")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Spotlight on mavrilimumab for the treatment of rheumatoid arthritis: evidence to date
Authors Crotti C , Raimondo MG, Becciolini A , Biggioggero M , Favalli EG
Received 3 October 2016
Accepted for publication 17 December 2016
Published 13 January 2017 Volume 2017:11 Pages 211—223
DOI https://doi.org/10.2147/DDDT.S104233
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Georgios Panos
Chiara Crotti,1 Maria Gabriella Raimondo,1 Andrea Becciolini,2 Martina Biggioggero,1 Ennio Giulio Favalli2
1Department of Clinical Sciences and Health Community, University of Milan, Division of Rheumatology, Gaetano Pini Institute, 2Department of Rheumatology, Gaetano Pini Institute, Milan, Italy
Abstract: The introduction of biological therapies into clinical practice has dramatically modified the natural history of chronic inflammatory diseases, such as rheumatoid arthritis (RA). RA is a systemic autoimmune disease that causes articular damage and has a great negative impact on patients’ quality of life. Despite the wide spectrum of available biological treatments, ~30% of RA patients are still unresponsive, resulting in high disability and increased morbidity and mortality. In the last few decades, the scientific knowledge on RA pathogenesis vastly improved, leading to the identification of new proinflammatory molecules as potential therapeutic targets. Several in vitro and in vivo studies showed that granulocyte-macrophage colony-stimulating factor (GM-CSF), known to be a hematopoietic factor, is also one of the proinflammatory cytokines involved in macrophage activation, crucial for the pathogenic network of RA. Mavrilimumab, a human monoclonal antibody targeting the subunit α of GM-CSF receptor, was recently developed as a competitive antagonist of GM-CSF pathway and successfully adopted in human trials for mild to moderate RA. Mavrilimumab phase I and phase II studies reported an overall good efficacy and safety profile of the drug, and these encouraging results promoted the initiation of worldwide phase III studies. In particular, 158-week results of phase II trials did not show long-term lung toxicity, addressing the major concern about this target of pulmonary alveolar proteinosis development. However, further clinical studies conducted in larger RA populations are needed to confirm these promising results. This review summarizes the biological role of GM-CSF in RA and the preclinical and clinical data on mavrilimumab and other monoclonal antibodies targeted on this pathway as an alternative therapeutic option in RA patients who are unresponsive to conventional biological drugs.
Keywords: rheumatoid arthritis, GM-CSF, mavrilimumab, monoclonal antibody, biologic drugs
Introduction
Rheumatoid arthritis (RA) is an autoimmune heterogeneous disease of unknown etiology affecting ~0.5%–1% of the general population.1 It is associated primarily with articular inflammation, synovial joint damage, and increasing disability over time, but it has been more recently recognized as a broader syndrome that includes psychological impairment, increased cardiovascular morbidity, osteoporosis, and risk of cancer.2 In the last two decades, the introduction of targeted biologic disease-modifying antirheumatic drugs (bDMARDs) has revolutionized the treatment of RA, improving the application of novel strategic management approaches that have made remission or low disease activity the target of therapy.3,4 Tumor necrosis factor inhibitors (TNFis) were the first biotherapies to be developed for rheumatologic disorders and, over the last decade, have become the most frequently prescribed class of bDMARDs for the treatment of RA patients who failed synthetic disease-modifying antirheumatic drugs (sDMARDs). Moreover, the increasing knowledge about RA pathways has drawn the attention on other potential targets involved in the complex pathogenesis of the disease, leading to the licensing of biologics with different mechanisms of action, such as interleukin −6 (IL-6) blockade (tocilizumab), B-cell depletion (rituximab), and T-cell costimulation inhibition (abatacept). Despite a so wide therapeutic armamentarium, inhibition of any single cytokine or cellular subset cannot control the disease in whole RA population, and in randomized controlled trials (RCTs) ~30%–40% of treated patients show a primary null response to bDMARDs, fail to maintain over time an initially good response, or experience adverse events (AEs) leading to treatment withdrawal.5–7 Moreover, biologic agents are usually even less effective in daily clinical practice8 than in RCTs because of the heterogeneity in disease activity and clinical characteristics of patients encountered in clinical practice.9 In this scenario, the right choice of the first-line biologic agent and the strategy for treating bDMARD failures still remains a crucial unmet need in the management of RA, and investigations of other cytokines or cellular mechanisms aimed at the development of new therapeutic options are mandatory in order to improve the opportunity of treatment customization.
Granulocyte-macrophage colony-stimulating factor (GM-CSF, also known as colony-stimulating factor 2 [CSF2]) is a cytokine preferentially acting as hematopoietic white cell growth factor, used therapeutically as an alternative to G-CSF for the treatment of chemotherapy-induced neutropenia.10 Moreover, besides the hematologic effect, GM-CSF has been demonstrated to be a modulator of immune/inflammatory cascade able to influence the functions of myeloid cells such as macrophages in the pathogenesis of inflammatory diseases as RA.11 Therefore, GM-CSF blockade could be expected to hamper autoimmune inflammation through decreasing leukocyte activation, even if targeted therapies against this cytokine or its receptor could also be complicated by potential AEs, such as neutropenia or pulmonary alveolar proteinosis.12 Mavrilimumab, a human monoclonal antibody targeted on GM-CSF receptor α, is a competitive antagonist of GM-CSF signaling that showed very promising results and a favorable safety profile in preclinical and clinical studies conducted in RA patients.13
In this review, moving from the pathogenic rationale for GM-CSF blockade in autoimmune diseases, we summarize the findings that have led to the development of mavrilimumab as a possible treatment of RA.
