Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 4
STAT3 as an emerging molecular target in pancreatic cancer
Authors Sharma N, Shankar S, Srivastava R
Received 21 March 2014
Accepted for publication 15 May 2014
Published 25 August 2014 Volume 2014:4 Pages 115—122
DOI https://doi.org/10.2147/GICTT.S48993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Narinder Kumar Sharma,1 Sharmila Shankar,2 Rakesh K Srivastava1
1Department of Pharmacology, Toxicology and Therapeutics, and Medicine, University of Kansas Medical Center, Kansas City, KS, USA; 2Kansas City VA Medical Center, Kansas City, MO, USA
Abstract: Pancreatic cancer is the fourth leading cause of cancer related deaths. Although, surgical resection of pancreatic cancer may provide the best chance for cure and long-term survival, due to the late onset of symptoms only 15% to 20% of patients have resectable tumors. Most of the pancreatic tumors have mutations in the K-ras gene, followed by mutations in tumor suppressor genes such as p53 and SMAD4. In addition, there is growing evidence for the potential involvement of signal transducer and activator of transcription 3 (STAT3) in malignant transformation of pancreatic cancer. STAT3 plays critical roles in regulating many physiological functions in normal and malignant tissues, such as inflammation, survival, proliferation, differentiation, and angiogenesis. STAT3 is activated by a wide variety of cytokines, growth factors, and other stimuli. Unlike other members of the STAT family, ablation of STAT3 leads to embryonic lethality and conditional loss of STAT3 protein in adult tissues, leading to a variety of abnormalities, confirming that STAT3 participates in a wide variety of physiological processes. Constitutive activation of STAT3 is implicated in a wide range of human cancers; therefore, STAT3 has been identified as a novel target to treat and prevent cancers. Several STAT3 inhibitors display antitumor effectiveness, and data supporting the use of STAT3 inhibitors are emerging. Different approaches used for the inhibition of activated STAT3 include modulating upstream positive or negative regulators or directly targeting its different domains. These approaches have been used in the inhibition of STAT3 in different cancers, but in this review, we will focus specifically on the inhibition of STAT3 in pancreatic cancer.
Keywords: pancreatic cancer, Kras, oncogene, angiogenesis, apoptosis, STAT3
Introduction
Pancreatic cancer is a significant threat to life worldwide due to its high mortality. It encompasses both exocrine and endocrine tumors of the pancreas. Since more than 90% of pancreatic tumors originate from ductal epithelium of the pancreas, the term pancreatic ductal adenocarcinoma (PDAC) is commonly referred to as pancreatic cancer. The incidence of pancreatic cancer increases with age, and the majority of cases are diagnosed above the age of 65 years. The incidence rates of pancreatic cancer are steadily increasing in Western countries. Various risk factors for pancreatic cancer include smoking, obesity, and high consumption of processed meat. Although the 5-year survival rates for most cancers have been improved in the last few decades, there is little improvement in the survival rate for pancreatic cancer due to various factors including its silent nature in early stages, aggressive tumor biology, low scope of surgical management, and lack of effective systemic therapies.1–3
Inflammation plays a critical role in the initiation of pancreatic cancer and malignant progression. In fact, pancreatitis is a well-known risk factor for pancreatic cancer.4 Cystic fibrosis is also associated with an increased risk of developing pancreatic cancer.5 In the majority of pancreatic tumors, the K-ras gene is mutated to K-rasG12D (oncogenic form of K-ras).6 With age, people acquire oncogenic mutations in K-ras in the organs, including lung, pancreas, colon, and other tissues. However, this mutation in K-ras does not serve as a good biomarker because it also appears in noncancer cells. In addition to activation of oncogene K-ras, inactivation of several tumor suppressor genes, such as p16, p53, and SMAD4, was found to play an important role in the development of pancreatic cancer. Mutation frequencies for p16, p53, and K-ras in pancreatic cancer are approximately 80%, 70%, and 90%, respectively.7
Surgical removal of tumor is the primary option for treatment of pancreatic cancer. If the tumor is not resectable, treatment mainly aims to increase survival. Systemic chemotherapy may be used at any stage of pancreatic cancer, with the goal to minimize the patient’s disease-related symptoms and to prolong survival.
