")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Spotlight on Givosiran as a Treatment Option for Adults with Acute Hepatic Porphyria: Design, Development, and Place in Therapy
Authors Majeed CN , Ma CD, Xiao T, Rudnick S, Bonkovsky HL
Received 15 January 2022
Accepted for publication 23 April 2022
Published 16 June 2022 Volume 2022:16 Pages 1827—1845
DOI https://doi.org/10.2147/DDDT.S281631
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Chaudry Nasir Majeed,1 Christopher D Ma,2 Ted Xiao,3 Sean Rudnick,1 Herbert L Bonkovsky1
1Department of Internal Medicine, Section on Gastroenterology and Hepatology, Wake Forest School of Medicine, Winston-Salem, NC, USA; 2Wake Forest University School of Medicine, Winston-Salem, NC, USA; 3Department of Internal Medicine, Internal Medicine Residency Program, Wake Forest School of Medicine, Winston-Salem, NC, USA
Correspondence: Chaudry Nasir Majeed, Department of Internal Medicine, Section on Gastroenterology and Hepatology, Wake Forest School of Medicine, Medical Center Boulevard, Winston-Salem, NC, 27157, USA, Tel +1 (336) 713-7311, Fax +1 (336) 713-7322, Email [email protected]
Abstract: Small interfering ribonucleic acids [siRNAs] are short ribonucleic acid (RNA) fragments cleaved from double-stranded RNA molecules that target and bind to specific sequences on messenger RNA (mRNA), leading to their destruction. Therefore, the siRNA down-regulates the formation of selected mRNAs and their protein products. Givosiran is one such siRNA that uses this mechanism to treat acute hepatic porphyrias. Acute hepatic porphyrias are a group of rare, inherited metabolic disorders, characterized by acute potentially life-threatening attacks as well as chronic symptoms with a negative impact on quality of life. It has four types, each associated with distinct enzyme defects in the heme biosynthesis pathway in the liver. By targeting the expression of hepatic 5-aminolevulinic acid [ALA] synthase-1 [ALAS1], givosiran can down-regulate levels of toxic metabolites, leading to biochemical and clinical improvement. Givosiran selectively targets hepatocytes due to its linkage to N-acetylgalactosamine (GalNac) leading to its selective uptake via asialoglycoprotein receptors (ASGPR). We provide an up-to-date literature review regarding givosiran in the context of a clinical overview of the porphyrias, an overview of siRNAs for therapy of human disorders, the design and development of givosiran, key clinical trial results of givosiran for prevention of acute porphyric attacks, emerging concerns regarding chronic use of givosiran, and the overall management of acute hepatic porphyrias. These insights are important not only for the management of acute hepatic porphyrias but also for the emerging field of siRNAs and their role in novel therapies for various diseases.
Keywords: givosiran, small interfering RNA’s, siRNAs, 5- (or δ)-aminolevulinic acid synthase 1 (ALAS1) expression inhibitors, porphyrias
Clinical Overview of Porphyrias
Porphyria refers to a group of inherited or acquired disorders in the enzymes catalyzing one of the eight steps in heme synthesis [see Figure 1]. Each enzyme dysfunction may lead to accumulation and excretion of the precursor molecules that precede the defective step. The prevalence of the porphyrias varies by specific disease and by country or region of the world. In the case of acute intermittent porphyria [AIP], the major form of acute hepatic porphyrias [AHP], the prevalence of disease-associated mutations recently has been found to be more common than previously believed, approximately 1 in 1700 among Europeans and European-Americans.1 Because the prevalence of clinical disease is far lower, we now estimate that the clinical penetrance of AIP is only about 1–2%, although in first-degree relatives of known patients, the clinical penetrance is ~20%.2 We will review different types of porphyria with an emphasis on AHPs as summarized in Table 1.3 Elevation of urinary ALA and porphobilinogen (PBG) may occur in acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP) while elevation of urinary ALA but not PBG is characteristic of delta-aminolevulinic acid dehydratase porphyria (ADP). Elevation of ALA, in particular, may be associated with acute attacks of disease with diverse neurovisceral symptoms.
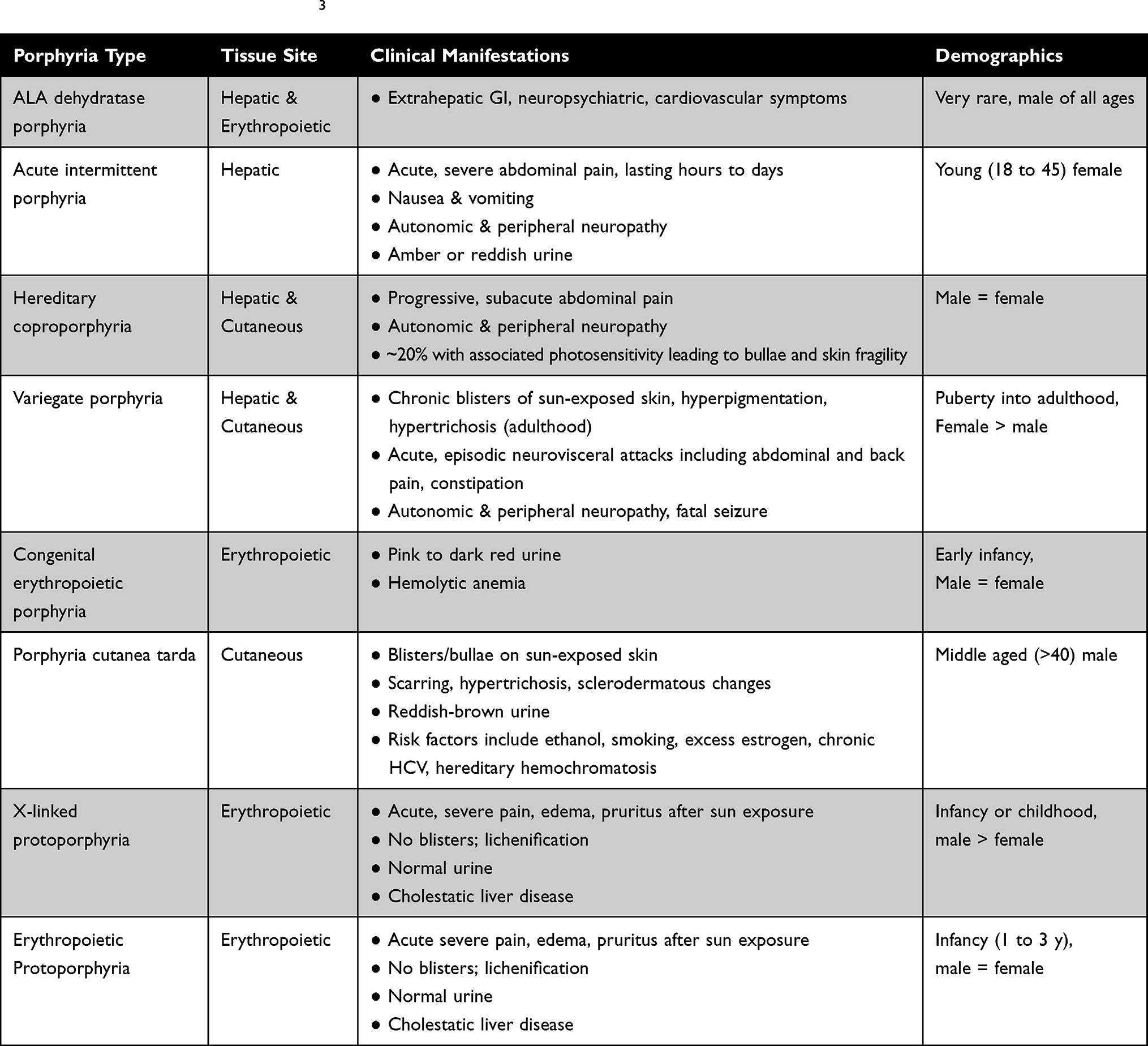 |
Table 1 Porphyria Clinical Summary3 |
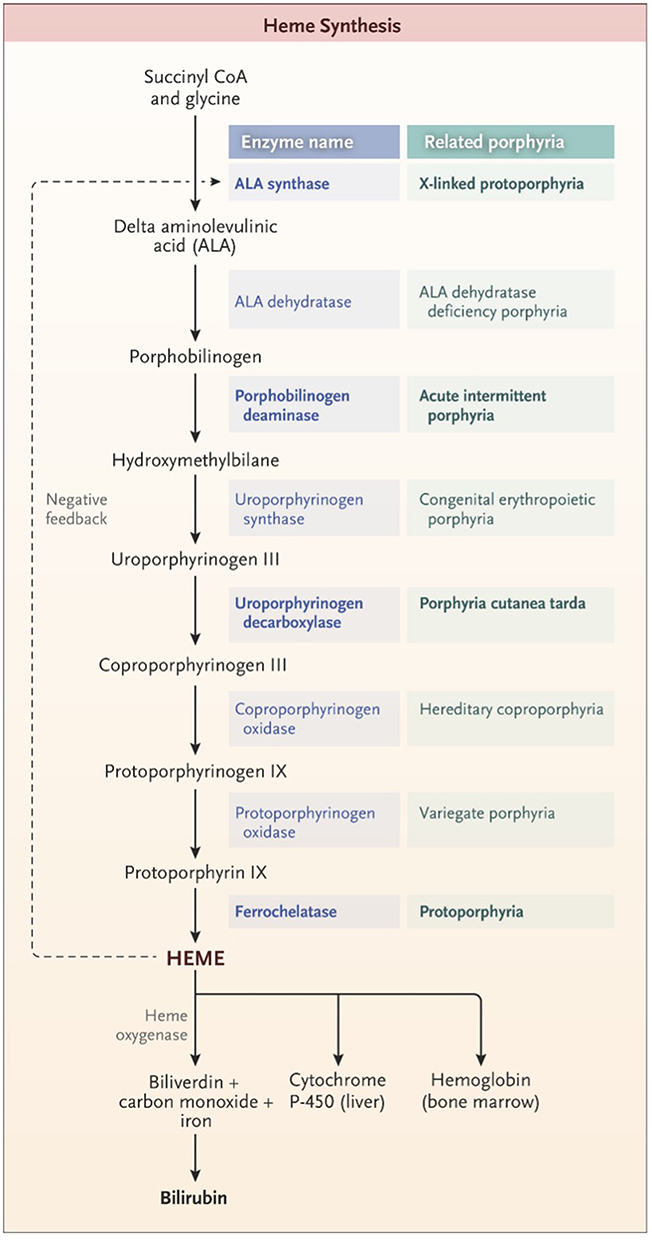 |
Figure 1 The pathway of heme synthesis, showing pathway intermediates and end-product regulation by heme. The eight steps of heme synthesis (left column) are shown with the enzyme (middle column) that catalyzes each step. The enzymes in bold type are the clinically most prevalent porphyrias. From The New England Journal of Medicine, Bissell DM, Anderson KE, Bonkovsky HL, Porphyria, 377(9), 862–872. Copyright © (2017) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.3 |
Acute intermittent porphyria (AIP) is the most common among AHPs. It is an autosomal dominant disease caused by a deficiency in porphobilinogen (PBG) deaminase, also called hydroxymethylbilane synthase [HMBS]. When clinically active, AIP is characterized by episodes of progressively worsening, unexplained abdominal pain with normal examination findings. An acute attack commonly presents with incapacitating abdominal pain in reproductive age women. Seizures can occur in 10–20% of patients.4 Diagnosis is made with an elevated urinary PBG test confirmed by genetic testing. Excess delta-aminolevulinic acid (ALA) production in the liver causes chronic neural, hepatic, and renal injuries.5,6 Treatment involves trigger avoidance and intravenous (IV) heme infusion (hydroxyheme [Panhematin] or heme arginate [Normosang]) for acute attacks. Givosiran is a novel therapy that has shown to have significant efficacy in prevention of frequent recurrent attacks of AIP in recent clinical trials.7,8
The other AHPs include delta-aminolevulinic acid dehydratase porphyria (ADP), hereditary coproporphyria (HCP), and variegate porphyria (VP). ADP is a very rare condition due to severe ALA dehydratase [also called PBG synthase] deficiency. It is an autosomal recessive disorder that is highly heterogeneous with various mutant alleles, thus far, for unknown reasons, found only in males.9 Typical findings on evaluation of ADP include markedly deficient activity (<10% of normal) of ALA dehydratase in erythrocytes, elevation in urinary ALA and coproporphyrin with no increase in urinary PBG. However, a definitive diagnosis is made by genetic testing that typically reveals disease-associated mutations in both alleles of the ALA dehydratase gene.10
