Back to Journals » The Application of Clinical Genetics » Volume 12
Spotlight on Warsaw Breakage Syndrome
Authors Pisani FM
Received 25 September 2019
Accepted for publication 13 November 2019
Published 5 December 2019 Volume 2019:12 Pages 239—248
DOI https://doi.org/10.2147/TACG.S186476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Francesca M Pisani
Istituto di Biochimica e Biologia Cellulare, Consiglio Nazionale delle Ricerche, Naples 80131, Italy
Correspondence: Francesca M Pisani
Istituto di Biochimica e Biologia Cellulare, Consiglio Nazionale delle Ricerche, Via P. Castellino, 111, Naples, Italy
Tel +39 – 0816132292
Fax +39 0816132277
Email [email protected]
Abstract: Warsaw breakage syndrome (WABS) is a very rare recessive hereditary disease caused by mutations in the gene coding for the DNA helicase DDX11, involved in genome stability maintenance and sister cohesion establishment. Typical clinical features observed in WABS patients include growth retardation, facial dysmorphia, microcephaly, hearing loss due to cochlear malformations and, at cytological level, sister chromatid cohesion defects. Molecular bases of WABS have not yet been elucidated, due to lack of disease animal model systems and limited knowledge of the DDX11 physiological functions. However, WABS is considered to belong to the group of cohesinopathies, genetic disorders due to mutations of subunits or regulators of cohesin, the protein complex responsible for tethering sister chromatids from the time of their synthesis till they separate in mitosis. Recent evidences suggest that cohesin and its regulators have additional key roles in chromatin organization by promoting the formation of chromatin loops. This “non-canonical” function of cohesin is expected to impact gene transcription during cell differentiation and embryonic development and its dis-regulation, caused by mutation/loss of genes encoding cohesin subunits or regulators, could originate the developmental defects observed in cohesinopathies. Ethiopathogenesis of WABS is discussed in line with these recent findings and evidence of a possible role of DDX11 as a cohesin regulator.
Keywords: cohesinopathies, developmental disorders, Warsaw breakage syndrome, DDX11, genome stability, sister chromatid cohesion
Introduction
Warsaw breakage syndrome (WABS) is a very rare autosomal recessive disease due to bi-allelic mutations of the gene coding for the DNA helicase DDX11.1,2 Cardinal clinical features observed in WABS patients include severe pre- and post-natal growth retardation, microcephaly, sensorineural hearing loss, cochlear anomalies, facial dysmorphia and sister chromatid cohesion defects. DDX11 (also named ChlR1, being related to the Saccharomyces cerevisiae chromosome loss 1, Chl1, protein) is an ATP-dependent DNA helicase with 5′ to 3′ directionality that belongs to the DNA helicase super-family 2 (SF2). The presence of an iron–sulfur cluster (Fe–S) domain classifies DDX11 as a member of the subgroup of Fe–S DNA helicases. This latter also includes the Xeroderma pigmentosum group D (XPD) protein, FANCJ and RTEL1, which all have key roles in genome maintenance pathways and are linked to rare genetic syndromes and cancer predisposition.3,4
Here, I review what is known about WABS in terms of clinical reports, diagnostic tools, disease animal model systems and ethiopathogenesis in light of the most recent discoveries of DDX11 physiological roles.2
Molecular Properties and Cellular Functions of DDX11
The biochemical and enzymatic properties of human DDX11 were investigated in many laboratories in the last two decades.1,2,5–9 DDX11 DNA helicase was reported to preferentially unwinds forked duplex DNA substrates with non-complementary 5ʹ- and 3ʹ-single-stranded arms; whereas, DNA molecules having blunt ends or only a 3ʹ-tail are not unwound by DDX11 in enzymatic assays carried out in vitro. Additional substrates of the DDX11 helicase are three-stranded D-loops with an invading 3ʹ-end, bi-molecular anti-parallel G-quadruplex (G4) with two 5ʹ-tails and DNA molecules containing triple-stranded (triplex) structures with a 5ʹ-single-stranded overhang on the third strand. In contrast, unimolecular G4- and Holliday junction-containing DNA molecules are not resolved by DDX11. Besides, DDX11 was proposed to displace proteins bound to DNA, as it was found to be able to disrupt the high-affinity streptavidin:biotin interaction in a helicase protein concentration- and ATP-dependent manner in assays where biotinylated oligonucleotides bound to streptavidin were used as substrates.7 A similar protein displacement activity was also demonstrated for the Fe–S DNA helicase FANCJ, whereas human RECQ1, Werner and Bloom DNA helicases do not display this enzymatic function.10 Nonetheless, the physiological relevance of DDX11 substrate preference is not completely understood. It was reported that DDX11-depleted U2OS cells were resistant to treatment with Telomestatin, a G4 DNA-binder, and did not display increased DNA damage (using γ-H2AX foci formation as a readout) upon treatment with this compound.8 On the other hand, exposure of DDX11-downregulated HeLa cells to a triplex DNA-stabilizing agent (benzoquinoquinoxaline, BQQ) caused a remarkable increase of triplex-DNA structures and DNA damage (as detected by immuno-fluorescence with an anti-triplex DNA and anti-γ-H2AX specific antibodies, respectively).9 These cellular analyses suggest that DDX11 may have a more prominent role in counteracting the formation of triplex-DNA structures than in untangling unimolecular G-quartets, in line with the substrate preference displayed in vitro.