Gemcitabine has been the reference regimen since its approval following a Phase III trial in 1997 by Burris et al, in which 126 patients with advanced pancreatic cancer received either gemcitabine or bolus 5-fluorouracil (5-FU).8 Clinical response was experienced in 23.8% of gemcitabine-treated patients compared with 4.8% of 5-FU-treated patients. The 1-year survival rate was 18% and 2% for the gemcitabine and 5-FU groups, respectively. This was the pivotal study highlighting the use of gemcitabine as first-line treatment for patients with advanced pancreatic cancer. 5-FU treatment compared with no chemotherapy provided a survival advantage of 33 weeks for the treated group compared with 15 weeks in the untreated group for pancreatic cancer patients. Fluoropyrimidine has been developed in order to potentiate the antitumor activity of 5-FU and reduce gastrointestinal toxicity.
Another drug, capecitabine, had shown antitumor activity in combination with gemcitabine in Phase II trials in chemotherapy-naive patients with locally advanced or metastatic pancreatic cancer. However, two Phase III trials are not encouraging.9 Efforts have been made to develop a combination using one or more other drugs with gemcitabine for pancreatic cancer patients to produce a significant overall survival benefit.
STAT3 – normal physiological functions and oncogenic role
Signal transducer and activator of transcription 3 (STAT3) belongs to a family of transcription factors comprising STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. In humans, this is encoded by the STAT3 gene. Structurally, STAT3 comprises several distinct functional domains including the NH2-terminal, coiled-coil, DNA binding, linker, SH2, and C-terminal transactivation domains. These domains play critical roles from cytokine- or growth factor-receptor binding to activation of STAT3, which involves phosphorylation events, dimerization, and nuclear transfer. Initially, STAT3 appeared to be an additional member of the growing STAT family that can control a limited set of target genes in response to interleukin (IL)-6. But later, through subsequent studies in cell culture, the list of possible functions of STAT3 grew, showing that it is activated in a wide variety of signaling systems and mediates diverse responses. STAT3 is activated by many cytokines including IL-6, IL-9, IL-10, IL-27, tumor necrosis factor α, monocyte chemotactic protein-1 and transmits the signals from the cell membrane to the nucleus to regulate gene expression for cell development, differentiation, proliferation, survival, and angiogenesis (Figure 1).10–12 In addition, STAT3 can also be activated by various growth factors such as epidermal growth factor, platelet-derived growth factor, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor. One of the major roles of activated STAT3 is inflammation, which is regulated by cytokines such as IL-6 and a proinflammatory enzyme, cyclooxygenase 2. Other critical functions of activated STAT3 include cell proliferation by upregulating the transcription of cyclin D1 and cyclin B1, and apoptosis inhibition by upregulating the expression of Bcl-2, Bcl-xL, and Mcl-1. It also regulates metabolism by regulating the expression of Hsp70, Hsp90, and Cdc2. Activated STAT3 plays a critical role in various aspects of tumorigenesis, including angiogenesis, by regulating the transcription of vascular endothelial growth factor (VEGF), FGF, and HIF1 α, and invasion and metastasis by regulating the expression of MMP2, MMP9, TWIST, and ICAM1.
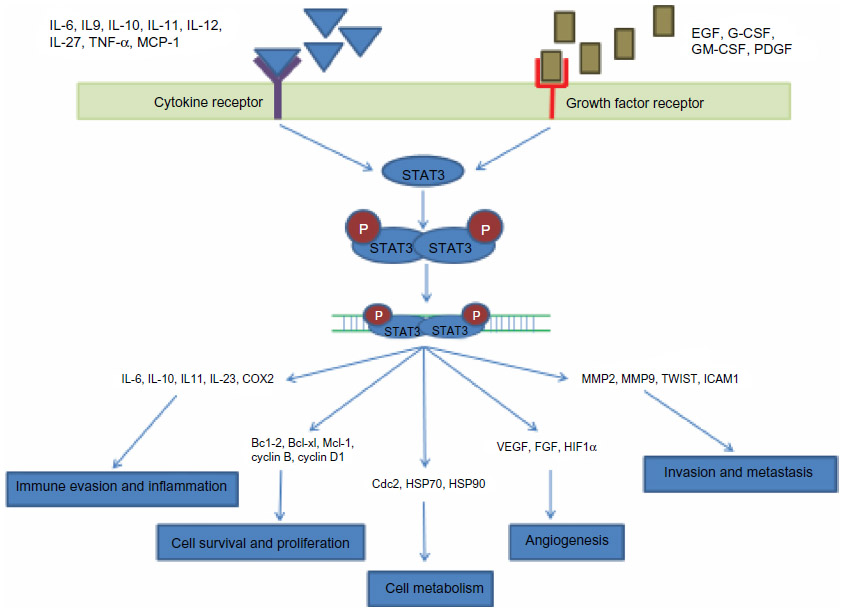 | Figure 1 Schematic model for the role of STAT3 pathway in various physiological processes. |
Biological effects of STAT3 have been assessed by targeted gene ablation in knockout mice. Although, ablation of other STAT family genes produced viable mice with relatively limited phenotypes, the ablation of STAT3 led to early embryonic lethality in mice. Moreover, loss of STAT3 is also lethal in embryonic stem cells. Mouse embryos with homozygous STAT3-null genotype degenerate rapidly after blastocyst implantation (between 6.5 and 7.5 days of embryogenesis). However, STAT3 ablation in adult tissues leads to surprisingly mild phenotypes with normal tissue growth although their responses to particular cytokines are impaired without STAT3.
