Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Sputum Streptococcus pneumoniae is reduced in COPD following treatment with benralizumab
Authors George L ![]() , Wright A
, Wright A ![]() , Mistry V, Sutcliffe A
, Mistry V, Sutcliffe A ![]() , Chachi L, Haldar K, Ramsheh MY, Richardson M, van der Merwe R, Martin U
, Chachi L, Haldar K, Ramsheh MY, Richardson M, van der Merwe R, Martin U ![]() , Newbold P, Brightling CE
, Newbold P, Brightling CE
Received 14 December 2018
Accepted for publication 29 March 2019
Published 5 June 2019 Volume 2019:14 Pages 1177—1185
DOI https://doi.org/10.2147/COPD.S198302
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Leena George,1 Adam Wright,1 Vijay Mistry,1 Amanda Sutcliffe,1 Latifa Chachi,1 Koirobi Haldar,1 Mohammadali Yavari Ramsheh,1 Matthew Richardson,1 René van der Merwe,2 Ubaldo Martin,3 Paul Newbold,3 Christopher E Brightling1
1Department of Respiratory Sciences, Institute for Lung Health, National Institute for Health Research Biomedical Research Centre, University of Leicester, Leicester, UK; 2Clinical Respiratory Management, MedImmune Ltd., Cambridge, UK; 3Global Medical Affairs, AstraZeneca, Gaithersburg, MD, USA
Abstract: We hypothesized whether the reduction in eosinophilic airway inflammation in patients with chronic obstructive pulmonary disease (COPD) following treatment with benralizumab, a humanized, afucosylated, monoclonal antibody that binds to interleukin-5 receptor α, increases the airway bacterial load. Analysis of sputum samples of COPD patients participating in a Phase II trial of benralizumab indicated that sputum 16S rDNA load and Streptococcus pneumoniae were reduced following treatment with benralizumab. However, in vitro, eosinophils did not affect the killing of the common airway pathogens S. pneumoniae or Haemophilus influenzae. Thus, benralizumab may have an indirect effect upon airway bacterial load.
Keywords: COPD, benralizumab, IL-5, bacterial load, S. pneumoniae, H. influenzae
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by irreversible airflow obstruction and airway inflammation. Although typically neutrophilic, COPD is eosinophil-predominant in 10%‒40% of cases.1–3 Increased airway or blood eosinophil counts are associated with a good response to corticosteroids in stable COPD3 and during exacerbations.4 Interleukin-5 (IL-5) binds with high affinity to the IL-5 receptor (R) alpha (IL-5Rα) subunit and plays a pivotal role in the differentiation and maturation of eosinophils in the bone marrow and their survival in tissue.1 In a 1-year randomized placebo-controlled trial of benralizumab,5 a humanized, afucosylated, monoclonal antibody that inhibits IL-5Rα activation and promotes antibody-dependent cell-mediated cytotoxicity (leading to near complete eosinophil depletion), improvements in lung function and symptoms and reduction in exacerbations were observed in patients with eosinophilic inflammation. However, in non-eosinophilic COPD patients, exacerbation frequency increased following benralizumab treatment vs placebo. Likewise, in a 6-month trial, the IL-5 neutralizing monoclonal antibody mepolizumab6 reduced exacerbations vs placebo in COPD patients with an increased blood eosinophil count but resulted in a greater exacerbation frequency in those with a low blood eosinophil count. This finding contrasts with that for asthma for which absence of eosinophilic inflammation is associated with neither benefit nor harm to anti‒IL-5(R).1 Interestingly, the airway microbiome is distinct between COPD patients with vs those without eosinophilic inflammation.7 Corticosteroid therapy alters the airway microbiome7 and consequently might hinder recovery during exacerbations in patients without eosinophilic inflammation.4 Whether this exacerbation relationship to low eosinophil count is genuine and these effects are partly because of attenuation of eosinophilic inflammation remain unknown. We, therefore, hypothesized that reduction in eosinophilic airway inflammation following benralizumab treatment increases airway bacterial load.
