vanharanta S, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150(1):165–178. doi:10.1016/j.cell.2012.04.042
87. Yu PF, Huang Y, Han YY, et al. TNFalpha-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2(+) neutrophils. Oncogene. 2017;36(4):482–490. doi:10.1038/onc.2016.217
88. Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4(3):206–216. doi:10.1016/j.stem.2009.02.001
89. Eggenhofer E, Luk F, Dahlke MH, Hoogduijn MJ. The life and fate of mesenchymal stem cells. Front Immunol. 2014;5:148. doi:10.3389/fimmu.2014.00148
90. Almalki SG, Agrawal DK. Effects of matrix metalloproteinases on the fate of mesenchymal stem cells. Stem Cell Res Ther. 2016;7(1):129. doi:10.1186/s13287-016-0393-1
91. Lozito TP, Jackson WM, Nesti LJ, Tuan RS. Human mesenchymal stem cells generate a distinct pericellular zone of MMP activities via binding of MMPs and secretion of high levels of TIMPs. Matrix Biol. 2014;34:132–143. doi:10.1016/j.matbio.2013.10.003
92. Ho IA, Chan KY, Ng WH, et al. Matrix metalloproteinase 1 is necessary for the migration of human bone marrow-derived mesenchymal stem cells toward human glioma. Stem Cells. 2009;27(6):1366–1375. doi:10.1002/stem.50
93. Klein G, Schmal O, Aicher WK. Matrix metalloproteinases in stem cell mobilization. Matrix Biol. 2015;44–46:175–183. doi:10.1016/j.matbio.2015.01.011
94. Fu X, Liu G, Halim A, Ju Y, Luo Q, Song AG. Mesenchymal stem cell migration and tissue repair. Cells. 2019;8(8):784. doi:10.3390/cells8080784
95. Li Z, Yang A, Yin X, et al. Mesenchymal stem cells promote endothelial progenitor cell migration, vascularization, and bone repair in tissue-engineered constructs via activating CXCR2-Src-PKL/Vav2-Rac1. FASEB J. 2018;32(4):2197–2211. doi:10.1096/fj.201700895R
96. Xinaris C, Morigi M, Benedetti V, et al. A novel strategy to enhance mesenchymal stem cell migration capacity and promote tissue repair in an injury specific fashion. Cell Transplant. 2013;22(3):423–436. doi:10.3727/096368912X653246
97. Szekanecz Z, Koch AE. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol. 2016;12(1):5–13. doi:10.1038/nrrheum.2015.157
98. Rudan I, Sidhu S, Papana A, et al. Prevalence of rheumatoid arthritis in low- and middle-income countries: a systematic review and analysis. J Glob Health. 2015;5(1):010409. doi:10.7189/jogh.05.010409
99. Sun Y, Deng W, Geng L, et al. Mesenchymal stem cells from patients with rheumatoid arthritis display impaired function in inhibiting Th17 cells. J Immunol Res. 2015;2015:284215. doi:10.1155/2015/284215
100. Szekanecz Z, Vegvari A, Szabo Z, Koch AE. Chemokines and chemokine receptors in arthritis. Front Biosci. 2010;2:153–167. doi:10.2741/s53
101. Elemam NM, Hannawi S, Maghazachi AA. Role of chemokines and chemokine receptors in rheumatoid arthritis. Immunotargets Ther. 2020;9:43–56. doi:10.2147/ITT.S243636
102. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7(6):429–442. doi:10.1038/nri2094
103. Khang D, Choi J, Im YM, et al. Role of subnano-, nano- and submicron-surface features on osteoblast differentiation of bone marrow mesenchymal stem cells. Biomaterials. 2012;33(26):5997–6007. doi:10.1016/j.biomaterials.2012.05.005
104. Toupet K, Maumus M, Luz-Crawford P, et al. Survival and biodistribution of xenogenic adipose mesenchymal stem cells is not affected by the degree of inflammation in arthritis. PLoS One. 2015;10(1):e0114962. doi:10.1371/journal.pone.0114962