Role of STAT3 in pancreatic cancer
In recent years, evidence has accumulated supporting the critical role of activated STAT3 in various cancers.11–13 In fact, STAT3 activation has been detected in different experimental malignancies, and its abolition by use of inhibitors or antisense oligonucleotides has led to reversal of the malignant phenotype. STAT3 can be constitutively activated by upstream signaling components, including increased cytokine (IL-6 and IL-10) production, activated receptor (cytokine receptors, epidermal growth factor receptor) and nonreceptor tyrosine kinases (including Janus kinases [JAKs] and Src). Recently, mutated hyperactive forms of STAT3 have been detected in tumors, with the majority of mutations found in the SH2 domain of STAT3.14–15 Constitutive STAT3 activation has also been reported in different studies from mouse models and specimens from human malignancies including head and neck cancers, breast cancer, and multiple myelomas. STAT3 activation has been described in nearly 70% of solid and hematological tumors.16
STAT3 is found to be constitutively activated in PDAC by phosphorylation of Tyr705, as reported in human tumor specimens as well as in various PDAC cell lines. Moreover, the STAT3 pathway has been shown to have a role in the proliferation of some PDAC cell lines in vitro and in the induction of tumors in PDAC xenografts.17–20 However, studies in conditional knockout mice demonstrate that the STAT3 pathway is inactive in normal pancreas, and it is not required for any vital process related to pancreatic development and homeostasis.21
Further, it has been demonstrated that STAT3 is obligatory for the development of the acinar-to-ductal metaplasia process, an early event in PDAC pathogenesis, which is mediated by ectopic expression of the Pdx1 transcription factor, a key regulator in early pancreatic development.19 STAT3 is shown to be activated and overexpressed in ductal carcinoma cells as compare to the ducts from chronic pancreatitis.17 Functional inactivation of STAT3 in a subset of pancreatic cancer cell lines leads to significant inhibition in cell proliferation in vitro and reduced tumor growth in vivo. Inhibition of activated STAT3 resulted in the delay in G1/S-phase progression due to inhibition of cyclin-dependent kinase 2 activity because of increased expression of p21WAF1. Cells treated with tyrphostin, a JAK2 inhibitor, also resulted in growth arrest because of delayed G1/S-phase progression and higher expression of p21WAF1. Overall, this study clearly showed that with malignant transformation, activated STAT3 promotes proliferation of cells by modulating G1/S-phase progression and supports the malignant phenotype of human pancreatic cancer.
IL-6 transsignaling and suppressor of cytokine signaling (SOCS)3 are involved in the progression of pancreatic intraepithelial neoplasia (PanIN), a well-known precursor to PDAC development.22 The myeloid cells in the pancreas induce STAT3 activation by releasing IL-6, which promotes PanIN progression and PDAC development. The inactivation of classical IL-6 signaling, IL-6 transsignaling, and the STAT3 pathway by genetic manipulation resulted in impaired PanIN progression and PDAC development. STAT3 was also found to be activated in PDAC due to homozygous deletion of SOCS3, which in turn accelerates PanIN progression and PDAC development.
Expression of the angiogenic protein VEGF has been associated with progression of pancreatic cancer. Studies have shown that overexpression of VEGF coincides with constitutive STAT3 activation in human pancreatic cancer specimens.20 Various human pancreatic cancer cell lines were shown to express constitutively activated STAT3, which is correlated to the elevated level of VEGF expression. With the blockade of activated STAT3, VEGF expression was also significantly suppressed. This resulted in the inhibition of angiogenesis, tumor growth, and metastasis in vivo. A putative STAT3-responsive element on the VEGF promoter was identified using a protein–DNA binding assay. Therefore, it has been suggested that STAT3 directly regulates VEGF expression and hence angiogenesis, growth, and metastasis of human pancreatic cancer.