Hereditary coproporphyria (HCP) is a rare inherited type of porphyria due to a mutation in the coproporphyrinogen oxidase gene [CPOX]. Acute HCP attacks present with progressive, subacute abdominal pain and peripheral weakness if not treated. Photosensitivity can occur in ~20% with acute attacks.11 Variegate porphyria (VP) is caused by a mutation in protoporphyrinogen oxidase [PPOX]. VP has a chronic cutaneous and an acute porphyria component. Patients develop blistering lesions in sun-exposed skin, facial hyperpigmentation, and hypertrichosis. Acute neurovisceral attacks present with abdominal and back pain, constipation, motor neuropathy, and potentially fatal seizures.12
Porphyria cutanea tarda (PCT) is likely the most common porphyria worldwide. It is due to an inherited or acquired deficiency of hepatic uroporphyrinogen decarboxylase. Excessive circulating porphyrins cause skin friability and blisters on sun-exposed areas. The most common risk factors for PCT in the US are ethanol use, smoking, excess estrogen, chronic hepatitis C virus (HCV) infection, and hereditary hemochromatosis.3 Diagnosis is made by elevation in urine or plasma uroporphyrin and heptacarboxylporphyrin. Trigger avoidance along with iron reduction via therapeutic phlebotomy or iron chelation, and/or low-dose hydroxychloroquine are effective treatments. They decrease ongoing overproduction of uro- and heptacarboxyl-porphyrins by the liver and enhance mobilization and urinary excretion of porphyrins from hepatocytes and other sites of deposition. Normalization of urine uroporphyrin level signals that it is safe for treatment cessation. Due to the risk of recurrence, especially among patients with continuing alcohol and tobacco use, chronic hepatitis C, and/or hereditary hemochromatosis, an annual check of urine or plasma uroporphyrin levels is recommended for early detection of recurrence.13 Such monitoring is best done in the early spring prior to the sunny seasons of the year. Among patients with PCT and chronic hepatitis C, both disorders usually can be treated with direct-acting antivirals that cure HCV infection.14
Protoporphyria is caused by the overproduction of protoporphyrin in the bone marrow. Ferrochelatase deficiency leads to erythropoietic protoporphyria whereas gain-of-function mutations in ALA synthase 2 lead to X-linked protoporphyria. The clinical presentation consists of acute pain, redness, and swelling of sun-exposed skin. With repeat injury, mild hyperkeratosis and lichenification may develop, especially notable over the knuckles and on the face. The screening test for protoporphyria is a significant elevation in blood protoporphyrin. Beta-carotene has not proven very effective in ameliorating acute photosensitivity, whereas afamelanotide, an analog of melanocyte-stimulating hormone that leads to increased eumelanin pigmentation, has shown significant benefits in clinical trials.15
Overview of Small Interfering RNA (siRNAs) for Therapy of Human Disorders
The concept of ribonucleic acid (RNA) interference was first proven in Caenorhabditis elegans in 1998, and this technology has evolved to be utilized for many applications, including the development of novel therapies for various mammalian disease.16–18 There are various methods to administer RNA-based gene silencing therapies.19 Short hairpin RNAs, which are 19–22 base pairs (bp) molecules, for example, can be delivered via plasmid DNA or viral vectors, which are then subsequently transcribed endogenously and converted into siRNAs in the cytoplasm via the enzyme dicer.20 Micro-RNA molecules, which are approximately 22 nucleotide molecules, are dicer-independent and can be processed directly by another enzyme Argonaute 2 (Ago2).21 When a 20–30 bp siRNA-based therapy is administered and enters a eukaryotic cell, generally, dicer, which is a protein complex with other RNA-binding proteins, such as TAR-RNA-binding protein, PACT, and Argonaute-2 [Ago-2], interacts with the 2-nucleotide overhang on the 3’ end of the modified siRNA and transfers it to the RNA-induced silencing complex [RISC].22,23 During this hand-off, Ago-2 separates the two siRNA strands; the more thermodynamically stable “guide” strand stays bound to and activates RISC, allowing this complex to recognize the target mRNA via base pairing.24–28 Once bound to the target RNA, the PIWI domain of Ago-2 precisely targets a phosphodiester linkage and creates a nick. The degradative process is then completed by cellular exonucleases, fragmenting the target mRNA, freeing the RISC/siRNA complex, and allowing the complex to find other target mRNAs in the cytosol.29–31 Through this mechanism of action, siRNAs can theoretically target and down-regulate almost all genes of interest.
Although siRNAs appear to have spectacular promise, multiple barriers must be overcome during the drug development process. The most obvious barrier is RNA degradation via endonucleases and exonucleases, which can quickly degrade naked and unmodified siRNAs, leading to poor stability and pharmacokinetic behavior. Other barriers include poor bioavailability, poor clearance, inappropriate recognition by innate immunity, and off-target effects if the siRNA does not successfully localize to its target tissue.32,33 Over decades, however, multiple strategies have been developed to mitigate these issues, including the use of different carriers, such as lipids, polymers, and nanoparticles, to protect and deliver siRNAs to their desired target organs; chemical modifications to the base, ribose, or phosphonate groups of the siRNAs; and the use of longer RNA hairpin transcripts, which can be subsequently processed intracellularly to their siRNA effector forms.32,34,35
Even with these modifications, it is still difficult to translate promising in vivo efficacy, pharmacokinetics, and pharmacodynamics results to human clinical trials. For example, the first clinical application of RNA interference was for the treatment of age-related macular degeneration using vascular endothelial growth factor [VEGF] A-targeting bevasiranib and VEGFR1-targeting AGN211745, but these siRNAs triggered significant induction of toll-like receptor 3, interleukin-12, and interferon-gamma.36–38 Thus, it is important for researchers to continue to optimize and maximize treatment potency while avoiding or minimizing these undesirable effects.