105. Haringman JJ, Oostendorp RL, Tak PP. Targeting cellular adhesion molecules, chemokines and chemokine receptors in rheumatoid arthritis. Expert Opin Emerg Drugs. 2005;10(2):299–310. doi:10.1517/14728214.10.2.299
106. Brylka LJ, Schinke T. Chemokines in physiological and pathological bone remodeling. Front Immunol. 2019;10:2182. doi:10.3389/fimmu.2019.02182
107. Kanbe K, Takagishi K, Chen Q. Stimulation of matrix metalloprotease 3 release from human chondrocytes by the interaction of stromal cell-derived factor 1 and CXC chemokine receptor 4. Arthritis Rheum. 2002;46(1):130–137. doi:10.1002/1529-0131(200201)46:1<130::AID-ART10020>3.0.CO;2-D
108. Maurer M, von Stebut E. Macrophage inflammatory protein-1. Int J Biochem Cell Biol. 2004;36(10):1882–1886. doi:10.1016/j.biocel.2003.10.019
109. Rosengren S, Corr M, Boyle DL. Platelet-derived growth factor and transforming growth factor beta synergistically potentiate inflammatory mediator synthesis by fibroblast-like synoviocytes. Arthritis Res Ther. 2010;12(2):R65. doi:10.1186/ar2981
110. Ponte AL, Marais E, Gallay N, et al. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25(7):1737–1745. doi:10.1634/stemcells.2007-0054
111. Ullah I, Subbarao RB, Rho GJ. Human mesenchymal stem cells - current trends and future prospective. Biosci Rep. 2015;35(2). doi:10.1042/BSR20150025
112. Gao J, Zhang G, Xu K, et al. Bone marrow mesenchymal stem cells improve bone erosion in collagen-induced arthritis by inhibiting osteoclasia-related factors and differentiating into chondrocytes. Stem Cell Res Ther. 2020;11(1):171. doi:10.1186/s13287-020-01684-w
113. Kim JH, Lee YT, Oh K, Cho J, Lee DS, Hwang YI. Paradoxical effects of human adipose tissue-derived mesenchymal stem cells on progression of experimental arthritis in SKG mice. Cell Immunol. 2014;292(1–2):94–101. doi:10.1016/j.cellimm.2014.10.005
114. Zhou B, Yuan J, Zhou Y, et al. Administering human adipose-derived mesenchymal stem cells to prevent and treat experimental arthritis. Clin Immunol. 2011;141(3):328–337. doi:10.1016/j.clim.2011.08.014
115. Mao F, Xu WR, Qian H, et al. Immunosuppressive effects of mesenchymal stem cells in collagen-induced mouse arthritis. Inflamm Res. 2010;59(3):219–225. doi:10.1007/s00011-009-0090-y
116. Parolini O, Souza-Moreira L, O’Valle F, et al. Therapeutic effect of human amniotic membrane-derived cells on experimental arthritis and other inflammatory disorders. Arthritis Rheumatol. 2014;66(2):327–339. doi:10.1002/art.38206
117. Hodgkinson CP, Gomez JA, Mirotsou M, Dzau VJ. Genetic engineering of mesenchymal stem cells and its application in human disease therapy. Hum Gene Ther. 2010;21(11):1513–1526. doi:10.1089/hum.2010.165
118. Nowakowski A, Walczak P, Lukomska B, Janowski M. Genetic engineering of mesenchymal stem cells to induce their migration and survival. Stem Cells Int. 2016;2016:4956063. doi:10.1155/2016/4956063
119. Varkouhi AK, Monteiro APT, Tsoporis JN, Mei SHJ, Stewart DJ, Dos Santos CC. Genetically modified mesenchymal stromal/stem cells: application in critical illness. Stem Cell Rev Rep. 2020;16(5):812–827. doi:10.1007/s12015-020-10000-1
120. Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release. 2006;116(2):255–264. doi:10.1016/j.jconrel.2006.06.024
121. Lian T, Ho RJ. Trends and developments in liposome drug delivery systems. J Pharm Sci. 2001;90(6):667–680. doi:10.1002/jps.1023
122. Somiari S, Glasspool-Malone J, Drabick JJ, et al. Theory and in vivo application of electroporative gene delivery. Mol Ther. 2000;2(3):178–187. doi:10.1006/mthe.2000.0124