Potential for targeting STAT3 in pancreatic cancer
Complex signaling pathways of STATs and involvement of different molecules may also contribute to the resistance of pancreatic tumor to chemotherapies. For example, multidrug resistance-associated proteins (MRPs) are found to be overexpressed in many tumors, including pancreatic cancer, and a correlation of MRP expression with tumor grading and survival rate has been described in different studies.23,24 Further, it has been found that addition of interferon (IFN)-α to chemotherapy treatments leads to the restoration of chemosensitivity in an orthotopic mouse model of pancreatic cancer. The mechanism behind this involves the downregulation of MRPs.25 Therefore, efforts to develop a strategy to target pancreatic cancer should also consider increasing chemosensitivity to different drugs that are being tested.
As discussed above, increasing evidence supports the role of aberrant STAT3 activation in dysregulated growth and survival of cancer cells, promotion of angiogenesis, and invasion metastasis. Therefore, STAT3 serves as a potential therapeutic target to inhibit tumorigenesis, and a number of studies have confirmed STAT3 as a potential drug target in cancer. In the following section, we discuss the strategies and studies targeting STAT3 in pancreatic cancer cell lines, including in vivo studies in mouse models.
Since tyrosine protein kinases are involved in the activation of STAT3, inhibitors of tyrosine kinase indirectly inhibit STAT3 and, therefore, have antitumor activity. However, STAT3 can also be activated when suppressors of the STAT signaling pathway are inactivated. These suppressors include the SOCS, protein inhibitor of activated STATs, protein tyrosine phosphatases, and peroxisome proliferator-activated receptor-γ (PPAR-γ).13,26 Therefore, STAT3 can be targeted by activation of molecules that inhibit the activity of STAT3. However, inhibition of upstream positive or negative regulators of STAT3 is not the only option to inhibit the activity of STAT3, and direct targeting of STAT3 becomes a relevant strategy for antitumor treatments. Table 1 summarizes the use of various agents for targeting STAT3 in pancreatic cancer cell lines.
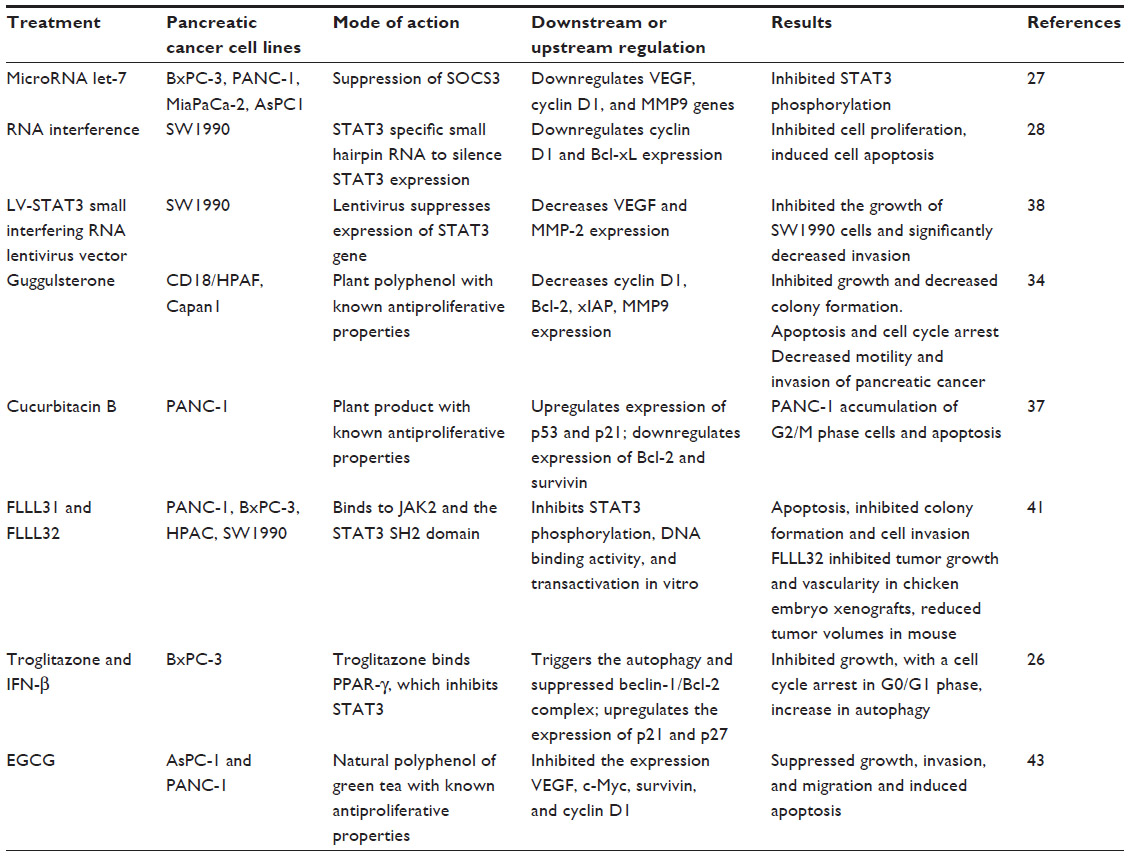 | Table 1 Different agents which directly or indirectly inhibit STAT3 activity in pancreatic cancer cell lines |
Recently, a study has demonstrated that reexpression of let-7 in poorly-differentiated PDAC cell lines reduced the phosphorylation or activation of STAT3, which resulted in reduction of growth and migration potential of these cells.27 However, this study has shown that that let-7 reexpression did not inhibit the expression of total STAT3 as well as its activator, cytokine IL-6. Reexpression of let-7 leads to increased expression of SOCS3, which in turn inhibits the activation/phosphorylation of STAT3. Overall, this study suggests that let-7 expression controls STAT3 activity in pancreatic cancer cells and, therefore, has good therapeutic potential.
