Back to Journals » OncoTargets and Therapy » Volume 16
Spotlight on New Therapeutic Opportunities for MYC-Driven Cancers
Authors D'Avola A, Kluckova K, Finch AJ, Riches JC ![]()
Received 4 March 2023
Accepted for publication 2 June 2023
Published 7 June 2023 Volume 2023:16 Pages 371—383
DOI https://doi.org/10.2147/OTT.S366627
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Annalisa D’Avola,1 Katarina Kluckova,1 Andrew J Finch,2 John C Riches1
1Centre for Haemato-Oncology, Barts Cancer Institute, Queen Mary University of London, Charterhouse Square, London, EC1M 6BQ, UK; 2Centre for Tumour Biology, Barts Cancer Institute, Queen Mary University of London, Charterhouse Square, London, EC1M 6BQ, UK
Correspondence: John C Riches, Centre for Haemato-Oncology, Barts Cancer Institute, Queen Mary University of London, Charterhouse Square, London, EC1M 6BQ, UK, Tel +44 20 7882 3825, Email [email protected]
Abstract: MYC can be considered to be one of the most pressing and important targets for the development of novel anti-cancer therapies. This is due to its frequent dysregulation in tumors and due to the wide-ranging impact this dysregulation has on gene expression and cellular behavior. As a result, there have been numerous attempts to target MYC over the last few decades, both directly and indirectly, with mixed results. This article reviews the biology of MYC in the context of cancers and drug development. It discusses strategies aimed at targeting MYC directly, including those aimed at reducing its expression and blocking its function. In addition, the impact of MYC dysregulation on cellular biology is outlined, and how understanding this can underpin the development of approaches aimed at molecules and pathways regulated by MYC. In particular, the review focuses on the role that MYC plays in the regulation of metabolism, and the therapeutic avenues offered by inhibiting the metabolic pathways that are essential for the survival of MYC-transformed cells.
Keywords: MYC, cancer, inhibitor, metabolism, target, therapeutics
Introduction
Cancer results from alterations in critical regulatory genes (called oncogenes) that typically control cell proliferation, differentiation, and survival. The first human oncogene identified was RAS, which has been implicated in the pathogenesis of approximately 20% of all human malignancies, including 50% of colon and 25% of lung carcinoma. The RAS oncogene is not present in normal cells; rather, it is generated in tumor cells as a consequence of point-mutations resulting in single amino acid substitutions during the development of cancer. These mutations have the effect of making the RAS protein constitutively active and able to transduce mitogenic signals from a variety of growth factor receptors.1 Another example of an oncogene is the SRC gene, which encodes for a tyrosine kinase. Dysregulation of SRC is implicated in the development of approximately 50% of human cancers, including neuroblastoma, small-cell lung, colon and breast carcinoma, and rhabdomyosarcoma.2 SRC acts to phosphorylate specific tyrosine residues in other tyrosine kinases, thereby regulating several pathways involved in cell growth survival and invasion.
The oncoprotein MYC (also known as c-MYC) is transcription factor that regulates the expression of thousands of genes involved in multiple cellular processes.3 The start of MYC’s discovery began in 1911 from studies by Peyton Rous on the importance of retroviruses in cancer. He demonstrated that chicken sarcomas and leukemia could be transmitted between individuals through the use of cell-free filtrates that also remove bacteria (Berkefeld filters).4 This finding established that cancers could be transmitted by a virus (in this case the Rous sarcoma virus), a finding which was confirmed in many different animal tumors over the following 50 years. In the 1960s and 1970s, a number of different retroviruses were isolated from avian neoplasms, including the avian myelocytomatosis virus MC29. The name given to the genetic element responsible for the neoplastic transformation was v-myc from viral myelocytomatosis.5 Later, a gene homologue of v-myc in uninfected mammalian cells was found and called c-myc, with the human MYC gene finally being cloned and characterized in 1982.6,7
MYC is vital for normal development, with its loss being lethal to the developing embryo beyond day 10 due to abnormalities in organogenesis and tissue growth.8 In addition, it is also vital for the normal functioning of many adult tissues with conditional deletion of MYC in fibroblasts, keratinocytes and B cells blocking cell-cycle progression from G0 to S phase upon stimulation.9,10 As a result, its expression is tightly regulated under physiological conditions, with dysregulation leading to aberrant cell-cycle progression and carcinogenesis.11–13 MYC along with L-MYC and N-MYC comprise the family of MYC transcription factors. The three variants share 40% sequence homology and display very similar functions, albeit with different patterns of expression.14,15 Although L-MYC and N-MYC are implicated in the development of cancer, it is c-MYC that is the most commonly affected variant in human malignancies, being dysregulated in up to 70% of cancers.16,17 In light of this, this is the one that we will focus on in this review.
MYC is an important transcription factor as it controls the expression of 2000–4000 genes, representing 10–15% of the genome as a “master regulator” of multiple cellular processes including proliferation, differentiation, cell cycle, metabolism, and apoptosis.18,19 Increased expression of MYC is required for many cells to proliferate, as it acts as a switch to initiate the wide range of molecular programs required to support cell division.20 One of the most intriguing aspects of MYC biology is that a cell can have “too much of a good thing” in terms of MYC, with overexpression promoting apoptosis in normal cells. As excess MYC is oncogenic, this pro-apoptotic tendency invariably needs to be accompanied by secondary mutations in the apoptosis machinery during tumorigenesisfor example, in DNA-damage response pathways or B-cell lymphoma 2 (BCL2) family members, to allow the nascent cancer cells to survive.21
The Mechanism and Consequences of MYC Overexpression in Cancer
When targeting MYC in cancer, it is important to understand how MYC is overexpressed or “activated” in tumors, and the consequences of this activation. Broadly, MYC can be dysregulated by multiple mechanisms, including genetic, epigenetic, signaling and post-translational alterations. Approximately 20–30% of cancers have a primary genetic lesion driving MYC overexpression.22,23 The commonest genetic aberration is amplification of the MYC gene that is seen in a wide variety of solid malignancies including breast and liver cancer.24–26 In contrast, in B-cell lymphomas, such as Burkitt lymphoma or diffuse large B-cell lymphoma, chromosomal translocation brings the MYC gene under the control of an immunoglobulin gene promoter, most commonly the immunoglobulin heavy chain loci on chromosome 14 (~85%), but also the light chain loci on chromosomes 2 and 22 (~15%), resulting in constitutive activation of MYC in germinal center B cells.27,28 The MYC gene can also be translocated into one of the T-cell receptor loci in some cases of T-cell acute leukemia.29 More recently, translocations of MYC into one of the immunoglobulin genes have been found in multiple myeloma.25 In contrast to many other oncogenes, MYC is rarely mutated. However, mutations in MYC do occur, for example on the T58 residue resulting in a switch of threonine to alanine in the resultant MYC protein reducing its degradation.30,31 Finally, MYC expression can also be increased through the insertion of upstream enhancers by retroviruses, recapitulating the early observations by Rous.32
While about 20–30% of cancers have genetic lesions of MYC, another 30–40% of malignancies have other forms of MYC dysregulation resulting in overexpression of MYC protein and activation of MYC-driven pathways. An example of this is when pathways that normally induce or stabilize MYC under physiological conditions can themselves become the target of activating mutations in cancers (eg, Phosphoinositide 3-kinase (PI3K), Wnt, RAS-MEK-ERK, NOTCH) leading to uncontrolled signaling that increases MYC expression.33–35 A further mechanism is that alterations in the stability of MYC mRNA or protein, or post-translational modifications of MYC protein can all affect the level and kinetics of MYC expression. MYC protein normally has a short-half life that is tightly regulated by phosphorylation and degradation.23 For example, signaling through the RAS-MEK-ERK pathway can increase phosphorylation at the serine 62 (phospho-S62) residue in the MYC box I (MBI) domain of the molecule increasing MYC stability.36 Cancers can also downregulate protein phosphatase 2A (PP2A), a phosphatase that dephosphorylates phospho-S62, which will also increase MYC stability.37 As mentioned above, mutations in threonine 58 residue result in reduced phosphorylation at this residue (reduced phospho-T58).30,31 Normally, phosphorylation of threonine 58 is recognized by the E3 ligase FBW7 which ubiquitinates MYC leading to proteasomal degradation.38 Hence, mutations in T58, loss of PP2A or inactivation of FBW7, another feature of many cancers, all result in high levels of MYC due to reduced ubiquitination and increased stability.23 Many cancers exhibit a combination of these alterations, which act together to raise and sustain MYC levels.
