Back to Journals » OncoTargets and Therapy » Volume 16
Spotlight on GOT2 in Cancer Metabolism
Authors Kerk SA ![]() , Garcia-Bermudez J, Birsoy K, Sherman MH, Shah YM, Lyssiotis CA
, Garcia-Bermudez J, Birsoy K, Sherman MH, Shah YM, Lyssiotis CA
Received 5 May 2023
Accepted for publication 29 July 2023
Published 22 August 2023 Volume 2023:16 Pages 695—702
DOI https://doi.org/10.2147/OTT.S382161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Samuel A Kerk,1 Javier Garcia-Bermudez,2 Kivanc Birsoy,3 Mara H Sherman,4 Yatrik M Shah,5 Costas A Lyssiotis5
1Doctoral Program in Cancer Biology, University of Michigan, Ann Arbor, MI, USA; 2Children’s Medical Center Research Institute, University of Texas Southwestern Medical Center, Dallas, TX, USA; 3Laboratory of Metabolic Regulation and Genetics, The Rockefeller University, New York, NY, USA; 4Cancer Biology & Genetics Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA; 5Department of Molecular & Integrative Physiology, University of Michigan, Ann Arbor, MI, USA
Correspondence: Costas A Lyssiotis, Email [email protected]
Abstract: GOT2 is at the nexus of several critical metabolic pathways in homeostatic cellular and dysregulated cancer metabolism. Despite this, recent work has emphasized the remarkable plasticity of cancer cells to employ compensatory pathways when GOT2 is inhibited. Here, we review the metabolic roles of GOT2, highlighting findings in both normal and cancer cells. We emphasize how cancer cells repurpose cell intrinsic metabolism and their flexibility when GOT2 is inhibited. We close by using this framework to discuss key considerations for future investigations into cancer metabolism.
Keywords: transaminase, mitochondria, tumor microenvironment, nucleotides, redox, pancreatic cancer
Introduction
Tumors rewire cellular metabolism to sustain aberrant growth and survival.1–3 Cancer cells do not create new metabolic pathways, but rather re-purpose inherent metabolic tendencies of the cells and tissues of origin.4 Thus, understanding cellular metabolism under homeostatic conditions can provide insight into the metabolic underpinnings of cancer cells. Furthermore, unique aspects of cancer metabolism present vulnerabilities that can be exploited therapeutically without disrupting non-malignant cell function.5
Here, we discuss these concepts through the lens of glutamate-oxaloacetate transaminase 2 (GOT2), a mitochondrial enzyme that has recently garnered considerable interest in cancer metabolism. GOT2 is a mitochondrial aminotransferase that reversibly produces aspartate and α-ketoglutarate from glutamate and oxaloacetate, while the cytosolic isoform GOT1 catalyzes the converse reaction. GOT2 is most well known as a member of the malate-aspartate shuttle (MAS), a cytosolic-mitochondrial pathway that transfers reducing equivalents into the mitochondria to support oxidative phosphorylation.6 In addition to, and as a part of this function, GOT2 also participates in several critical metabolic functions: nucleotide synthesis, redox homeostasis, fueling the tricarboxylic acid (TCA) cycle, fatty acid transport, nitrogen balance, and sulfur catabolism (Figure 1). The following sections will dissect how each of these GOT2-related pathways have been investigated under both cellular homeostasis and cancer metabolism.
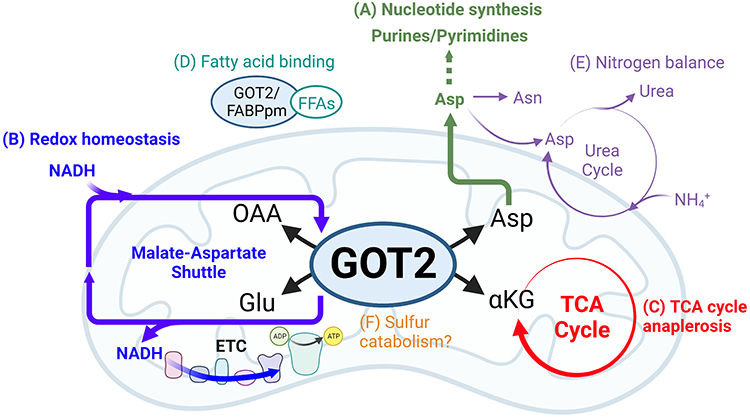 |
Figure 1 Cellular metabolism involving GOT2. (A) Asp produced by GOT2 provides carbon and nitrogen for synthesis of purine and pyrimidine nucleotide bases. (B) As part of the malate-aspartate shuttle, GOT2 facilitates the transfer of electrons generated from cytosolic glucose oxidation and carried by membrane-impermeable NADH into the mitochondria, where NADH is regenerated and electrons are deposited into the ETC to drive ATP production through oxidative phosphorylation. (C) GOT2 fuels anabolic TCA cycling through production of the intermediate αKG. (D) Cytosolic GOT2, also known as FABPpm, binds to free fatty acids with implications for FFA transport, oxidation, and signaling. (E) Asp from GOT2 is further utilized to synthesize the amino acid Asn and for incorporation into the urea cycle, which detoxifies cellular NH4+ through urea excretion or recycling of nitrogen for numerous anabolic pathways. (F) Some evidence suggests promiscuous GOT2 activity is involved in the catabolism of sulfur-containing metabolites, though this mechanism remains to be thoroughly investigated. Created with BioRender.com. Abbreviations: GOT2, glutamate-oxaloacetate transaminase 2; OAA, oxaloacetate; Glu, glutamate; Asp, aspartate; αKG, α-ketoglutarate; NADH, reduced nicotinamide adenine dinucleotide; ETC, electron transport chain; ADP, adenosine diphosphate; ATP, adenosine triphosphate; FFA, free fatty acid; Asn, asparagine; NH4+, ammonium; TCA, tricarboxylic acid. |