123. Moiani A, Paleari Y, Sartori D, et al. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J Clin Invest. 2012;122(5):1653–1666. doi:10.1172/JCI61852
124. Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther. 2021;6(1):53. doi:10.1038/s41392-021-00487-6
125. Voysey M, Clemens SAC, Madhi SA, et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021;397(10269):99–111. doi:10.1016/S0140-6736(20)32661-1
126. Keeler AM, Flotte TR. Recombinant adeno-associated virus gene therapy in light of luxturna (and zolgensma and glybera): where are we, and how did we get here? Annu Rev Virol. 2019;6(1):601–621. doi:10.1146/annurev-virology-092818-015530
127. Cho SW, Kim S, Kim Y, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–141. doi:10.1101/gr.162339.113
128. Kim TK, Eberwine JH. Mammalian cell transfection: the present and the future. Anal Bioanal Chem. 2010;397(8):3173–3178. doi:10.1007/s00216-010-3821-6
129. Leung RK, Whittaker PA. RNA interference: from gene silencing to gene-specific therapeutics. Pharmacol Ther. 2005;107(2):222–239. doi:10.1016/j.pharmthera.2005.03.004
130. Lau TT, Wang DA. Stromal cell-derived factor-1 (SDF-1): homing factor for engineered regenerative medicine. Expert Opin Biol Ther. 2011;11(2):189–197. doi:10.1517/14712598.2011.546338
131. Takano T, Li YJ, Kukita A, et al. Mesenchymal stem cells markedly suppress inflammatory bone destruction in rats with adjuvant-induced arthritis. Lab Invest. 2014;94(3):286–296. doi:10.1038/labinvest.2013.152
132. Won YW, Patel AN, Bull DA. Cell surface engineering to enhance mesenchymal stem cell migration toward an SDF-1 gradient. Biomaterials. 2014;35(21):5627–5635. doi:10.1016/j.biomaterials.2014.03.070
133. Marquez-Curtis LA, Gul-Uludag H, Xu P, Chen J, Janowska-Wieczorek A. CXCR4 transfection of cord blood mesenchymal stromal cells with the use of cationic liposome enhances their migration toward stromal cell-derived factor-1. Cytotherapy. 2013;15(7):840–849. doi:10.1016/j.jcyt.2013.02.009
134. Wang Y, Fu W, Zhang S, et al. CXCR-7 receptor promotes SDF-1alpha-induced migration of bone marrow mesenchymal stem cells in the transient cerebral ischemia/reperfusion rat hippocampus. Brain Res. 2014;1575:78–86. doi:10.1016/j.brainres.2014.05.035
135. Kim DS, Kim JH, Lee JK, et al. Overexpression of CXC chemokine receptors is required for the superior glioma-tracking property of umbilical cord blood-derived mesenchymal stem cells. Stem Cells Dev. 2009;18(3):511–519. doi:10.1089/scd.2008.0050
136. Kim SM, Kim DS, Jeong CH, et al. CXC chemokine receptor 1 enhances the ability of human umbilical cord blood-derived mesenchymal stem cells to migrate toward gliomas. Biochem Biophys Res Commun. 2011;407(4):741–746. doi:10.1016/j.bbrc.2011.03.093
137. Hara-Chikuma M, Verkman AS. Aquaporin-1 facilitates epithelial cell migration in kidney proximal tubule. J Am Soc Nephrol. 2006;17(1):39–45. doi:10.1681/ASN.2005080846
138. Meng F, Rui Y, Xu L, Wan C, Jiang X, Li G. Aqp1 enhances migration of bone marrow mesenchymal stem cells through regulation of FAK and beta-catenin. Stem Cells Dev. 2014;23(1):66–75. doi:10.1089/scd.2013.0185
139. Maijenburg MW, Gilissen C, Melief SM, et al. Nuclear receptors Nur77 and Nurr1 modulate mesenchymal stromal cell migration. Stem Cells Dev. 2012;21(2):228–238. doi:10.1089/scd.2011.0076
140. Maijenburg MW, Gilissen C, Veltman JA, et al. Molecular signature of migratory human mesenchymal stromal cells; influence of the cell cycle. Blood. 2009;114(22):587. doi:10.1182/blood.V114.22.1450.1450