The participation of human DDX11 in DNA repair pathways was underlined by the finding that DDX11-knockdown HeLa cells are highly sensitive to cisplatinum and bleomycin, a radio-mimetic compound that induces the formation of DNA double-stranded breaks.11 The role of DDX11 in DNA repair appears to be evolutionarily conserved, as the budding yeast DDX11 ortholog, Chl1, was shown to preserve genome integrity against exposure to genotoxic agents, such as methylmethane sulfonate (MMS) or ultraviolet (UV) rays.12,13 Recently, the Branzei group demonstrated that in DT40 chicken cell DDX11 is required to repair DNA bulky lesions induced by MMS and to promote DNA trans-lesion synthesis through abasic sites in concert with the 9-1-1 checkpoint clamp and its loader subunit, Rad17, mainly in a post-replicative manner. Besides, avian DDX11 was found to be involved in the resolution of DNA inter-strand cross-links in a pathway that is parallel and subsidiary to the Fanconi anemia one.14
In a study carried out in collaboration by the Pisani and Brosh groups, it was shown that DDX11 cooperates with Timeless, a component of the replication fork-protection complex, in assisting smooth progression of replisomes in stressful conditions in HeLa cells. DDX11 and Timeless were found to act in concert to promote efficient rescue of stalled replication forks after treatment of HeLa cells with hydroxyurea.15 However, the precise mechanism by which DDX11 and Timeless counteract replication stress has not yet been clarified. In a subsequent recent work, Pisani and colleagues reported that the direct interaction between DDX11 and Timeless was critical for promoting sister chromatid cohesion establishment in HeLa cells and for stable binding of cohesin, the protein complex responsible for tethering sister chromatids, to the replication machinery during S phase.16 In this same study purified recombinant DDX11 and cohesin were demonstrated to directly interact in vitro. Besides, DDX11 down-regulation was found to remarkably reduce the association of cohesin to the ongoing replisomes, as revealed to co-immunoprecipitation experiments in cell extracts using an antibody directed against Cdc45. This latter is a component of the replicative DNA helicase (the Cdc45/Mcm2-7/GINS, CMG, complex) that associates to chromatin by directly binding Mcm and GINS only during S phase. Analogously, a key role in coupling DNA replication with chromosomal cohesion is played by the S. cerevisiae Chl1 protein, which was demonstrated to be anchored to the replication forks through a direct interaction with the replication factor Ctf4.17 Nevertheless, the molecular mechanisms underlying the functional connection between the DNA replication machinery and the cohesin network have not yet been completely elucidated either in yeast or in mammalian cells. Besides, while roles in DNA repair and sister chromatid cohesion were proposed to be separable functions of yeast Chl1,17 it is not clear if the same paradigm is also valid for human DDX11. Addressing this issue would be relevant for our understanding of the molecular basis of WABS and would probably require the identification of any DDX11 separation-of-function mutant proficient in DNA repair, but unable to promote chromosomal cohesion. However, finding that WABS derived cells combine increased sensitivity to genotoxic agents together with sister chromatid cohesion defects (and chromatin structure alterations) indicates that anomalies of both DDX11 cellular functions underlie the observed pathological phenotypes.
Warsaw Breakage Syndrome, a Cohesinopathy
Analysis of cultured T lymphocytes and immortalized B lymphoblasts or fibroblasts from WABS patients revealed sister chromatid cohesion anomalies. These chromosome morphological defects resemble the ones observed in metaphase lymphoblasts from individuals affected by Roberts syndrome (RBS), another rare autosomal recessive disease due to mutations of the ESCO2 gene.18 ESCO2 is an acetyl-transferase required for chromosomal cohesion establishment during S phase and is one of the two human orthologs of budding yeast ECO1.19,20 RBS patients show various skeletal malformations and various degree of intellectual disability.18 Besides, most cases of RBS result in spontaneous