GM-CSF and its biological function
GM-CSF (or CSF2) is a small 127-amino acid soluble cytokine, secreted by a variety of cells, such as activated T and B cells, macrophages, endothelial and epithelial cells, chondrocytes, fibroblasts, osteoblasts, and microglia.14 GM-CSF production is the result of the stimulating effect of proinflammatory cytokines, including IL-1, IL-2, TNFα, prostaglandin E, and interferon γ, and the suppressive influence of IL-10 and transforming growth factor β.15,16 As suggested by its name, the main function of GM-CSF is stimulating the proliferation of myeloid cells from bone marrow progenitors.17
Besides the well-known hematopoietic effect, GM-CSF has been proven to modulate innate immunity and several inflammatory processes, such as chemotaxis, cell adhesion, phagocytosis, and secretion of proinflammatory cytokines, especially by regulating functions of monocyte/macrophage lineage. In vitro studies demonstrated that, when combined with other proinflammatory agents, GM-CSF polarizes macrophages into a phlogistic M1-like phenotype, which can produce a wide variety of inflammatory cytokines, such as IL-6, IL-12p17, IL-23, and TNF.18 In contrast, in vivo studies showed that GM-CSF may have also been associated with a Th2 immunity, so to an M2 polarization, especially in the lung,19 suggesting that the M1/M2 paradigm linked to GM-CSF should be further investigated.20 Moreover, through the upregulation of toll-like receptor 4 and CD14, GM-CSF is able to activate resident macrophage-like microglia and induce central nervous system inflammation.21 GM-CSF can also upregulate the integrin CD11b expression by mature neutrophils, increasing their adhesion to vascular endothelium.22
Epithelial cells of pulmonary mucosa are major producers of GM-CSF, which has a pivotal role in the alveolar macrophages maturation, helping to clear surfactant lipids and proteins from lung surface.12 In murine model, GM-CSF or its receptor deficiency caused mice death because of lung pathology, characterized by accumulation of surfactant-like proteins leading to inflammatory peribronchovascular infiltrate and increased susceptibility to microbial infection.23 These findings support a GM-CSF function in lung physiology maintenance and local resistance to infections.24 Moreover, GM-CSF effects on myeloid-lineage embrace also osteoclasts and dendritic cells (DCs). In vitro studies demonstrated that GM-CSF may influence osteoclast development on one side by stimulating the differentiation of osteoclast precursors and on the other side by stopping their late-stage maturation in cooperation with M-CSF and receptor activator of nuclear factor κB ligand.25 In addition, GM-CSF showed several functions on mature DCs, such as increasing uptake capacity26 and cross-presentation,27 and promoting nonlymphoid tissue DC homeostasis.28
GM-CSF interacts with a transmembrane heterodimeric receptor (GM-CSF-R), consisting in a specific ligand-binding α-chain (GM-CSF-Rα) and a common signal-transducing β-chain (GM-CSF-Rβ), shared with IL-3 and IL-5 receptors (Figure 1).22 High-affinity interaction between GM-CSF and its receptor activates multiple downstream transducing pathways including Janus kinase 2 (JAK2), activator of transcription-suppressor of cytokine signaling (JAK-STAT-SOCS), phosphatidylinositol 3 kinase, mitogen-activated protein kinase, and nuclear factor κB.22,29
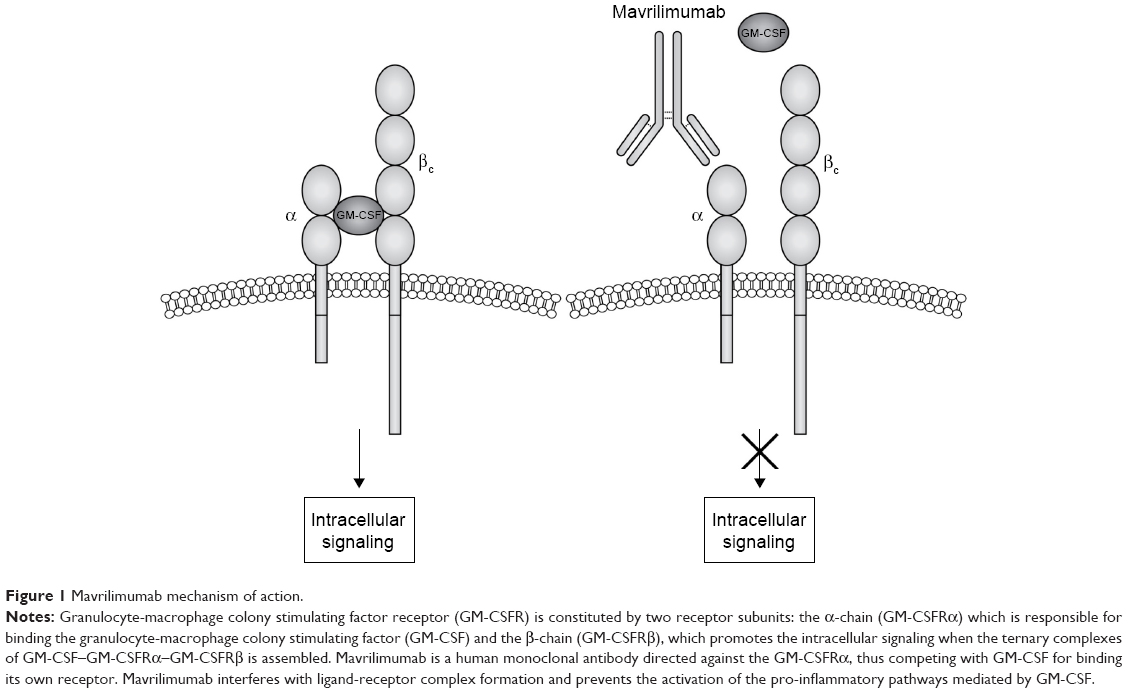 | Figure 1 Mavrilimumab mechanism of action. |
Target identification: GM-CSF pathway blockade in RA
Given the broad effect on macrophage and neutrophil maturation and activation, GM-CSF may be considered as a key mediator in the interface between innate and adaptive immunity and a regulator of the proinflammatory network in the context of autoimmune diseases.30 The existence at inflammation sites of a “CSF network,” involving bidirectional communication through CSF family members (GM-CSF, macrophage CSF [M-CSF], and granulocyte CSF [G-CSF]) between resident tissue cell types (endothelial cells, fibroblasts, chondrocytes, osteoblasts, epithelial cells, and keratinocytes) and activated macrophages has been proposed. The latter may produce pro-phlogistic cytokines, such as IL-1 and TNF, which, in turn, can activate the neighboring nonhematologic cells, leading to an increased release of GM-CSF, M-CSF, and G-CSF in a sort of paracrine/autocrine loop.31 This network may be important in the pathogenesis of an autoimmune disease such as RA, in which synovial tissue macrophages have been proven to actively participate in the inflammatory cascade responsible for joint damage and to be a potential biomarker for treatment response in patients with RA.32,33 The major in vitro and in vivo studies investigating the role of GM-CSF in RA are reported in Table 1.
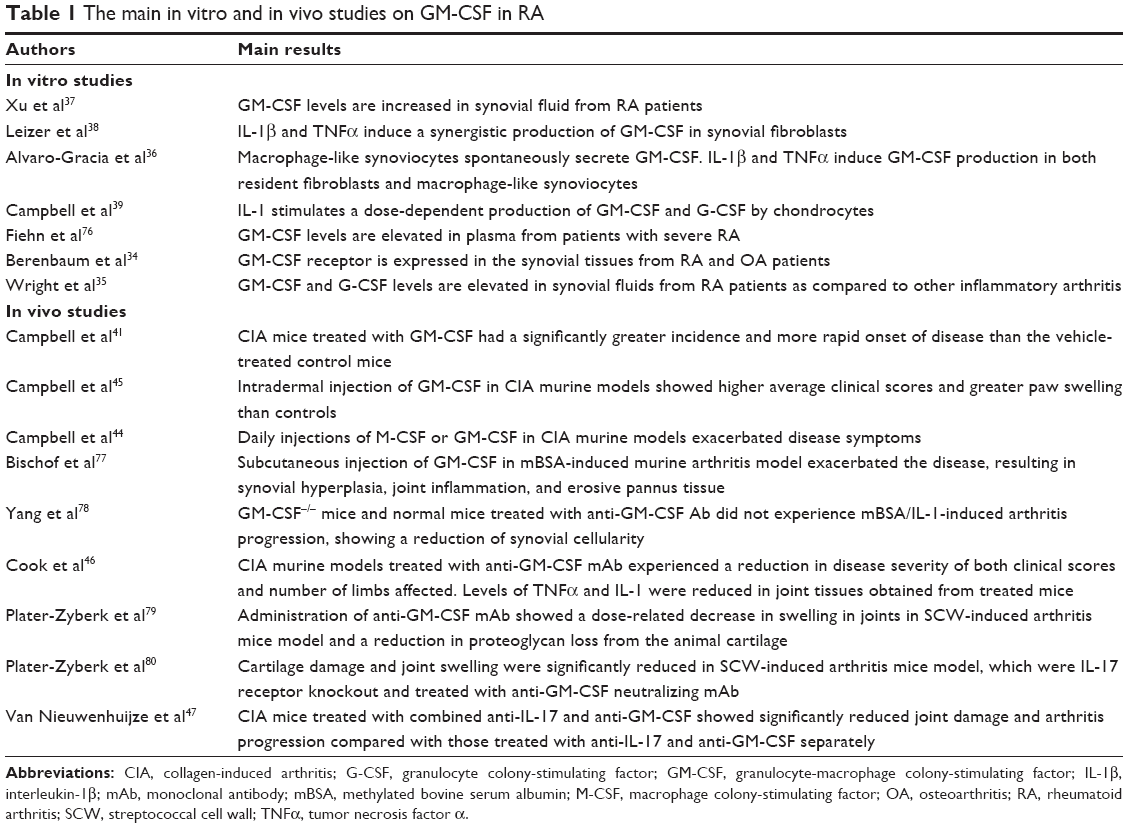 | Table 1 The main in vitro and in vivo studies on GM-CSF in RA |
As a demonstration, GM-CSF levels were found elevated in inflamed synovial membrane and fluid from patients with RA when compared with control group.34,35 Moreover, in vitro studies reported a significant TNFα/IL-1-induced GM-CSF production by resident fibroblast, macrophage-like synoviocytes, and chondrocytes,36–39 and a similar overexpression of M-CSF by synovial endothelial cells from rheumatoid joints.40 In addition, GM-CSF and M-CSF administration exacerbated experimental arthritis in mice models,41 and flares of RA were described in patients receiving recombinant GM-CSF as growth factor after chemotherapy and in patients affected by Felty’s syndrome.42,43 Mice models of experimental arthritis provided the first confirmation that GM-CSF and M-CSF blockade could produce a therapeutic effect in autoimmune diseases.44 In a collagen-induced arthritis (CIA) model, GM-CSF knockout rodents did not develop arthritis despite a normal humoral response to the arthritogenic stimulation.45 Moreover, GM-CSF and M-CSF receptor blockade by a monoclonal antibody reduces overall progression of clinical scores, macrophage number in the synovial tissue, and development of joint erosions.46 Finally, other in vitro studies suggest a role for GM-CSF pathway inhibition in TNFi-resistant arthritis.47
Overall, the results of previously described in vitro and in vivo studies supported the hypothesis that GM-CSF may play a crucial role in the pathogenesis of RA and that the inhibition of its activity might represent a novel therapeutic approach for the treatment of the disease,13 leading to the development of GM-CSF blockers targeted on both the cytokine (nalimumab [MT203], MOR103, lenzilumab [KB003], and MORAb-022) or its receptor (mavrilimumab).