The importance of these increases in MYC expression is due to MYC’s role as a transcription factor. MYC typically exerts its impact by binding to its partner protein MYC-associated protein X (MAX) to form a heterodimer that binds to Enhancer-boxes (E-boxes), portions of DNA with the CACGTG consensus sequence to initiate gene transcription.39–41 When bound, the MYC-MAX dimer recruits other molecules such as histone acetyltransferases that acetylate histones, thereby increasing the accessibility of chromatin to RNA polymerases, facilitating transcription.42 MYC can also recruit transcription factors and elongation factors, such as positive transcription elongation factor b (P-TEFb) that interact with RNA polymerase directly to activate transcription. In addition, MYC can bind repressive factors such as MYC-interacting-zinc finger protein 1 (MIZ1) which causes histone methylation, reducing the accessibility of chromatin to RNA polymerases thereby blocking transcription.41,43 Therefore, MYC acts both to recruit RNA polymerase directly while simultaneously opening up chromatin to facilitate expression of a wide variety of genes. The more MYC that is around, the more DNA binding, with more pronounced changes in transcription and gene expression.44
As previously stated, the fact that MYC binds and regulates approximately 10–15% of the genome, means that its upregulation has a profound impact on the transcriptional state of the cell. MYC regulates virtually all of the pathways regarded as “hallmarks of cancer”, including growth, metabolism, apoptosis, stemness, immune evasion and migration, to the point where MYC dysregulation itself has been referred to as a hallmark of cancer in its own right.12 MYC drives cellular proliferation by activation of cyclin/CDK expression including CDK4, cyclins D, E and inhibition of cell cycle checkpoints.45,46 MYC induction of cyclin-dependent kinases (CDKs) CDK4/6-cyclinD and CDK2-cyclinE promotes retinoblastoma (Rb) hyperphosphorylation, enabling the release of the transcription factor E2F from Rb promoting S-phase entry.45,47 In addition to driving cells into cycle, MYC also promotes many of the processes needed by cells to grow and divide, including nucleotide and protein synthesis, ribosome biogenesis and the metabolic reprogramming required to facilitate these changes.48 For example, MYC recruits RNA polymerase I to activate synthesis of ribosomal RNA, regulates RNA polymerase II-dependent transcription of messenger RNA and ribosomal protein genes, and activates synthesis of 5S ribosomal RNA and transfer RNA through RNA polymerase III.49 In addition to activating components of the ribosome, MYC regulates translation initiation factors, such as eIF4E, eIF4A and eIF4G.50,51 MYC also induces mTOR-dependent phosphorylation and inhibition of eukaryotic translation initiation factor 4E (eIF4E) and binding protein (4E-BP), a repressor of eIF4E, to facilitate cap-dependent mRNA translation.
Extensive metabolic rewiring must occur in order for cells to acquire sufficient nutrients to support cell growth and to deal with the redox challenges that arise from anabolic metabolism.52 MYC directly regulates many of the genes involved in glucose metabolism including glucose transporter 1 (GLUT1), hexokinase 2 (HK2), phosphofructokinase (PFKM), enolase (ENO1), and lactate dehydrogenase (LDHA).22 By regulating these genes, MYC contributes directly to the Warburg effect (aerobic glycolysis) – the ability of highly proliferating cells to convert glucose to lactate even in the presence of oxygen.53 In particular, MYC activation of the RNA-binding proteins heterogeneous nuclear ribonucleoproteins (hnRNPs) A1 and A2, and polypyrimidine tract-binding protein, was shown to regulate the alternative splicing of pyruvate kinase (PKM) to promote the expression of PKM2 directly contributing to the Warburg effect.54 MYC also plays an important role in mitochondrial biogenesis by inducing nuclear-encoded mitochondrial genes. Moreover, MYC has been shown to bind to the promoters of genes encoding proteins involved in mitochondrial function, including those involved in the synthesis of acetyl-CoA, which in turn enables increases in histone acetylation and fatty acid biosynthesis in proliferating cells.55 Finally, MYC overexpression is involved in the expression of genes necessary for nucleotide and amino acid metabolism, regulating many of the enzymes involved in glutamine metabolism and the de novo serine synthesis pathway.22,56,57
MYC can also modulate cellular differentiation depending on the cell context and stage of development. Down-regulation of MYC expression is associated with maturation of B cells into non-proliferative, terminally differentiated plasma cells.58,59 In line with this, MYC overexpression blocks differentiation into various cell types, such as keratinocytes in vitro and in vivo.60,61 MYC can also promote differentiation of the stem cell compartmentfor example, by promoting the exit of hematopoietic stem cells from the stem cell niche and by initiating the terminal differentiation of human epidermal stem cells.62,63 A further aspect of MYC biology is its ability to sensitize cells to different apoptotic stimuli rather than directly inducing apoptosis by itself. Among the main mechanisms adopted by MYC to induce apoptosis are the disruption of the equilibrium between pro-apoptotic BH3-only protein and anti-apoptotic proteins BCL-2 and BCL-X, and the activation of the ARF-MDM2-p53 axis during tumorigenesis.64–66 The ability of MYC to drive both programmed cell death and proliferation in a well-balanced way is a safeguarding mechanism to prevent uncontrolled growth and tumor onset.