In another study, the potential use of small hairpin RNAs to silence the expression of STAT3 was determined in the human pancreatic cancer cell line SW1990.28 By using the recombinant plasmid, a SW1990-RNA interference (RNAi) cell line was generated, which resulted in significantly decreased STAT3 expression. The altered expression of STAT3 results in a change in growth characteristics of human SW1990 cells, both in vitro and in vivo. STAT3 silencing by RNAi inhibited SW1990 cell proliferation, inhibited anchorage-independent growth ability, arrested the cell cycle at G0/G1 phase, and increased cell apoptosis. This study also demonstrated that in vivo STAT3 silencing inhibited tumorigenicity and tumor growth potential of SW1990 cells in athymic nude mouse. The reduced SW1990 cell growth, both in vitro and in vivo, was due to impaired STAT3 expression, which is mediated by the downregulation of the expression of cyclin D1 and Bcl-xL. Therefore, inhibition of STAT3 activation by RNAi may be a potential useful therapeutic option in the treatment of pancreatic adenocarcinoma.
Nam et al29 has studied the effects of E738, a derivative of indirubin and dual inhibitor of JAKs and SFKs on pancreatic cell lines. It inhibits the inhibiting tyrosyl phosphorylation of STAT3 through inactivation of upstream JAKs and SFKs and induces apoptosis in pancreatic cancer cells. Induction of apoptosis was associated with the decreased expression of antiapoptotic proteins Mcl-1 and survivin.
A plant-derived polyphenol bioactive phytochemical, guggulsterone (GS), has been used in traditional medicines, and studies show the antiproliferative activity of GS against several human cancers, including head and neck, prostate, lung, and ovarian cancer.30,31 The inhibitory effects of GS in cancer cells are mediated by inhibiting the activation of transcription factors NF-kappaB and STAT3.32,33 Recently, GS has also been reported to induce apoptosis and inhibit proliferation, motility, and invasion in pancreatic cancer cells.34 This study suggests that antitumor effects of GS possibly involve multiple networks including inhibition of FAK, Src, and JAK/STAT signaling, and down-regulation of MUC4.
Another natural plant product, cucurbitacin B (CuB), is a member of the triterpenoid family and has shown antiproliferative effects on cells from various cancers.35–37 It can suppress the activation of STAT3 and regulate downstream genes involved in cell cycle regulation and apoptosis. The antiproliferative effects of CuB have been assessed in the human pancreatic cancer cell line PANC-1, which has constitutively activated STAT3.37 CuB exerts its inhibitory effect on PANC-1 cells through inhibition of STAT3 activation/phosphorylation, which is completely suppressed after 24 hours of treatment.
Another study has investigated the potential use of RNAi by targeting STAT3 invasion properties of human pancreatic cancer cells.38 Lentivirus LV-STAT3 small interfering RNA inhibits the expression of the STAT3 gene in SW1990 cells. RNAi of STAT3 significantly inhibited the growth of SW1990 cells and their invasive ability and decreased the expression of VEGF and MMP-2. This study suggested that small interfering RNA targeting of STAT3 mRNA via a lentivirus effectively sustains knockdown of STAT3 gene expression in SW1990 cells.