Preclinical development of anti-GM-CSF-R treatment
As discussed, GM-CSF-R is a heterodimer constituted by α (GM-CSF-α) and β-chains (GM-CSF-β). While the latter guides the intracellular signaling through JAK2/STAT3/STAT5, GM-CSF-α is responsible for binding the cytokine with high specificity and low affinity, representing a good target for the antibody-based approach adopted in human clinical trials.22,48,49 Thus, the identification and cloning of the gene encoding for GM-CSF-α – CSF2RA – led to the development of mavrilimumab (CAM-3001), a competitive antagonist of GM-CSF signaling.48 This human IgG4 monoclonal antibody (isolated by phage display) has low ability of complement activation because of its IgG4 Fc isotype, while it binds GM-CSF-α with high affinity that, in turn, results in a potent antagonism of GM-CSF.50 In Greven et al’s preclinical study, mavrilimumab was adopted as a detection agent. The immunohistochemistry analysis showed an increased expression of GM-CSF-α in CD68+ and CD163+ synovial macrophages resident in tissue samples from RA and psoriatic arthritis patients as compared with samples from osteoarthritis patients and from healthy controls.51 In the same study, an analogous antibody – CAM-3003 – which targets mouse GM-CSF-α was developed and adopted in an in vivo experimental model. CAM-3003 dose-dependently inhibited both GM-CSF–induced upregulation of CD11b on mouse granulocytes in vitro and margination response to GM-CSF of peripheral blood monocytes and neutrophils in vivo. In the CIA model, the administration of CAM-3003 at the onset of arthritis inhibited disease progression with reduced synovial inflammation and joint destruction. CAM-3003-treated mice had less cellular infiltrations, cartilage damage, and bone erosion, and a significant reduction in F4/80 (adhesion G-protein–coupled receptor E1) positive macrophages. These findings are consistent with the inhibition of inflammatory cell trafficking to the inflamed synovium and might be the result of reduced survival and differentiation of these cells within the joint.51 In another study, mavrilimumab was internalized along with GM-CSF-R, a process that might reduce both receptor recycling and expression of the receptor itself on the cell surface.52 In vitro data showed that GM-CSF stimulated peripheral blood mononuclear cells to produce TNF in a dose-dependent manner, whereas mavrilimumab inhibited the production of proinflammatory cytokines, such as IL-8, IL-6, and TNF.50 Moreover, mavrilimumab demonstrated analog potency in GM-CSF inhibition in human and cynomolgus monkeys as reported by Ryan et al in the toxicology evaluation of the drug.53 Given the well-known role of GM-CSF as a key molecule in the regulation of pulmonary surfactant homeostasis, major concerns have been raised about lung toxicity secondary to the use of agents that target the GM-CSF pathway, such as mavrilimumab.24 The nonclinical safety of mavrilimumab was evaluated with comprehensive toxicology studies performed on cynomolgus monkeys. These animals were selected for the similar binding affinity of the monoclonal antibody to monkeys’ GM-CSF-Rα compared to the human receptor. According to the toxicology evaluation, the repeated intravenous administration of mavrilimumab for 4–26 weeks showed minimal changes, as in the microscopic findings, which were reversible and considered not adverse. However, at the highest dose (>30 mg/kg each week), accumulation of lung foreign material, cholesterol clefts, and observation of granulomatous inflammation were reported in some of the studies. Despite these findings, the study supported the wide clinical safety margins of mavrilimumab compared with the doses that cause adverse modifications in lung.53 More recently, another study was aimed to model systemic versus pulmonary pharmacodynamics of an anti-GM-CSF-Rα antibody to better understand the pharmacology that contributes to the therapeutic margin of mavrilimumab.54 Mice received intraperitoneal anti-GM-CSF-Rα antibody, and pharmacodynamics bioassays for GM-CSF-Rα blockade was performed on blood and bronchoalveolar lavage cells to quantify the effect in the circulation and lung, respectively. Partial inhibition of GM-CSF-Rα function on cells from the bronchoalveolar lavage was only observed after administration of 30 mg/kg for 5 or 7 consecutive days, 10-fold higher than the proposed therapeutic dose for mavrilimumab in RA.54
Overall, these data supported the development process of mavrilimumab toward in-human studies conducted in RA patients, and are summarized in Table 2.
 | Table 2 Clinical trials of mavrilimumab in rheumatoid arthritis |
Clinical efficacy of mavrilimumab in RA
The earliest evidence of the clinical efficacy of mavrilimumab in RA has been shown in a phase I, randomized, double-blind, placebo-controlled, clinical study conducted in 32 RA patients enrolled to receive a single intravenous dose of mavrilimumab (n=27; 0.01, 0.03, 0.1, 0.3, 1.0, 3.0, or 10.0 mg/kg) or placebo (n=5).55 The half-life of mavrilimumab was proportional to the administered dose, being ~15 days in the 10 mg/kg subgroup. Moreover, the GM-CSF-stimulated expression of suppressor of cytokine signaling 3 (SOCS3) mRNA was analyzed in placebo, 0.3, 1, and 3 mg/kg subgroups. In the latter two groups, the induction of SOCS3 mRNA by GM-CSF was suppressed from 4 to 336 hours after the administration. Although the aim of the trial was to evaluate safety, tolerability, and pharmacokinetic and pharmacodynamic profile of mavrilimumab, the authors observed in the active treatment group a significant proportion (33%) of subjects achieving a disease activity score of 28 (DAS28) remission at week 4. Moreover, all patients treated with mavrilimumab with high baseline C-reactive protein (CRP) showed a significant reduction for 4 weeks.55
Subsequently, mavrilimumab has been extensively tested in the EARTH development program including three main RCTs (Table 3). In the first one, the EARTH study, an Eastern Europe multicenter, randomized, double-blind, placebo-controlled phase IIa trial, 233 RA patients (with at least moderate activity as defined by DAS28-CRP >3.2) were enrolled to receive placebo (n=79) or ascending subcutaneous mavrilimumab (10, 30, 50, or 100 mg every other week [eow], n=158) in association with methotrexate (MTX).56 The peak serum concentration was observed 3 days after the subcutaneous injection of 100 mg mavrilimumab with a half-life of ~13 days, whereas the pharmacokinetic steady state was reached by day 57. Treatment with mavrilimumab was associated with a significantly higher proportion of subjects achieving a ≥1.2-point reduction in DAS28-CRP when compared to placebo (55.7% vs 34.7%, P=0.003). Furthermore, a EULAR moderate or good response was observed more frequently in mavrilimumab versus placebo group (67.7% vs 50.7%, P=0.025). By week 2, in the 50 and 100 mg mavrilimumab group, DAS28-CRP mean change from baseline was significantly different when compared to placebo (P=0.013 and P=0.003, respectively), whereas swollen and tender joints count showed significant improvement since week 4. At the end of the treatment period (week 12), 100 mg mavrilimumab-treated patients showed a higher clinical response according to the American College of Rheumatology criteria (ACR20, ACR50, and ACR70) when compared to placebo (69.2% vs 40%, P=0.005; 30.8% vs 12%, P=0.021; 17.9% vs 4%, P=0.030, respectively).