abortion, stillbirth, or neonatal death. Both WABS and RBS are classified as cohesinopathies, genetic diseases caused by mutations in genes involved in the sister chromatid cohesion process. Other cohesinopathies include Cornelia de Lange syndrome (CdLS), caused by mutations in genes encoding cohesin structural components (SMC1A, SMC3 and RAD21) and regulators (NIPBL and HDAC8) and Chronic Atrial and Intestinal Dysrhythmia (CAID) syndrome, linked to mutations of the SGOL1 gene encoding Shugoshin.21–23 The sister chromatid cohesion anomalies observed in cells derived from WABS (and also RBS) patients consist in a high proportion of metaphase chromosomes displaying a “railroad” morphology of the paired sisters in each cell: the centromere constriction appears loosened (premature centromere division, PCD), a cytogenetic feature likely due to repulsion of the corresponding heterochromatic regions. Besides, in a not negligible percentage of metaphase chromosome spreads, all the sister chromatid pairs are completely separated, a phenotype known as premature chromatid separation (PCS). Of note, the percentage of patient-derived cells displaying these chromosomal cohesion anomalies (PCD and PCS) is substantially increased after treatment of the cultures with genotoxic agents (such as, mitomycin C, MMC, a DNA cross-linking agent).24 The sister chromatid cohesion defects, observed in cells derived from WABS probands, are in line with the findings that DDX11 and Chl1 play a key role in the chromosomal cohesion process in human and S. cerevisiae cells, respectively.25–27 In contrast, sister chromatid cohesion anomalies are not found in cells from individuals affected by other cohesinopathies (CdLS or CAID).21–23
Diagnostic Overlap Between WABS and Fanconi Anemia
WABS and RBS are very rare diseases with a prevalence estimated to be less than 1/1×106. However, it is likely that the incidence of WABS is underestimated. First of all, it should be pointed out that the compound heterozygosity condition of the DDX11 gene could give rise to a lower penetrance phenotype in individuals that could have not been even diagnosed with WABS due to very mild clinical features (see Section 5). Moreover, it should be also taken into account a possible diagnostic overlap of WABS with Fanconi anemia (FA). This latter is a recessive genetic disorder characterized by a variety of clinical manifestations including growth retardation, microcephaly, skeletal malformations, progressive bone marrow failure and a pronounced cancer predisposition.28 Because of this symptom diversity, FA diagnosis is based on the analysis of increased chromosome breakage in cultured patient lymphocytes, following treatment with DNA cross-linking agents, such as diepoxybutane (DEB) and MMC. Since cells from FA patients exhibit an extraordinarily sensitive response to these genotoxic agents, this test has been considered the gold standard for the ultimate diagnosis of this disease. Nonetheless, a high sensitivity to these same compounds was also detected in cells derived from individuals suffering from other chromosomal instability disorders, including WABS, RBS and Nijmegen breakage syndrome. Therefore, the use of the chromosomal breakage test could lead to a misdiagnosis of FA, also due to other common clinical manifestations of all these genetic syndromes. In particular, a high percentage of FA-patients of the Complementation Group A display short stature and anomalous skin pigmentation; whereas a small proportion of FA patients belonging to the Complementation Group A, B, D1 and D2 has limb (radial and/or thumb) anomalies. All these congenital defects are commonly found in WABS-affected individuals (see Table 1). Therefore, in order to avoid mistakes in diagnosis, clinicians are advised to analyze metaphase chromosome spreads for spontaneously occurring morphological anomalies (PCD and PCS), which are typically found in WABS- and RBS-affected individuals and not in FA or CdLS patients.28
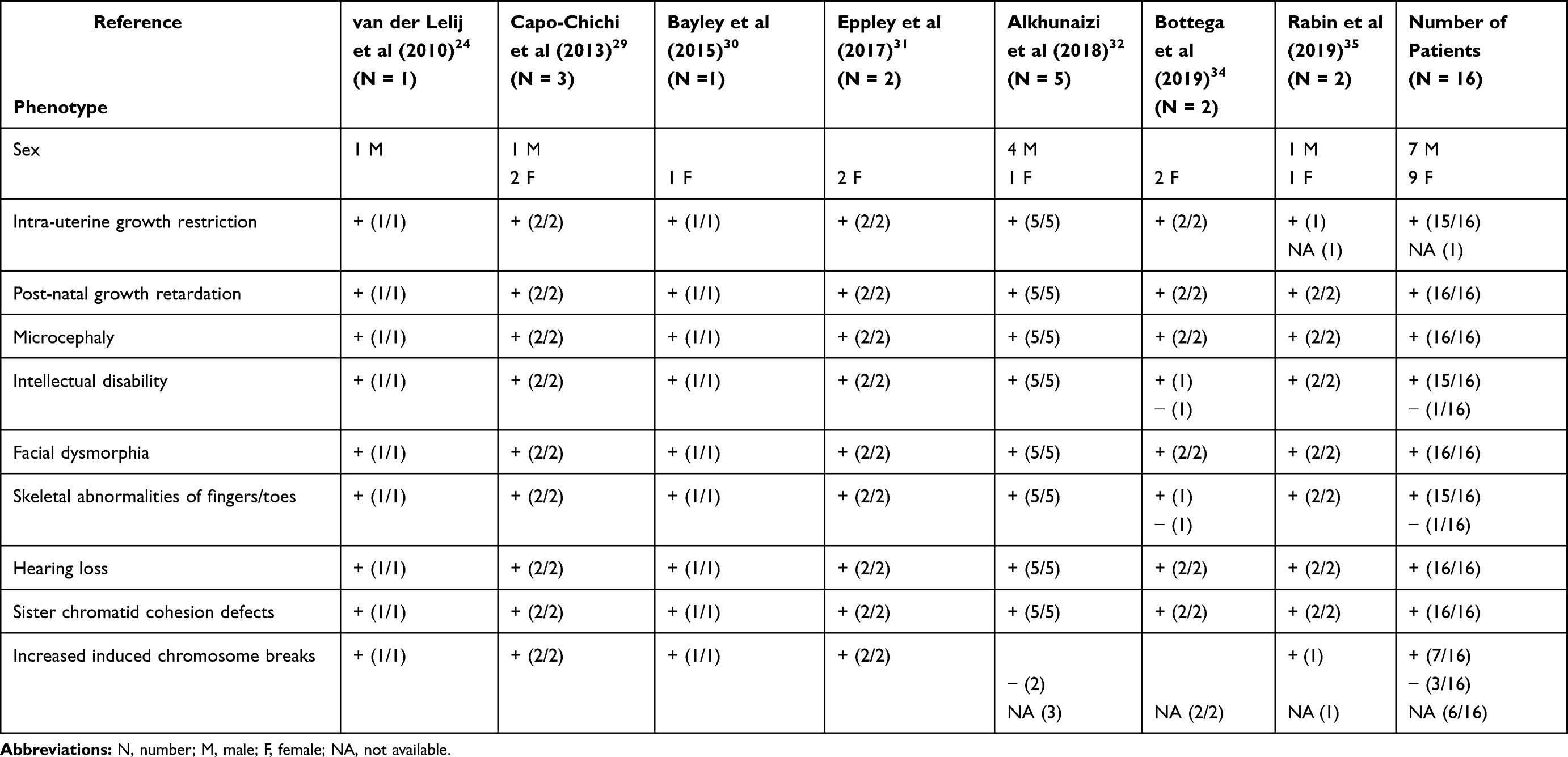 |
Table 1 Most Common Clinical Features Observed in WABS Patients |
History of WABS Clinical Reports
To date, a total of 16 cases of patients suffering from WABS were described in the medical literature with different gene mutations (see Figure 1).