Although there are several promising siRNA therapeutics in clinical studies, such as fitusiran for hemophilia,39 and tivanisiran for Sjogren Syndrome,40 only four siRNA therapies are currently FDA-approved, namely, patisiran for transthyretin-mediated amyloidosis,41 lumasiran for primary hyperoxaluria type 1,42 inclisiran for hypercholesterolemia,43 and givosiran for acute hepatic porphyrias (AHP’s).8 These FDA-approved siRNAs show remarkable efficacy in treating their respective target diseases with favorable safety profiles, although effects of long-term therapy are not yet known. For this review, only givosiran will be further discussed.
Design and Development of Givosiran
Givosiran (Figure 2), which was developed by Alnylam Pharmaceuticals (Cambridge, MA), is a siRNA that decreases hepatic delta-aminolevulinic acid synthase 1 (ALAS1) expression to prevent recurrent acute attacks of AIP or other acute porphyrias.44 There have been many reports that show associations between basal hepatic ALAS1 mRNA levels and AIP attacks, indicating that decreasing ALAS1 mRNA expression is a critical target for therapy.7 Givosiran was initially designed based on the nucleotide sequence, NM_020559.2 [National Center for Biotechnology Information Reference Sequence], in murine ALAS1.45 From there, Alnylam investigators worked to improve the delivery system of givosiran to hepatocytes by conjugating a trivalent N-acetyl-galactosamine [GalNac] to the siRNA.44–46 Human hepatocytes express high levels of asialoglycoprotein receptors [ASGPR], which are transmembrane receptors with high affinity and specificity for GalNac. Thus, conjugation of givosiran to GalNAC achieved greater silencing of hepatocytic ALAS1 with fewer off-target effects.47 In absence of the triGalNAc ligand, no activity is observed with siRNA in liver.47,48 Givosiran was further modified to protect it from nuclease digestion by incorporating either a 2’-deoxy-2’-fluoro or a 2’-O-methyl group in the ribose sugar moieties, and phosphorothioate linkages at the 5’ end of the siRNA strands.44,49 After uptake into cell, it is unclear how siRNA escapes from the trafficking pathway. One hypothesis is that small amounts are released during fission/fusion events of endolysosomes.48 Upon uptake, the GalNAc ligand rapidly degrades in the endolysosomes. It is thought that siRNA degradation is largely intracellular by nucleases and a smaller portion is removed through urine and bile.50 It is believed that slower onset of activity with conjugate siRNAs is due to time it takes for it to escape from subcellular compartments and reach RNA-induced silencing complex (RISC) for RNAi activity. It also likely provides the depot for long duration of activity.48 Both in vitro and in vivo studies were conducted to investigate the metabolic stability and metabolite profile of givosiran. The metabolic stability of givosiran was evaluated in serum and liver S9-fractions obtained from mouse, rat, monkey, and human. The in vivo metabolism of givosiran, assessed by analyzing plasma and liver samples after administration of givosiran in rats and monkeys, was generally similar to that observed in vitro. Metabolic profiling was performed in human plasma and urine. No human unique metabolites were detected. An in vitro study conducted with human liver S9 fraction confirmed that both sense and antisense strands of givosiran were stable, and no change was observed with and without nicotinamide adenine dinucleotide phosphate hydrogen (NADPH), suggesting that cytochrome P450 (CYP) isozymes are not involved in the metabolism of givosiran. The proposed biotransformation and breakdown products are described in more detail in Figure 3.51 Before human trials, the rodents trials of weekly subcutaneous injections were encouraging and showed dose dependent ALAS1 gene silencing with a sustained response of over 9 months with no adverse effects.47
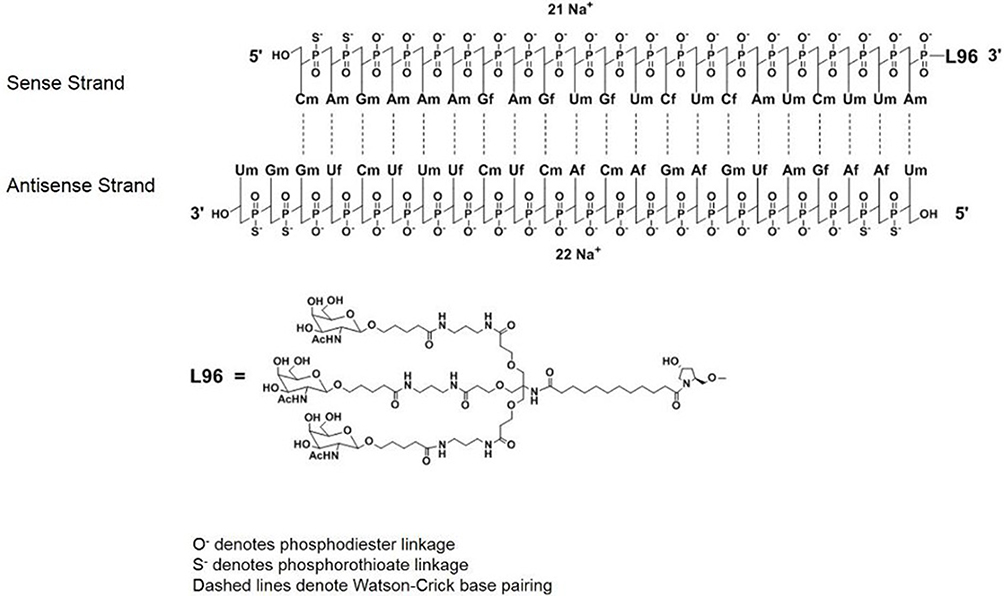 |
Figure 2 Chemical structure of givosiran. Note: Reprinted from FDA. Highlights of prescribing information; 2019. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/0212194s000lbl.pdf.67Abbreviations: Af, adenine 2’-F ribonucleoside; Cf, cytosine 2’-F ribonucleoside; Uf, uracil 2’-F ribonucleoside; Am, adenine 2’-OMe ribonucleoside; Cm, cytosine 2’-OMe ribonucleoside; Gf, guanine 2’-F ribonucleoside; Gm, guanine 2’-OMe ribonucleoside; Um, uracil 2’-OMe ribonucleoside; L96, triantennary GalNAc (N-acetylgalactosamine). |
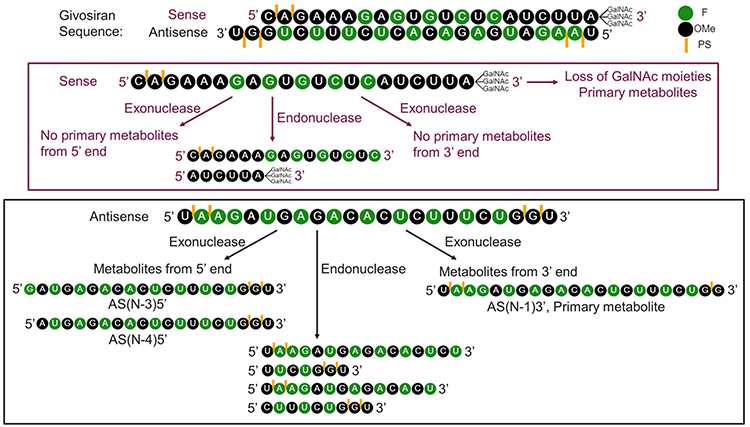 |
Figure 3 Metabolism of givosiran. Note: Li J, Liu J, Zhang X, et al. Nonclinical pharmacokinetics and absorption, distribution, metabolism, and excretion of givosiran, the first approved N-Acetylgalactosamine-conjugated RNA interference therapeutic. Drug Metab Dispos. 2021;49(7):572–580.51 |
Givosiran’s mechanism of action is very similar to that of any siRNA as outlined in the “Overview of siRNAs for therapy of human disorders” section. The steps that are unique to givosiran are the initial interaction of the GalNac moieties to asialoglycoprotein receptor (ASGPR) on hepatocytes, allowing internalization of the siRNA, and givosiran subsequently complexing with both dicer and HIV1 transactivation response RNA-binding protein, catalyzing the separation of the two RNA strands (Figure 4).44,46,52,53
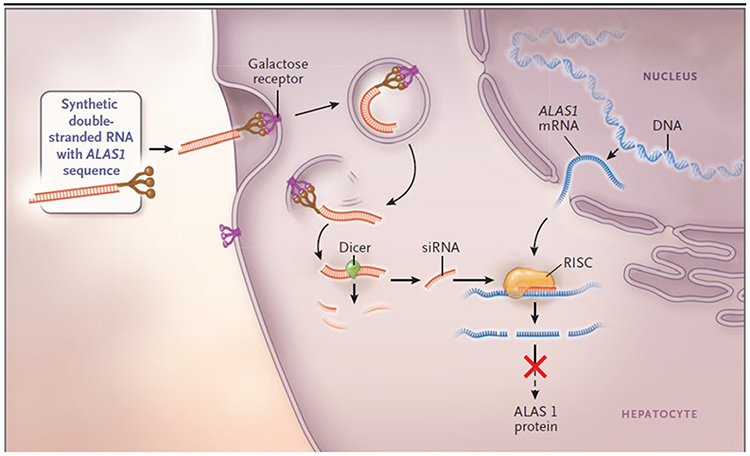 |