Metabolic Functions of GOT2
Aspartate Requirement for Nucleotide Synthesis
The deregulated cell growth and division in cancer cells imposes significant requirements for RNA and DNA. Perhaps, the most well-studied product of the GOT2-catalyzed reaction is the amino acid aspartate, which is a rate-limiting metabolite for cancer cells due to its contribution of both carbon and nitrogen to nucleotide synthesis7–10 (Figure 1A). In vitro studies demonstrated that impaired proliferation of pancreatic cancer cells due to loss of GOT2 could be rescued by aspartate supplementation. In vivo, however, pancreatic tumors overcome loss of GOT2 by acquisition of aspartate from the tumor microenvironment using macropinocytosis.11 In acute myelogenous leukemia, disruption of vitamin B6 synthesis, a co-factor in the GOT2 reaction, disrupted aspartate synthesis and impaired cancer cell growth. Supplementation with aspartate was sufficient to rescue decreased GOT2 activity due to chemical and genetic inhibition of the vitamin B6 pathway.12 Furthermore, GOT2 expression is increased in triple negative breast cancer and overexpression of GOT2 in these cells increased intracellular aspartate levels and promoted proliferation.13,14 Interestingly, VHL-deficient clear-cell renal clear cell renal carcinoma tumors have decreased expression of GOT2,15,16 suggesting alternative strategies to generate or acquire aspartate, like macropinocytosis, though those mechanisms remain to be identified.
Aspartate has also been reported to play important roles in metabolism in healthy cells. For example, chondrocytes depend on GOT2-derived aspartate for differentiation and to carry out bone remodeling functions.17 Similarly, perturbations in hematopoietic stem cells (HSCs) that elevate intracellular aspartate levels, such as overexpression of the mitochondrial aspartate transporter or deletion of GOT1, the cytosolic isoform that consumes aspartate, boosted HSC number and activity. Deletion of both GOT1 and GOT2 was lethal for HSCs as they were unable to synthesize nucleotides or another product of aspartate metabolism, the amino acid asparagine.18
Redox Balance
Disruption of the MAS induces “reductive stress”, characterized by increased levels of NADH that interfere with redox sensitive metabolic pathways. Patients with germline loss-of-function GOT2 variants present with neurological defects including epilepsy, and fibroblasts derived from these patients demonstrated impaired serine biosynthesis (a redox coupled pathway). Proper function could be restored through supplementation with serine or pyruvate, the latter being a metabolite that accepts electrons from NADH to resolve reductive stress.19 Similarly, in vitro studies in pancreatic cancer demonstrated that loss of GOT2 induced reductive stress due to NADH accumulation, which was alleviated through NADH oxidizing mechanisms.20 Furthermore, impaired mitochondrial metabolism and reduced expression of GOT2 in neurons have been identified in several neurological pathologies.21–24 This suggests that GOT2 is involved in sustaining oxidative metabolism to generate energy for highly active neuronal subtypes. Dietary, chemical, or genetic interventions that alleviate reductive stress could be effective therapies for defects in the MAS, including GOT2 inactivation.25
Maintenance of NADPH/NADP+ pools is also essential to protect cells against damaging levels of reactive oxygen species (ROS). GOT2 consumes glutamate, therefore downregulation of GOT2 expression may lead to an increased availability of glutamate for production of glutathione (GSH), a tripeptide of glycine, glutamate, and cysteine and the most abundant endogenous antioxidant.26 In ischemic hearts, hypoxia-inducible factor 1α expression, the primary cellular factor involved in the hypoxic response, is inversely correlated with GOT2 expression and coincides with a concomitant increase in GSH.