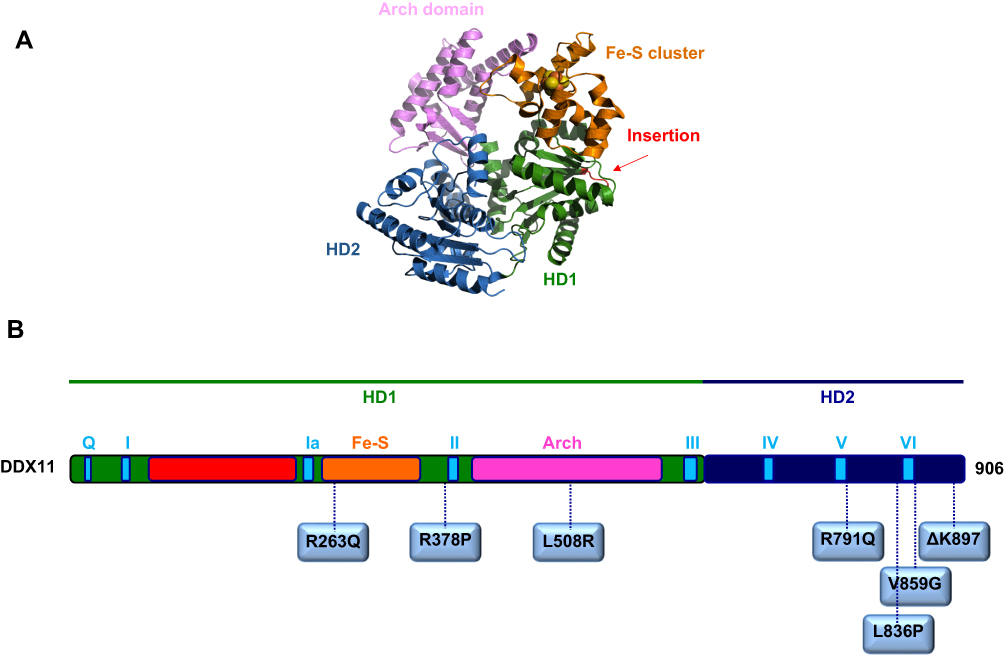 |
Figure 1 (A) A homology model for the human DDX11 helicase based on an archaeal XPD X-ray structure (PDB ID: 4A15). (B) Schematic representation of the human DDX11 polypeptide chain showing location of the amino-acid substitutions described in WABS-affected individuals (see text for details). The domain color-coded is as follows: RecA homology domain 1, HD1, green, and 2, HD2, dark blue; conserved helicase motifs, light blue; Fe-S cluster domain, orange; Arch domain, pink; partially unstructured insertion between motifs I and Ia, red. |
The first case was reported in 2010 by de Winter group in Amsterdam in a male individual from Warsaw (Poland), inspiring the disease name.24 Sequence analysis of the patient genomic DNA revealed the presence of a splice-site mutation in intron 22 of the maternal DDX11 gene allele (leading to deletion of the last 10 bps of exon 22 and to a premature stop codon in exon 23), and a 3-bp deletion in exon 22 of the paternal allele (leading to deletion of the triplet coding for the highly conserved residue K897). Immunoblotting revealed hardly detectable DDX11 protein levels in lymphoblasts and fibroblasts from the affected individual suggesting that the mutant protein was highly unstable.24 In a subsequent biochemical study by Brosh and colleagues, DDX11 ΔK897 mutant protein was produced in recombinant form, purified and found to be able to bind ATP, completely unable to hydrolyze it and to bind and unwind forked DNA duplexes in vitro.7
Three years later, in 2013, a second case of WABS was reported.29 Two brothers and one sister from the same Lebanese consanguineous family were identified as bearing a novel DDX11 bi-allelic homozygous mutation that resulted in the substitution of a conserved Arginine with Glutamine (R263Q) in the protein Fe–S cluster domain.29 The DDX11 mutant protein (DDX11 R263Q) was produced in recombinant form, purified and found to be almost completely unable to bind and unwind either a forked DNA duplex or a two-stranded anti-parallel G-4 DNA substrate. This latter was tested in this study, as it was found to be the best G4 DNA substrate for DDX11 (see Section 2).8 It should be pointed out that the expression levels of the DDX11 protein were not evaluated in the cell lines derived from these WABS patients and, thus, it is not known if the R263Q amino acid change impaired the protein stability, as it was found for other DDX11 pathogenic missense mutations. Of note, the three Lebanese WABS-affected siblings showed a severe intellectual disability, in addition to growth defects, microcephaly, deafness and facial dysmorphia. However, this latter clinical manifestation is quite likely due to the specific peculiar genetic background of these individuals, as it was not found in all WABS cases described so far (see Table 1), and it can not be excluded that it might be due to additional recessive mutations caused by the family inbreeding or to a large amount of runs of homozygosity in intellectual disability patients.