In another study, specimens from human pancreatic cancer tissues and a subset of cell lines were found to constitutively express activated STAT3. Overall, this study showed that JAK/STAT3 signaling supports the growth of human pancreatic cancer via regulation of genes related with cell cycle progression, angiogenesis, and antiapoptosis. Inhibition of activated STAT3 by JAK kinase inhibitor AG490 significantly inhibited the growth of pancreatic cancer cell lines. The antiproliferative effect of AG490 included inhibition of angiogenesis and induction of apoptosis in pancreatic cancer.39
Background benzyl isothiocyanate (BITC) is a naturally occurring anticancer agent from cruciferous vegetables, which has shown anticancer properties and inhibits growth of human pancreatic cancer cells. It has been demonstrated that BITC induces apoptosis in human pancreatic cancer cells, which is associated with substantial reductions in the levels of both phospho-STAT3 and total STAT3 protein.40 BITC suppresses the expression of STAT3 in both a dose- and time-dependent manner. The inhibition of STAT3 by BITC was associated with decreased STAT3-mediated DNA binding activity and decreased transcription of the genes for Mcl-1 and Bcl-2, which are downstream targets of STAT3 activation. In addition, this study also demonstrated that feeding BITC to athymic nude mice significantly suppressed the growth of BxPC-3 pancreatic tumor xenografts, which was associated with increased apoptosis and decreased STAT3 expression.
Two novel small molecule STAT3 inhibitors, FLLL31 and FLLL32, derived from curcumin (bioactive compound of turmeric) are able to bind selectively to JAK2 and the STAT3 SH2 domain, which has important roles in STAT3 dimerization and signal transduction.41 FLLL31 and FLLL32 inhibit STAT3 phosphorylation, its DNA binding ability, and its transactivation in vitro, which results in the impairment of multiple oncogenic processes such as colony formation, cell invasion, and the induction of apoptosis in pancreatic cancer cell lines. FLLL32 can also inhibit tumor growth in chicken embryo xenografts as well as in mouse xenografts.
Troglitazone, a member of the thiazolidinediones, is an antidiabetic and anti-inflammatory drug, which is prescribed for patients with type 2 diabetes. Troglitazone targets the PPAR-γ, which is a member of the nuclear receptor superfamily and regulates fatty acid storage and glucose metabolism. Troglitazone and other PPAR-γ agonists were also reported to have antitumor activity.42 Vitale et al have studied the effects of IFN-β and troglitazone in the pancreatic cancer cell line BxPC-3.26 The combination of IFN-β and troglitazone induced a synergistic effect on growth inhibition, with a cell cycle arrest in the G0/G1 phase. The combination of these agents triggered autophagy and suppressed beclin-1/Bcl-2 complex formation. Troglitazone inhibited STAT3 activity, and its combination with IFN-β upregulated the expression of p21 and p27, which resulted in cell cycle perturbations in BxPC-3 cells.
Epigallocatechin gallate (EGCG) is a major catechin from green tea, which suppresses growth, induces apoptosis, and inhibits invasion and migration of pancreatic cancer cells by interfering with the STAT3 signaling pathway. EGCG inhibited the expression of STAT3 (phospho-STAT3 and total STAT3), its transcription and activation, and the expression of STAT3-regulated genes. This resulted in the induction of apoptosis and the inhibition of cell motility, cell migration, and invasion in pancreatic cancer cell lines AsPC-1 and PANC-1.43 This study also demonstrated the synergistic effects of EGCG with gemcitabine in inducing apoptosis in both cell lines. Moreover, the combination of EGCG and gemcitabine had additive effects on inhibiting the expression of VEGF, c-Myc, survivin, and cyclin D1 in both the cell lines. EGCG and CP690550 (JAK3 inhibitor) also showed synergy in inhibiting the expression of VEGF, c-Myc, survivin, and cyclin D1 in both cell lines. This study suggests that EGCG can enhance the therapeutic potential of gemcitabine and CP690550 in pancreatic cancer cells by inhibiting STAT3. However, most of these inhibitors are only at the experimental stage, and more studies are required before they may be considered for clinical use.