In cancer cells, the maintenance of a redox state that supports sustained proliferation is paramount. Because the MAS sustains redox balance between the cytosol and the mitochondria (Figure 1B), its activity is modulated in various cancer types. For instance, a rewired MAS in PDAC fuels production of NADPH through malic enzyme 1 (ME1) in the cytosol to regenerate GSH and protect cells from ROS.27 Several enzymes in this pathway are overexpressed in pancreatic tumors compared to normal pancreas.28 Loss of GOT2 also induced senescence mediated by decreased NADPH synthesis and ROS accumulation.29 Furthermore, inhibition of glutamine breakdown (or glutaminolysis) coupled with therapies that generated ROS-induced redox catastrophe in pancreatic cancer cells both in vitro and in vivo.28 Conversely, in hepatocellular carcinoma, GOT2 is downregulated in response to insulin signaling,30 resulting in increased glutamate availability for GSH synthesis.31
The Dysregulated Cancer TCA Cycle
The net activity of the MAS does not consume carbon or nitrogen. However, when provided substrates in excess of MAS activity, GOT2 can generate surplus α-ketoglutarate and aspartate. The aspartate can be used as a biosynthetic precursor, as described above. Production of α-ketoglutarate from GOT2 provides a major carbon input that sustains anaplerotic TCA cycling in proliferating cells (Figure 1C). When functioning in this mode, the TCA cycle generates both the reducing equivalents needed to produce energy and provide biosynthetic precursors.32 Pancreatic cancer cells in culture utilize glutamine-derived α-ketoglutarate through GOT2 as the primary anaplerotic input to support biosynthesis and proliferation.27 Similarly, GOT2 overexpression in diffuse large B cell lymphoma drives elevated glutaminolysis to support anaplerosis through α-ketoglutarate production.33
The ratios of TCA cycle intermediates have also been implicated in cell fate.32 For example, paragangliomas and pheochromocytomas, rare neurological tumors, are unique in that they harbor loss of function mutations in the MAS components MDH2, SLC25A11, and a gain of function mutation in GOT2.34 Indeed, a whole-exome sequencing study of 11 patients revealed a patient with a germline GOT2 gain of function mutation, while two other tumors had increased GOT2 activity.35 This increased GOT2 activity elevated both the succinate/fumarate ratio and the available aspartate, due to increased anaplerosis.35,36 Epigenetic modifying enzymes, such as histone and DNA demethylases, consume α-ketoglutarate and produce succinate. Therefore, while the TCA cycle is important for biosynthesis and energy production, it is necessary to consider how disruption of the α-ketoglutarate/succinate ratio through increased GOT2 activity has broader implications on cell state and differentiation programs in these and other tumors, as well as in normal physiology.
Emerging GOT2 Pathways in Cancer
GOT2 has been reported to have additional functions, outside of the classical roles described above, such as fatty acid binding, cysteine/sulfur breakdown, and nitrogen balance (Figure 1D–F). For example, several studies have proposed GOT2 is a plasma membrane-bound fatty acid-binding protein and have provided the alternate protein name FABPpm. In this role, GOT2/FABPpm is responsible for the import of free fatty acids under conditions of cellular stress, organ injury, or fatty acid accumulation, potentially influencing fatty acid oxidation.37–46 Despite considerable literature examining the role of GOT2 as a plasma-membrane bound fatty acid transporter in several tissues, this capacity has only recently been examined in cancer. One provocative study confirmed GOT2 as a fatty acid-binding protein in pancreatic cancer47 (Figure 1D). GOT2 free fatty acid-binding activity in syngeneic allografts in immune competent mice promoted anti-inflammatory PPARδ activity, ultimately blocking cytotoxic T cell infiltration, and promoting tumor growth.47 These findings indicate more remains to be investigated regarding the role of GOT2 in fatty acid metabolism in cancer.
GOT2 is also involved in nitrogen balance through production/consumption of the non-essential amino acids aspartate and glutamate. Regarding aspartate, it can enter the urea cycle, the primary mechanism by which cells either recycle or dispose of nitrogen (Figure 1E). Human patients with inborn errors of GOT2 metabolism displayed dysregulated levels of serum ammonia.19,25 As such, further work is needed to identify mechanisms by which GOT2 is involved in removing toxic levels of ammonia or recovering nitrogen for incorporation into amino acids and nucleotides.
In colorectal cancer, GOT2 is essential in nitrogen balance through production of amino acids and the urea cycle via a HIF1a-SOX12-GOT2 axis that drives asparagine production from aspartate (Figure 1E). Inhibition of this pathway impairs cell growth.48 Additionally, in Kaposi’s sarcoma-associated herpesvirus cells, GOT2-derived nitrogen is essential for both nucleotide and amino acid production.49 Cells detoxify ammonia through the urea cycle and aspartate incorporation is a critical node in fueling this cycle. Cancer cells often cells downregulate urea cycle enzymes to maintain aspartate pools for nucleotide production.50 However, how cancer cells tune aspartate flux between the urea cycle or nucleotide synthesis remains unclear.