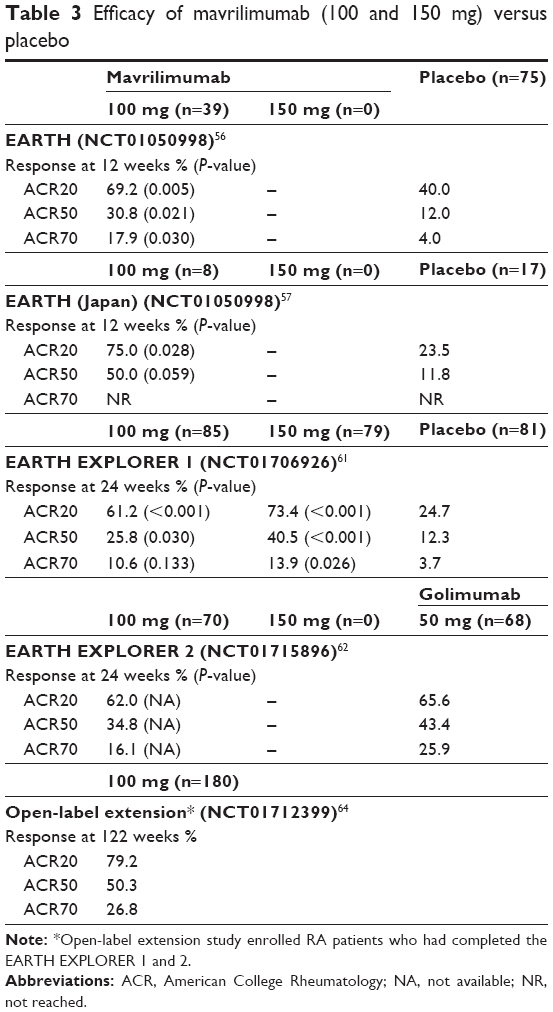 | Table 3 Efficacy of mavrilimumab (100 and 150 mg) versus placebo |
The EARTH study was separately conducted with the same design in Japan also, enrolling 51 RA patients treated with placebo (n=17) or ascending subcutaneous mavrilimumab (10, 30, 50, or 100 mg eow, n=34) in combination with MTX. DAS28-CRP response (>1.2-point reduction) at week 12 was observed more frequently, although not significantly, in subjects receiving mavrilimumab as compared to placebo (50.0% vs 23.5%, P=0.081).57
To facilitate the design of a phase IIb trial, an exposure–response relationship for mavrilimumab was evaluated by clinical simulations through a pharmacokinetics-efficacy dropout model based on the described efficacy results (DAS28-CRP, ACR20/50) of the phase IIa studies. This explorative analysis indicated a priori that the maximum efficacy may be reached at the 150 mg eow dosage, although this mavrilimumab regimen was not evaluated in EARTH studies.58 According to these findings, in the subsequent 24-week randomized, double-blind, placebo-controlled phase IIb EARTH EXPLORER 1 trial, 326 sDMARD-insufficient responder RA patients were randomized to receive three different mavrilimumab eow doses including 150 mg (30 mg, n=81; 100 mg, n=85; or 150 mg, n=79) or placebo (n=81) in association with MTX. As early as week 1 all mavrilimumab doses showed significant DAS28-CRP reduction when compared to placebo (P<0.001).59 Treatment benefit appeared to improve till week 12. Patients treated with mavrilimumab 150 mg showed the highest improvements on DAS28-CRP.60 Moreover, at 24 weeks, a significantly greater proportion of all mavrilimumab-treated patients met the ACR20 co-primary end point versus placebo (50.6%, 61.8%, and 73.4% for 30, 100, and 150 mg, respectively, against 24.7%; P<0.001 for all values). Similarly, ACR50 response was significantly lower in the placebo-treated patients (12.3%) compared with each mavrilimumab arm (28.4% [P=0.013], 25.8% [P=0.03], and 40.5% [P<0.001], respectively), whereas an evident, better ACR70 response was found only in the mavrilimumab 150 mg subgroup (13.9% vs 3.7%, P<0.026).61
Finally, the development program has been completed by the EARTH EXPLORER 2 trial, an explorative, 24-week phase IIb, randomized, double-blind, comparative study, including 138 RA patients IR (insufficient responder) to at least one sDMARD and/or one TNFis, randomized to receive mavrilimumab 100 mg eow (n=70) or golimumab 50 mg every 4 weeks (n=68) on top of MTX.62 Although the study was not powered to demonstrate statistical significance between mavrilimumab and golimumab, in sDMARD-IR subgroup, 24-week ACR20, ACR50, and ACR70 response rates seem to be apparently lower in the mavrilimumab-treated patients compared with golimumab (53.8%, 35.9%, and 10.3% vs 69.4%, 44.4%, and 27.8%, respectively).62 However, the authors argued that the dose adopted in this study might be suboptimal as compared to the one (150 mg eow) predicted as most effective by the previously described exposure–response relationship simulation58 and subsequently confirmed by the results of EARTH EXPLORER 1 study.61 This argument is reasonable because ACR20, ACR50, and ACR70 response rates in the 150 mg group of EARTH EXPLORER 1 study seem to be higher (73.4%, 40.5%, and 13.9%, respectively) compared with the 100 mg group of EARTH EXPLORER 2 study (53.8%, 35.9%, and 10.3%, respectively) and comparable with the golimumab group of EARTH EXPLORER 2 study (69.4%, 44.4%, and 27.8%, respectively). Moreover, despite the suboptimal dose, ACR20, ACR50, and ACR70 response rates were similar between mavrilimumab and golimumab in the TNFi-IR group of EARTH EXPLORER 2 study (72.3%, 33.5%, and 23.5% vs 61.2%, 42.2%, and 24.2%, respectively).62
Interestingly, a subanalysis of the EARTH EXPLORER 2 study pointed out that patients treated with mavrilimumab showed suppressed serum levels of CCL22 and CCL17, whereas patients receiving golimumab showed suppressed levels of CXCL13 and ICAM1, thus suggesting different specific pathogenic pathways affected by the two bDMARDs.63 Moreover, it appeared that in the TNFi-IR group, only mavrilimumab was able to induce a permanent suppression of inflammatory markers (such as CRP, SAA, MMP1, MMP3, IL6, VEGF, IL2R, and CD163) and extracellular matrix markers (C1M, C3M, and P4NP7S), whereas golimumab only induced a transient change,63 even if the clinical response was similar in the two subgroups. These findings might be explained by a hypothetic, more upstream, and widespread effect of GM-CSF modulation compared with TNF blockade, especially in RA irresponsive to a previous TNFi, but should be better clarified by further future analyses.