Conclusion
STAT3 has many critical functions in normal human cells and tissues such as differentiation, proliferation, survival, angiogenesis, and regulation of the immune system. However, constitutively activated STAT3 has been identified as abnormal and oncogenic and is associated with a wide range of human cancers. STAT3 plays a pivotal role in the initiation and progression of cancers, involving proliferation, antiapoptosis, invasion, and angiogenesis. Therefore, targeting STAT3 using a specific inhibitor is a useful cancer treatment approach, with the potential for a broad clinical impact.
PDAC is one of the most aggressive human cancer diseases, with early metastasis and high resistance to conventional chemotherapy. Throughout the last decade, a number of studies have shown aberrant activation of STAT3 in pancreatic cancer; therefore, efforts have been poured into the discovery of novel STAT3 inhibitors. Among a number of reported STAT3 inhibitors, a few show promising activity in terms of in vitro inhibition of STAT3 biological functions and the associated antitumor effects, as well as the inhibition of xenografted tumor growth in mouse models. However, the ultimate question is, which inhibitors will be available for clinical use and how well will patients respond to these inhibitors. The survival rate in patients cannot be addressed until a suitable anti-STAT3 agent becomes available for use in patients. Therefore, there is an urgent need for suitable and effective STAT3 inhibitors for clinical application in pancreatic cancer patients.
Importantly, it is evident from specimens of human PDAC that in addition to the STAT3, other signaling pathways are also often upregulated in pancreatic cancer; therefore, a combinational treatment seems even more attractive. In fact, the complexity of pancreatic tumor, vascular involvement, stromal cells, and the heterogeneity in the genetic alterations associated with PDAC create a formidable challenge for targeting a single component. Therefore, it is necessary to explore the use of STAT3 inhibitors along with the conventional therapies and/or inhibitors of other singling components involved in the PDAC.
Acknowledgments
We acknowledge our laboratory members for critical reading of the manuscript, insightful discussions, and valuable advice. The project was funded through the National Institutes of Health (RKS) and the VA Merit Award (SS).
Disclosure
The authors report no conflicts of interest in this work.
References
Neesse A, Krug S, Gress TM, Tuveson DA, Michl P. Emerging concepts in pancreatic cancer medicine: targeting the tumor stroma. Onco Targets Ther. 2013;7:33–43. | |
Bardou M, Le Ray I. Treatment of pancreatic cancer: a narrative review of cost-effectiveness studies. Best Pract Res Clin Gastroenterol. 2013;27(6):881–892. | |
Kolodecik T, Shugrue C, Ashat M, Thrower EC. Risk factors for pancreatic cancer: underlying mechanisms and potential targets. Front Physiol. 2013;4:415. | |
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. | |
Maisonneuve P, Marshall BC, Lowenfels AB. Risk of pancreatic cancer in patients with cystic fibrosis. Gut. 2007;56(9):1327–1328. | |
di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144(6):1220–1229. | |
Rozenblum E, Schutte M, Goggins M, et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57(9):1731–1734. | |
Burris HA, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. | |
Herrmann R, Bodoky G, Ruhstaller T, et al; Swiss Group for Clinical Cancer Research; Central European Cooperative Oncology Group. Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J Clin Oncol. 2007;25(16):2212–2217. | |
Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282(28):20059–20063. | |
Bar-Natan M, Nelson EA, Xiang M, Frank DA. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAKSTAT. 2012;1(2):55–64. | |
Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18(1):45–56. | |
Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007;251(2):199–210. | |
Pilati C, Amessou M, Bihl MP, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med. 2011;208(7):1359–1366. | |
Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905–1913. | |
Mankan AK, Greten FR. Inhibiting signal transducer and activator of transcription 3: rationality and rationale design of inhibitors. Expert Opin Investig Drugs. 2011;20(9):1263–1275. | |
Scholz A, Heinze S, Detjen KM, et al. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology. 2003;125(3):891–905. | |