Promiscuous GOT2 transaminase activity has also been proposed to act on cysteine sulfinate to produce 3-sulfinylpyruvate, which is further catabolized to sulfur dioxide and pyruvate, presenting a potential link between GOT2 and taurine biosynthesis51 (Figure 1F). Downregulation of GOT2 and the sulfur dioxide pathway is also implicated in myocardial injury.52 Furthermore, serum levels of GOT2 correlated with increased cardiovascular disease,45 and this was proposed to be due to increased sulfur dioxide production. This pathway has not yet been evaluated in cancer cells.
GOT2 in the Tumor Microenvironment
Tumors are dynamic pseudo-organs in which malignant and non-transformed cell types work in concert to support growth and survival.53,54 As such, the metabolism of immune and stromal cells can have a strong influence on cancer cells.55 Aside from its myriad functions in cancer cells, GOT2 is also expressed, and potentially important, in several of these non-cancer tumor cells. For instance, in an autochthonous model of pancreatic tumorigenesis with loss of Got2, while immunohistochemistry (IHC) confirmed knockdown of Got2 in transformed, epithelial lesions, the surrounding stroma retained strong expression of Got2.20 Indeed, single-cell RNA-sequencing of mouse and human pancreatic tumors also detected significant GOT2 expression in several compartments including macrophages, fibroblasts, T cells, and endothelial cells.56 Furthermore, CAR-T cells engineered to express GOT2 demonstrated enhanced cytotoxic activity against liver cancer both in vitro and in vivo.57 An in-silico analysis of clear cell renal carcinoma detected GOT2 expression in both immune and stromal cell types.16 In sum, these findings suggest that understanding the role of GOT2 in the tumor as whole is important when evaluating GOT2 as a potential target in cancer.
Regulation of GOT2 in Cancer
The regulation of metabolic enzyme activity enables cells to meet the dynamic requirements imposed by both the external environment and intracellular signaling. Many cancer types induce GOT2 activity through epigenetic, post-transcriptional, or post-translational mechanisms. For example, in breast cancer, the BRCA1/ZBRK1 complex represses GOT2 promoter activity,14 suggesting that loss of BRCA1 induces GOT2 expression. As mentioned earlier, VHL-deficient clear cell renal carcinoma tumors display decreased GOT2 expression.15 This correlates with hypermethylation at the GOT2 promoter.16 However, in CRC, hypoxic HIF1 activity induces SOX12 binding to the promoters of several metabolic enzymes, including GOT2, to promote expression.48 These two studies suggest differential activity between cancer cells from different tissue types, as well as between hypoxia versus loss of VHL.
At the post-transcriptional level, some evidence in non-small cell lung cancer indicates that GOT2 transcript levels could be regulated by miRNAs.58 Post-translationally, one study posited that GOT2 acetylation promoted complex formation with MDH2 to enhance activity of the MAS.59 A second study found that inflammatory signaling in the liver-induced acetylation of GOT2 to promote its activity and drive the MAS.60 However, the gamut of GOT2 post-translational modifications remains to be characterized. Lastly, GOT2 expression and activity can be modulated according to external stimuli. For instance, in diffuse large B cell lymphoma, STAT3 and NF-kB inflammatory signaling increased GOT2 expression through c-Myc.33 Further work into the regulatory underpinnings of GOT2 could provide a better understanding for differential metabolic activities and dependencies between cancer cell types and environments.
Conclusion: Key Considerations When Interrogating Cancer Metabolism
Cumulatively, the body of work involving GOT2 highlights several key points in tumor metabolism. First, cancer cells exhibit dynamic metabolism to support malignant functions. Second, the metabolic pathways utilized by cancer cells are dictated by tissue of origin, genomic profile, environmental nutrient composition, and external signaling. Third, investigating cancer metabolism in the proper context is essential for identifying therapeutically tractable targets.
A major challenge in targeting cancer metabolism is the ability of cancer cells to rapidly adapt to metabolic pathway blockade.61 As it relates to the MAS, GOT2 inhibition leads to an elevated NADH/NAD+ ratio, and this reductive stress is detrimental to rapidly proliferating cells. Several essential metabolic pathways depend on this ratio, and NADH accumulation decreases flux through these pathways. Yet, while loss of GOT2 in PDAC cell lines led to reductive stress and impaired proliferation, xenograft tumors grew unimpeded in the absence of GOT2. It was discovered that pancreatic cancer-associated fibroblasts (CAFs) release sufficient pyruvate to ameliorate NADH accumulation through reduction to lactate20 (Figure 2A). In addition to alleviating reductive stress, PDAC cells can acquire aspartate through macropinocytosis in the absence of GOT2 (Figure 2A). PDAC tumors are rich in extracellular matrix with a prominent stromal compartment secreting numerous proteins. These proteins can be non-specifically scavenged and broken down into their amino acid components to replenish aspartate pools in the absence of GOT2 in vivo.11 Additionally, GOT1 can compensate for the loss of GOT2, and double GOT1/GOT2 knockout slowed pancreatic cancer xenograft growth11 (Figure 2A). Lastly, a recent study has demonstrated how the MAS can reverse its direction to correct mitochondrial redox imbalance imposed by inhibition of the ETC.62 Collectively, these studies emphasize both cell-intrinsic and -extrinsic mechanisms can compensate for loss of GOT2.