Methods
Sputum samples were collected from COPD patients participating in the Phase II trial5 of benralizumab at 28 days before baseline, at baseline, and 57 and 255 days after receiving benralizumab or placebo. Written informed consent was provided by all patients. The study was conducted in accordance with the Declaration of Helsinki and was approved by the national and local ethics committees (Table S1). Supernatants were stored at −80 °C. For those patients who provided adequate sputum supernatant (>100 μL) and ≥1 sample before and after therapy, DNA was extracted using QIAamp DNA mini kit (QIAGEN, Hilden, Germany) as per the manufacturer’s protocol. Hydrolysis-based TaqMan assays were used to quantify 16S rDNA gene load (Integrated DNA Technologies [IDT], Coralville, Iowa) and Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae by targeting the FucP, CopB (Applied Biosystems Life Technologies), and pneumolysin (IDT) genes, respectively. Quantification was determined relative to prepared standard curves for each bacterial strain by using Stratagene Mx3000P (Stratagene; La Jolla, CA) (Table S2).
Ex-vivo peripheral blood eosinophils and neutrophils of >95% purity from healthy individuals were isolated as previously described.8 In vitro, Escherichia coli (strain PA360), H. influenzae (NCTC11872), and S. pneumoniae (D39), grown to late-log phase, were incubated alone or with blood eosinophils or neutrophils in triplicate for 1 h at 37 °C in the presence of 0.1% (E. coli, H. influenzae) or 10% (S. pneumoniae) non-heat inactivated human serum. We chose serum concentrations that were sublethal for nontypeable H. influenzae and E. coli based on titration experiments. Colony forming units were enumerated for each condition to determine the bacterial killing effect of each granulocyte.
Statistical analysis was performed using R 3.4.1 (The R Foundation for Statistical Computing) and PRISM (GraphPad; La Jolla, CA). Change in bacterial load over time within the treatment or placebo group was performed by fitting a generalized linear mixed model for each of 16S rDNA, H. influenzae, and S. pneumoniae. The dependent variables were time, treatment group, and an interaction term, time*treatment group. A random intercept and a random effect for time were included. Correlations were undertaken between change in bacterial load, forced expiratory volume in 1 s, and symptoms and health status. Comparisons were made between conditions for bacterial killing experiments by 2-way analysis of variance.
Results
Sputum supernatant samples were assessed from 14 benralizumab-treated and 15 placebo-treated patients. Clinical characteristics were similar between the groups (Table S3). The 16S rDNA load decreased following benralizumab treatment but remained unchanged with placebo (Figure 1A and B). The quantity of S. pneumoniae in the benralizumab and placebo groups decreased significantly, with the reduction numerically and statistically greater in the benralizumab group (Figure 1C and D). The reduction in 16S rDNA load was associated with a reduction in the quantity of S. pneumoniae in the benralizumab group but not in the placebo group (Figure 1C and D). However, there were no significant changes in the quantity of H. influenzae (Figure 1E and F) or M. catarrhalis (data not shown) in the 2 treatment groups. The reduction in total bacterial load, or quantity of S. pneumoniae in the sputum supernatants in benralizumab-treated patients, was not associated with either baseline blood eosinophil count or change in lung function, symptoms or health status following treatment with benralizumab. In contrast with ex-vivo neutrophils, ex-vivo blood eosinophils did not kill H. influenzae or S. pneumoniae in vitro (Figure 2A‒D).