A further important aspect of the role of MYC in cancer is its role on the tumor microenvironment. It has long been recognized that tumorigenesis is not only affected by events altering the biology of tumor cells themselves, but is also impacted upon by the microenvironment in which they reside. This tumor microenvironment comprises a variety of cell types including immune cells, endothelial cells, stromal cells and fibroblasts in addition to the tumor cells themselves. All of these types of cells are involved in various tumor-promoting processes, such as immune response evasion, angiogenesis, metabolic changes, invasion and metastasis. Several studies demonstrate that MYC impacts on the tumor microenvironment through the effects on both innate and adaptive immune effector cells. Specifically, MYC promotes the expression of immunosuppressive factors and inhibits the expression of immune activation regulators.67 MYC expression in cancer cells can influence host endothelial cells by up-regulating vascular endothelial growth factor (VEGF) and hypoxia-inducible factor 1-alpha (HIF-1α), two critical elements involved in promoting tumor growth.67 MYC also plays a critical role in immune evasion, the mechanism through which cancer cells avoid host immune surveillance mechanisms, by a variety of mechanisms. For example, increased expression of MYC limits the host immune response by upregulating multiple immune checkpoints, such as programmed death-ligand 1 (PD-L1) and CD47.23,68–70 In addition to inhibiting immune responses, MYC expression has been shown to help tumor cells evade detection by suppressing the expression of major histocompatibility complex class 1 molecules, something that has been observed in several cancers including neuroblastoma, melanoma and lymphoma.71–73 Furthermore, as well as these direct effects on immune-cell interactions, MYC has also been demonstrated to regulate the immune microenvironment through its impact on metabolism. The voracious appetite of cancer cells for glucose and other nutrients means that the microenvironment within tumors can become glucose/nutrient depleted, affecting the metabolism of tumor-infiltrating lymphocytes to impede functional immune responses.23
MYC as a Therapeutic Target
Decades of research have provided a good understanding of the mechanisms that regulate MYC expression and the impact that MYC has on numerous cellular processes. Therefore, this offers us two main ways to target MYC-driven cancers: to target MYC directly with the aim of reducing its expression or blocking its function, or to target the one or more of the many mechanisms by which MYC promotes tumorigenesis. Despite the critical role of MYC in cancer, targeting it has proven to be challenging. One reason is that MYC is a transcription factor that functions through protein–protein and DNA–protein interactions rather than an enzyme, making it intrinsically more difficult to “inhibit”. This is compounded by the fact that MYC has a disordered structure with no fixed 3-dimensional conformation on which to base the development of conventional small-molecule inhibitors.74 Furthermore, MYC is necessary for the growth and function of normal cells, resulting in concerns about “on-target off-cancer” effects on normal tissues.75 Despite this, numerous pre-clinical observations that inactivation of MYC is sufficient to induce cancer regression have emphasized the importance of “oncogene addiction”, meaning that a safe and effective treatment to directly block MYC remains one of the “holy grails” of cancer therapy.76 As a result, numerous drugs have been developed that target MYC in a variety of ways, including aiming to reduce expression by targeting MYC transcription/mRNA translation, reducing the stability of MYC mRNA or protein, or by blocking MYC-function by targeting its interaction with MAX (Figure 1).
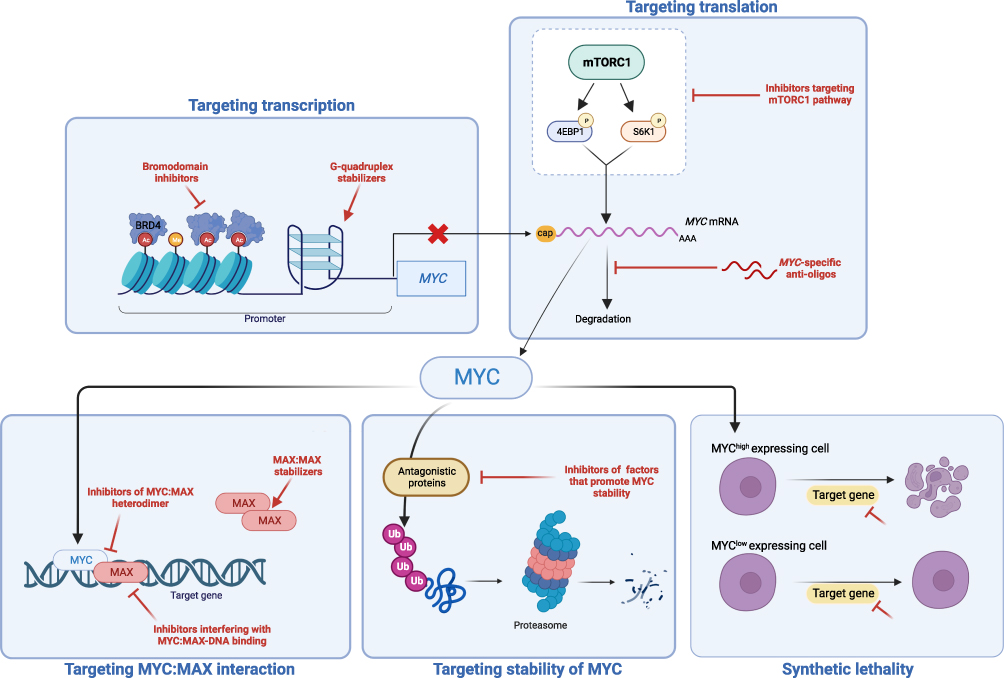 |
Figure 1 Approaches to therapeutically target MYC-driven cancers. MYC expression can be targeted directly by blocking transcription through the use of bromodomain inhibitors or G-quadruplex stabilizers, by blocking translation through mTOR inhibition, or by promoting MYC degradation. MYC function can also be blocked by targeting its interaction with MAX, and/or by interfering with the ability of MYC-MAX heterodimers to bind to DNA. The effects of MYC on cancer cells can also be targeted by inhibiting genes and pathways that are essential for their proliferation and survival. |
Targeting MYC Expression
Targeting MYC Transcription
A widely investigated approach for targeting MYC involves blocking its transcription. One such approach involves the use of compounds that stabilize G quadruplex (G4) motifs in the MYC promoter.20 G-quadruplexes are so-called as they are four-stranded DNA structures formed in guanine-rich regions of DNA.77 They can act as silencers to repress the transcription of proximal genes, and can also act as gene activators under certain circumstances.74 The MYC promoter contains one of these structures, and several studies have shown that small molecules can bind to and stabilize G4s to downregulate MYC. The G4-stabilizer APTO-253 had entered clinical development, undergoing testing in acute myeloid leukemia and high-risk myelodysplastic syndrome. However, clinical development was terminated in 2021 after it failed to demonstrate efficacy in a Phase 1 clinical trial.78 A second approach is to use inhibitors of bromodomain and extraterminal domain (BET) proteins. BET proteins regulate transcription by associating with acetylated chromatin to facilitate transcription factor recruitment. One of the members of the BET family of proteins, BRD4, has been demonstrated to bind to the MYC promoter and regulate its transcription. BRD4 inhibitors, such as JQ1, have been shown to downregulate MYC, blocking tumor growth in animal models of MYC-driven cancer activation.20 Despite this, one of the major issues with this class of drugs is that they may affect many other genes besides MYC, such as the signaling protein ERK1, limiting their clinical utility.79
Targeting MYC mRNA Translation
Another approach to interfere with MYC protein expression is to block its translation. There has been some interest in targeting MYC mRNA directly through the use of anti-oligonucleotides and short interfering and short hairpin RNAs, albeit with limited clinical success to date. Other strategies have focused on blocking translation directly, by inhibiting the mammalian target of rapamycin (mTOR) pathway to block 5′ cap-dependent and internal ribosome entry site (IRES)-dependent translation of MYC RNA.74,80 Despite, the fact that multiple mTOR inhibitors are already approved for clinical use, they suffer from the same limitations as BRD4 inhibitors, in that MYC is only one of their targets, making it difficult to quantify whether any effect is specific to MYC.80
Targeting Regulators of MYC Protein Stability
As discussed above, the expression of MYC is short-lived and tightly regulated by a complex interaction of ubiquitination and phosphorylation. However, this does offer potential therapeutic opportunities to increase MYC degradation to nullify its effects. MYC is ubiquitinated by a number of E3 ligases, with FBW7 and S-Phase kinase-associated protein 2 (Skp2) being the most characterized. Therefore, one approach would be to trigger proteasomal degradation of MYCfor example, by the use of oridonin, a natural plant diterpenoid, which induces FBW7-mediated MYC breakdown.81,82 Conversely, a similar effect could be achieved by inhibiting de-ubiquitinases that help stabilize MYC, such as USP28, with triazolo-pyrimidine derivatives.80 A further strategy, more specific to N-MYC, is to target Aurora-A kinase, as it can complex with N-MYC to allow it to escape from proteasomal degradation.83 While the first-in-class Aurora-A inhibitor, MLN8054, was withdrawn from clinical testing due to side effects, the second generation compound, alisertib, continues to be evaluated in clinical trials for a number of different cancers.74 A further area of potential development is the use of specific protein degraders or proteolysis-targeting chimeras (PROTACs). These are based upon on cereblon-mediated protein degradation, originally discovered as being the molecular target of immunomodulatory drugs used for myeloma, such as thalidomide and pomalidomide.84 While these may provide a new way to directly target MYC for proteasomal degradation, it is not yet clear whether the challenges that have plagued the development of direct MYC inhibitors will also be a factor with these so-called “degraders”.