Figure 4 The mechanism of small interfering RNA (siRNA) therapy. Synthetic double-stranded RNA containing an ALAS1-specific sequence is derivatized with N-acetylgalactosamine to target the asialoorosomucoid (galactose) receptor, which is expressed nearly exclusively on hepatocytes. Within the hepatocyte, the RNA is processed into approximately 20-bp fragments by a cellular enzyme (dicer), and then separated into single strands. The strand that is complementary to ALAS1 (the guide strand) binds to cellular ALAS1 messenger RNA (mRNA) and enters the RNA-induced silencing complex (RISC), where the new double-stranded RNA is cleaved by a group of factors that include argonaute, a ribonuclease. The result is a reduction in the level of delta ALA synthase 1 protein and decreased production of ALA. From The New England Journal of Medicine, Bissell DM, Anderson KE, Bonkovsky HL, Porphyria, 377(9), 862–872. Copyright © (2017) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.3 |
Due to givosiran’s high specificity for the liver, its pharmacokinetics and absorption, distribution, elimination, metabolism, and excretion profiles were all favorable in rats (Table 2). In this pharmacokinetic study, the givosiran concentration was found to be 11 times higher in the liver than in the kidneys. Givosiran was found in multiple organs, including the heart, lungs, and adrenal glands, at significantly lower concentrations, ranging from 1/100 to 1/800 than of the concentrations found in the liver. Givosiran was not detected in the brain, and the degraded RNA fragments were excreted through the urine.54–56 Different dosing of givosiran produced rapid and sustained, dose-dependent reductions in serum ALAS1 mRNA and urinary levels of δ-ALA and PBG. The maximum reductions in serum ALAS1 mRNA from baseline ranged from 49% to 74% based on dosage (2.5 or 5 mg/kg) and frequency (monthly or quarterly). Givosiran exhibits linear plasma pharmacokinetics over a dose range of 0.35 to 2.5 mg/kg. At 2.5 mg/kg, givosiran exhibited a time to maximum plasma concentration of 2.07 hours, steady state apparent volume of distribution of 10.4 liters, elimination half-life of 4 to 40 hours, and fraction of renal clearance over 24 h of 6.59%. These results were consistently observed after stratification based on age, gender, race, kidney function.56 Although ASGPRs are also expressed in peripheral monocytes, peritoneal macrophages, endometrial cells, renal tubular cells and other cell types, givosiran has a selective preference for localization in the liver, which limits the likelihood of off-target effects.57–60 Perhaps, related to some degree of givosiran’s localization in the kidneys, both the clinical trials of its use and a recent study reported an increase in serum creatinine (Cr) and a decrease in estimated glomerular filtration rate [eGFR].; long-term adverse effects of givosiran on renal function cannot be excluded.61
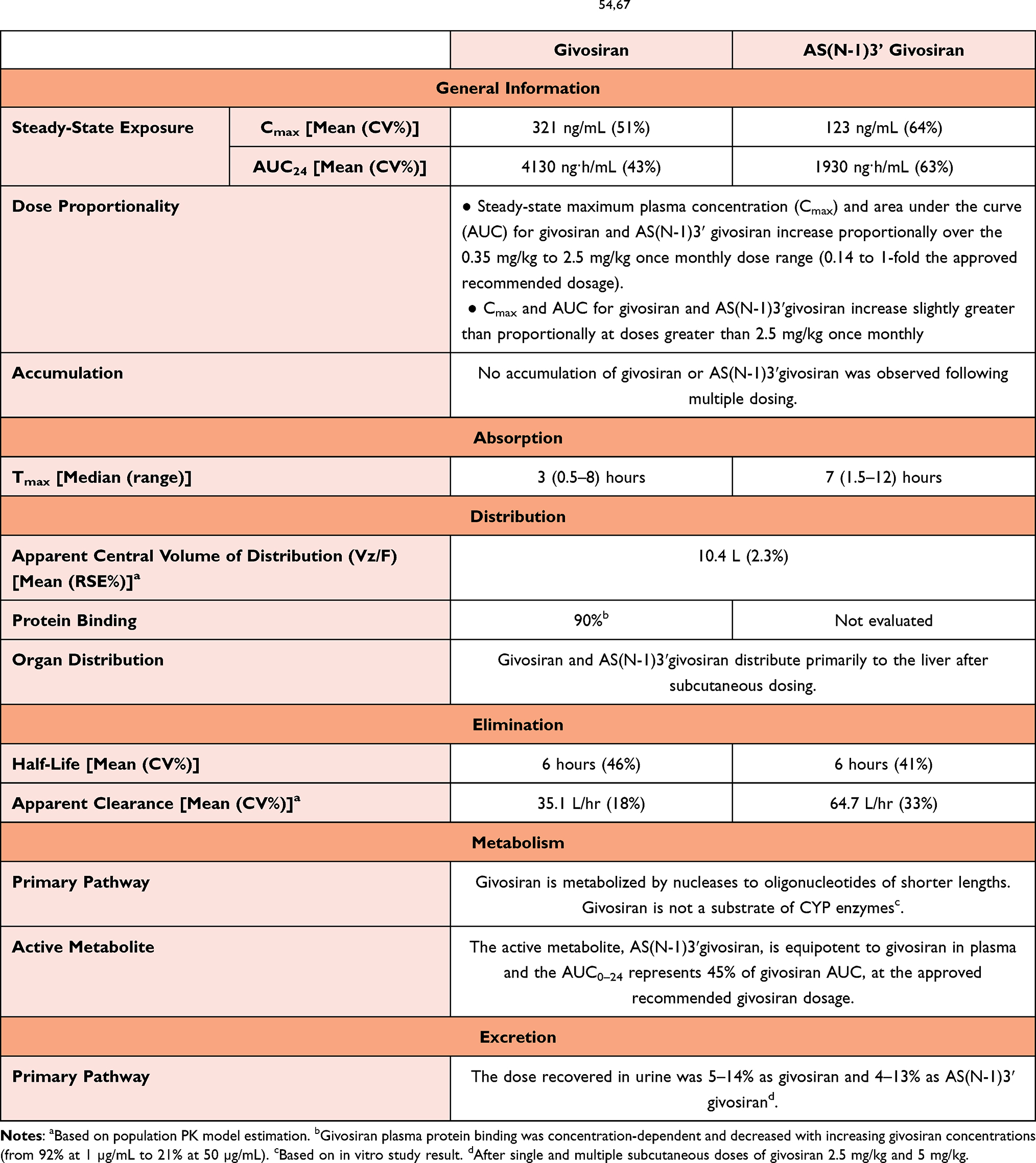 |
Table 2 Pharmacokinetic Parameters of Givosiran and Its Active Metabolite54,67 |
Prior to the approval of givosiran, weekly IV heme infusions were, and perhaps still are, the most widely used prescription for prophylaxis of frequent acute attacks of AHP. Due to its instability in aqueous solution and its risk for painful phlebitis, IV heme is better administered bound to human serum albumin and through a high-flow large peripheral vein or central line. Frequent heme infusions also increase the risk of iron overload.3,62–64 These issues do not exist with givosiran, but there are adverse effects of givosiran that have been reported as outlined in the “Emerging concerns regarding chronic use of givosiran” section.
Key Clinical Trial Results for Givosiran for Prevention of Acute Porphyric Attacks
A Phase I trial (NCT02452372) was performed to investigate the safety, pharmacokinetic, pharmacodynamic, and efficacy of givosiran in confirmed AIP patients.7 This trial enrolled a total of 40 patients with mutation-confirmed AIP. Parts A and B of the trial investigated patients without attacks in the past 6 months (N=23). Part A of the trial investigated selected dosages of givosiran, given as a single subcutaneous [sc] injection, while part B investigated once-monthly injections for two injections. Higher doses of givosiran were associated with lower expression of urinary ALA and PBG levels. Part C of the trial investigated patients with recurrent attacks (N=17) at two different dosages (2.5 or 5.0 mg/kg) and dosing frequencies (monthly vs quarterly). The 5.0 mg/kg once-monthly group led to the greatest reduction in ALAS1 mRNA level at −74 ± 6%. The placebo groups in this study were administered sterile normal saline.
In conclusion, the pharmacokinetic profile indicated dose-proportional characteristics of givosiran with no drug accumulation after repeated injections. Once-monthly treatment in the patient population with recurrent attacks resulted in low-grade adverse events (AE), maximal reduction in ALAS1 mRNA levels, near normalization of neurotoxic intermediates, a 48% reduction of annualized hemin administration, and a 79% lower attack rate compared to the placebo group. The study limitations included a short intervention period (12 weeks) and small patient populations (N = 23 and 17). This trial confirmed an association between lower biochemical activities among the givosiran patient population and reduction in annualized attack rate (ARR) and reduced rate of as-needed hemin use.