The long-term efficacy of mavrilimumab has been evaluated in the open-label extensions of both EARTH EXPLORER 1 and 2 studies, including 397 patients treated with mavrilimumab 100 mg eow. Two hundred seventy-nine RA patients were assessed for efficacy at week 74, 231 at week 98, and 180 at week 122. Overall, ACR20, ACR50, and ACR70 response rates at 74, 98, and 122 weeks were 75.6%, 47.3%, and 27.6%; 80.1%, 50.2%, and 30.3%; and 79.2%, 50.3%, and 26.8%, respectively.64 Moreover, at 74 weeks, the mean (±standard deviation [SD]) change from baseline in modified-total Sharp score (mTSS) was 1.3 (±6.8) and 67.9% of patients showed no radiographic progression (<0.5 change in mTSS) when compared to baseline (mean [±SD] mTSS 36.2 [±2.7]).65
More recently, the existence of biomarkers predictive of clinical response to mavrilimumab has been suggested. Specifically, the presence of antibodies directed to peptidyl arginine deiminase 4 in serum of patients treated with mavrilimumab (150 mg) was associated with a worst clinical response as compared to patients who were negative for those antibodies.66 However, further investigations need to confirm these interesting preliminary data.
Safety profile of mavrilimumab in RA
The safety profile of mavrilimumab in RA patients has been evaluated in several phase I/II trials (Table 4). Overall, these RCTs did not highlight unexpected safety issues or specific organ-related toxicities. A well-known possible concern regarding GM-CSF blockade is pulmonary alveolar proteinosis caused by the abnormal accumulation in the alveoli of lung surfactant proteins as the result of an impaired clearance by alveolar macrophages,24 as demonstrated in GM-CSF-deficient mice.23 However, no clinically significant changes in serum surfactant D and KL-6 levels – which are established biomarkers for lung damage – were found in mavrilimumab-exposed patients in the EARTH study.56 Moreover, no serious lung toxicities were observed in the overall population exposed to GM-CSF blockade in the development program of mavrilimumab (625 RA patients treated for up to 158 weeks).55–57,62,65 In particular, a recent comprehensive review of pulmonary safety has been conducted in the phase IIb clinical program of mavrilimumab (EARTH EXPLORER 1 and 2 double-blind and open-label extensions), including 442 RA-treated patients over a 158-week follow-up period (~900 patient-years with a median [min–max] exposure of 2.5 [0.1–3.3] years).67 Pulmonary monitoring included chest X-rays, standardized lung function testing (spirometry and diffusing capacity of lung carbon monoxide [DLCO]), and assessments of dyspnea and pulmonary AEs. Mean forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), and DLCO were mostly maintained within 5% of baseline values and only <7% patients showed mostly transient clinically significant (>20% from baseline and <80% predicted) decreases in predicted FEV1 and FVC. Overall, only 83 patients (9.24/100 patient-years) reported ≥1 pulmonary AE (mainly bronchitis [3.78/100 patient-years] and respiratory tract infection [1.56/100 patient-years]), with a stable incidence over time and no significant difference compared to golimumab comparator arm. No pulmonary-related deaths, no suspected/confirmed pulmonary alveolar proteinosis cases, and only one treatment-related pulmonary serious AE (acute bronchitis) were reported.67
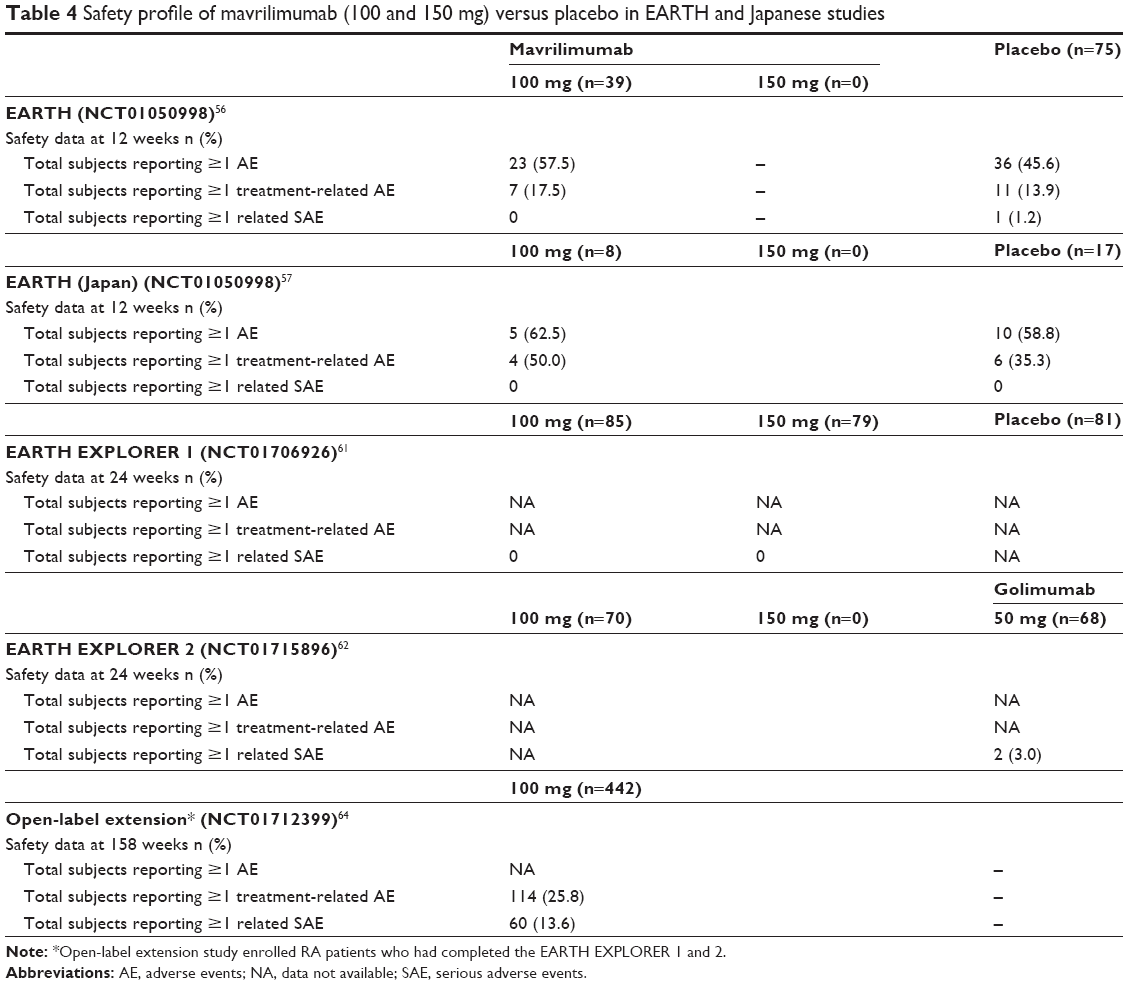 | Table 4 Safety profile of mavrilimumab (100 and 150 mg) versus placebo in EARTH and Japanese studies |
The majority of AEs observed in patients with RA treated with mavrilimumab were mild to moderate in severity. In the EARTH study, five serious AEs were reported: one (worsening of RA) in the placebo group and four (one intervertebral disc disorder, one spontaneous abortion, one fracture of the humerus, and one fracture of the patella) in the mavrilimumab group, and none considered treatment related. Low-titer and transient anti-mavrilimumab antibodies were observed in 23 subjects (3 of 75 in the placebo and 20 of 158 in the active treatment groups) and they did not impact the pharmacokinetics. High-titer antidrug antibodies were reported in 1 placebo (1.3%) subject and 10 mavrilimumab (6.3%; 3 at 10 mg, 4 at 30 mg, 1 at 50 mg, and 2 at 100 mg) subjects, with no correlation with tolerability and tachyphylaxis.56 Similarly, in the Japanese EARTH study, 22 treatment-emergent adverse events (TEAEs) were reported (6 [35.3%] in the placebo group and 16 [47.1%] in the mavrilimumab group). Only one serious AE (pneumonia) was reported, leading to mavrilimumab discontinuation, whereas no antidrug antibodies were detected.57 However, in this Japanese cohort study, duration was shorter (12 weeks) and sample size smaller than in the European one, thus limiting the broader extrapolation of data. Moreover, methods for detection of antidrug antibodies were different between the two studies (electrochemiluminescence assay in the Japanese population and a two-step approach including electrochemiluminescence screening plus a conformatory immunoassay inhibition in the European one), potentially explaining the different incidence of antidrug antibodies development reported in Europe and Japan.