Targeting MYC–MAX Interaction to Block MYC Function
As MYC exerts most of its functional effects by forming a heterodimer with MAX, this interaction has been targeted for drug development. A variety of approaches have been tried, including blocking the interaction between MYC and MAX, stabilizing MAX-MAX heterodimers to reduce the available MAX pool, or to block the ability of the MYC-MAX heterodimer to bind to DNA.23 While a number of molecules have been developed to target these interactions, the “lead” compound is omoMYC. OmoMYC is a peptide that mimics the basic helix-loop-helix leucine zipper (bHLHLZ) domain of C-MYC, but with mutations that alter its dimerization properties.85 This altered binding both promotes binding to MYC leading to the formation of omoMYC-MYC heterodimers that sequester MYC away from MAX leading to the down-regulation of MYC target genes.75 Other examples of agents that interfere with MYC function are the MAX-MAX stabilizer KI-MS2, MYC-MAX complex disruptors MYCi361 and MYCi975, and the MYC-MAX DNA binding inhibitor ME47.23 Despite all of this research interest, omoMYC remains the only agent to have begun clinical testing.
Targeting the Consequences of MYC Dysregulation
An alternative approach to develop new treatments for MYC-driven cancers is to target the pathways activated by the expression of this oncogene. Given that MYC regulates 10–15% of the genome there should be a myriad number of potential targets that could be exploited. One concept that has received a great deal of attention is that of synthetic lethality. In the context of aberrant MYC expression, the hypothesis is that overexpression of MYC reprograms the cells in such a way that they become dependent on certain genes, proteins or pathways for their continued survival. Identification of such targets would then highlight particular molecules for further investigation and ultimately potential clinical testing. This approach potentially offers a couple of advantages. Firstly, the specific dependence of these targets on MYC overexpression should theoretically create a “therapeutic window” for an agent that preferentially kills the cancer cells while sparing normal tissue. Secondly, some novel synthetic lethal targets may already have a clinically approved inhibitor, or at least may be more “druggable” than MYC itself. As with targeting MYC directly, this approach also has the potential to be applied across a range of cancers where MYC is implicated.
Putative targets for a synthetic-lethal interaction with MYC can identified in two main ways.76 Firstly, the multitude of existing descriptions regarding the role of MYC in cancer-cell biology can be used as the basis for hypothesis-driven investigation of the impact of targeting these pathways. Secondly, a synthetic-lethal screening approach could be used to analyze the impact of a wider variety of targetsfor example, by RNA interference (RNAi) or by pharmacological treatment. These approaches have already identified several potential synthetic-lethal “hits”. One study employing the RNA library approach in fibroblasts modified to over-express MYC uncovered 40 such targets, including the chromatin regulator CECR2, known to play a role in breast cancer metastasis.86,87 Similarly, an siRNA screen performed in U2OS cells revealed that MYC overexpression rendered cells dependent upon the kinase ARK5/NUAK1.88 Given observations that oncogenic levels of MYC can promote apoptosis, targeting apoptotic pathways has also been a natural avenue for investigation. This can be achieved by either activating pro-apoptotic pathways such as death receptor 5 (DR5), or by inhibiting anti-apoptotic molecules such as Myeloid leukemia 1 (MCL-1).89,90 Another family of molecules that have been repeatedly identified as synthetic-lethal candidates, also perhaps unsurprisingly given MYC’s role in driving cells into cycle, are the cyclin-dependent kinases (CDKs). Inhibition of CDK1, a small protein essential for progression through mitosis, has been demonstrated to block progression of MYC-driven lymphomas in murine models.91 Two other members of the CDK family, CDK7 and CDK9, which modulate RNA transcription by phosphorylation of the C-terminal domain of RNA polymerase II, have also been identified as targets in MYC-driven cancers. CDK9 activity has been demonstrated to be critical for the survival of MYC overexpressing hepatocellular cells with a CDK9 inhibitor, AZ5576, now in clinical trials.76,92 Inhibition of CDK7 was shown to disrupt the transcription of amplified N-MYC in neuroblastoma cells, resulting in downregulation of this onco-protein, suppression of N-MYC-driven gene expression, and tumor regression in a neuroblastoma murine model.93,94 Many other synthetic lethal targets have been identified including SUMO-activating enzyme and PIM1 kinase.95,96
Targeting Metabolism in MYC-Driven Cancers
Of particular interest to our laboratories is the potential of tackling MYC-driven cancers by targeting metabolism. As discussed above, uncontrolled expression of MYC drives cells to enter the cell cycle and proliferate, whilst also activating the anabolic and energy producing pathways required to support the necessary cell growth and division. MYC is well known to regulate the expression of multiple metabolic pathways including glycolysis, glutaminolysis, nucleotide biosynthesis and one-carbon metabolism. Therefore, dysregulated MYC expression increases metabolic demand for nutrients that may not be met in the tumor microenvironment. In addition to restricting cell growth through reduction in anabolic processes, insufficient nutrient supply can trigger tumor suppressive responses. For example, withdrawal of glutamine from MYC-overexpressing primary cells triggers apoptosis, indicating that oncogenic transformation could confer vulnerabilities to the inhibition of glutamine metabolism pathways.97,98 This observation promoted the idea that targeting glutaminase (GLS), the aminohydrolase that converts glutamine to glutamate, may be an attractive cancer therapeutic approach for MYC-driven cancers.99,100 However, compounds inhibiting GLS1 (eg, BPTES, CB-839) have demonstrated limited therapeutic potential, primarily due to the activity of several other enzymes that can convert glutamine to glutamate.101 Our recent work has demonstrated that it is the elevated demand for energy to fuel MYC-induced biosynthesis that renders cells dependent on glutamine and the tricarboxylic acid (TCA) cycle to maintain viability.102 Accordingly, inhibition of oxoglutarate dehydrogenase (OGDH), the TCA cycle enzyme that converts α-ketoglutarate to succinyl-coA, induced tumor suppression in MYC-driven lymphoma in mice.102 These studies demonstrate the importance of targeting non-redundant enzymes in metabolic pathways to reduce the metabolic plasticity that often confounds metabolic cancer therapies.