A phase I/II open-label extension (OLE) study (NCT02949830) of givosiran enrolled all patients from phase I part C.65 The study had a mean intervention duration of 22.8 months, and up to 30.9 months of total treatment. The results of the OLE study demonstrated maintenance of clinical activity with continuous monthly dosing of givosiran at 2.5 mg/kg. There was consistent reduction of urinary ALA and PBG by ≥80% at 12 months and >85% at 18 months. The mean annualized attack rate was reduced by 96% and annualized hemin use was reduced by 98%. Continued givosiran dosing maintained the reduced of mean attack rate in patients out to 30 months.
Based upon the encouraging results of the phase I and II trials, the pivotal Phase III ENVISION trial (NCT03338816) was performed with participation from 36 sites.8 For a 6-month period, AIP patients (N=94) were randomly assigned to receive once monthly (QM) givosiran at 2.5 mg/kg vs placebo (sterile normal saline). The study investigated annualized attack rate, urinary ALA and PBG levels, annualized hemin use, and daily worst pain scores. The givosiran group showed a lower attack rate, reduction in biochemical markers, fewer hemin infusions, and reduction in physical pain and limitations. The mean annualized attack rate over 6 months was 3.2 in the givosiran group vs 12.5 in the placebo group, a 74% reduction. Fifty percent of patients in the givosiran group had no attacks during the intervention period compared to 17% in the placebo group. Urinary ALA level was reduced by 86% and PBG level reduced by 91% after 6 months of givosiran (Figure 5). Fifty-four percent of patients in the givosiran group did not require hemin use compared to 23% in the placebo group. This trial demonstrated a similar pharmacokinetic profile and efficacy of givosiran in a larger population for a longer duration of treatment period.
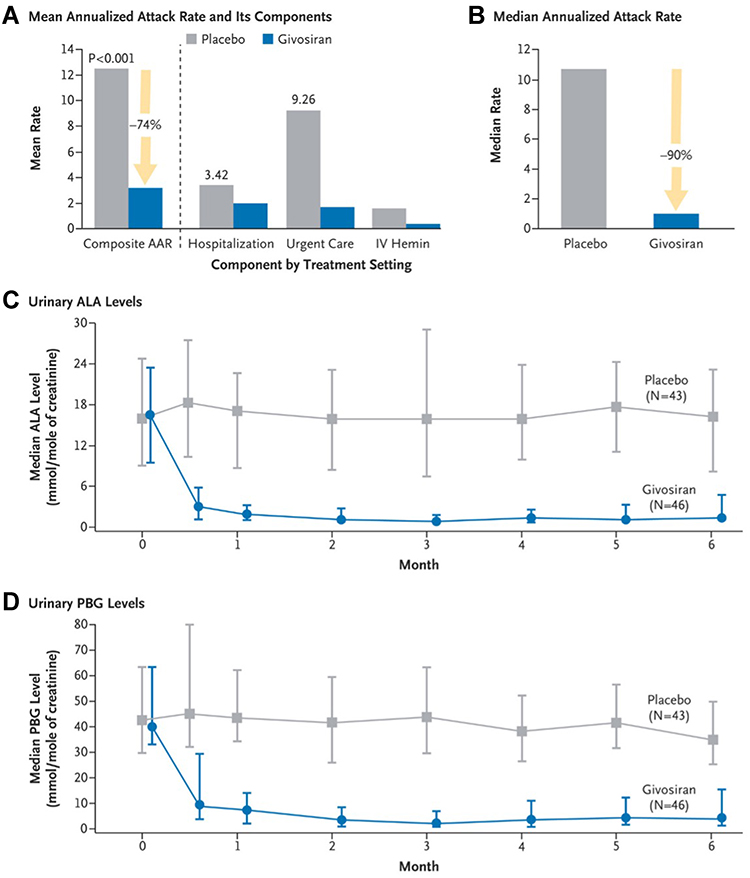 |
Figure 5 Annualized attack rate (AAR) and urinary levels of neurotoxic heme intermediates in patients with acute intermittent porphyria. (A) shows the mean annualized rate of composite porphyria attacks (the primary end point) among the 89 patients with acute intermittent porphyria who received either givosiran or placebo. A composite porphyria attack was defined as an attack that resulted in hospitalization, an urgent health care visit, or intravenous administration of hemin at home. IV denotes intravenous. (B) shows the median annualized attack rate, which was calculated from the individual patients’ annualized attack rates. Also shown are the median levels of urinary delta-aminolevulinic acid (ALA) ((C) and porphobilinogen (PBG) ((D) in patients with acute intermittent porphyria. In (C and D), the I bars denote the interquartile range. From The New England Journal of Medicine, Balwani M et al, Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria, 382 (24), 2289–2301. Copyright © (2020) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.8 |
With the statistically significant results in the phase III ENVISION study an OLE study was performed for a 24-month interval on all eligible patients from the ENVISION study for a total of 30 months.66 The givosiran group from the ENVISION study continue to receive givosiran 2.5 mg/kg while the placebo group from ENVISION study crossed over and received givosiran 1.25 mg/kg. Givosiran treatment led to sustained lowering of median ALA levels to near normal and PBG levels by >75% through month 24 as seen in Figure 6. The median number of attacks during the OLE period was 0 for the continuous givosiran group and 1.35 (87% reduction) in the placebo crossover group. In addition, a higher proportion of patients in the givosiran group (83%) was attack-free compared to the placebo crossover group (76%). The median annualized hemin use remained at 0 in the continuous givosiran group during the OLE period and 0.71 (95% reduction) in the placebo crossover group. In addition, a higher proportion of patients in the givosiran group (68%) required no hemin use compared to the patients in the crossover group (49%). Patient-reported outcomes reflected improvements across all domains, including activities of daily living, satisfaction with treatment, and living a more normal life.
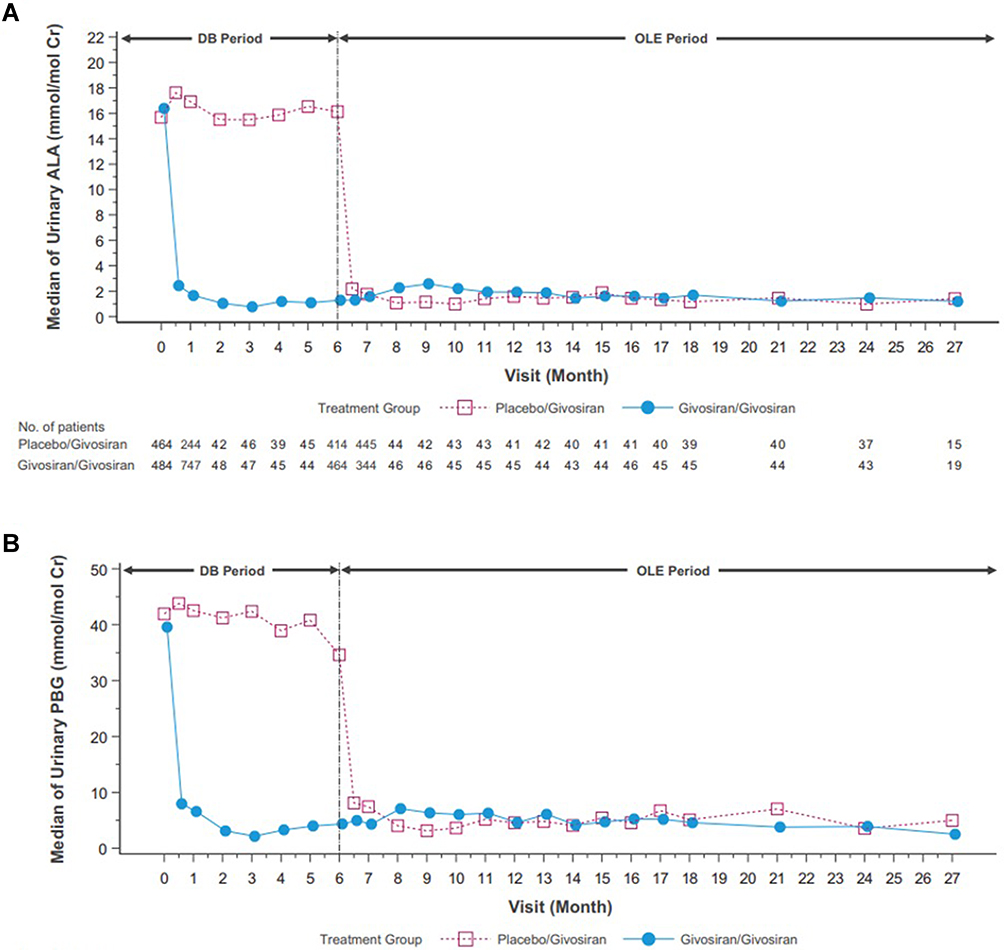 |
Figure 6 Phase 3 open label extension study mean levels of urinary ALA (A) and PBG (B) of the placebo/givosiran crossover group and the givosiran/givosiran group. DB denotes the double-blind phase of study. Note: Reprinted from Ventura P, Bonkovsky HL, Gouya L, et al. Efficacy and safety of givosiran for acute hepatic porphyria: 24-month interim analysis of the randomized phase 3 ENVISION study. Liver Int. 2022;42(1):161–172. © 2021 The Authors. Liver International published by John Wiley & Sons Ltd. Creative Commons license and disclaimer available from: http://creativecommons.org/licenses/by/4.0/legalcode.66 |
Emerging Concerns Regarding Chronic Use of Givosiran
Overall, givosiran was well tolerated during the trials. In Phase I, the side effects were mostly mild to moderate in severity and were mostly GI-related (nausea, abdominal pain, diarrhea) and nasopharyngitis.7,8 Part A of phase I had two patients with severe abdominal pain while part B had one patient with a spontaneous abortion at seven weeks but considered unlikely to be related to givosiran.7 Of note, givosiran administration at maternally toxic doses in an embryo-fetal development study in pregnant rabbits (1.5 mg/kg/day which is five times the maximum recommended human dose) resulted in increased post-implantation loss. With one time dose of 20 mg/kg, an increased incidence of skeletal variations of the sternebrae was observed. In a combined fertility and embryo-fetal development study in female rats, givosiran administration at nine times the normalized maximum recommended human dose was associated with a skeletal variation (incomplete ossification of pubes) and produced maternal toxicity. As per FDA, there is not enough data of givosiran in pregnant women to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Thus, the current package insert for givosiran recommends to assess risk vs benefits of potential effects on fetus and mother while prescribing givosiran to pregnant patients.67
In part C, there were five serious adverse events (SAE) reported with one patient developing influenza A infection and another developing opioid-induced bowel dysfunction responsive to the treatment. A single patient had 3 serious adverse events (including hemorrhagic pancreatitis) while receiving 5 mg/kg monthly injections of givosiran, but that patient had confounding factors, including a complex medical history and acute pulmonary embolism.7 Eighteen percent of treatment groups developed a serious adverse event. During, Phase I/II, 100% (16/16) patients reported at least 1 adverse event. Six patients developed serious adverse events, but only one with an anaphylactic reaction was deemed related to givosiran. There were no clinically significant laboratory changes, including in liver chemistries.