The EARTH EXPLORER 1 and 2 studies confirmed the overall good safety profile of mavrilimumab. The most common TEAEs reported were headache, nasopharyngitis, and upper respiratory tract infections.61,62,68 In the first study, two cases of pneumonia were observed (one in the mavrilimumab 30 mg eow group and one in the placebo group),61 whereas in the second study, one pneumocystis pneumonia and one lung disorder were observed in the golimumab group.68 During the open-label long-term extension of both phase IIb studies, the most common TEAEs were bronchitis (4.8%), respiratory tract infection (2%), cholelithiasis (1%), cough (0.8%), and neutropenia (0.8%).65
GM-CSF pathway blockade in RA beyond mavrilimumab
As previously described, four monoclonal antibodies targeting GM-CSF have been developed: lenzilumab (KB003), MORAb-022, MOR103, and namilumab (MT203; Table 5). The only one study conducted with lenzilumab in RA was early terminated upon completion of safety run-in because of the development program refocus.69 MORAb-022 has been evaluated in a phase I study completed in July 2014, but up to date no results have been published yet.70 MOR103 was analyzed in phase Ib/IIa clinical trials with safety end points, involving 96 RA patients with moderately active disease (DAS28 ≤5.1) randomized to receive MOR103 (0.3, 1.0, or 1.5 mg/kg intravenously) or placebo.71 TEAEs were mild or moderate in intensity and reported in similar frequencies among the four groups, with nasopharyngitis being the most common one. Moreover, subjects in the MOR103 1.0 and 1.5 mg/kg groups showed significant improvements in DAS28 scores when compared with subjects receiving placebo.71 Namilumab has been evaluated in a phase Ia study72 and in the PRIORA trial, a phase Ib study involving 24 patients with mild-to-moderate RA randomized to receive three single injections of namilumab (150 or 300 mg) or placebo eow in combination with MTX, with a follow-up period of 12 weeks.73 Nasopharyngitis, exacerbation/worsening of RA, and musculoskeletal pain were the most commonly reported TEAEs, with a similar incidence among the three groups of treatment. Greater improvement in DAS28-CRP was observed in both the namilumab groups compared with placebo from day 27 until the end of the study.73 Two phase II studies (NEXUS74 and TELLUS75) have been initiated in patients with moderate-to-severe RA, but no results have been reported yet.
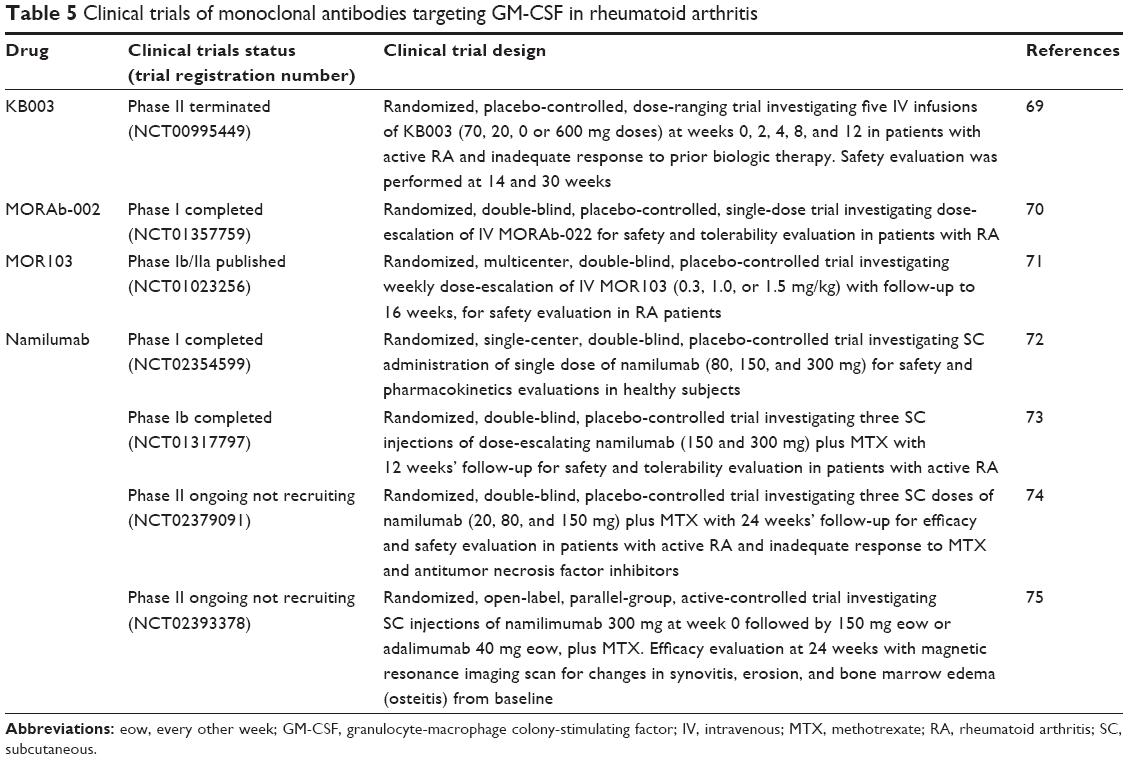 | Table 5 Clinical trials of monoclonal antibodies targeting GM-CSF in rheumatoid arthritis |
Taken together, data on anti-GM-CSF monoclonal antibodies seem to be promising, but further future analyses are required to better clarify the role of this drug class in the treatment of RA.
Conclusion
The introduction of biological therapies has completely transformed the paradigm of RA treatment, improving the real-life application of treat-to-target approach. However, ~30% of RA patients are unresponsive to available bDMARDs, resulting in high disability and increased morbidity and mortality. In the last few decades, the scientific knowledge on RA pathogenesis vastly improved, leading to the identification of new proinflammatory molecules and new therapeutic targets. Several in vitro and in vivo studies showed that GM-CSF, besides a hematopoietic factor, is one of the proinflammatory cytokines involved in myeloid cells (mainly macrophages) activation in autoimmune processes, such as RA. Mavrilimumab, a human monoclonal antibody targeting the subunit α of CM-CSF-R, was recently developed as a competitive antagonist of GM-CSF and adopted in human trials for moderate RA. The very first experiences with mavrilimumab demonstrated an overall good safety profile of this novel bDMARD. In particular, no cases of pulmonary alveolar proteinosis have been observed in over 600 patients exposed to mavrilimumab, similar to that observed in the preclinical studies. Moreover, phase II studies demonstrated a very rapid clinical response (since week 1 after administration) with a progressive improvement through week 12, and highlighted a dose-dependent successful clinical outcome, probably with mavrilimumab 150 mg eow being the most effective. Interestingly, in the EARTH EXPLORER 2 study, mavrilimumab 100 mg eow showed an overall good efficacy in TNFi failures when compared to golimumab. Although the observed clinical response is comparable to available bDMARDs, the novel mode of action of GM-CSF blockade may provide an alternative treatment option for patients resistant to other biologics. However, these findings should be confirmed by ongoing worldwide phase III trials on mavrilimumab in RA.
Disclosure
The authors report no conflicts of interest in this work.