The third case of WABS was reported in 2015 in a British girl, born from non-consanguineous parents.30 Two heterozygous mutations, which introduced frame-shifts in the DDX11 open reading frame, were identified by DNA sequencing analysis. In addition to other common WABS symptoms, the patient presented a chronic skin rash with telangiectasia on her legs. Of note, skin lesions were also observed in the first Polish WABS proband.24 However, since this clinical feature was not found in all patients, it can not be considered as a characteristic of the WABS phenotype (see Table 1).
In 2017, two sisters were diagnosed with WABS: in addition to the typical clinical features, they were reported to have bilateral small thumbs and the younger sister had small fibulae.31 However, these malformations, which revealed a further clinical overlap with FA and RBS, were not observed in all WABS individuals (see Table 1). Both sisters had a compound mutation of the DDX11 gene: a missense mutation (leading to the L508R amino acid change) in the maternal allele and a mutation predicted to affect splicing in the paternal allele. The L508 amino acid residue is predicted to reside in the putative DDX11 Arch domain, for which a role was proposed in DNA binding (see Figure 1).2 However, since a biochemical characterization of the DDX11 L508R mutant protein was not reported, it is not known if this amino acid substitution has any effect on the enzyme catalytic functions.
Very recently, five novel additional WABS patients were described bearing five novel bi-allelic DDX11 gene variants.32 Two of these novel mutations introduce frame-shifts in the DDX11 coding sequence leading to prematurely truncated proteins; three of them are missense variants (R378P, R791Q and V859G). Since DDX11 amino acid residues R791 and V859, both map in the vicinity of the conserved helicase box VI, their mutation is expected to be highly deleterious for the enzyme functionality. Of note, the DDX11 gene mutation that gives rise to the V859G amino acid change was found to be a novel Saudi founder variant.32 The fifth novel identified mutation is homozygous and caused R378P substitution in the DDX11 protein. This mutation was initially classified as a variant of unknown significance (VOUS) because R378 does not map inside or in very close proximity to any conserved helicase box of DDX11. However, biochemical analysis of the recombinant DDX11 R378P mutant protein revealed that this amino acid change tremendously affected protein stability. In fact, level of this DDX11 mutant protein in HEK 293T cells, transiently transfected with the over-expressing plasmid, was estimated to be reduced to half compared to the wild type DDX11 protein. When the transfected cells were treated with the proteasome inhibitor MG132, a difference in the expression level between the DDX11 wild type and R378P mutant proteins was not anymore observed. This result suggested that the mutant protein underwent degradation through the ubiquitin-proteasome pathway, quite likely because of a non-correct folding of its polypeptide chain.32 Structural predictions revealed that the DDX11 R378 amino acid residue resides in a putative α-helix, which in the X-ray structure of the sequence-related Thermoplasma acidophilum XPD helicase was shown to directly contact a co-crystallized short single-stranded DNA oligonucleotide.33 It seems probable that substitution of DDX11 R387 with Proline, an amino acid residue known to be not compatible with an α-helical structure, would lead to protein destabilization in WABS cells. Of note, blood cells derived from two of the patients described in this study (one bearing the DDX11 allele mutation leading to the R378P change combined with the frame-shift mutation in the other allele; and the other one characterized by the homozygous mutation leading to DDX11 V859G substitution) did not display increased chromosomal fragmentation after exposure to MMC, as observed in the other patients described in this same study as well as in the first two reported WABS cases.28,29 In contrast, sister chromatid cohesion anomalies (railroad chromosomes and PCS) were detected in all WABS cases described so far (see Table 1). In view of these findings, it was proposed to rename the disease “Warsaw syndrome”, eliminating reference to the chromosomal breakage phenotype.32
More recently, a novel DDX11 mutation (leading to L836P amino acid change; Figure 1) was identified in two Italian young sisters showing a mild phenotype with no major physical or intellectual disabilities.34 Both sisters had intra-uterine growth retardation, pronounced microcephaly, facial dysmorphia and hearing defects. Of note, they did not display intellectual disability. Sensitivity to MMC of lymphoblastoid cells from both sisters was of intermediate level compared to the one observed in FA patients with mutation of the FANCA gene. Karyotype analysis revealed characteristic railroad chromosomes in immortalized lymphoblastoid cells of both sisters, treated or not with MMC. Sequencing of the DDX11 gene and segregation analysis revealed the presence of a missense mutation (leading to the L836P amino acid substitution) and a deletion of 14 nucleotides (giving rise to a frame-shift and premature stop codon) in the DDX11 paternal and maternal allele, respectively, of both sisters. Multiple sequence alignments among SF2 Fe–S cluster DNA helicases revealed that L836 is conserved in the vertebrate DDX11 orthologs. Bioinformatic analyses suggested that these residues are part of an α-helix, located in close proximity to the helicase motif VI. Moreover, L836 was predicted to sit at the interface between homology domain 1 and 2 (HD1 and HD2) of DDX11, and possibly close to a putative DNA-binding region (see Figure 1).2,34 All that considered, it is not surprising that DDX11 expression level in cells derived from these WABS probands was found to be remarkably diminished compared to healthy individuals. In addition, the recombinant DDX11 L836P mutant protein displayed reduced stability when over-expressed using the baculovirus/insect cell system and underwent extensive proteolytic degradation during purification, quite likely as a consequence of protein misfolding. Besides, it was found that the L836P DDX11 mutant had a remarkably reduced DNA helicase activity compared to the wild type protein (Bottega et al, manuscript in preparation).