We have also recently demonstrated that inhibition of another metabolic pathway regulated by MYC, the serine synthesis pathway, is able to induce apoptosis in Burkitt lymphoma cells and to block cancer progression in the Eµ-Myc murine model of B-cell lymphoma. Notably, while inhibition of phosphoglycerate dehydrogenase (PHGDH), the first and rate-limiting enzyme in this pathway, inhibited the proliferation of normal B cells resulting in attenuation of germinal center formation, PHGDH blockade did not induce apoptosis in these cells. In contrast, inhibition of PHGDH did induce apoptosis in MYC-transformed germinal center B cells, providing further evidence that the metabolic demands placed on cells by uncontrolled MYC expression could open a therapeutic window.56 Furthermore, other work has also identified a closely related metabolic enzyme, serine hydroxymethyltransferase 2 (SHMT2), which catalyzes the conversion of serine to glycine, as a metabolic vulnerability in Burkitt lymphoma.103
This approach may also provide therapeutic opportunities in malignancies without genomic aberrations of MYC. One such example is chronic lymphocytic leukemia (CLL), where lesions of MYC are rare. However, CLL cells do retain features of mature B-cells, including an ability to signal through the B-cell receptor (BCR) signaling pathway and enter the cell cycle in “proliferation centers” in lymph nodes. Whilst basal expression of MYC is low in CLL cells, it can be induced by BCR-activation in vitro, with increased expression of MYC being observed in these proliferation centers.104 We have also observed that inhibition of BCR-signaling or use of the BET inhibitor, JQ1, are able to block the upregulation of glycolysis induced by MYC, with inhibitors of BCR-signaling able to act synergistically with glycolytic inhibitors such as 2-deoxyglucose.105 A number of reports have investigated the potential of inhibiting MYC targets LDHA and monocarboxylate transporter 1 (MCT1), which are involved in the production and export of lactate, respectively. Overexpression of MYC and LDHA has been shown to correlate with stage and tumor size and predict for a poor prognosis in pancreatic cancer, with inhibition of LDHA blocking tumor growth and metastasis.106 The LDH inhibitor, galloflavin, has been shown to slow the proliferation of Burkitt lymphoma cells due to a reduction in the NAD+:NADH ratio, which in turn results in decreased levels of sirtuin-1 and MYC.107 Inhibition of MCT1 has been demonstrated to lead to accumulation of intracellular lactate in Eµ-Myc B cells, which results in disruption of glycolysis and tumor growth, along with a reduction in glutathione levels, increased hydrogen peroxide and cell death.108 A further report identified that the glycolytic inhibitor 3-bromopyruvate efficiently kills human cancer cells with high levels of MYC, while sparing cells with low levels of this oncoprotein.109 Interestingly, this differential effect was due to MYC’s upregulation of MCT1 and MCT2, which enable the uptake of 3-bromopyruvate by the cancer cells.109 This suggests that while high levels of MYC promote lactate generation and export to maintain glycolytic flux and redox balance, the increased expression of monocarboxylate transporters sensitizes cells to metabolic inhibitors that are trafficked by these molecules.
A further area of interest is the impact of MYC on fatty acid metabolism. One study noted a dramatic upregulation of intermediates of fatty acid oxidation (FAO) in a MYC-driven model of triple negative breast cancer (TNBC).110 Pharmacologic inhibition of FAO with etomoxir blocked energy metabolism in MYC-overexpressing TNBC cells and reduced tumor growth in MYC-driven transgenic TNBC models and patient-derived xenografts. A similar dependence on FAO was observed in Burkitt lymphoma cells. Treatment of Burkitt lymphoma cells with ST1326, an inhibitor of carnitine-palmitoyl transferase 1A (CPT1A), the rate-limiting enzyme for mitochondrial fatty acid import, led to reduced availability of cytosolic acetyl-coA and blocked tumor growth.111 Interestingly, the impact of MYC on fatty acid metabolism may be tissue dependent as other reports have highlighted the importance of fatty acid synthesis for the progression of hepatocellular carcinoma T cell acute lymphoblastic leukemia (T-ALL), renal cell carcinoma (RCC), and lung carcinoma.112,113 The metabolic pathways that are responsive to MYC dysregulation and inhibitors that have been tested MYC-driven cancer models are summarized in Figure 2 and Table 1.
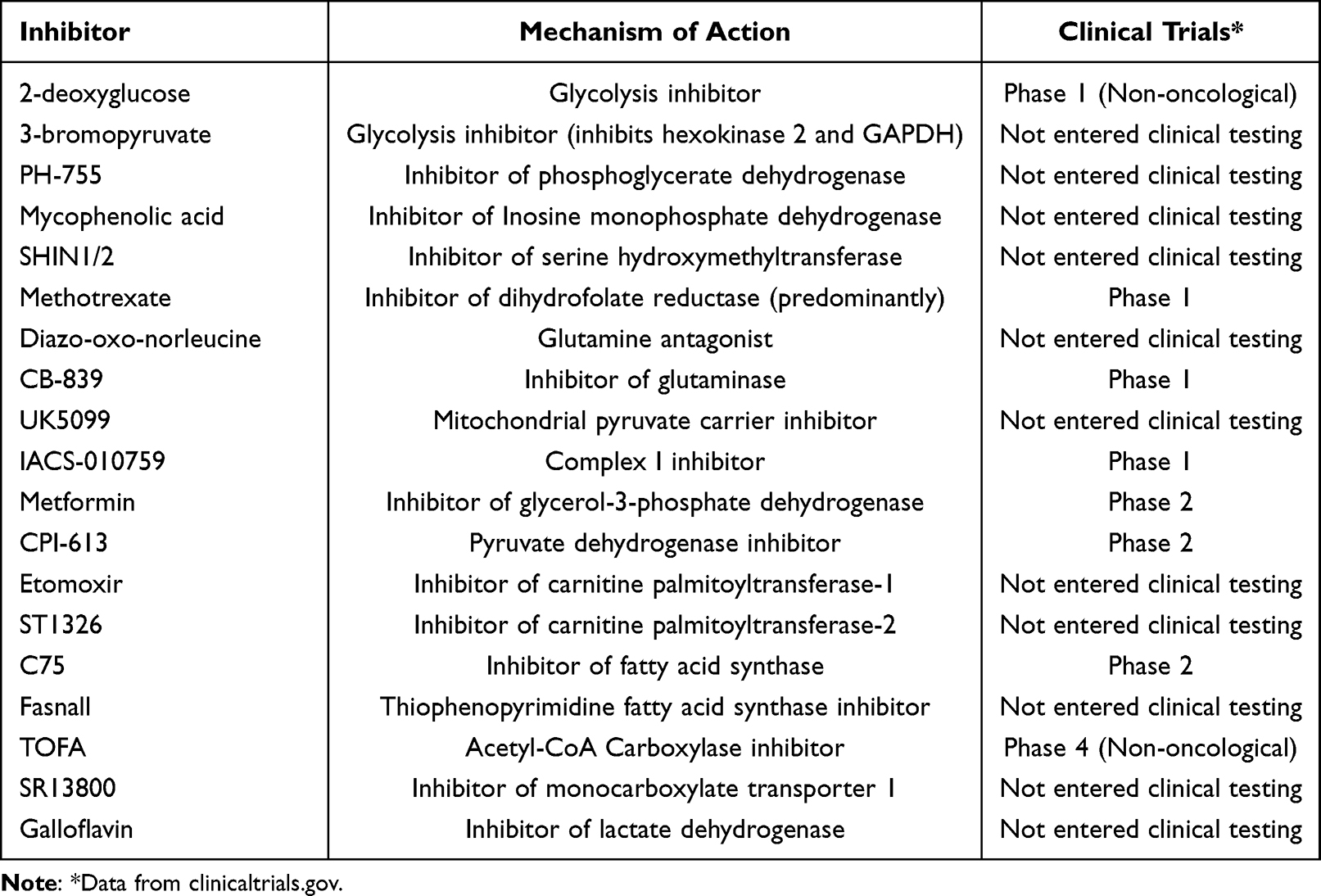 |
Table 1 Details Regarding the Mechanism of Action and Clinical Testing of Inhibitors of Metabolic Pathways Regulated by MYC |
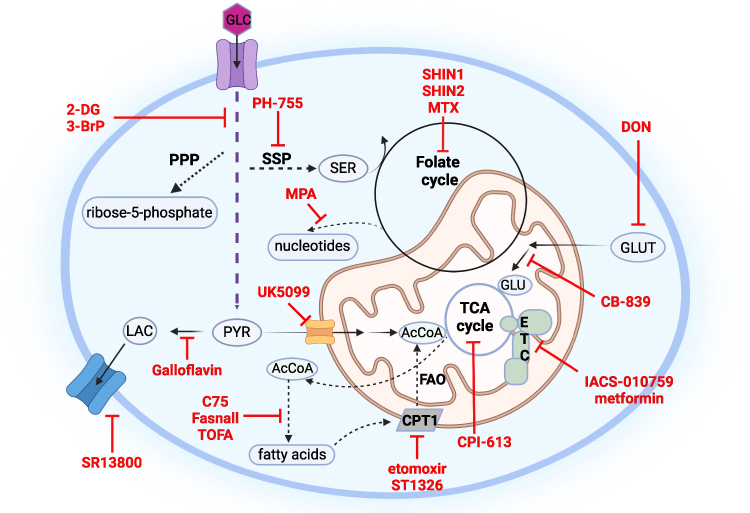 |
Figure 2 Metabolic pathways responsive to MYC deregulation and their inhibitors in MYC-driven cancer models. MYC positively regulates glucose and glutamine metabolism including other related pathways using their carbons, such as pentose phosphate pathway (PPP), serine synthesis pathway (SSP), folate cycle, tricarboxylic acid cycle (TCA), fatty acid synthesis as well as fatty acid oxidation (FAO), pyruvate (PYR) conversion to lactate (LAC) as well as its mitochondrial oxidation; depending on the tissue and cancer model. Other abbreviations used: acetyl coenzyme A (AcCoA), serine (SER), glutamine (GLUT), glutamate (GLU), electron transport chain (ETC), carnitine palmitoyltransferase 1 (CPT1), 5-tetradecyloxy-2-furoic acid (TOFA), 6-diazo-5-oxo-L-norleucine (DON), mycophenolic acid (MPA), methotrexate (MTX), 2-deoxy-D-glucose (2DG), 3-bromopyruvate (3-BrP). |
Conclusion