References
Mcinnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. | ||
Firestein GS. The disease formerly known as rheumatoid arthritis. Arthritis Res Ther. 2014;16(3):114. | ||
Smolen JS, Breedveld FC, Burmester GR, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis. 2016;75(1):3–15. | ||
Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492–509. | ||
Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340(4):253–259. | ||
Keystone EC, Kavanaugh A, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5):1400–1411. | ||
Lipsky PE, van der Heijde DM, Clair EWS, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343(22):1594–1602. | ||
Favalli EG, Pregnolato F, Biggioggero M, et al. Twelve-year retention rate of first-line tumor necrosis factor inhibitors in rheumatoid arthritis: real-life data from a local registry. Arthritis Care Res (Hoboken). 2016;68(4):432–439. | ||
Kievit W, Fransen J, Oerlemans AJ, et al. The efficacy of anti-TNF in rheumatoid arthritis, a comparison between randomised controlled trials and clinical practice. Ann Rheum Dis. 2007;66(11):1473–1478. | ||
Metcalf D. The colony-stimulating factors and cancer. Cancer Immunol Res. 2013;1(6):351–356. | ||
Cornish AL, Campbell IK, McKenzie BS, Chatfield S, Wicks IP. G-CSF and GM-CSF as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):554–559. | ||
Avci AB, Feist E, Burmester GR. Targeting GM-CSF in rheumatoid arthritis. Clin Exp Rheumatol. 2016;34(4 Suppl 98):39–44. | ||
Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. 2016;12(1):37–48. | ||
Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178(8):5245–5252. | ||
Hamilton JA. GM-CSF as a target in inflammatory/autoimmune disease: current evidence and future therapeutic potential. Expert Rev Clin Immunol. 2015;11(4):457–465. | ||
Quill H, Gaur A, Brown D, Infante AJ, Phipps RP. Synergistic activation of granulocyte-macrophage colony-stimulating factor production by IL-1 and IL-2 in murine Th1 cells. J Immunol. 1989;143(7):2242–2247. | ||
Burgess AW, Metcalf D. The effect of colony stimulating factor on the synthesis of ribonucleic acid by mouse bone marrow cells in vitro. J Cell Physiol. 1977;90(3):471–483. | ||
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. | ||
Cates AM, Holden VI, Myers EM, Smith CK, Kaplan MJ, Kahlenberg JM. Interleukin 10 hampers endothelial cell differentiation and enhances the effects of interferon α on lupus endothelial cell progenitors. Rheumatology (Oxford). 2015;54(6):1114–1123. | ||
Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol. 2009;86(2):411–421. | ||
Parajuli B, Sonobe Y, Kawanokuchi J, et al. GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflammation. 2012;9(1):268. | ||
Hansen G, Hercus TR, McClure BJ, et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. 2008;134(3):496–507. | ||
Stanley E, Lieschke GJ, Grail D, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A. 1994;91(12):5592–5596. | ||
Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol. 2009;21(5):514–521. | ||
Lari R, Fleetwood AJ, Kitchener PD, et al. Macrophage lineage phenotypes and osteoclastogenesis–complexity in the control by GM-CSF and TGF-beta. Bone. 2007;40(2):323–336. | ||
Daro E, Pulendran B, Brasel K, et al. Polyethylene glycol-modified GM-CSF expands CD11b(high)CD11c(high) but notCD11b(low)CD11c(high) murine dendritic cells in vivo: a comparative analysis with Flt3 ligand. J Immunol. 2000;165(1):49–58. | ||
Zhan Y, Vega-Ramos J, Carrington EM, Villadangos JA, Lew AM, Xu Y. The inflammatory cytokine, GM-CSF, alters the developmental outcome of murine dendritic cells. Eur J Immunol. 2012;42(11):2889–2900. | ||
Greter M, Helft J, Chow A, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36(6):1031–1046. | ||
Shi Y, Liu CH, Roberts AI, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res. 2006;16(2):126–133. | ||
Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nature Publishing Group. 2008;8(7):533–544. | ||
Hamilton JA. Rheumatoid arthritis: opposing actions of haemopoietic growth factors and slow-acting anti-rheumatic drugs. Lancet. 1993;342(8870):536–539. | ||
Wijbrandts CA, Vergunst CE, Haringman JJ, Gerlag DM, Smeets TJM, Tak PP. Absence of changes in the number of synovial sublining macrophages after ineffective treatment for rheumatoid arthritis: implications for use of synovial sublining macrophages as a biomarker. Arthritis Rheum. 2007;56(11):3869–3871. | ||
Haringman JJ, Gerlag DM, Zwinderman AH, et al. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64(6):834–838. | ||
Berenbaum F, Rajzbaum G, Amor B, Toubert A. Evidence for GM-CSF receptor expression in synovial tissue. An analysis by semi-quantitative polymerase chain reaction on rheumatoid arthritis and osteoarthritis synovial biopsies. Eur Cytokine Netw. 1994;5(1):43–46. | ||
Wright HL, Bucknall RC, Moots RJ, Edwards SW. Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatology (Oxford). 2012;51(3):451–459. | ||
Alvaro-Gracia JM, Zvaifler NJ, Brown CB, Kaushansky K, Firestein GS. Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL-1 and tumor necrosis factor-alpha. J Immunol. 1991;146(10):3365–3371. | ||
Xu WD, Firestein GS, Taetle R, Kaushansky K, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. II. Granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J Clin Invest. 1989;83(3):876–882. | ||
Leizer T, Cebon J, Layton JE, Hamilton JA. Cytokine regulation of colony-stimulating factor production in cultured human synovial fibroblasts: I. Induction of GM-CSF and G-CSF production by interleukin-1 and tumor necrosis factor. Blood. 1990;76(10):1989–1996. | ||
Campbell IK, Novak U, Cebon J, Layton JE, Hamilton JA. Human articular cartilage and chondrocytes produce hemopoietic colony-stimulating factors in culture in response to IL-1. J Immunol. 1991;147(4):1238–1246. | ||
Nakano K, Okada Y, SAITO K, et al. Rheumatoid synovial endothelial cells produce macrophage colony-stimulating factor leading to osteoclastogenesis in rheumatoid arthritis. Rheumatology (Oxford). 2007;46(4):597–603. | ||
Campbell IK, Bendele A, Smith DA, Hamilton JA. Granulocyte-macrophage colony stimulating factor exacerbates collagen induced arthritis in mice. Ann Rheum Dis. 1997;56(6):364–368. | ||
de Vries EG, Willemse PH, Biesma B, Stern AC, Limburg PC, Vellenga E. Flare-up of rheumatoid arthritis during GM-CSF treatment after chemotherapy. The Lancet. 1991;338(8765):517–518. | ||
Hazenberg BP, Van Leeuwen MA, van Rijswijk MH, Stern AC, Vellenga E. Correction of granulocytopenia in Felty’s syndrome by granulocyte-macrophage colony-stimulating factor. Simultaneous induction of interleukin-6 release and flare-up of the arthritis. Blood. 1989;74(8):2769–2770. | ||
Campbell IK, Rich MJ, Bischof RJ, Hamilton JA. The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J Leukoc Biol. 2000;68(1):144–150. | ||
Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol. 1998;161(7):3639–3644. | ||
Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. 2001;3(5):293–298. | ||
van Nieuwenhuijze AEM, van de Loo FA, Walgreen B, et al. Complementary action of granulocyte macrophage colony-stimulating factor and interleukin-17A induces interleukin-23, receptor activator of nuclear factor-κB ligand, and matrix metalloproteinases and drives bone and cartilage pathology in experimental arthritis: rationale for combination therapy in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):1094. | ||
Gearing DP, King JA, Gough NM, Nicola NA. Expression cloning of a receptor for human granulocyte-macrophage colony-stimulating factor. EMBO J. 1989;8(12):3667–3676. | ||