The last report of WABS describes two novel unrelated patients of Ashkenazi Jewish descent, both homozygous for the same DDX11 variant (c.1763-1G>C).35 RNA sequencing analyses revealed that this mutation caused altered messenger RNA splicing giving rise to a frame-shift of the DDX11 open reading frame with production of an abnormal protein. The two patients were found to share growth retardation, microcephaly, intellectual disability and deafness with the previously reported WABS probands (see Table 1). Besides, as previously described only for one WABS patient,30,32 both of them displayed congenital hypothyroidism and seizure disorder.35 Screening of the Ashkenazi Jewish population revealed that the c.1763-1G>C variant in the DDX11 gene has a high carrier frequency (1 in 68 individuals, corresponding to 1.47%). Given this high carrier frequency and limited number of WABS patients, it can be postulated that homozygosity of this DDX11 allele may lead to spontaneous abortions and, at the same time, that the heterozygosity condition may be responsible for reduced fertility of carrier individuals. It is interesting that family history of one of these novel Jewish WABS patients was characterized by four first trimester miscarriages of unknown etiology.35 Besides, spontaneous abortions were also reported in the family of other WABS individuals.24,32 However, additional studies are required to assess whether homozygosity of certain DDX11 variants is truly linked to embryonic lethality.
WABS Animal Model Systems and Pathogenesis
No vertebrate animal models of WABS were described so far. Early embryonic lethality was observed at day E10.5 in DDX11-null mice with homozygous deletion of the DDX11 helicase motif I, likely due to placental malformations.36 Furthermore, N-ethyl-N-nitrosourea (ENU)-induced mutagenesis in mice produced an amino acid substitution (L743P) in DDX11 helicase motif V that also caused embryonic lethality at day E8.5.37 More recently, it was shown that DDX11-knockdown in zebrafish embryos caused growth retardation and vertebral and craniofacial anomalies, analogous to the malformations observed in WABS patients. In these animals, the epigenetic status of ribosomal DNA gene cluster was changed from euchromatic to heterochromatic and recruitment of RNA polymerase I to these loci was significantly reduced suggesting that a nucleolar dysfunction, due to DDX11 loss, could have given rise to the above developmental defects.38
The molecular bases of WABS have not yet been elucidated, mainly because of the lack of disease animal models and limited knowledge of the exact roles played by human DDX11 in various genome stability maintenance pathways. However, the finding that WABS and other cohesinopathies (CdLS and RBS) display some common clinical manifestations (growth retardation, microcephaly, intellectual disability, skeletal anomalies) indicates that dis-regulation of gene expression during embryonic development underlies all these genetic diseases.39 In fact, it is now well established that cohesin, together with its regulator network, has a “non-canonical” role in promoting the formation and/or stabilization of chromatin loops that regulate gene transcription developmental programs.40 A “transcriptome disruption model” has been recently proposed for cohesinopathies and other related diseases, due to mutation of genes/proteins that are involved in different aspects of the transcriptional regulation process (including epigenetic chromatin modifications). Of note, many of these hereditary diseases display common clinical features (e.g. CdLS, RBS, WABS, Coffin-Siris, CHOPS syndromes and others). Thus, to classify all these genetic disorders in a single group, the term “transcriptomopathies” or “disorders of transcriptional regulation” has been recently proposed.41,42 In this context, it is of note that DDX11 was reported to be involved in chromosome architecture maintenance, since its down-regulation in mammalian cells not only caused sister chromatid cohesion defects but also altered the chromatin condensation status in metaphase.43 Besides, studies carried out in DDX11-depleted HeLa and in DDX11-knockout mouse embryo-derived cells revealed an important role of DDX11 in heterochromatin formation by targeting HP1α factor to proper sites in pericentric regions and at telomeres.38 Interestingly, the proposal that WABS (and other cohesinopathies) is caused by dis-regulation of gene expression during embryonic development is consistent with a recent finding that DDX11 promoted cohesin loading onto chromatin in HeLa cells16 and a previous report that Chl1 was required for association of the loader complex subunit Scc2 to chromatin in S. cerevisiae.43
An alternative theory about the cohesinopathy etiology envisages that defective protein translational mechanisms could underlie altered embryonic development in these hereditary disorders.45,46 This hypothesis is based on the observation that nucleolar anomalies were caused by mutation of cohesin complex subunits and ribosomal RNA synthesis and ribosome assembly were remarkably reduced in the absence of chromosomal cohesion.47,48 Besides, it was recently reported that DDX11 localizes at the nucleolus in HeLa cells, preferentially binding to hypomethylated active ribosomal DNA gene loci, and its depletion was found to suppress ribosomal RNA synthesis, inhibiting cell proliferation.38 In the same study, as previously pointed out, the developmental defects detected in DDX11-knockdown zebrafish embryos were proposed to be linked, at least partially, to a nucleolar dysfunction.38
Conclusions and Perspectives
In this review, I have tried to summarize the biomedical literature concerning Warsaw breakage syndrome (WABS), a very rare genetic syndrome due to bi-allelic mutations of the genes coding for DDX11, a SF2 Fe-S cluster DNA helicase, whose roles in DNA repair and sister chromatid cohesion have yet been not fully elucidated. The recent discovery that DDX11 is responsible for anchoring cohesin to the ongoing replisomes in human16 and in budding yeast17 cells paves the way for a deeper understanding of the molecular mechanisms underlying chromosomal cohesion establishment and cohesin loading onto chromatin. Besides, DDX11 is believed to have a role in regulating cohesin non-canonical functions (formation and stabilization of chromatin loops) that may affect gene transcription during development. Thus, creation of different WABS animal models (fly, zebrafish and mouse) could be instrumental in pinpointing evolutionarily conserved developmental programs that could be dis-regulated due to DDX11 mutation/loss. Besides, biophysical analyses at the level of single molecules using reconstituted proteins and protein complexes will allow to analyze different possible DNA-binding modes of cohesin and how these are modulated by cohesin regulators (including DDX11). The results of these complementary in vitro and in vivo studies will help unveil the molecular bases of WABS and other related cohesinopathies.