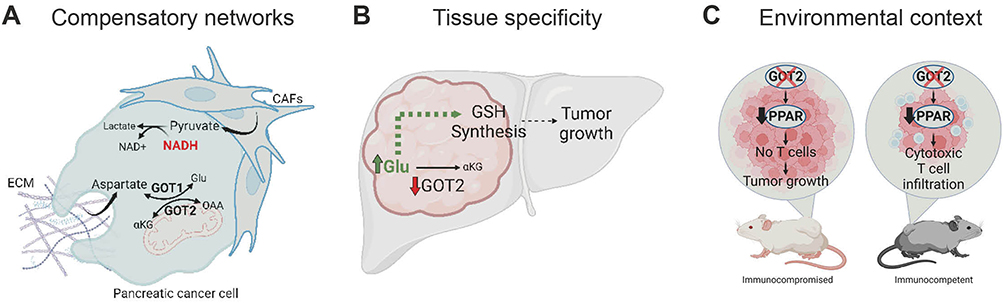 |
Figure 2 Context-specific GOT2 metabolism in cancer. (A) Pancreatic cancer cells compensate for loss of GOT2 via several mechanisms. CAF- and environment-derived pyruvate can be utilized to reduce pyruvate to lactate and alleviate NADH accumulation. Aspartate can either be produced by compensatory cytosolic GOT1 activity or through macropinocytosis and breakdown of ECM components. (B) GOT2 is tumor promoting or restraining depending on tumor type. Hepatocellular carcinoma downregulates GOT2. This increases Glu availability for GSH synthesis and sustained tumor growth. (C) GOT2 impairment induces different phenotypes in immune competent or compromised mouse models. A decrease in PPARδ anti-inflammatory pathways caused by loss of GOT2 fatty acid binding activity lifts the blockade on cytotoxic T cells, leading to increased infiltration and cancer cell killing. Created with BioRender.com. Abbreviations: GOT2, glutamate-oxaloacetate transaminase 2; OAA, oxaloacetate; Glu, glutamate; αKG, α-ketoglutarate; NADH, reduced nicotinamide adenine dinucleotide; GSH, glutathione; PPAR, peroxisome proliferator-activated receptor. |
The tissue of origin plays a critical role in the tumor-promoting or -restraining function of GOT2 in cancer metabolism.4 For instance, in HCC, downregulation of GOT2 is associated with more aggressive disease and worse outcomes. Indeed, overexpressing GOT2 in HCC cell lines restrains tumor growth. One potential explanation is that loss of GOT2 increases pools of the GOT2 substrate glutamate, increasing its incorporation in glutathione31 (Figure 2B). Conversely, in PDAC GOT2 is part of the rewired MAS utilized to synthesize NADPH and reduce glutathione.27 Perhaps, tumors arising in the liver versus the pancreas utilize distinct pathways for glutathione production, which then dictate whether GOT2 expression is detrimental or beneficial for tumor metabolism.
Lastly, the pre-clinical models utilized can influence metabolic findings, even in the same cancer type. Two studies found that loss of GOT2 in human pancreatic cancer cell lines in vitro impaired proliferation, while human xenografts in immunocompromised mice progressed unimpeded in the absence of GOT2. As discussed, cell-intrinsic rewiring and metabolic crosstalk within the TME could both contribute to this compensation.11,20,61 However, intriguingly, a third study reported the opposite phenotype. Loss of GOT2 had no effect on in vitro cellular proliferation but significantly inhibited the growth of murine syngeneic allografts in immunocompetent mice. GOT2 loss induced cytotoxic T cell infiltration due to decreased fatty acid binding and PPARδ signaling in cancer cells, which was absent in the models using human PDAC cells in immunocompromised mice47 (Figure 2C). To complicate matters further, deletion of Got2 in an autochthonous model of pancreatic tumorigenesis did not affect the number or severity of lesions.20 However, these two studies compared rapidly growing, established allografts to gradually developing neoplastic lesions. Future work examining the role of GOT2 in advanced mouse models that more accurately recapitulate human disease are warranted to fully understand the role of GOT2 in pancreatic cancer. Even still, differences between mouse and human tissue will have to be considered. In sum, the model systems used to investigate metabolism can impact cancer metabolism, and careful study across several model systems is needed to accurately assess the metabolic vulnerabilities within a cancer type.