As an oncogene product of central importance in cancer, MYC has been the subject of many studies of mechanistic and therapeutic importance. Targeting MYC itself has proven highly challenging, but as we have described here, advances have been made in this area with some novel agents entering clinical trials. Targeting the consequences of MYC activation opens up many more avenues for the development of therapies for MYC-driven tumors, reflecting the widespread effects of MYC overexpression on tumorigenesis. Interfering with the function of direct MYC target genes is one example of this approach that has yielded promising results in various models of tumorigenesis. Furthermore, targeting the consequences of MYC activation irrespective of target genesfor example, through interfering with metabolic pathways that are required to support MYC-overexpressing cancer cells, is a further therapeutic option of increasing interest. This could enable the development of rational combinations of drugs that directly interfere with MYC expression/function with agents that target pathways regulated by this molecule. As with all studies in cancer research, there is no single “correct” approach and it remains likely that all of these strategies will contribute to clinical management of MYC-driven cancers going forward. An important aspect of this will be the need to design clinical trials to correlate efficacy with molecular information regarding genetic lesions/overexpression of MYC, to optimize future therapeutic interventions.
Acknowledgments
This work was supported by grant G-002148 from Barts Charity (JCR) and Core Service Grant at Barts Cancer Institute (Core Award C16420/A18066). The figures were created in Biorender.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–774. doi:10.1038/nrc3106
2. Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19(49):5636–5642. doi:10.1038/sj.onc.1203912
3. Chakraborty AA, Scuoppo C, Dey S, et al. A common functional consequence of tumor-derived mutations within c-MYC. Oncogene. 2015;34(18):2406–2409. doi:10.1038/onc.2014.186
4. Rous P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med. 1911;13(4):397–411. doi:10.1084/jem.13.4.397
5. Mladenov Z, Heine U, Beard D, Beard JW. Strain MC29 avian leukosis virus. Myelocytoma, endothelioma, and renal growths: pathomorphological and ultrastructural aspects. J Natl Cancer Inst. 1967;38(3):251–285.
6. Vennstrom B, Sheiness D, Zabielski J, Bishop JM. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol. 1982;42(3):773–779. doi:10.1128/JVI.42.3.773-779.1982
7. Sheiness D, Bishop JM. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J Virol. 1979;31(2):514–521. doi:10.1128/JVI.31.2.514-521.1979
8. Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7(4):671–682. doi:10.1101/gad.7.4.671
9. de Alboran IM, O’Hagan RC, Gartner F, et al. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001;14(1):45–55. doi:10.1016/s1074-7613(01)00088-7
10. Rabbitts PH, Watson JV, Lamond A, et al. Metabolism of c-myc gene products: c-myc mRNA and protein expression in the cell cycle. EMBO J. 1985;4(8):2009–2015. doi:10.1002/j.1460-2075.1985.tb03885.x
11. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127–1133. doi:10.1038/ng.2762
12. Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4(6):a014241–a014241. doi:10.1101/cshperspect.a014241
13. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8(12):976–990. doi:10.1038/nrc2231
14. Xu L, Morgenbesser SD, DePinho RA. Complex transcriptional regulation of myc family gene expression in the developing mouse brain and liver. Mol Cell Biol. 1991;11(12):6007–6015. doi:10.1128/mcb.11.12.6007-6015.1991
15. Bull JJ, Muller-Rover S, Patel SV, Chronnell CM, McKay IA, Philpott MP. Contrasting localization of c-Myc with other Myc superfamily transcription factors in the human hair follicle and during the hair growth cycle. J Invest Dermatol. 2001;116(4):617–622. doi:10.1046/j.1523-1747.2001.12771234.x
16. Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16(4):253–264. doi:10.1016/j.semcancer.2006.07.014
17. Kalkat M, De Melo J, Hickman KA, et al. MYC deregulation in primary human cancers. Genes. 2017;8(6):151. doi:10.3390/genes8060151
18. Sabo A, Kress TR, Pelizzola M, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014;511(7510):488–492. doi:10.1038/nature13537
19. Walz S, Lorenzin F, Morton J, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511(7510):483–487. doi:10.1038/nature13473
20. Duffy MJ, O’Grady S, Tang M, Crown J. MYC as a target for cancer treatment. Cancer Treat Rev. 2021;94:102154. doi:10.1016/j.ctrv.2021.102154
21. Schuster C, Berger A, Hoelzl MA, et al. The cooperating mutation or “second hit” determines the immunologic visibility toward MYC-induced murine lymphomas. Blood. 2011;118(17):4635–4645. doi:10.1182/blood-2010-10-313098
22. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18(20):5546–5553. doi:10.1158/1078-0432.CCR-12-0977
23. Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, Hansen AS, Gouw AM, Felsher DW. The MYC oncogene - The grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol. 2022;19(1):23–36. doi:10.1038/s41571-021-00549-2
24. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi:10.1038/nature08822
25. Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001;20(40):5595–5610. doi:10.1038/sj.onc.1204595
26. Joshua DE, Bryant C, Dix C, Gibson J, Ho J. Biology and therapy of multiple myeloma. Med J Aust. 2019;210(8):375–380. doi:10.5694/mja2.50129
27. Janz S. Myc translocations in B cell and plasma cell neoplasms. DNA Repair. 2006;5(9–10):1213–1224. doi:10.1016/j.dnarep.2006.05.017
28. Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20(40):5580–5594. doi:10.1038/sj.onc.1204640
29. Erikson J, Finger L, Sun L, et al. Deregulation of c-myc by translocation of the alpha-locus of the T-cell receptor in T-cell leukemias. Science. 1986;232(4752):884–886. doi:10.1126/science.3486470
30. Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–2995. doi:10.1093/emboj/16.11.2985
31. Madden SK, de Araujo AD, Gerhardt M, Fairlie DP, Mason JM. Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc. Mol Cancer. 2021;20(1):3. doi:10.1186/s12943-020-01291-6
32. Dudley JP, Mertz JA, Rajan L, Lozano M, Broussard DR. What retroviruses teach us about the involvement of c-Myc in leukemias and lymphomas. Leukemia. 2002;16(6):1086–1098. doi:10.1038/sj.leu.2402451
33. Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3(2):169–179. doi:10.1016/s1097-2765(00)80308-1
34. He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. doi:10.1126/science.281.5382.1509
35. Palomero T, Lim WK, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103(48):18261–18266. doi:10.1073/pnas.0606108103
36. Farrell AS, Joly MM, Allen-Petersen BL, et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat Commun. 2017;8(1):1728. doi:10.1038/s41467-017-01967-6
37. Ruvolo PP. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016;6:87–99. doi:10.1016/j.bbacli.2016.08.002
38. Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18(3):717–726. doi:10.1093/emboj/18.3.717
39. Luscher B, Larsson LG. The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: function and regulation. Oncogene. 1999;18(19):2955–2966. doi:10.1038/sj.onc.1202750
40. Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251(4998):1211–1217. doi:10.1126/science.2006410
41. Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22(20):2755–2766. doi:10.1101/gad.1712408
42. Cole MD, McMahon SB. The Myc oncoprotein: a critical evaluation of transactivation and target gene regulation. Oncogene. 1999;18(19):2916–2924. doi:10.1038/sj.onc.1202748
43. Herkert B, Eilers M. Transcriptional repression: the dark side of myc. Genes Cancer. 2010;1(6):580–586. doi:10.1177/1947601910379012
44. Scafuro M, Capasso L, Carafa V, Altucci L, Nebbioso A. Gene transactivation and transrepression in MYC-driven cancers. Int J Mol Sci. 2021;22(7):3458. doi:10.3390/ijms22073458
45. Hermeking H, Rago C, Schuhmacher M, et al. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A. 2000;97(5):2229–2234. doi:10.1073/pnas.050586197
46. Yang W, Shen J, Wu M, et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001;20(14):1688–1702. doi:10.1038/sj.onc.1204245
47. Santoni-Rugiu E, Falck J, Mailand N, Bartek J, Lukas J. Involvement of Myc activity in a G(1)/S-promoting mechanism parallel to the pRb/E2F pathway. Mol Cell Biol. 2000;20(10):3497–3509. doi:10.1128/MCB.20.10.3497-3509.2000
48. Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98(6):779–790. doi:10.1016/s0092-8674(00)81512-3
49. Hsieh AL, Walton ZE, Altman BJ, Stine ZE, Dang CV. MYC and metabolism on the path to cancer. Semin Cell Dev Biol. 2015;43:11–21. doi:10.1016/j.semcdb.2015.08.003
50. Rosenwald IB, Rhoads DB, Callanan LD, Isselbacher KJ, Schmidt EV. Increased expression of eukaryotic translation initiation factors eIF-4E and eIF-2 alpha in response to growth induction by c-myc. Proc Natl Acad Sci U S A. 1993;90(13):6175–6178. doi:10.1073/pnas.90.13.6175
51. Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421(6920):290–294. doi:10.1038/nature01327
52. Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. 2019;20(7):436–450. doi:10.1038/s41580-019-0123-5
53. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi:10.1126/science.1160809
54. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463(7279):364–368. doi:10.1038/nature08697
55. Morrish F, Noonan J, Perez-Olsen C, et al. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J Biol Chem. 2010;285(47):36267–36274. doi:10.1074/jbc.M110.141606
56. D’Avola A, Legrave N, Tajan M, et al. PHGDH is required for germinal center formation and is a therapeutic target in MYC-driven lymphoma. J Clin Invest. 2022;132(9). doi:10.1172/JCI153436
57. Bott AJ, Peng IC, Fan Y, et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015;22(6):1068–1077. doi:10.1016/j.cmet.2015.09.025
58. Schuhmacher M, Staege MS, Pajic A, et al. Control of cell growth by c-Myc in the absence of cell division. Curr Biol. 1999;9(21):1255–1258. doi:10.1016/s0960-9822(99)80507-7
59. Bacon TA, Wickstrom E. Daily addition of an anti-c-myc DNA oligomer induces granulocytic differentiation of human promyelocytic leukemia HL-60 cells in both serum-containing and serum-free media. Oncogene Res. 1991;6(1):21–32.
60. Lin KI, Lin Y, Calame K. Repression of c-myc is necessary but not sufficient for terminal differentiation of B lymphocytes in vitro. Mol Cell Biol. 2000;20(23):8684–8695. doi:10.1128/MCB.20.23.8684-8695.2000
61. Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8(1):51–56. doi:10.1038/nrc2274
62. Gandarillas A, Watt FM. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997;11(21):2869–2882. doi:10.1101/gad.11.21.2869
63. Laurenti E, Varnum-Finney B, Wilson A, et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell. 2008;3(6):611–624. doi:10.1016/j.stem.2008.09.005
64. Eischen CM, Woo D, Roussel MF, Cleveland JL. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol. 2001;21(15):5063–5070. doi:10.1128/MCB.21.15.5063-5070.2001
65. Oster SK, Ho CS, Soucie EL, Penn LZ. The myc oncogene: marvelouslY complex. Adv Cancer Res. 2002;84:81–154. doi:10.1016/s0065-230x(02)84004-0
66. Hemann MT, Bric A, Teruya-Feldstein J, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436(7052):807–811. doi:10.1038/nature03845
67. Meskyte EM, Keskas S, Ciribilli Y. MYC as a multifaceted regulator of tumor microenvironment leading to metastasis. Int J Mol Sci. 2020;21(20):7710. doi:10.3390/ijms21207710
68. Sun L, Wang Q, Chen B, et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death Dis. 2018;9(9):928. doi:10.1038/s41419-018-0988-9
69. Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352(6282):227–231. doi:10.1126/science.aac9935
70. Kharma B, Baba T, Matsumura N, et al. STAT1 drives tumor progression in serous papillary endometrial cancer. Cancer Res. 2014;74(22):6519–6530. doi:10.1158/0008-5472.CAN-14-0847
71. Bernards R, Dessain SK, Weinberg RA. N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell. 1986;47(5):667–674. doi:10.1016/0092-8674(86)90509-x
72. Braun J, Felsher DW, Goodglick LA. c-myc, MHCI, and NK resistance in immunodeficiency lymphomas. Ann N Y Acad Sci. 1992;651:467–469. doi:10.1111/j.1749-6632.1992.tb24647.x
73. Versteeg R, Noordermeer IA, Kruse-Wolters M, Ruiter DJ, Schrier PI. c-myc down-regulates class I HLA expression in human melanomas. EMBO J. 1988;7(4):1023–1029. doi:10.1002/j.1460-2075.1988.tb02909.x
74. Whitfield JR, Soucek L. The long journey to bring a Myc inhibitor to the clinic. J Cell Biol. 2021;220(8). doi:10.1083/jcb.202103090
75. McKeown MR, Bradner JE. Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med. 2014;4(10):a014266–a014266. doi:10.1101/cshperspect.a014266
76. Donati G, Amati B. MYC and therapy resistance in cancer: risks and opportunities. Mol Oncol. 2022;16(21):3828–3854. doi:10.1002/1878-0261.13319
77. Bahls B, Aljnadi IM, Emídio R, Mendes E, Paulo A. G-quadruplexes in c-MYC promoter as targets for cancer therapy. Biomedicines. 2023;11(3):969. doi:10.3390/biomedicines11030969
78. Ohanian M, Arellano ML, Levy MY, et al. A Phase 1a/b dose escalation study of the MYC repressor Apto-253 in patients with relapsed or refractory AML or high-risk MDS.
79. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. 2017;28(8):1776–1787. doi:10.1093/annonc/mdx157
80. Wang C, Zhang J, Yin J, et al. Alternative approaches to target Myc for cancer treatment. Signal Transduct Target Ther. 2021;6(1):117. doi:10.1038/s41392-021-00500-y
81. Whitfield JR, Beaulieu ME, Soucek L. Strategies to inhibit Myc and their clinical applicability. Front Cell Dev Biol. 2017;5:10. doi:10.3389/fcell.2017.00010
82. Huang HL, Weng HY, Wang LQ, et al. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol Cancer Ther. 2012;11(5):1155–1165. doi:10.1158/1535-7163.MCT-12-0066
83. Otto T, Horn S, Brockmann M, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15(1):67–78. doi:10.1016/j.ccr.2008.12.005
84. Riches JC, Gribben JG. Mechanistic and clinical aspects of lenalidomide treatment for chronic lymphocytic leukemia. Curr Cancer Drug Targets. 2016;16(8):689–700. doi:10.2174/1568009616666160408145741
85. Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene. 1998;17(19):2463–2472. doi:10.1038/sj.onc.1202199
86. Toyoshima M, Howie HL, Imakura M, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A. 2012;109(24):9545–9550. doi:10.1073/pnas.1121119109
87. Zhang M, Liu ZZ, Aoshima K, et al. CECR2 drives breast cancer metastasis by promoting NF-kappaB signaling and macrophage-mediated immune suppression. Sci Transl Med. 2022;14(630):eabf5473. doi:10.1126/scitranslmed.abf5473
88. Liu L, Ulbrich J, Muller J, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483(7391):608–612. doi:10.1038/nature10927
89. Kelly GL, Grabow S, Glaser SP, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014;28(1):58–70. doi:10.1101/gad.232009.113
90. Wang Y, Engels IH, Knee DA, Nasoff M, Deveraux QL, Quon KC. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell. 2004;5(5):501–512. doi:10.1016/s1535-6108(04)00113-8
91. Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med. 2007;13(7):820–827. doi:10.1038/nm1606
92. Huang CH, Lujambio A, Zuber J, et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014;28(16):1800–1814. doi:10.1101/gad.244368.114
93. Chipumuro E, Marco E, Christensen CL, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159(5):1126–1139. doi:10.1016/j.cell.2014.10.024
94. Kwiatkowski N, Zhang T, Rahl PB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511(7511):616–620. doi:10.1038/nature13393
95. Horiuchi D, Camarda R, Zhou AY, et al. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat Med. 2016;22(11):1321–1329. doi:10.1038/nm.4213
96. Kessler JD, Kahle KT, Sun T, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335(6066):348–353. doi:10.1126/science.1212728
97. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178(1):93–105. doi:10.1083/jcb.200703099
98. Yuneva MO, Fan TW, Allen TD, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15(2):157–170. doi:10.1016/j.cmet.2011.12.015
99. Dang CV. Therapeutic targeting of Myc-reprogrammed cancer cell metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:369–374. doi:10.1101/sqb.2011.76.011296
100. Le A, Lane AN, Hamaker M, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110–121. doi:10.1016/j.cmet.2011.12.009
101. Mendez-Lucas A, Lin W, Driscoll PC, et al. Identifying strategies to target the metabolic flexibility of tumours. Nat Metab. 2020;2(4):335–350. doi:10.1038/s42255-020-0195-8
102. Edwards-Hicks J, Su H, Mangolini M, et al. MYC sensitises cells to apoptosis by driving energetic demand. Nat Commun. 2022;13(1):4674. doi:10.1038/s41467-022-32368-z
103. Wilke AC, Doebele C, Zindel A, et al. SHMT2 inhibition disrupts the TCF3 transcriptional survival program in Burkitt lymphoma. Blood. 2022;139(4):538–553. doi:10.1182/blood.2021012081
104. Krysov S, Dias S, Paterson A, et al. Surface IgM stimulation induces MEK1/2-dependent MYC expression in chronic lymphocytic leukemia cells. Blood. 2012;119(1):170–179. doi:10.1182/blood-2011-07-370403
105. Kluckova K, Clear AJ, D’Avola A, et al. B-cell receptor signaling induced metabolic alterations in chronic lymphocytic leukemia can be partially bypassed by TP53 Abnormalities. Hemasphere. 2022;6(6):e722. doi:10.1097/HS9.0000000000000722
106. He TL, Zhang YJ, Jiang H, Li XH, Zhu H, Zheng KL. The c-Myc-LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med Oncol. 2015;32(7):187. doi:10.1007/s12032-015-0633-8
107. Vettraino M, Manerba M, Govoni M, Di Stefano G. Galloflavin suppresses lactate dehydrogenase activity and causes MYC downregulation in Burkitt lymphoma cells through NAD/NADH-dependent inhibition of sirtuin-1. Anticancer Drugs. 2013;24(8):862–870. doi:10.1097/CAD.0b013e328363ae50
108. Doherty JR, Yang C, Scott KE, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014;74(3):908–920. doi:10.1158/0008-5472.CAN-13-2034
109. Gan L, Xiu R, Ren P, et al. Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporters. Oncogene. 2016;35(23):3037–3048. doi:10.1038/onc.2015.360
110. Camarda R, Zhou AY, Kohnz RA, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22(4):427–432. doi:10.1038/nm.4055
111. Pacilli A, Calienni M, Margarucci S, et al. Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis. J Natl Cancer Inst. 2013;105(7):489–498. doi:10.1093/jnci/djt030
112. Jia J, Che L, Cigliano A, et al. Pivotal role of fatty acid synthase in c-MYC driven hepatocarcinogenesis. Int J Mol Sci. 2020;21(22):8467. doi:10.3390/ijms21228467
113. Gouw AM, Margulis K, Liu NS, et al. The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metab. 2019;30(3):556–572 e5. doi:10.1016/j.cmet.2019.07.012
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
JAK-STAT Signaling in Autoimmunity and Cancer
Parveen S, Fatma M, Mir SS, Dermime S, Uddin S
ImmunoTargets and Therapy 2025, 14:523-554
Published Date: 11 May 2025
Integrating Machine Learning Algorithms to Construct a Triaptosis-Related Prognostic Model in Melanoma
Xie J, Zhang M, Qi M
Cancer Management and Research 2025, 17:1127-1141
Published Date: 13 June 2025
Immunotargets and Therapy for Systemic Lupus Erythematosus
Mok CC
ImmunoTargets and Therapy 2025, 14:605-629
Published Date: 24 June 2025