Acknowledgments
FMP has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 859853; from Consorzio CNCCS (“Progetto B - Ricerca di nuovi farmaci per malattie rare trascurate e della povertà”); from Regione Campania (POR-FESR 2014-2010, “Progetto SATIN”).
Disclosure
The author reports no conflicts of interest in this work.
References
1. Bharti SK, Khan I, Banerjee T, Sommers JA, Wu Y, Brosh RM Jr. Molecular functions and cellular roles of the ChlR1 (DDX11) helicase defective in the rare cohesinopathy Warsaw breakage syndrome. Cell Mol Life Sci. 2014;71:2625–2639. doi:10.1007/s00018-014-1569-4
2. Pisani FM, Napolitano E, Napolitano LMR, Onesti S. Molecular and cellular functions of the Warsaw breakage syndrome DNA helicase DDX11. Genes. 2018;9(11):E564. doi:10.3390/genes9110564
3. Wu Y, Brosh RM Jr. DNA helicase and helicase-nuclease enzymes with a conserved iron-sulfur cluster. Nucleic Acids Res. 2012;40:4247–4260. doi:10.1093/nar/gks039
4. Brosh RM Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer. 2013;13:542–558. doi:10.1038/nrc3560
5. Hirota Y, Lahti JM. Characterization of the enzymatic activity of hChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000;28:917–924. doi:10.1093/nar/28.4.917
6. Farina A, Shin JH, Kim DH, et al. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J Biol Chem. 2008;283:20925–20936. doi:10.1074/jbc.M802696200
7. Wu Y, Sommers JA, Khan I, de Winter JP, Brosh RM Jr. Biochemical characterization of Warsaw breakage syndrome helicase. J Biol Chem. 2012;287:1007–1021. doi:10.1074/jbc.M111.276022
8. Bharti SK, Sommers JA, George F, et al. Specialization among iron–sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J Biol Chem. 2013;288:28217–28229. doi:10.1074/jbc.M113.496463
9. Guo M, Hundseth K, Ding H, et al. A distinct triplex DNA unwinding activity of ChlR1 helicase. J Biol Chem. 2015;290:5174–5189. doi:10.1074/jbc.M114.634923
10. Sommers JA, Rawtani N, Gupta R, et al. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J Biol Chem. 2009;284:7505–7517. doi:10.1074/jbc.M809019200
11. Shah N, Inoue A, Lee SW, Beishline K, Lahti JM, Noguchi E. Roles of Chlr1 DNA helicase in replication fork recovery from DNA damage. Exp Cell Res. 2013;319:2244–2253. doi:10.1016/j.yexcr.2013.06.005
12. Laha S, Das SP, Hajra S, Sau S, Sinha P. The budding yeast protein Chl1p is required to preserve genome integrity upon DNA damage in S-phase. Nucleic Acids Res. 2006;34:5880–5891. doi:10.1093/nar/gkl749
13. Ogiwara H, Ohuchi T, Ui A, Tada S, Enomoto T, Seki M. Ctf18 is required for homologous recombination-mediated double-strand break repair. Nucleic Acids Res. 2007;35:4989–5000. doi:10.1093/nar/gkm523
14. Abe T, Ooka M, Kawasumi R, et al. Warsaw breakage syndrome DDX11 helicase acts jointly with RAD17 in the repair of bulky lesions and replication through abasic sites. Proc Natl Acad Sci U S A. 2018;115:8412–8417. doi:10.1073/pnas.1803110115
15. Calì F, Bharti SK, Di Perna R, Brosh RM Jr, Pisani FM. Tim/Timeless, a member of the replication fork protection complex, operates with the Warsaw breakage syndrome DNA helicase DDX11 in the same fork recovery pathway. Nucleic Acids Res. 2016;44:705–717. doi:10.1093/nar/gkv1112
16. Cortone G, Zheng G, Pensieri P, et al. Interaction of the Warsaw breakage syndrome DNA helicase DDX11 with the replication fork-protection factor Timeless promotes sister chromatid cohesion. PLoS Genet. 2018;14:e1007622. doi:10.1371/journal.pgen.1007622
17. Samora CP, Saksouk J, Goswami P, et al. Ctf4 links DNA replication with sister chromatid cohesion establishment by recruiting the Chl1 helicase to the replisome. Mol Cell. 2016;63:371–384. doi:10.1016/j.molcel.2016.05.036
18. Vega H, Waisfisz Q, Gordillo M, et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet. 2005;37:468–470. doi:10.1038/ng1548
19. Skibbens RV, Corson LB, Koshland D, Hieter P. Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev. 1999;13:307–319. doi:10.1101/gad.13.3.307
20. Tóth A, Ciosk R, Uhlmann F, Galova M, Schleiffer A, Nasmyth K. Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev. 1999;13:320–333. doi:10.1101/gad.13.3.320
21. Liu J, Krantz ID. Cohesin and human disease. Annu Rev Genomics Hum Genet. 2008;9:303–320. doi:10.1146/annurev.genom.9.081307.164211
22. Krantz ID. Cohesin embraces new phenotypes. Nat Genet. 2014;46:1157–1158. doi:10.1038/ng.3123
23. Chetaille P, Preuss C, Burkhard S, et al. Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm. Nat Genet. 2014;46:1245–1249. doi:10.1038/ng.3113
24. van der Lelij P, Chrzanowska KH, Godthelp BC, et al. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–266. doi:10.1016/j.ajhg.2010.01.008
25. Parish JL, Rosa J, Wang X, Lahti JM, Doxsey SJ, Androphy E. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J Cell Sci. 2006;119:4857–4865. doi:10.1242/jcs.03262
26. Holloway SL. CHL1 is a nuclear protein with an essential ATP binding site that exhibits a size-dependent effect on chromosome segregation. Nucleic Acids Res. 2000;28:3056–3064. doi:10.1093/nar/28.16.3056
27. Skibbens RV. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi:10.1534/genetics.166.1.33
28. van der Lelij P, Oostra AB, Rooimans MA, Joenje H, de Winter JP. Diagnostic overlap between Fanconi anemia and the cohesinopathies: roberts syndrome and Warsaw Breakage syndrome. Anemia. 2010;2010:565268.