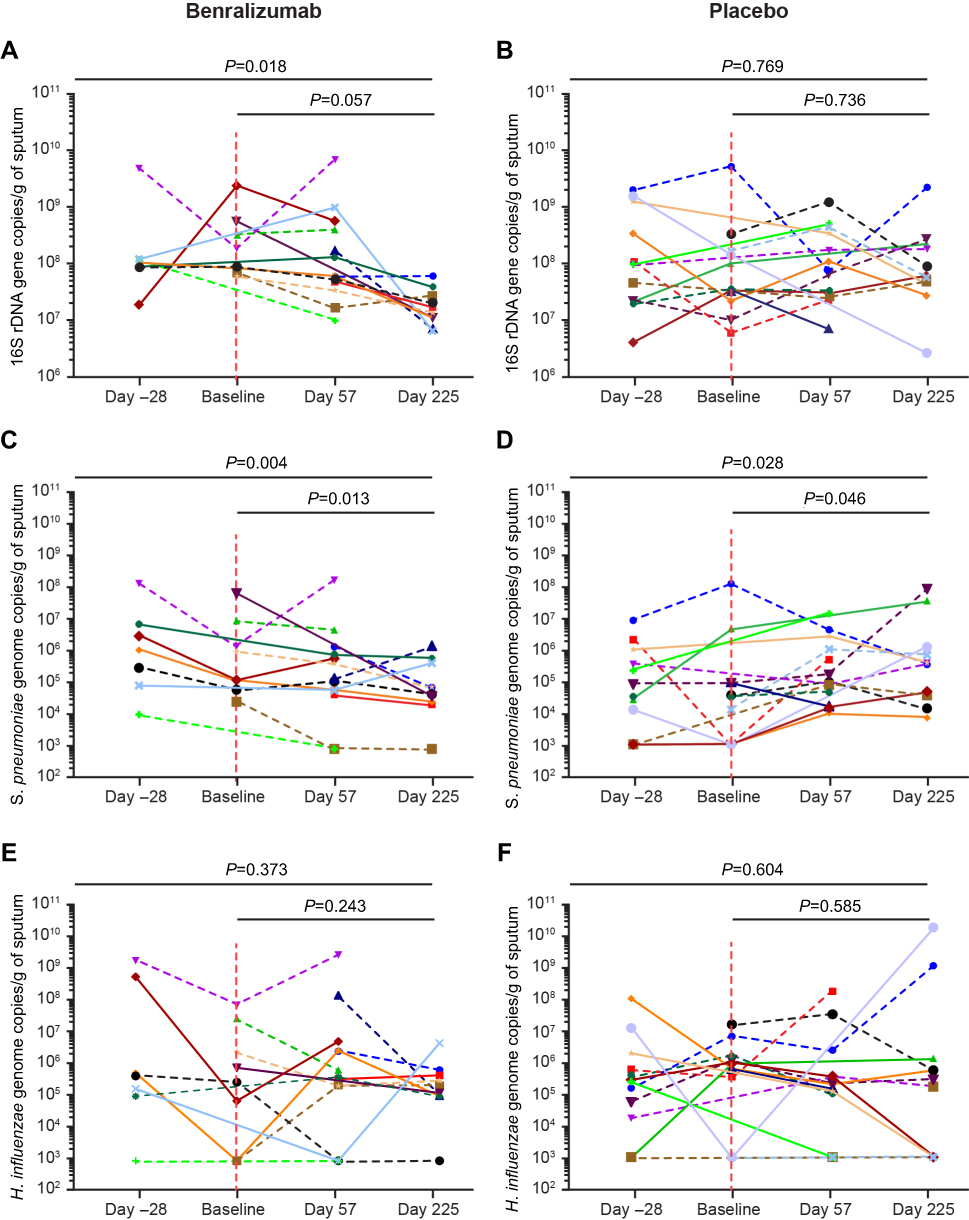 | Figure 1 Sputum bacterial load in response to benralizumab vs placebo. Plots for (A and B) total bacterial load, (C and D) Streptococcus pneumoniae, and (E and F) Haemophilus influenzae for individual patients from Day −28 to Day 225 in response to benralizumab vs placebo. Vertical dotted lines delineate first treatment dose. Horizontal dotted lines represent patients with a baseline sputum eosinophil count ≥3%. P-values are provided for the mixed models, including all time points, but excluding Day −28. |
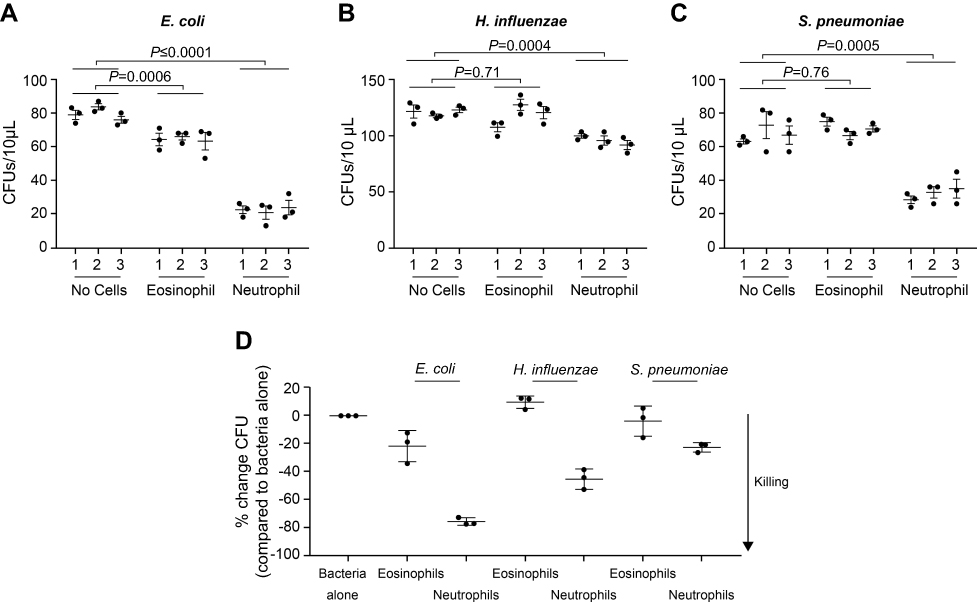 | Figure 2 Ex-vivo blood eosinophils do not kill Haemophilus influenzae or Streptococcus pneumoniae in vitro. Escherichia coli (A), H. influenzae (B), and S. pneumoniae (C) were incubated alone or with purified blood eosinophils or neutrophils, as indicated in triplicate (x-axis), for 1 h at 37 °C, with shaking at 200 rpm in U-bottom plates. Bacteria and cells were incubated in a ratio of ~1:100 in the presence of either 0.1% (A and B) or 10% (C) non-heat inactivated AB-human serum supplemented with RPMI. By removing 3×10 µL aliquots from each well and incubating overnight on agar, colony forming units were determined in each 10 µL suspension. Data in A, B, and C represent data from one individual. Data in D represents cumulative data from three separate healthy donors showing % change in CFU/10 µL for each bacterium tested in the presence of eosinophils or neutrophils relative to bacteria alone. A, B, and C bars indicate mean (SEM), with statistics calculated using a 2-way ANOVA (Dunnett’s multiple comparison test). Abbreviations: ANOVA, analysis of variance; CFU, colony forming unit; rpm, revolutions per minute; RPMI, Roswell Park Memorial Institute medium; SEM, standard error of the mean. |
Discussion
Contrary to our hypothesis, we found that 16S rDNA load and the quantity of the common pathogen S. pneumoniae decreased following benralizumab treatment. However, this was not associated with clinical outcomes. There was also a small decrease in the quantity of S. pneumoniae in the placebo group. In vitro, blood eosinophils did not affect bacterial killing of S. pneumoniae and H. influenzae, while small effects were observed on E. coli, similar to previously published reports.9,10 Therefore, the effects observed in vivo are more likely an indirect effect of benralizumab attenuating eosinophilic inflammation. Macrophage efferocytosis of eosinophils8 might further impair macrophage phagocytosis of bacteria; therefore, their reduction may improve bacterial clearance. Similarly, reduction in eosinophilic inflammation is likely to result in a greater percentage of neutrophils that might consequently enhance bacterial clearance. Intriguingly, although these possible mechanisms might explain the reduction in bacterial load following benralizumab treatment, they do not provide a rationale for the apparent increased exacerbation risk in non-eosinophilic COPD patients, as observed in the Phase IIa study, following anti‒IL-5Rα treatment. Further investigation is needed to assess whether the clinical findings are real and what the underlying cause may be.