141. Boehm M, Nabel EG. Cell cycle and cell migration: new pieces to the puzzle. Circulation. 2001;103(24):2879–2881. doi:10.1161/01.CIR.103.24.2879
142. Auria-Soro C, Nesma T, Juanes-Velasco P, et al. Interactions of nanoparticles and biosystems: microenvironment of nanoparticles and biomolecules in nanomedicine. Nanomaterials. 2019;9(10):1365. doi:10.3390/nano9101365
143. Kim SW, Park JY, Lee S, Kim SH, Khang D. Destroying deep lung tumor tissue through lung-selective accumulation and by activation of caveolin uptake channels using a specific width of carbon nanodrug. ACS Appl Mater Interfaces. 2018;10(5):4419–4428. doi:10.1021/acsami.7b16153
144. Meng H, Wang M, Liu H, et al. Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. Acs Nano. 2015;9(4):3540–3557. doi:10.1021/acsnano.5b00510
145. Di Mauro GM, Hardin NZ, Ramamoorthy A. Lipid-nanodiscs formed by paramagnetic metal chelated polymer for fast NMR data acquisition. Biochim Biophys Acta Biomembr. 2020;1862(9):183332. doi:10.1016/j.bbamem.2020.183332
146. Zein R, Sharrouf W, Selting K. Physical properties of nanoparticles that result in improved cancer targeting. J Oncol. 2020;2020:5194780. doi:10.1155/2020/5194780
147. Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat Nanotechnol. 2020;15(4):313–320. doi:10.1038/s41565-020-0669-6
148. Ke X, Howard GP, Tang H, et al. Physical and chemical profiles of nanoparticles for lymphatic targeting. Adv Drug Deliv Rev. 2019;151–152:72–93. doi:10.1016/j.addr.2019.09.005
149. Wang S, Dormidontova EE. Nanoparticle design optimization for enhanced targeting: Monte Carlo simulations. Biomacromolecules. 2010;11(7):1785–1795. doi:10.1021/bm100248e
150. Miller-Kleinhenz JM, Bozeman EN, Yang L. Targeted nanoparticles for image-guided treatment of triple-negative breast cancer: clinical significance and technological advances. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7(6):797–816. doi:10.1002/wnan.1343
151. Narain A, Asawa S, Chhabria V, Patil-Sen Y. Cell membrane coated nanoparticles: next-generation therapeutics. Nanomedicine. 2017;12(21):2677–2692. doi:10.2217/nnm-2017-0225
152. Wang LT, Ting CH, Yen ML, et al. Human mesenchymal stem cells (MSCs) for treatment towards immune- and inflammation-mediated diseases: review of current clinical trials. J Biomed Sci. 2016;23(1):76. doi:10.1186/s12929-016-0289-5
153. Pasto A, Giordano F, Evangelopoulos M, Amadori A, Tasciotti E. Cell membrane protein functionalization of nanoparticles as a new tumor-targeting strategy. Clin Transl Med. 2019;8(1):8. doi:10.1186/s40169-019-0224-y
154. Wang M, Xin Y, Cao H, et al. Recent advances in mesenchymal stem cell membrane-coated nanoparticles for enhanced drug delivery. Biomater Sci. 2021;9(4):1088–1103. doi:10.1039/D0BM01164A
155. Freese C, Gibson MI, Klok HA, Unger RE, Kirkpatrick CJ. Size- and coating-dependent uptake of polymer-coated gold nanoparticles in primary human dermal microvascular endothelial cells. Biomacromolecules. 2012;13(5):1533–1543. doi:10.1021/bm300248u
156. Fu SY, Wang Y, Xia XH, Zheng JLC. Exosome engineering: current progress in cargo loading and targeted delivery. Nanoimpact. 2020;20:100261. doi:10.1016/j.impact.2020.100261
157. Liang Y, Duan L, Lu J, Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11(7):3183–3195. doi:10.7150/thno.52570
158. Vinas JL, Spence M, Gutsol A, et al. Receptor-ligand interaction mediates targeting of endothelial colony forming cell-derived exosomes to the kidney after ischemic injury. Sci Rep. 2018;8(1):16320. doi:10.1038/s41598-018-34557-7