Toyonaga T, Nakano K, Nagano M, et al. Blockade of constitutively activated Janus kinase/signal transducer and activator of transcription-3 pathway inhibits growth of human pancreatic cancer. Cancer Lett. 2003;201(1):107–116. | |
Miyatsuka T, Kaneto H, Shiraiwa T, et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006;20(11):1435–1440. | |
Wei D, Le X, Zheng L, et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene. 2003;22(3):319–329. | |
Lee JY, Hennighausen L. The transcription factor Stat3 is dispensable for pancreatic beta-cell development and function. Biochem Biophys Res Commun. 2005;334(3):764–768. | |
Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(4):456–469. | |
König J, Hartel M, Nies AT, et al. Expression and localization of human multidrug resistance protein (ABCC) family members in pancreatic carcinoma. Int J Cancer. 2005;115(3):359–367. | |
Lu Z, Kleeff J, Shrikhande S, et al. Expression of the multidrug-resistance 1 (MDR1) gene and prognosis in human pancreatic cancer. Pancreas. 2000;21(3):240–247. | |
Hoffmann K, Mehrle S, Schmidt J, Büchler MW, Märten A. Interferon-alpha restitutes the chemosensitivity in pancreatic cancer. Anticancer Res. 2008;28(3A):1499–1507. | |
Vitale G, Zappavigna S, Marra M, et al. The PPAR-γ agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-β treated pancreatic cancer cells. Biotechnol Adv. 2012;30(1):169–184. | |
Patel K, Kollory A, Takashima A, Sarkar S, Faller DV, Ghosh SK. MicroRNA let-7 downregulates STAT3 phosphorylation in pancreatic cancer cells by increasing SOCS3 expression. Cancer Lett. 2014;347(1):54–64. | |
Huang C, Yang G, Jiang T, Cao J, Huang KJ, Qiu ZJ. Down-regulation of STAT3 expression by vector-based small interfering RNA inhibits pancreatic cancer growth. World J Gastroenterol. 2011;17(25):2992–3001. | |
Nam S, Wen W, Schroeder A, et al. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol Oncol. 2013;7(3):369–378. | |
Leeman-Neill RJ, Wheeler SE, Singh SV, et al. Guggulsterone enhances head and neck cancer therapies via inhibition of signal transducer and activator of transcription-3. Carcinogenesis. 2009;30(11):1848–1856. | |
Shishodia S, Aggarwal BB. Guggulsterone inhibits NF-kappaB and IkappaBalpha kinase activation, suppresses expression of anti-apoptotic gene products, and enhances apoptosis. J Biol Chem. 2004;279(45):47148–47158. | |
Lv N, Song MY, Kim EK, Park JW, Kwon KB, Park BH. Guggulsterone, a plant sterol, inhibits NF-kappaB activation and protects pancreatic beta cells from cytokine toxicity. Mol Cell Endocrinol. 2008; 289(1–2):49–59. | |
Macha MA, Matta A, Chauhan SS, Siu KW, Ralhan R. Guggulsterone (GS) inhibits smokeless tobacco and nicotine-induced NF-κB and STAT3 pathways in head and neck cancer cells. Carcinogenesis. 2011;32(3):368–380. | |
Macha MA, Rachagani S, Gupta S, et al. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013;341(2):166–177. | |
Wakimoto N, Yin D, O’Kelly J, et al. Cucurbitacin B has a potent antiproliferative effect on breast cancer cells in vitro and in vivo. Cancer Sci. 2008;99(9):1793–1797. | |
Yin D, Wakimoto N, Xing H, et al. Cucurbitacin B markedly inhibits growth and rapidly affects the cytoskeleton in glioblastoma multiforme. Int J Cancer. 2008;123(6):1364–1375. | |
Zhang M, Sun C, Shan X, Yang X, Li-Ling J, Deng Y. Inhibition of pancreatic cancer cell growth by cucurbitacin B through modulation of signal transducer and activator of transcription 3 signaling. Pancreas. 2010;39(6):923–929. | |
Yang G, Huang C, Cao J, Huang KJ, Jiang T, Qiu ZJ. Lentivirus-mediated shRNA interference targeting STAT3 inhibits human pancreatic cancer cell invasion. World J Gastroenterol. 2009;15(30):3757–3766. | |
Huang C, Cao J, Huang KJ, et al. Inhibition of STAT3 activity with AG490 decreases the invasion of human pancreatic cancer cells in vitro. Cancer Sci. 2006;97(12):1417–1423. | |
Sahu RP, Srivastava SK. The role of STAT-3 in the induction of apoptosis in pancreatic cancer cells by benzyl isothiocyanate. J Natl Cancer Inst. 2009;101(3):176–193. | |
Lin L, Hutzen B, Zuo M, et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010;70(6):2445–2454. | |
Motomura W, Nagamine M, Tanno S, et al. Inhibition of cell invasion and morphological change by troglitazone in human pancreatic cancer cells. J Gastroenterol. 2004;39(5):461–468. | |
Tang SN, Fu J, Shankar S, Srivastava RK. EGCG enhances the therapeutic potential of gemcitabine and CP690550 by inhibiting STAT3 signaling pathway in human pancreatic cancer. PLoS One. 2012;7(2):e31067. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.