While during the ENVISION trial, givosiran generally showed mild adverse effects (nausea, fatigue, and injection site rash), there were reports of adverse events related to the liver and kidneys including increased serum alanine aminotransferase (ALT), worsening chronic kidney disease (CKD), decreased estimated glomerular filtration rate (eGFR).8
Serum ALT elevation up to >3 times the upper limit of normal was more common in the givosiran-treated group than in the placebo group (7 vs. 1 patient).8 Generally, ALT elevations occurred between 3 and 6 months after starting treatment and resolved with continued givosiran treatment, suggesting possible adaptation by the liver.8 One patient developed serum ALT elevation 9.9 times the upper limit of normal without symptoms of hepatitis or elevation of serum total bilirubin, which led to permanent discontinuation of givosiran and subsequently ALT normalization after 5 months.8 In another patient with elevated ALT, the givosiran was interrupted according to the protocol and then resumed at a lower dose of (1.25 mg/kg) with no signs of ALT elevation.8 The other patients with elevations in their aminotransferases had eventual resolution of their ALT elevations with continued dosing of givosiran at 2.5 mg/kg. It appeared that elevations of serum aminotransferases were seen at similar rates in patients with or without elevated aminotransferases at baseline.8 Thus, despite increases in ALT levels, most of these elevations were transient and/or resolved with decreased monthly doses.
Patients receiving givosiran also demonstrated adverse renal events (15% in the givosiran group compared to 7% in the placebo group) manifested by increased serum creatinine (median increase at 3 months, was 0.07 mg/dL) and a corresponding decrease in the eGFR. Five patients in the givosiran group had onset of worsening CKD.8 On renal biopsies, two of these patients’ renal adverse events were related to worsening of their underlying CKD (hypertensive and porphyria-associated nephropathy). Overall, the rise in creatinine observed with givosiran use was noted early in the treatment course and was mainly reversible.8
During the OLE, the safety profile of givosiran remained acceptable with long-term treatment associated with the same common adverse events. Mild to moderate elevations in liver chemistries occurred in 17 patients. Renal adverse events were reported in 21 patients; however, none required treatment discontinuation.66
There are concerns that the decrease in ALAS1 could eventually cause hepatic heme deficiency and would also impact heme-dependent processes in hepatocytes. There have been reports of elevated homocysteine levels in AHP patients that are being treated with givosiran,66,68 especially in patients with variants that reduce activity of the methylene-tetrahydrofolate reductase [MTHFR] gene.69,70 Several mechanisms have been proposed for this finding, including cystathionine beta-synthase (CBS) impairment caused by low heme availability in the liver. Altered ALAS1 activity can also influence pyridoxine (B6) availability and impact other pyridoxine-dependent enzymes involved in homocysteine metabolism leading to hyperhomocysteinemia. In a study of 15 patients, high doses of vitamin B6 (pyridoxine) resulted in lowering homocysteine and methionine plasma levels.68 Hyperhomocysteinemia can lead to venous thrombosis, atherosclerotic cardiovascular disease, and pancreatitis.70–72 In a recent French study, some additional side effects were noted in patients with homocysteine level over 150 μmol/L.73 This included quadriceps myoclonus and/or hand tremors in three (12%) patients and an increase in hair loss in five (20%) patients. The mechanism[s] for such adverse effects remains unclear. Hyperhomocysteinemia was previously associated with myoclonus in a child with methylene tetrahydrofolate reductase deficiency (MTHFR).74 Another patient with plasma homocysteine level 187 μmol/L was admitted with pulmonary embolism without any identifiable risk factors and found to have moderate protein C deficiency. Homocystinuria is a similar disease that is characterized by involvement of the eye, skeletal system, vascular system, and central nervous system with frequent developmental delay. Thromboembolism is the major cause of morbidity and early death. Thus, perhaps, patients who are considering or are taking givosiran should be tested for MTHFR polymorphisms and vitamins B6, B9 (folate) and B12 (cobalamin) deficiency. Supplementation of these vitamins if deficient is appropriate and has been associated with decreases in plasma levels of homocysteine, albeit not always into the reference range.70,73 With ongoing use of givosiran, the possible impact of heme deficiency on other heme-dependent processes should be considered. One such concern is possible down-regulation of the cytochrome P450 (CYP) super-family of hemoproteins,75 which can influence the metabolism of many endogenous and exogenous compounds, including many drugs.8 In a mouse study, givosiran reduced ALAS1 mRNA substantially by approximately 75% but did not affect CYP2E1 activity or the degree of heme saturation of tryptophan 2,3 dioxygenase [formerly known as tryptophan pyrrolase].45 In a short term, one-month drug–drug interaction study in normal human volunteers, givosiran had moderate down-regulatory effect (CYP1A2 and CYP2D6), weak effect (CYP2C19 and CYP3A4), and no effect (CYP2C9) on these selected CYP enzymes.76 While this suggests that givosiran does not have a large effect on heme-dependent CYP enzyme activity in the liver, this was a short-term study; thus, long-term effects of givosiran on hepatic hemoprotein levels in persons with partial defects in normal heme synthesis, such as those with AHP, cannot be ruled out.
Across the givosiran clinical program, two cases of pancreatitis have been reported in givosiran-treated patients, both considered unlikely related to givosiran, but rather due to the presence of gallbladder sludge or gallstones. Elevations in lipase and amylase were observed in both givosiran and placebo treatment groups, with a higher-grade severity observed more frequently in the placebo group than in the givosiran group. Considering confounding factors, the potential role of givosiran in the development of pancreatitis has not been established. The cases of pancreatitis can be related to increased incidence of elevations in amylase or lipase, acute and chronic pancreatitis, and a higher frequency of gallstones naturally seen in patients with AHP.66,73,77
In a retrospective study of twenty-five patients in France, a personalized medicine approach was suggested by which timing of givosiran was titrated based on close surveillance of heme precursor levels (ALA) without a reduction in biological or clinical efficacy. Patients were divided in two groups: one required givosiran every 3 months or less and the second group received givosiran at intervals greater than once every 3 months. Thus, givosiran dosing frequency was decreased in patients with a prolonged response to treatment leading to a decrease in the dosing frequency in most patients and a presumed decreased risk of adverse events. The probability of dose reduction was higher in patients with the shortest disease course with shorter mean time since disease onset, suggesting a probable benefit to initiate givosiran early in the course of the disease.73 It must also be acknowledged that such reduced dosing of givosiran is “off label” use, perhaps, exposing prescribers to adverse exposure for deviation from approved use of the drug in the event of possible adverse effects associated with such use.
One approach to therapeutic monitoring can be checking for MTHFR polymorphisms and vitamins B6, B9, and B12 deficiencies before the start of treatment and assessing liver function tests (LFTs), plasma homocysteine, blood urea nitrogen, creatinine, eGFR, and urinalysis monthly for first three months. If the results are stable, a reasonable plan is then to repeat lab studies every 3 months for the first year, every 4 months for the second year, and every 6 months after that. We also recommend checking random urine samples for ALA, PBG, total porphyrins, and creatinine at each follow-up visit.54
Management of AHP
In AIP, a disease-causing mutation in porphobilinogen deaminase in combination with precipitating factors are needed to manifest clinical signs and symptoms. In fact, only a small percentage of patients (<5%) with disease-causing mutations will develop acute attacks.78 Common precipitating factors of acute attacks in patients with AIP include excess alcohol, certain medications, rapid weight loss, acute illness/physiologic stress, and changes in hormone levels (Table 3). Avoiding identified triggers in individuals may minimize, but not eliminate, the risk of precipitating an acute attack.