Funding
This work was supported by F99CA264414 (S.A.K.), Cancer Prevention and Research Institute of Texas (CPRIT) award (RR210059) and NIH/NCI (4R00CA248838-02) (J.G-B.), (K.B.), MSK Cancer Center Support Grant/Core Grant P30CA008748 (M.H.S.), R01CA148828 and R01CA245546 (Y.M.S.), and R01CA248160 and R01CA244931 (C.A.L.).
Disclosure
C.A.L. has received consulting fees from Astellas Pharmaceuticals, Odyssey Therapeutics, and T-Knife Therapeutics, and is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting the GOT1-pathway as a therapeutic approach (US Patent No: 2015126580-A1, 05/07/2015; US Patent No: 20190136238, 05/09/2019; International Patent No: WO2013177426-A2, 04/23/2015). K.B. reports personal fees from Atavistik, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi:10.1126/sciadv.1600200
2. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. doi:10.1016/j.cmet.2015.12.006
3. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi:10.1126/science.1160809
4. Kerk SA, Papagiannakopoulos T, Shah YM, Lyssiotis CA. Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nat Rev Cancer. 2021;21(8):510–525. doi:10.1038/s41568-021-00375-9
5. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671–684. doi:10.1038/nrd3504
6. Borst P. The malate-aspartate shuttle (Borst cycle): how it started and developed into a major metabolic pathway. IUBMB Life. 2020;72(11):2241–2259. doi:10.1002/iub.2367
7. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162(3):540–551. doi:10.1016/j.cell.2015.07.016
8. Garcia-Bermudez J, Baudrier L, La K, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. 2018;20(7):775–781. doi:10.1038/s41556-018-0118-z
9. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 2015;162(3):552–563. doi:10.1016/j.cell.2015.07.017
10. Sullivan LB, Luengo A, Danai LV, et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol. 2018;20(7):782–788. doi:10.1038/s41556-018-0125-0
11. Garcia-Bermudez J, Badgley MA, Prasad S, et al. Adaptive stimulation of macropinocytosis overcomes aspartate limitation in cancer cells under hypoxia. Nat Metab. 2022;4(6):724–738. doi:10.1038/s42255-022-00583-z
12. Chen -C-C, Li B, Millman SE, et al. Vitamin B6 addiction in acute myeloid leukemia. Cancer Cell. 2020;37(1):71–84.e77. doi:10.1016/j.ccell.2019.12.002
13. He Y, Deng F, Zhao S, et al. Analysis of miRNA-mRNA network reveals miR-140-5p as a suppressor of breast cancer glycolysis via targeting GLUT1. Epigenomics. 2019;11(9):1021–1036. doi:10.2217/epi-2019-0072
14. Hong R, Zhang W, Xia X, et al. Preventing BRCA1/ZBRK1 repressor complex binding to the GOT2 promoter results in accelerated aspartate biosynthesis and promotion of cell proliferation. Mol Oncol. 2019;13(4):959–977. doi:10.1002/1878-0261.12466
15. Meléndez-Rodríguez F, Urrutia AA, Lorendeau D, et al. HIF1α suppresses tumor cell proliferation through inhibition of aspartate biosynthesis. Cell Rep. 2019;26(9):2257–2265.e2254. doi:10.1016/j.celrep.2019.01.106
16. Ferreira WAS, de Oliveira EHC. Expression of GOT2 is epigenetically regulated by DNA methylation and correlates with immune infiltrates in clear-cell renal cell carcinoma. Curr Issues Mol Biol. 2022;44(6):2472–2489. doi:10.3390/cimb44060169
17. Stegen S, Rinaldi G, Loopmans S, et al. Glutamine metabolism controls chondrocyte identity and function. Dev Cell. 2020;53(5):530–544.e538. doi:10.1016/j.devcel.2020.05.001
18. Qi L, Martin-Sandoval MS, Merchant S, et al. Aspartate availability limits hematopoietic stem cell function during hematopoietic regeneration. Cell Stem Cell. 2021;28(11):1982–1999.e1988. doi:10.1016/j.stem.2021.07.011
19. van Karnebeek CDM, Ramos RJ, Wen XY, et al. Bi-allelic GOT2 mutations cause a treatable malate-aspartate shuttle-related encephalopathy. Am J Hum Genet. 2019;105(3):534–548. doi:10.1016/j.ajhg.2019.07.015