The retrospective study design limited the number of available samples, restricted analysis to sputum supernatants rather than whole sputum plugs, and limited us to targeted quantitative polymerase chain reactions rather than broader sequencing approaches. Future prospective studies, including whole sputum and microbiome sequencing, are required to further determine the effects of anti‒IL-5/anti‒IL-5Rα therapies on airway ecology.
Conclusions
Sputum 16S rDNA and S. pneumoniae bacterial load are reduced in COPD patients following benralizumab treatment. However, how biologics affect the airway microbiome in obstructive lung diseases warrants further investigation.
Abbreviation list
COPD, chronic obstructive pulmonary disease; IDT, Integrated DNA Technologies; IL-5, interleukin-5; IL-5Rα, IL-5 receptor alpha; R, receptor.
Acknowledgments
The authors thank Professor Peter Andrew and Dr Vitor Fernandes, of the University of Leicester for their assistance with the in vitro bacterial experiments. Editorial support was provided by Cactus Communications and by Michael A Nissen, ELS, of AstraZeneca. This support was funded by AstraZeneca. This work was funded in part by Airway Disease PRedicting Outcomes through Patient Specific Computational Modelling (AirPROM) project (funded through the European Union’s Seventh Framework Programme [FP7] grant), the National Institute for Health Research (NIHR) Leicester Respiratory Biomedical Centre, and MedImmune Ltd. This paper presents independent research funded by the NIHR. The views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR, or the Department of Health.
Author contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
RVDM is an employee of MedImmune LLC, the manufacturer of benralizumab. UM and PN are employees of AstraZeneca, the manufacturer of benralizumab. UM reports holding shares in AstraZeneca Pharmaceuticals. CEB report grants and personal fees from AstraZeneca/MedImmune LLC, during the conduct of the study. The authors report no other conflicts in this work.
References
1. George L, Brightling CE. Eosinophilic airway inflammation: role in asthma and chronic obstructive pulmonary disease. Ther Adv Chronic Dis. 2016;7:34–51. doi:10.1177/2040622315609251
2. Singh D, Kolsum U, Brightling CE, et al. Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J. 2014;44:1697–1700. doi:10.1183/09031936.00162414
3. Brightling CE, Monteiro W, Ward R, et al. Sputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2000;356:1480–1485. doi:10.1016/S0140-6736(00)02872-5
4. Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med. 2012;186:48–55. doi:10.1164/rccm.201108-1553OC
5. Brightling CE, Bleecker ER, Panettieri RA
6. Pavord ID, Chanez P, Criner GJ, et al. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. N Engl J Med. 2017;377:1613–1629. doi:10.1056/NEJMoa1708208
7. Wang Z, Bafadhel M, Haldar K, et al. Lung microbiome dynamics in COPD exacerbations. Eur Respir J. 2016;47:1082–1092. doi:10.1183/13993003.01406-2015
8. Eltboli O, Bafadhel M, Hollins F, et al. COPD exacerbation severity and frequency is associated with impaired efferocytosis of eosinophils. BMC Pulm Med. 2014;14:112. doi:10.1186/1471-2466-14-112
9. Yazdanbakhsh M, Eckmann CM, Bot AA, et al. Bactericidal action of eosinophils from normal human blood. Infect Immun. 1986;53:192–198.