159. Rana S, Yue S, Stadel D, Zoller M. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol. 2012;44(9):1574–1584. doi:10.1016/j.biocel.2012.06.018
160. de la Torre P, Perez-Lorenzo MJ, Alcazar-Garrido A, Flores AI. Cell-based nanoparticles delivery systems for targeted cancer therapy: lessons from anti-angiogenesis treatments. Molecules. 2020;25(3):715. doi:10.3390/molecules25030715
161. Yaman S, Chintapula U, Rodriguez E, Ramachandramoorthy H, Nguyen KT. Cell-mediated and cell membrane-coated nanoparticles for drug delivery and cancer therapy. Cancer Drug Resist. 2020;3:879–911. doi:10.20517/cdr.2020.55
162. Crist RM, Grossman JH, Patri AK, et al. Common pitfalls in nanotechnology: lessons learned from NCI’s Nanotechnology Characterization Laboratory. Integr Biol (Camb). 2013;5(1):66–73. doi:10.1039/c2ib20117h
163. Shvedova AA, Kagan VE, Fadeel B. Close encounters of the small kind: adverse effects of man-made materials interfacing with the nano-cosmos of biological systems. Annu Rev Pharmacol Toxicol. 2010;50:63–88. doi:10.1146/annurev.pharmtox.010909.105819
164. Yetisgin AA, Cetinel S, Zuvin M, Kosar A, Kutlu O. Therapeutic nanoparticles and their targeted delivery applications. Molecules. 2020;25(9):2193. doi:10.3390/molecules25092193
165. Liu Y, Luo J, Chen X, Liu W, Chen T. Cell membrane coating technology: a promising strategy for biomedical applications. Nano Micro Lett. 2019;11(1):100. doi:10.1007/s40820-019-0330-9
166. Liu Y, Zhao J, Jiang J, Chen F, Fang X. Doxorubicin delivered using nanoparticles camouflaged with mesenchymal stem cell membranes to treat colon cancer. Int J Nanomedicine. 2020;15:2873–2884. doi:10.2147/IJN.S242787
167. Harris JC, Scully MA, Day ES. Cancer cell membrane-coated nanoparticles for cancer management. Cancers. 2019;11(12):1836. doi:10.3390/cancers11121836
168. Thone MN, Kwon YJ. Extracellular blebs: artificially-induced extracellular vesicles for facile production and clinical translation. Methods. 2020;177:135–145. doi:10.1016/j.ymeth.2019.11.007
169. Mesland DA, Los G, Spiele H. Cytochalasin B disrupts the association of filamentous web and plasma membrane in hepatocytes. Exp Cell Res. 1981;135(2):431–435. doi:10.1016/0014-4827(81)90184-1
170. Gomzikova M, Kletukhina S, Kurbangaleeva S, Rizvanov A. Evaluation of cytochalasin B-induced membrane vesicles fusion specificity with target cells. Biomed Res Int. 2018;2018:7053623. doi:10.1155/2018/7053623
171. Peng LH, Zhang YH, Han LJ, et al. Cell membrane capsules for encapsulation of chemotherapeutic and cancer cell targeting in vivo. ACS Appl Mater Interfaces. 2015;7(33):18628–18637. doi:10.1021/acsami.5b05065
172. Zhai Y, Su J, Ran W, et al. Preparation and application of cell membrane-camouflaged nanoparticles for cancer therapy. Theranostics. 2017;7(10):2575–2592. doi:10.7150/thno.20118
173. Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. doi:10.3390/polym3031377
174. Mahapatro A, Singh DK. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J Nanobiotechnol. 2011;9:55. doi:10.1186/1477-3155-9-55
175. Khan I, Saeed K, Khan I. Nanoparticles: properties, applications and toxicities. Arab J Chem. 2019;12(7):908–931. doi:10.1016/j.arabjc.2017.05.011
176. Hou X, Tao Y, Pang Y, Li X, Jiang G, Liu Y. Nanoparticle-based photothermal and photodynamic immunotherapy for tumor treatment. Int J Cancer. 2018;143(12):3050–3060. doi:10.1002/ijc.31717