20. Kerk SA, Lin L, Myers AL, et al. Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. Elife. 2022;11:e73245.
21. Missailidis D, Sanislav O, Allan CY, Smith PK, Annesley SJ, Fisher PR. Dysregulated provision of oxidisable substrates to the mitochondria in ME/CFS lymphoblasts. Int J Mol Sci. 2021;22(4):2046. doi:10.3390/ijms22042046
22. Wesseling H, Chan MK, Tsang TM, et al. A combined metabonomic and proteomic approach identifies frontal cortex changes in a chronic phencyclidine rat model in relation to human schizophrenia brain pathology. Neuropsychopharmacology. 2013;38(12):2532–2544. doi:10.1038/npp.2013.160
23. Gao Y, Mu J, Xu T, et al. Metabolomic analysis of the hippocampus in a rat model of chronic mild unpredictable stress-induced depression based on a pathway crosstalk and network module approach. J Pharm Biomed Anal. 2021;193:113755. doi:10.1016/j.jpba.2020.113755
24. Honorat JA, Nakatsuji Y, Shimizu M, et al. Febuxostat ameliorates secondary progressive experimental autoimmune encephalomyelitis by restoring mitochondrial energy production in a GOT2-dependent manner. PLoS One. 2017;12(11):e0187215. doi:10.1371/journal.pone.0187215
25. Bölsterli BK, Boltshauser E, Palmieri L, et al. Ketogenic diet treatment of defects in the mitochondrial malate aspartate shuttle and pyruvate carrier. Nutrients. 2022;14(17):3605. doi:10.3390/nu14173605
26. Li K, Zheng Y, Wang X. The potential relationship between HIF-1α and amino acid metabolism after hypoxic ischemia and dual effects on neurons. Front Neurosci. 2021;15:676553. doi:10.3389/fnins.2021.676553
27. Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi:10.1038/nature12040
28. Chakrabarti G, Moore ZR, Luo X, et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ß-lapachone. Cancer Metab. 2015;3(1):12. doi:10.1186/s40170-015-0137-1
29. Yang S, Hwang S, Kim M, Seo SB, Lee JH, Jeong SM. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. 2018;9(2):55. doi:10.1038/s41419-017-0089-1
30. Honma K, Kamikubo M, Mochizuki K, Goda T. Insulin-induced inhibition of gluconeogenesis genes, including glutamic pyruvic transaminase 2, is associated with reduced histone acetylation in a human liver cell line. Metabolism. 2017;71:118–124. doi:10.1016/j.metabol.2017.03.009
31. Li Y, Li B, Xu Y, et al. GOT2 silencing promotes reprogramming of glutamine metabolism and sensitizes hepatocellular carcinoma to glutaminase inhibitors. Cancer Res. 2022;82(18):3223–3235. doi:10.1158/0008-5472.CAN-22-0042
32. Arnold PK, Finley LWS. Regulation and function of the mammalian tricarboxylic acid cycle. J Biol Chem. 2023;299(2):102838. doi:10.1016/j.jbc.2022.102838
33. Feist M, Schwarzfischer P, Heinrich P, et al. Cooperative STAT/NF-κB signaling regulates lymphoma metabolic reprogramming and aberrant GOT2 expression. Nat Commun. 2018;9(1):1514. doi:10.1038/s41467-018-03803-x
34. Buffet A, Burnichon N, Favier J, Gimenez-Roqueplo AP. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. 2020;34(2):101416. doi:10.1016/j.beem.2020.101416
35. Remacha L, Comino-Méndez I, Richter S, et al. Targeted exome sequencing of Krebs cycle genes reveals candidate cancer-predisposing mutations in pheochromocytomas and paragangliomas. Clin Cancer Res. 2017;23(20):6315–6324. doi:10.1158/1078-0432.CCR-16-2250
36. Dwight T, Kim E, Novos T, Clifton-Bligh RJ. Metabolomics in the diagnosis of pheochromocytoma and paraganglioma. Horm Metab Res. 2019;51(7):443–450. doi:10.1055/a-0926-3790
37. Heather LC, Cole MA, Lygate CA, et al. Fatty acid transporter levels and palmitate oxidation rate correlate with ejection fraction in the infarcted rat heart. Cardiovasc Res. 2006;72(3):430–437. doi:10.1016/j.cardiores.2006.08.020
38. Kalinowska A, Górski J, Harasim E, Harasiuk D, Bonen A, Chabowski A. Differential effects of chronic, in vivo, PPAR’s stimulation on the myocardial subcellular redistribution of FAT/CD36 and FABPpm. FEBS Lett. 2009;583(15):2527–2534. doi:10.1016/j.febslet.2009.07.008
39. Nie J, Ngokana LD, Kou J, et al. Low-dose ethanol intake prevents high-fat diet-induced adverse cardiovascular events in mice. Food Funct. 2020;11(4):3549–3562. doi:10.1039/C9FO02645B
40. Han -X-X, Chabowski A, Tandon NN. Metabolic challenges reveal impaired fatty acid metabolism and translocation of FAT/CD36 but not FABPpm in obese Zucker rat muscle. Am J Physiol Endocrinol Metab. 2007;293(2):E566–E575. doi:10.1152/ajpendo.00106.2007