10. Ahrén IL, Eriksson E, Egesten A, et al. Nontypeable haemophilus influenza activates human eosinophils through beta-glucan receptors. Am J Respir Cell Mol Biol. 2003;29:598–605. doi:10.1165/rcmb.2002-0138OC
Supplementary materials
 |  | 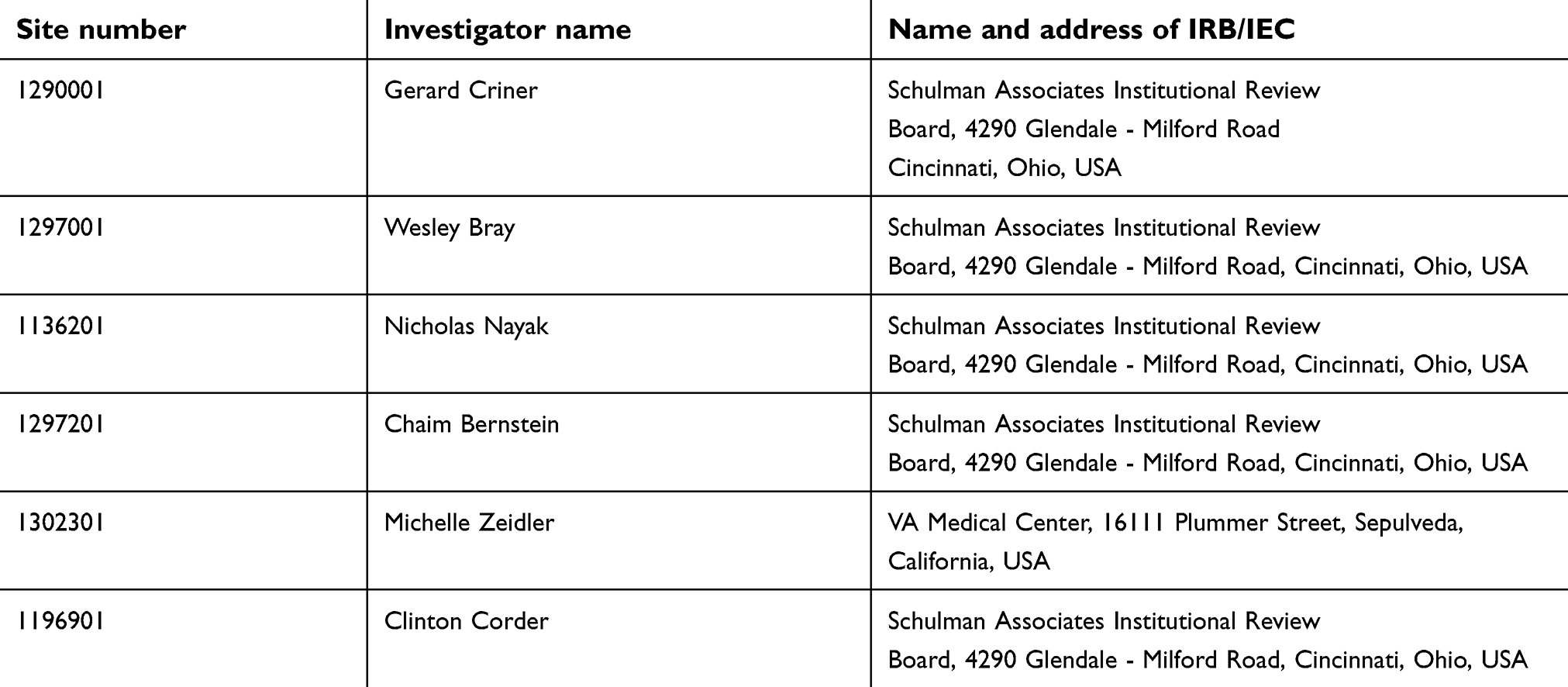 | Table S1 List of institutional review boards/independent ethics committees |
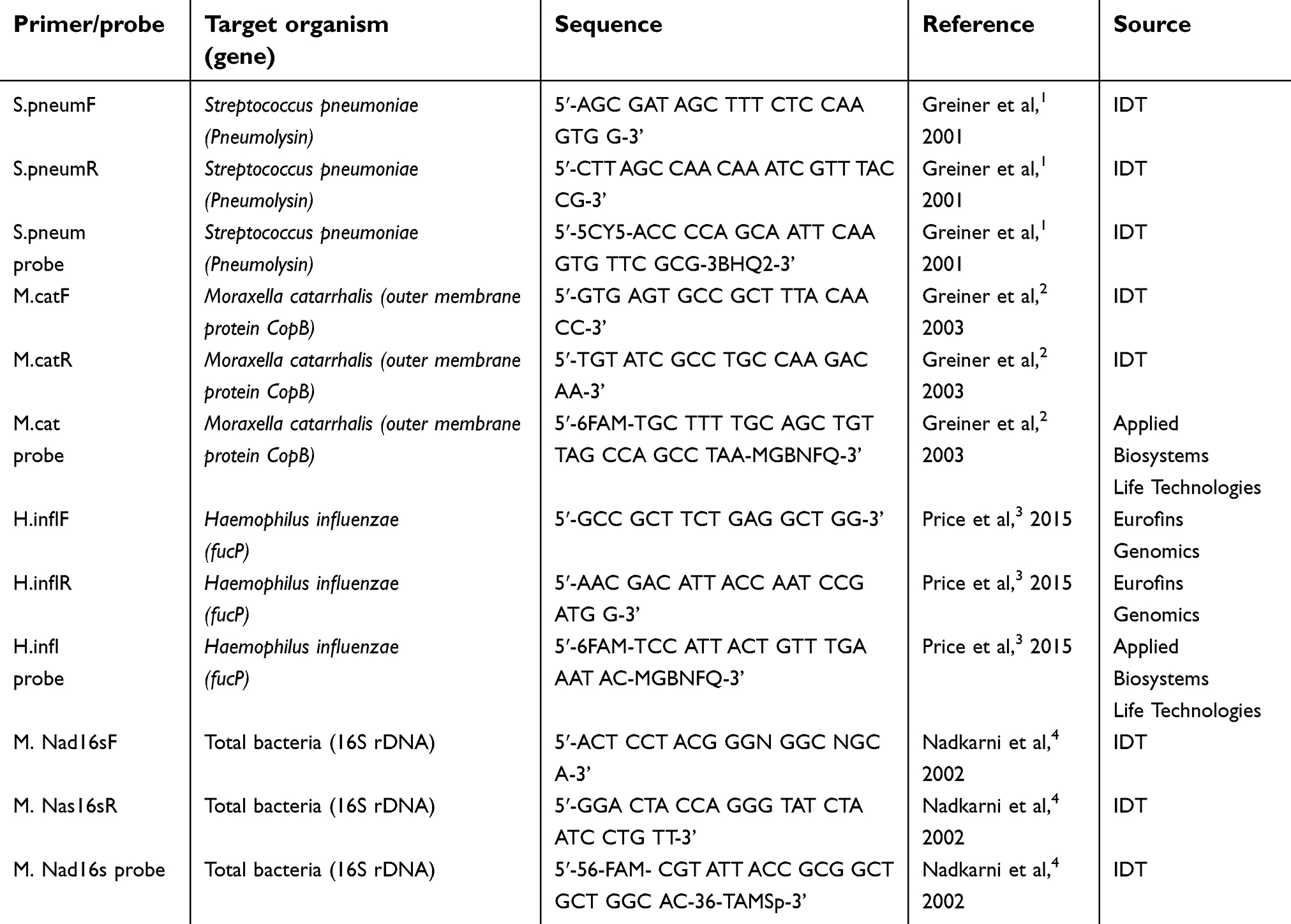 | Table S2 List of primers used in the study |