177. Vijayan V, Uthaman S, Park IK. Cell membrane-camouflaged nanoparticles: a promising biomimetic strategy for cancer theragnostics. Polymers. 2018;10(9):983. doi:10.3390/polym10090983
178. Bajracharya R, Song JG, Back SY, Han HK. Recent advancements in non-invasive formulations for protein drug delivery. Comput Struct Biotechnol J. 2019;17:1290–1308. doi:10.1016/j.csbj.2019.09.004
179. Yang N, Ding Y, Zhang Y, et al. Surface functionalization of polymeric nanoparticles with umbilical cord-derived mesenchymal stem cell membrane for tumor-targeted therapy. ACS Appl Mater Interfaces. 2018;10(27):22963–22973. doi:10.1021/acsami.8b05363
180. Gao C, Lin Z, Jurado-Sanchez B, Lin X, Wu Z, He Q. Stem cell membrane-coated nanogels for highly efficient in vivo tumor targeted drug delivery. Small. 2016;12(30):4056–4062. doi:10.1002/smll.201600624
181. Gao C, Lin Z, Wu Z, Lin X, He Q. Stem-cell-membrane camouflaging on near-infrared photoactivated upconversion nanoarchitectures for in vivo remote-controlled photodynamic therapy. ACS Appl Mater Interfaces. 2016;8(50):34252–34260. doi:10.1021/acsami.6b12865
182. Bose RJ, Kim BJ, Arai Y, et al. Bioengineered stem cell membrane functionalized nanocarriers for therapeutic targeting of severe hindlimb ischemia. Biomaterials. 2018;185:360–370. doi:10.1016/j.biomaterials.2018.08.018
183. Kim HY, Kim TJ, Kang L, et al. Mesenchymal stem cell-derived magnetic extracellular nanovesicles for targeting and treatment of ischemic stroke. Biomaterials. 2020;243:119942. doi:10.1016/j.biomaterials.2020.119942
184. Austin-Williams S, Hussain MT, Oggero S, Norling LV. Enhancing extracellular vesicles for therapeutic treatment of arthritic joints. Free Radic Biol Med. 2021;175:80–94. doi:10.1016/j.freeradbiomed.2021.08.235
185. Hosseinikhah SM, Barani M, Rahdar A, et al. Nanomaterials for the diagnosis and treatment of inflammatory arthritis. Int J Mol Sci. 2021;22(6):3092. doi:10.3390/ijms22063092
186. Ventimiglia LN, Alonso MA. Biogenesis and function of T cell-derived exosomes. Front Cell Dev Biol. 2016;4:84. doi:10.3389/fcell.2016.00084
187. Baek G, Choi H, Kim Y, Lee HC, Choi C. Mesenchymal stem cell-derived extracellular vesicles as therapeutics and as a drug delivery platform. Stem Cells Transl Med. 2019;8(9):880–886. doi:10.1002/sctm.18-0226
188. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:6478. doi:10.1126/science.aau6977
189. Li X, Corbett AL, Taatizadeh E, et al. Challenges and opportunities in exosome research-perspectives from biology, engineering, and cancer therapy. APL Bioeng. 2019;3(1):011503. doi:10.1063/1.5087122
190. Yang T, Martin P, Fogarty B, et al. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res. 2015;32(6):2003–2014. doi:10.1007/s11095-014-1593-y
191. Garcia-Manrique P, Matos M, Gutierrez G, Pazos C, Blanco-Lopez MC. Therapeutic biomaterials based on extracellular vesicles: classification of bio-engineering and mimetic preparation routes. J Extracell Vesicles. 2018;7(1):1422676. doi:10.1080/20013078.2017.1422676
192. Li SP, Lin ZX, Jiang XY, Yu XY. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol Sin. 2018;39(4):542–551. doi:10.1038/aps.2017.178
193. Zhang Y, Liu Y, Liu H, Tang WH. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9:19. doi:10.1186/s13578-019-0282-2
194. Akuma P, Okagu OD, Udenigwe CC. Naturally occurring exosome vesicles as potential delivery vehicle for bioactive compounds. Front Sustain Food S. 2019;3:23.