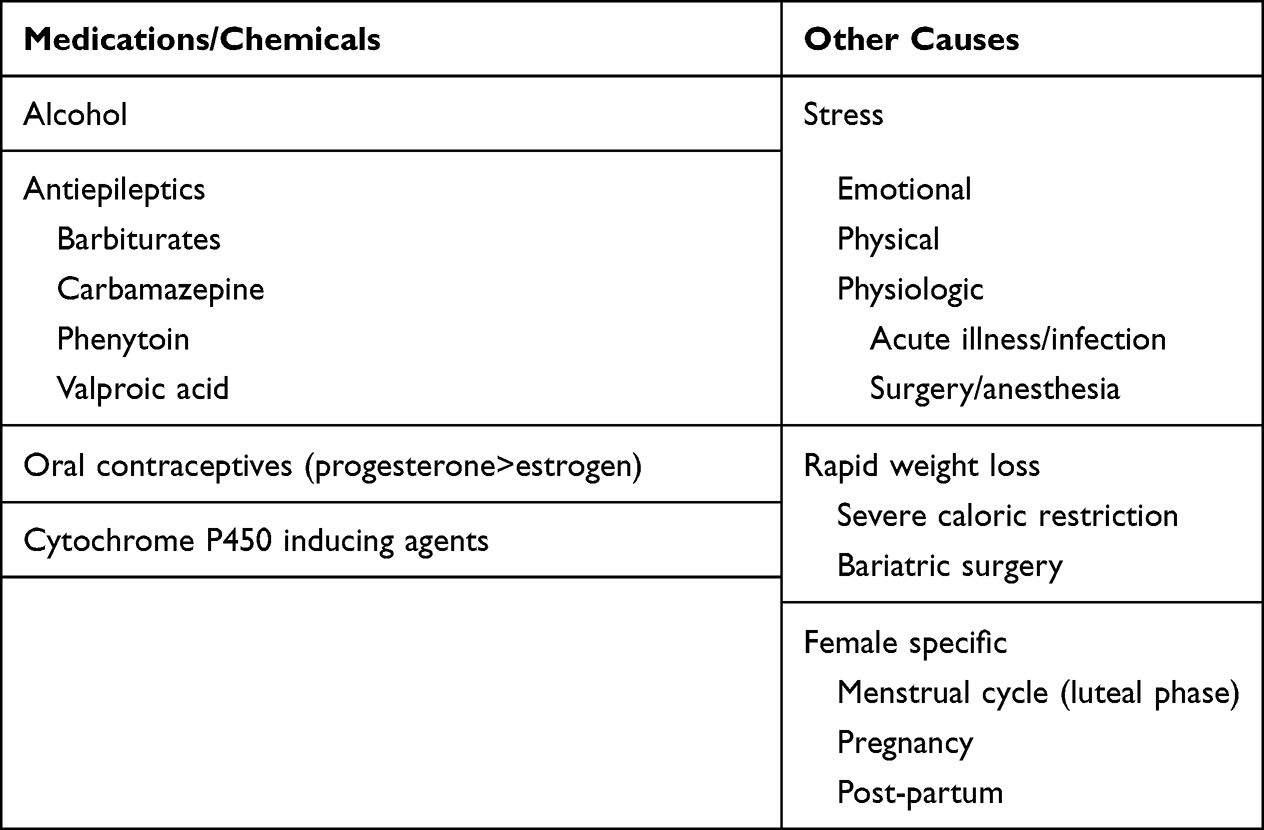 |
Table 3 Common Precipitants of Acute Porphyric Attacks |
Patients should be counseled regarding avoidance or cessation of alcohol and tobacco use. A unique nutritional aspect of AIP is that a diet to some extent high in carbohydrate intake (60–70% of total calories) is preferred, as carbohydrates act to down-regulate ALAS-1. Rapid weight loss either by crash dieting or starvation, and surgical weight loss may result in precipitation of an attack.79 Appropriate immunizations to avoid preventable infection, and prompt treatment of newly diagnosed infections may help prevent acute attacks. To the degree possible, patients and providers should develop strategies to avoid excess emotional or physical stress, with the understanding that these may be the most difficult precipitants to avoid. In women, attacks can be associated with the luteal phase of the menstrual cycle. In these patients, the use of gonadotropin-releasing hormone (GnRH) analogs to suppress ovulation may provide relief.
Awareness of the medications that are known to precipitate attacks is of particular importance to medical professionals. Antiepileptic medications deserve particular attention as patients may present with seizures, and the use of certain antiepileptic medications may further exacerbate an acute attack and lead to respiratory compromise (eg, phenytoin, valproic acid, carbamazepine). Treatment of seizures in a patient with suspected or known AIP should favor gabapentin, vigabatrin, or lorazepam. Additionally, medications that induce cytochrome P450 (eg, barbiturates, rifampin) are recognized as potentially detrimental. Multiple resources exist for the clinician to reference in regards to medication safety in patients with porphyria, including at the following web sites: www.drugs-porphyria.org (European) and www.porphyriafoundation.org/drugdatabase (the United States).
Once an acute attack is underway, general treatment strategies include management of pain, monitoring/treatment of electrolyte abnormalities and neurologic complications, and downregulation of ALAS-1.
Narcotic analgesics are typically required as initial therapy to decrease incapacitating pain, and the IV route is favored as patients in a severe acute attack can rarely tolerate oral intake. The authors favor the use of patient-controlled analgesia [PCA] in initial management. In those with significant nausea, antiemetics including chlorpromazine, promethazine, and ondansetron can be used safely. Typical electrolyte abnormalities during an acute attack include hyponatremia and hypomagnesemia, which should be managed appropriately.
As up-regulation in ALAS-1 activity drives the pathophysiology of acute attacks, its down-regulation provides the main therapeutic target. In the absence of infection, the use of carbohydrates (10% dextrose in 0.45% saline via continuous infusion) is a useful initial therapy and can be continued while other therapies are being utilized. The most effective therapy for acute attacks remains IV heme (Panhematin USA; Recordati; heme arginate [Normosang] in the EU and elsewhere), preferably administered with human serum albumin at 3–4 mg/kg/day.80,81 Down-regulation of ALAS-1 as measured by urinary ALA and PBG is typically observed within 4 days.75,82,83
For patients in whom preventative measures are unsuccessful and suffer recurrent attacks, possible preventative therapies include the use of IV heme and givosiran. In one study, the use of weekly IV heme (Panhematin) in patients with frequent recurrent attacks (defined as at least 3 attacks requiring treatment in the last 12 months) was associated with a decrease in acute attacks, inpatient admissions, emergency room visits, healthcare cost, and increased quality of life.84 The effectiveness of weekly IV heme arginate for prevention of recurrent acute attacks has recently been confirmed in a longitudinal observational study from Taiwan.85 Long-term use of hematin may be associated with adverse effects including hepatic iron overload, thrombocytopenia, phlebitis, and tachyphylaxis.64,86
Givosiran was studied in a similar patient population with a high disease burden; at least 2 attacks in the 6 months prior to study enrollment.8 Importantly, prophylactic hemin use was not permitted in the study population. Additionally, the overwhelming majority of patients included in the study (N=89) had AIP. There were 2 patients with VP, 1 patient with HCP, and 2 without identified mutations. Thus, although givosiran is indicated for the treatment of AHP in general, caution in generalizability is warranted given the small number of subjects with VP or HCP. Limitations notwithstanding, 70% of the patients treated with givosiran had fewer attacks compared to placebo. Furthermore, 50% of the patients treated with givosiran had zero attacks during the 6-month treatment period. Treatment with givosiran resulted in sustained reductions in ALA and PBG, linking the pathophysiologic mechanism to the observed clinical endpoints which suggest that givosiran is effective in preventing recurrent attacks in high-risk patients.
Givosiran is very expensive. For patients weighing 75.6 kg or less, the average annual cost of givosiran, 2.5 mg/kg =189 mg/1 mL dose × 12 months, is $575,000 per year, or $442,000 per year after expected discounts.87 For patients weighing more than 75.6 kg, a second dose each month, with double the cost, will be required, although it may be that such patients would respond adequately to doses a bit lower than the recommended 2.5 mg/kg body weight. A recent analysis of total costs for givosiran vs IV heme, either for treatment of acute attacks or for prophylaxis, assuming once weekly IV heme vs once monthly givosiran, showed that IV heme is much less expensive.88 Specifically, for all patients with AIP, the average annual total cost of care for IV heme therapy was $482,113 [78%] lower than for givosiran.
In severe cases of AIP [frequent attacks that require lengthy and repeated hospitalizations, etc], liver transplantation has been utilized as a curative therapy as it results in amelioration of the underlying enzymatic defect that is localized within the liver. In a study reviewing the European experience with liver transplantation for AIP, the 1-year and 5-year survival rates were similar to other metabolic diseases transplanted during the same time period.89 Neurologic and renal impairment were common pre-transplant patients, with advanced pre-transplant neurological impairment associated with increased mortality. Although improvement in renal function was uncommon, improvement or stabilization of neurologic symptoms was observed. Overall, liver transplantation for AHP should be reserved as a treatment of last resort in patients with a severe disease course not responding to other available therapies. With the availability of givosiran for prevention of recurrent attacks, we anticipate that liver transplantation will become less frequent in AHP.