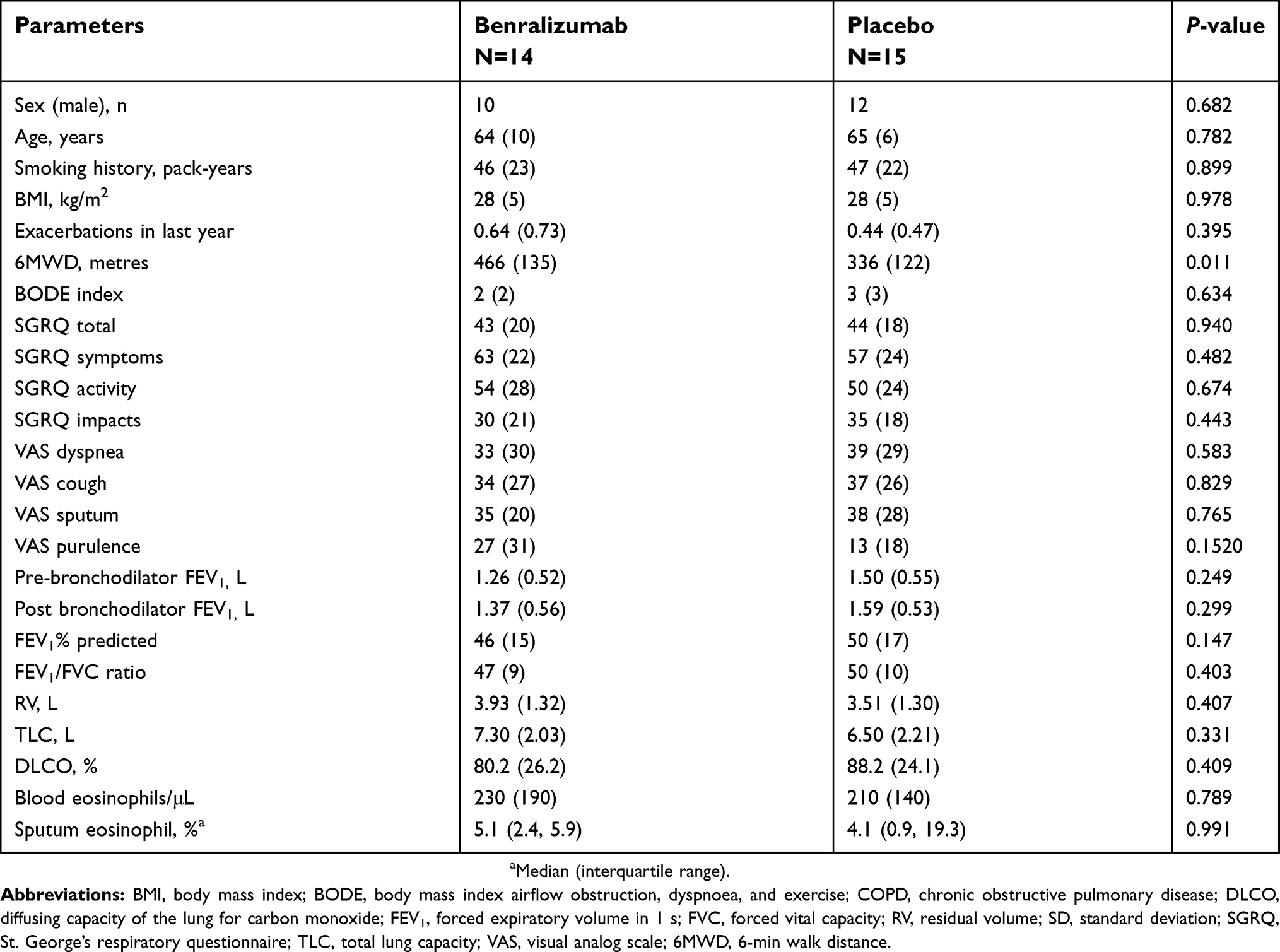 | Table S3 Clinical characteristics of COPD patients |
References
1. Greiner O, Day PJ, Bosshard PP, et al. Quantitative detection of Streptococcus pneumoniae in nasopharyngeal secretions by real-time PCR. J Clin Microbiol. 2001;39(9):3129–3134.
2. Greiner O, Day PJ, Altwegg M, et al. Quantitative detection of Moraxella catarrhalis in nasopharyngeal secretions by real-time PCR. J Clin Microbiol. 2003;41(4):1386–1390.
3. Price EP, Sarovich DS, Nosworthy E, et al. Haemophilus influenzae: using comparative genomics to accurately identify a highly recombinogenic human pathogen. BMC Genomics. 2015;16(1):641.
4. Nadkarni MA, Martin FE, Jacques NA, et al. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148(1):257–266.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.