195. Garcia-Manrique P, Gutierrez G, Blanco-Lopez MC. Fully artificial exosomes: towards new theranostic biomaterials. Trends Biotechnol. 2018;36(1):10–14. doi:10.1016/j.tibtech.2017.10.005
196. Antimisiaris SG, Mourtas S, Marazioti A. Exosomes and exosome-inspired vesicles for targeted drug delivery. Pharmaceutics. 2018;10(4):218. doi:10.3390/pharmaceutics10040218
197. Yoon J, Jo W, Jeong D, Kim J, Jeong H, Park J. Generation of nanovesicles with sliced cellular membrane fragments for exogenous material delivery. Biomaterials. 2015;59:12–20. doi:10.1016/j.biomaterials.2015.04.028
198. Peng H, Ji W, Zhao R, et al. Exosome: a significant nano-scale drug delivery carrier. J Mater Chem B. 2020;8(34):7591–7608. doi:10.1039/D0TB01499K
199. Katakowski M, Buller B, Zheng X, et al. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013;335(1):201–204. doi:10.1016/j.canlet.2013.02.019
200. Lou G, Song X, Yang F, et al. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J Hematol Oncol. 2015;8:122. doi:10.1186/s13045-015-0220-7
201. O’Brien KP, Khan S, Gilligan KE, et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene. 2018;37(16):2137–2149. doi:10.1038/s41388-017-0116-9
202. Pascucci L, Cocce V, Bonomi A, et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release. 2014;192:262–270. doi:10.1016/j.jconrel.2014.07.042
203. Bagheri E, Abnous K, Farzad SA, Taghdisi SM, Ramezani M, Alibolandi M. Targeted doxorubicin-loaded mesenchymal stem cells-derived exosomes as a versatile platform for fighting against colorectal cancer. Life Sci. 2020;261:118369. doi:10.1016/j.lfs.2020.118369
204. Kalani A, Chaturvedi P. Curcumin-primed and curcumin-loaded exosomes: potential neural therapy. Neural Regen Res. 2017;12(2):205–206. doi:10.4103/1673-5374.200799
205. Maia J, Caja S, Strano Moraes MC, Couto N, Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. 2018;6:18. doi:10.3389/fcell.2018.00018
206. Zhang B, Wang M, Gong A, et al. HucMSC-exosome mediated-wnt4 signaling is required for cutaneous wound healing. Stem Cells. 2015;33(7):2158–2168. doi:10.1002/stem.1771
207. Xin H, Li Y, Cui Y, Yang JJ, Zhang ZG, Chopp M. Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J Cereb Blood Flow Metab. 2013;33(11):1711–1715. doi:10.1038/jcbfm.2013.152
208. Akbarzadeh A, Rezaei-Sadabady R, Davaran S, et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):102. doi:10.1186/1556-276X-8-102
209. Senapati S, Mahanta AK, Kumar S, Maiti P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct Tar. 2018;3:1–9.
210. Patra JK, Das G, Fraceto LF, et al. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnol. 2018;16(1):71. doi:10.1186/s12951-018-0392-8
211. Elkhoury K, Kocak P, Kang A, Arab-Tehrany E, Ellis Ward J, Shin SR. Engineering smart targeting nanovesicles and their combination with hydrogels for controlled drug delivery. Pharmaceutics. 2020;12(9):849. doi:10.3390/pharmaceutics12090849
212. Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. doi:10.1016/j.addr.2015.09.012
213. Sato YT, Umezaki K, Sawada S, et al. Engineering hybrid exosomes by membrane fusion with liposomes. Sci Rep. 2016;6:21933. doi:10.1038/srep21933
214. Dang XTT, Kavishka JM, Zhang DX, Pirisinu M, Le MTN. Extracellular vesicles as an efficient and versatile system for drug delivery. Cells. 2020;9(10):2191. doi:10.3390/cells9102191
215. Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomed. 2006;1(3):297–315.