Rajotte D, Sadowski HB, Haman A, et al. Contribution of both STAT and SRF/TCF to c-fos promoter activation by granulocyte-macrophage colony-stimulating factor. Blood. 1996;88(8):2906–2916. | ||
Nair JR, Edwards SW, Moots RJ. Mavrilimumab, a human monoclonal GM-CSF receptor-α antibody for the management of rheumatoid arthritis: a novel approach to therapy. Expert Opin Biol Ther. 2012;12(12):1661–1668. | ||
Greven DEA, Cohen ES, Gerlag DM, et al. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Ann Rheum Dis. 2015;74(10):1924–1930. | ||
Vainshtein I, Roskos LK, Cheng J, Sleeman MA, Wang B, Liang M. Quantitative measurement of the target-mediated internalization kinetics of biopharmaceuticals. Pharm Res. 2015;32(1):286–299. | ||
Ryan PC, Sleeman MA, Rebelatto M, et al. Nonclinical safety of mavrilimumab, an anti-GMCSF receptor alpha monoclonal antibody, in cynomolgus monkeys: relevance for human safety. Toxicol Appl Pharmacol. 2014;279(2):230–239. | ||
Campbell J, Nys J, Eghobamien L, Cohen ES, Robinson MJ, Sleeman MA. Pulmonary pharmacodynamics of an anti-GM-CSFRα antibody enables therapeutic dosing that limits exposure in the lung. MAbs. 2016;8(7):1398–1406. | ||
Burmester GR, Feist E, Sleeman MA, Wang B, White B, Magrini F. Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-α, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann Rheum Dis. 2011;70(9):1542–1549. | ||
Burmester GR, Weinblatt ME, Mcinnes IB, et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis. 2013;72(9):1445–1452. | ||
Takeuchi T, Tanaka Y, Close D, Godwood A, Wu C-Y, Saurigny D. Efficacy and safety of mavrilimumab in Japanese subjects with rheumatoid arthritis: findings from a Phase IIa study. Mod Rheumatol. 2015;25(1):21–30. | ||
Jin DC, Wu C-Y, Roskos LK, Godwood A, Close D, Ben Wang. Exposure–efficacy analysis of mavrilimumab in rheumatoid arthritis: modeling and simulation of phase II clinical data [abstract]. Ann Rheum Dis. 2015;74(Suppl 2):S1043–S1043. | ||
Mcinnes IB, Burmester GR, Kremer JM, et al. Rapid onset of clinical benefit in patients with RA treated with mavrilimumab, a fully human monoclonal antibody targeting GM-CFSR-alpha: subanalysis of the phase IIb earth explorer 1 study[abstract]. Ann Rheum Dis. 2015;74(Suppl 2):S723–S724. | ||
Burmester GR, Mcinnes IB, Kremer JM, et al. Efficacy and safety/tolerability of mavrilimumab, a human GM-CSFRalfa monoclonal antibody in patients with rheumatoid arthritis [abstract]. Arthritis Rheumatol. 2014;66(Suppl 10):S1231–S1232. | ||
Burmester GR, McInnes IB, Kremer JM, et al. Efficacy and safety of mavrilimumab, A fully human Gm–CSFR-Alpha monoclonal antibody in patients with rheumatoid arthritis: primary results from the Earth Explorer 1 Study [abstract]. Ann Rheum Dis. 2015;74(Suppl 2):S78. | ||
Weinblatt ME, Mcinnes IB, Kremer J, et al. Earth explorer 2, a phase IIb exploratory study evaluating efficacy and safety of mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor-alpha monoclonal antibody, and the tumor necrosis factor antagonist golimumab in rheumatoid arthritis[abstract]. Ann Rheum Dis. 2016;75(Suppl 2):S717–S718. | ||
Guo X, Wang S, Bay-Jensen AC, et al. Sustained suppression of rheumatoid arthritis disease markers by mavrilimumab but not golimumab in anti-tumor necrosis factor-inadequate responders: an exploratory analysis in the phase IIb earth explorer 2 clinical trial[abstract]. Ann Rheum Dis. 2016;75(Suppl 2):S150–S151. | ||
Burmester G, McInnes I, Kremer J, et al. Mavrilimumab, a Fully Human Granulocyte-Macrophage Colony-Stimulating Factor Receptor-α Monoclonal Antibody: Long-Term Safety and Efficacy for up to 158 Weeks of Treatment in Patients with Rheumatoid Arthritis [abstract]. Arthritis Rheumatol. 2016;68(suppl 10). Abstract number 1616. | ||
Burmester GR, McInnes IB, Kremer J, et al. Long-term safety and efficacy of mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor-alpha monoclonal antibody, in patients with rheumatoid arthritis [abstract]. Ann Rheum Dis. 2016;75(Suppl 2):S510. | ||
Grant E, Schwickart M, Godwood A, et al. Lack of Autoantibodies to Peptidyl Arginine Deiminase 4 Predict Increased Efficacy of Mavrilimumab in Rheumatoid Arthritis [abstract]. Arthritis Rheumatol. 2016;68(suppl 10). Abstract number 2634. | ||
Burmester G, Michaels M, Close D, et al. Results of a Comprehensive Review of Pulmonary Function and Safety Data in a Phase IIb Clinical Program Testing Anti-GM-CSF Receptor Antagonist Mavrilimumab for Treatment of RA [abstract]. Arthritis Rheumatol. 2016;68(suppl 10). Abstract number 2653. | ||
Weinblatt M, McInnes I, Kremer J, et al. EARTH EXPLORER 2, a Phase IIb Exploratory Study Evaluating Efficacy and Safety of Mavrilimumab, a Fully Human Granulocyte-Macrophage Colony-Stimulating Factor Receptor-Alpha Monoclonal Antibody, and the Tumor Necrosis Factor Antagonist Golimumab in Rheumatoid Arthritis [abstract]. Arthritis Rheumatol. 2016;68(suppl 10). Abstract number 1619. | ||
Study of KB003 In biologics-inadequate rheumatoid arthritis. ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/results?term=NCT00995449. Accessed November 28, 2016. | ||
Safety and tolerability of MORAb-022 in healthy and rheumatoid arthritis subjects. ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/results?term=NCT01357759. Accessed November 28, 2016. | ||
Behrens F, Tak PP, Østergaard M, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2015;74(6):1058–1064. | ||
A Phase 1 MT203 Single-dose Study to Evaluate Safety, PK and PD. ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02354599. Accessed November 28, 2016. | ||
Huizinga TW, Batalov A, Yablanski K, et al. First-in-patient study of namilumab, an anti-GM-CSF monoclonal antibody, in active rheumatoid arthritis: results of the PRIORA phase Ib study. Ann Rheum Dis. 2015;74(Suppl 2):S733. | ||
Dose finding study of namilumab in combination with methotrexate in participants with moderate to severe rheumatoid arthritis (RA). ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02379091. Accessed November 28, 2016. | ||
Namilumab vs adalimumab in participants with moderate to severe early rheumatoid arthritis inadequately responding to methotrexate (TELLUS). ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02393378. Accessed November 28, 2016. | ||
Fiehn C, Wermann M, Pezzutto A, Hüfner M, Heilig B. Plasma GM-CSF concentrations in rheumatoid arthritis, systemic lupus erythematosus and spondyloarthropathy. Z Rheumatol. 1992;51(3):121–126. | ||
Bischof RJ, Zafiropoulos D, Hamilton JA, Campbell IK. Exacerbation of acute inflammatory arthritis by the colony-stimulating factors CSF-1 and granulocyte macrophage (GM)-CSF: evidence of macrophage infiltration and local proliferation. Clin Exp Immunol. 2000;119(2):361–367. | ||
Yang YH, Hamilton JA. Dependence of interleukin-1-induced arthritis on granulocyte-macrophage colony-stimulating factor. Arthritis Rheum. 2001;44(1):111–119. | ||
Plater-Zyberk C, Joosten LA, Helsen MM, Hepp J, Baeuerle PA, van den Berg WB. GM-CSF neutralisation suppresses inflammation and protects cartilage in acute streptococcal cell wall arthritis of mice. Ann Rheum Dis. 2007;66(4):452–457. | ||
Plater-Zyberk C, Joosten LA, Helsen MM, Koenders MI, Baeuerle PA, van den Berg WB. Combined blockade of granulocyte-macrophage colony stimulating factor and interleukin 17 pathways potently suppresses chronic destructive arthritis in a tumour necrosis factor alpha-independent mouse model. Ann Rheum Dis. 2009;68(5):721–728. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.