41. Chabowski A, Momken I, Coort SL, et al. Prolonged AMPK activation increases the expression of fatty acid transporters in cardiac myocytes and perfused hearts. Mol Cell Biochem. 2006;288(1–2):201–212. doi:10.1007/s11010-006-9140-8
42. Holloway GP, Lally J, Nickerson JG, et al. Fatty acid binding protein facilitates sarcolemmal fatty acid transport but not mitochondrial oxidation in rat and human skeletal muscle. J Physiol. 2007;582(1):393–405. doi:10.1113/jphysiol.2007.135301
43. Jeppesen J, Albers P, Luiken JJ, Glatz JF, Kiens B. Contractions but not AICAR increase FABPpm content in rat muscle sarcolemma. Mol Cell Biochem. 2009;326(1–2):45–53. doi:10.1007/s11010-008-0006-0
44. Nickerson JG, Alkhateeb H, Benton CR, et al. Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1 and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J Biol Chem. 2009;284(24):16522–16530. doi:10.1074/jbc.M109.004788
45. Sookoian S, Castaño GO, Scian R, et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and Krebs cycle level. Am J Clin Nutr. 2016;103(2):422–434. doi:10.3945/ajcn.115.118695
46. Challa TD, Straub LG, Balaz M, et al. Regulation of de novo adipocyte differentiation through cross talk between adipocytes and preadipocytes. Diabetes. 2015;64(12):4075–4087. doi:10.2337/db14-1932
47. Abrego J, Sanford-Crane H, Oon C, et al. A cancer cell-intrinsic GOT2-PPARd axis suppresses antitumor immunity. Cancer Discov. 2022;12(10):2414–2433. doi:10.1158/2159-8290.CD-22-0661
48. Du F, Chen J, Liu H, et al. SOX12 promotes colorectal cancer cell proliferation and metastasis by regulating asparagine synthesis. Cell Death Dis. 2019;10(3):239. doi:10.1038/s41419-019-1481-9
49. Zhu Y, Li T, Ramos da Silva S, et al. A critical role of glutamine and asparagine γ-nitrogen in nucleotide biosynthesis in cancer cells hijacked by an oncogenic virus. mBio. 2017;8(4). doi:10.1128/mBio.01179-17
50. Missiaen R, Anderson NM, Kim LC, et al. GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. 2022;34(8):1151–1167.e1157. doi:10.1016/j.cmet.2022.06.010
51. Jiang H, Stabler SP, Allen RH, Abman SH, Maclean KN. Altered hepatic sulfur metabolism in cystathionine β-synthase-deficient homocystinuria: regulatory role of taurine on competing cysteine oxidation pathways. FASEB J. 2014;28(9):4044–4054. doi:10.1096/fj.14-253633
52. Liang Y, Liu D, Ochs T, et al. Endogenous sulfur dioxide protects against isoproterenol-induced myocardial injury and increases myocardial antioxidant capacity in rats. Lab Invest. 2011;91(1):12–23. doi:10.1038/labinvest.2010.156
53. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017;27(11):863–875. doi:10.1016/j.tcb.2017.06.003
54. Li F, Simon MC. Cancer cells don’t live alone: metabolic communication within tumor microenvironments. Dev Cell. 2020;54(2):183–195. doi:10.1016/j.devcel.2020.06.018
55. Schwörer S, Vardhana SA, Thompson CB. Cancer metabolism drives a stromal regenerative response. Cell Metab. 2019;29(3):576–591. doi:10.1016/j.cmet.2019.01.015
56. Steele NG, Carpenter ES, Kemp SB, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nature Cancer. 2020;1(11):1097–1112.
57. Hickman TL, Choi E, Whiteman KR, et al. BOXR1030, an anti-GPC3 CAR with exogenous GOT2 expression, shows enhanced T cell metabolism and improved anti-cell line derived tumor xenograft activity. PLoS One. 2022;17(5):e0266980. doi:10.1371/journal.pone.0266980
58. Jin M, Shi C, Hua Q, et al. High circ-SEC31A expression predicts unfavorable prognoses in non-small cell lung cancer by regulating the miR-520a-5p/GOT-2 axis. Aging. 2020;12(11):10381–10397. doi:10.18632/aging.103264
59. Yang H, Zhou L, Shi Q, et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015;34(8):1110–1125. doi:10.15252/embj.201591041
60. Wang T, Yao W, Li J, He Q, Shao Y, Huang F. Acetyl-CoA from inflammation-induced fatty acids oxidation promotes hepatic malate-aspartate shuttle activity and glycolysis. Am J Physiol Endocrinol Metab. 2018;315(4):E496–E510. doi:10.1152/ajpendo.00061.2018
61. Biancur DE, Paulo JA, Małachowska B, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun. 2017;8(1):15965. doi:10.1038/ncomms15965
62. Altea-Manzano P, Vandekeere A, Edwards-Hicks J, et al. Reversal of mitochondrial malate dehydrogenase 2 enables anaplerosis via redox rescue in respiration-deficient cells. Mol Cell. 2022;82(23):4537–4547.e7. doi:10.1016/j.molcel.2022.10.005
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.