216. Goh WJ, Zou S, Lee CK, et al. EXOPLEXs: chimeric drug delivery platform from the fusion of cell-derived nanovesicles and liposomes. Biomacromolecules. 2018;19(1):22–30. doi:10.1021/acs.biomac.7b01176
217. Rayamajhi S, Nguyen TDT, Marasini R, Aryal S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019;94:482–494. doi:10.1016/j.actbio.2019.05.054
218. Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. 2012;64(8):739–748. doi:10.1016/j.addr.2011.06.010
219. Sharma RR, Pollock K, Hubel A, McKenna D. Mesenchymal stem or stromal cells: a review of clinical applications and manufacturing practices. Transfusion. 2014;54(5):1418–1437.
220. Ren G, Zhao X, Zhang L, et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol. 2010;184(5):2321–2328.
221. Cuesta-Gomez N, Graham GJ, Campbell JDM. Chemokines and their receptors: predictors of the therapeutic potential of mesenchymal stromal cells. J Transl Med. 2021;19(1):156. doi:10.1186/s12967-021-02822-5
222. Wang M, Yuan Q, Xie L. Mesenchymal stem cell-based immunomodulation: properties and clinical application. Stem Cells Int. 2018;2018:3057624. doi:10.1155/2018/3057624
223. Levy O, Kuai R, Siren EMJ, et al. Shattering barriers toward clinically meaningful MSC therapies. Sci Adv. 2020;6(30):eaba6884. doi:10.1126/sciadv.aba6884
224. Garcia-Bernal D, Garcia-Arranz M, Yanez RM, et al. The current status of mesenchymal stromal cells: controversies, unresolved issues and some promising solutions to improve their therapeutic efficacy. Front Cell Dev Biol. 2021;9:650664. doi:10.3389/fcell.2021.650664
225. Ocansey DKW, Pei B, Yan Y, et al. Improved therapeutics of modified mesenchymal stem cells: an update. J Transl Med. 2020;18(1):42. doi:10.1186/s12967-020-02234-x
226. Gao W, Zhang L. Coating nanoparticles with cell membranes for targeted drug delivery. J Drug Target. 2015;23(7–8):619–626. doi:10.3109/1061186X.2015.1052074
227. Vargason AM, Anselmo AC, Mitragotri S. The evolution of commercial drug delivery technologies. Nat Biomed Eng. 2021;5:951–967.
228. Jo W, Jeong D, Kim J, et al. Microfluidic fabrication of cell-derived nanovesicles as endogenous RNA carriers. Lab Chip. 2014;14(7):1261–1269. doi:10.1039/C3LC50993A
229. Wang L, Abhange KK, Wen Y, et al. Preparation of engineered extracellular vesicles derived from human umbilical cord mesenchymal stem cells with ultrasonication for skin rejuvenation. ACS Omega. 2019;4(27):22638–22645. doi:10.1021/acsomega.9b03561
230. Jo W, Kim J, Yoon J, et al. Large-scale generation of cell-derived nanovesicles. Nanoscale. 2014;6(20):12056–12064. doi:10.1039/C4NR02391A
231. He X, Dong Z, Cao Y, et al. MSC-derived exosome promotes M2 polarization and enhances cutaneous wound healing. Stem Cells Int. 2019;2019:7132708. doi:10.1155/2019/7132708
232. Wang Y, Zhang L, Li Y, et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int J Cardiol. 2015;192:61–69. doi:10.1016/j.ijcard.2015.05.020
233. Lv LL, Wu WJ, Feng Y, Li ZL, Tang TT, Liu BC. Therapeutic application of extracellular vesicles in kidney disease: promises and challenges. J Cell Mol Med. 2018;22(2):728–737. doi:10.1111/jcmm.13407
234. Lee C, Mitsialis SA, Aslam M, et al. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation. 2012;126(22):2601–2611. doi:10.1161/CIRCULATIONAHA.112.114173
235. Yuan Z, Kolluri KK, Gowers KH, Janes SM. TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy. J Extracell Vesicles. 2017;6(1):1265291. doi:10.1080/20013078.2017.1265291
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.