29. Capo-Chichi JM, Bharti SK, Sommers JA, et al. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Hum Mutat. 2013;34:103–107. doi:10.1002/humu.22226
30. Bailey C, Fryer AE, Greenslade M. Warsaw Breakage Syndrome–A further report, emphasising cutaneous findings. Eur J Med Genet. 2015;58:235–237. doi:10.1016/j.ejmg.2015.02.001
31. Eppley S, Hopkin RJ, Mendelsohn B, Slavotinek AM. Clinical report: Warsaw Breakage Syndrome with small radii and fibulae. Am J Med Genet A. 2017;173:3075–3081. doi:10.1002/ajmg.a.38382
32. Alkhunaizi E, Shaheen R, Bharti SK, et al. Warsaw breakage syndrome: further clinical and genetic delineation. Am J Med Genet A. 2018;176:2404–2418. doi:10.1002/ajmg.a.40482
33. Kuper J, Wolski SC, Michels G, Kisker C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2012;31:494–502. doi:10.1038/emboj.2011.374
34. Bottega R, Napolitano LMR, Carbone A, et al. Two further patients with Warsaw breakage syndrome. Is a mild phenotype possible? Mol Genet Genomic Med. 2019;7(5):e639. doi:10.1002/mgg3.639
35. Rabin R, Hirsch Y, Johansson MM, Ekstein J, Zeevi DA, Keena B, Zackai EH, Pappas J. Study of carrier frequency of Warsaw breakage syndrome in the Ashkenazi Jewish population and presentation of two cases. Am J Med Genet A. 2019;179:2144–215. doi:10.1002/ajmg.a.61284
36. Inoue A, Li T, Roby SK, et al. Loss of ChlR1 helicase in mouse causes lethality due to the accumulation of aneuploid cells generated by cohesion defects and placental malformation. Cell Cycle. 2007;6:1646–1654. doi:10.4161/cc.6.13.4411
37. Cota CD, Garcia-Garcia MJ. The ENU-induced cetus mutation reveals an essential role of the DNA helicase DDX11 for mesoderm development during early mouse embryogenesis. Dev Dyn. 2012;241:1249–1259. doi:10.1002/dvdy.v241.8
38. Sun X, Chen H, Deng Z, et al. The Warsaw breakage syndrome-related protein DDX11 is required for ribosomal RNA synthesis and embryonic development. Hum Mol Genet. 2015;24:4901–4915. doi:10.1093/hmg/ddv213
39. Skibbens RV, Colquhoun JM, Green MJ, et al. Cohesinopathies of a feather flock together. PLoS Genet. 2013;9:e1004036. doi:10.1371/journal.pgen.1004036
40. Zheng H, Xie W. The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol. 2019;20(9):535–550. doi:10.1038/s41580-019-0132-4
41. Yuan B, Pehlivan D, Karaca E, et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. 2015;125(2):636–651. doi:10.1172/JCI77435
42. Izumi K. Disorders of transcriptional regulation: an emerging category of multiple malformation syndromes. Mol Syndromol. 2016;7(5):262–273. doi:10.1159/000448747
43. Inoue A, Hyle J, Lechner MS, Lahti JM. Mammalian ChlR1 has a role in heterochromatin organization. Exp Cell Res. 2011;317:2522–2535. doi:10.1016/j.yexcr.2011.08.006
44. Rudra S, Skibbens RV. Chl1 DNA helicase regulates Scc2 deposition specifically during DNA replication in Saccharomyces cerevisiae. PLoS One. 2013;8:e75435. doi:10.1371/journal.pone.0075435
45. Gerton JL. Translational mechanisms at work in the cohesinopathies. Nucleus. 2012;3:520–525. doi:10.4161/nucl.22800
46. Xu B, Lu S, Gerton JL. Roberts syndrome: a deficit in acetylated cohesin leads to nucleolar dysfunction. Rare Dis. 2014;2:e27743. doi:10.4161/rdis.27743
47. Lu S, Lee KK, Harris B, et al. The cohesin acetyltransferase Eco1 coordinates rDNA replication and transcription. EMBO Rep. 2014;15:609–617. doi:10.1002/embr.201337974
48. Harris B, Bose T, Lee KK. Cohesion promotes nucleolar structure and function. Mol Biol Cell. 2014;25:337–346.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.