In the less common AHPs (ADP, VP, and HCP), IV heme can be utilized. In VP, in which cutaneous manifestations are common, sun exposure must be minimized.90 Dietary supplementation with vitamins C and E may help mitigate oxidative damage in VP,91 although the evidence for benefit is scant. Importantly, as the less common AHP presents with findings similar to other porphyrias with alternative etiologies/treatment (eg, VP presents similarly to PCT), accurate diagnosis is paramount to ensure appropriate therapy.
In women with frequent cyclic attacks associated with menstrual cycles, prophylactic GnRH, perhaps, with a low-dose estrogen supplement, is worthy of trial. Progestins are identified as the triggering agent; thus, oral contraceptives, hormone-releasing implants, and intrauterine devices should be avoided. GnRH treatment, if it proven effective (as it is in approximately 50% of women), is reevaluated after 6 to 12 months. Pregnant women should be evaluated and monitored by a high-risk obstetrician as acute attacks are more common during pregnancy and in the postpartum period.13
Summary/Conclusion
In summary, the acute hepatic porphyrias are rare, inherited inborn errors of normal heme synthesis. Clinically, they present first and foremost as diseases that cause recurrent and severe episodes of abdominal pain, mainly in women aged ~16–50 years. The key test for rapid diagnosis is measurement of ALA, PBG, and creatinine in a random urine sample. Up-regulation of hepatic ALAS1 is the key factor in pathogenesis. Acute attacks require early diagnosis, analgesia, anti-emetics, and removal of inciting factors, such as alcohol intake, starvation, drugs that induce CYPs. Treatment to down-regulate hepatic ALAS1 includes adequate nutrition, with at least 300 g/d of glucose or other similar metabolizable carbohydrates, and IV heme [3–4 mg/kg/d for 3–5 days]. Givosiran is an siRNA that is highly selective in down-regulating ALAS1 in hepatocytes and effective in decreasing the frequency of recurrent attacks. Givosiran has potential adverse effects, especially risk of hepatic and renal adverse effects and of hyperhomocysteinemia, the latter a risk especially in patients with defects in MTHFR activity. There is concern about possible long-term adverse effects of down-regulation of hepatic heme synthesis and a possible heme-deficient state, although thus far, 30-month follow-up in the OLE of the ENVISION trial has not unveiled major new adverse effects. Givosiran is very expensive, considerably more expensive than IV heme, either given for acute attacks or as weekly prophylaxis for prevention of recurrent attacks. Because of greater ease and speed of use, patients are likely to prefer once monthly sc givosiran to weekly IV heme-albumin, given by a peripheral vein inserted central catheter (PICC) line or into a central venous port, for prophylaxis against recurrent acute attacks. Currently, it seems reasonable for prescribers and insurers to restrict the chronic use of givosiran to those patients with well-documented biochemically and clinically active AHP and with a history of three or more acute attacks in the prior year.
Abbreviations
AAR, annualized attack rate; ADP, delta-aminolevulinic acid dehydratase porphyria; AE, adverse event; AHP, acute hepatic porphyria; AIP, acute intermittent porphyria; ALA, delta-aminolevulinic acid; ALAS1, delta-aminolevulinic acid synthase 1; ALT, alanine aminotransferase; Ago2, Argonaute 2; ASGPR, asialoglycoprotein receptor; AST, aspartate aminotransferase; B6, pyridoxine; B9, folate; B12, cobalamin; CBS, cystathionine beta-synthase; CKD, chronic kidney disease; CPOX, coproporphyrinogen oxidase gene; Cr, creatinine; eGFR, estimated glomerular filtration rate; CYP, cytochrome P450; GI, gastrointestinal; HCP, hereditary coproporphyria; GalNac, N-acetylgalactosamine; GnRH, gonadotropin releasing hormone; HCV, hepatitis C virus; HMBS, hydroxymethylbilane synthase; ISR, injection-site reaction; IV, intravenous; LFTs, liver function tests; mRNA, messenger RNA; MTHFR, methylene-tetrahydrofolate reductase; OLE, open-label extension; PBG, porphobilinogen; PCT, porphyria cutanea tarda; PICC, peripheral vein inserted central catheter; PPOX, protoporphyrinogen oxidase; QM, once monthly; RNA, ribonucleic acid; RISC, RNA-induced silencing complex; SAE, serious adverse event; SF-12v2, 12-Item Short Form Health Survey Version 2; siRNA, small interfering RNA; VP, variegate porphyria.
Disclosure
Dr Sean Rudnick reports personal fees from Alnylam Pharmaceuticals, outside the submitted work. Dr Herbert Bonkovsky reports grants, personal fees from Alnylam Pharma, grants from Gilead Sciences, grants from Mitsubishi-Tanabe NA, personal fees from Recordati Rare Chemicals, personal fees from Disc Medicine, personal fees from Protagonist Therapeutics, Chair liver-related adjudication committee for Eiger Biopharma, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Nordmann Y, Puy H, Da Silva V, et al. Acute intermittent porphyria: prevalence of mutations in the porphobilinogen deaminase gene in blood donors in France. J Intern Med. 1997;242(3):213–217. doi:10.1046/j.1365-2796.1997.00189.x
2. Lenglet H, Schmitt C, Grange T, et al. From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria. Hum Mol Genet. 2018;27(7):1164–1173. doi:10.1093/hmg/ddy030
3. Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017;377(9):862–872. doi:10.1056/NEJMra1608634
4. Innala E, Backstrom T, Bixo M, Andersson C. Evaluation of gonadotropin-releasing hormone agonist treatment for prevention of menstrual-related attacks in acute porphyria. Acta Obstet Gynecol Scand. 2010;89(1):95–100. doi:10.3109/00016340903390729
5. Bissell DM, Lai JC, Meister RK, Blanc PD. Role of delta-aminolevulinic acid in the symptoms of acute porphyria. Am J Med. 2015;128(3):313–317. doi:10.1016/j.amjmed.2014.10.026
6. Meyer UA, Schuurmans MM, Lindberg RL. Acute porphyrias: pathogenesis of neurological manifestations. Semin Liver Dis. 1998;18(1):43–52. doi:10.1055/s-2007-1007139
7. Sardh E, Harper P, Balwani M, et al. Phase 1 trial of an RNA interference therapy for acute intermittent porphyria. N Engl J Med. 2019;380(6):549–558. doi:10.1056/NEJMoa1807838
8. Balwani M, Sardh E, Ventura P, et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N Engl J Med. 2020;382(24):2289–2301. doi:10.1056/NEJMoa1913147
9. Mohan G, Madan A. Ala Dehydratase Deficiency Porphyria. Treasure Island (FL): StatPearls; 2022.
10. Phillips JD. Heme biosynthesis and the porphyrias. Mol Genet Metab. 2019;128(3):164–177. doi:10.1016/j.ymgme.2019.04.008
11. Wang B, Bissell DM. Hereditary coproporphyria. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews((R)). Seattle (WA); 1993.
12. Singal AK, Anderson KE. Variegate porphyria. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews((R)). Seattle (WA); 1993.
13. Balwani M, Wang B, Anderson KE, et al. Acute hepatic porphyrias: recommendations for evaluation and long-term management. Hepatology. 2017;66(4):1314–1322. doi:10.1002/hep.29313
14. Singal AK, Venkata KVR, Jampana S, Islam FU, Anderson KE. Hepatitis C treatment in patients with porphyria cutanea tarda. Am J Med Sci. 2017;353(6):523–528. doi:10.1016/j.amjms.2017.03.007
15. Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for erythropoietic protoporphyria. N Engl J Med. 2015;373(1):48–59. doi:10.1056/NEJMoa1411481
16. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. doi:10.1038/35888
17. Kim D, Rossi J. RNAi mechanisms and applications. Biotechniques. 2008;44(5):613–616. doi:10.2144/000112792
18. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. doi:10.1016/j.cell.2009.01.035
19. Kulkarni JA, Witzigmann D, Thomson SB, et al. The current landscape of nucleic acid therapeutics. Nat Nanotechnol. 2021;16(6):630–643. doi:10.1038/s41565-021-00898-0
20. Lundstrom K. Viral vectors applied for RNAi-based antiviral therapy. Viruses. 2020;12(9):45.
21. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). 2018;9. doi:10.3389/fendo.2018.00402
22. Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118(1):57–68. doi:10.1016/j.cell.2004.06.017
23. Kim K, Lee YS, Carthew RW. Conversion of pre-RISC to holo-RISC by Ago2 during assembly of RNAi complexes. RNA. 2007;13(1):22–29. doi:10.1261/rna.283207
24. Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123(4):607–620. doi:10.1016/j.cell.2005.08.044
25. Rand TA, Petersen S, Du F, Wang X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. 2005;123(4):621–629. doi:10.1016/j.cell.2005.10.020
26. Tomari Y, Zamore PD. Perspective: machines for RNAi. Genes Dev. 2005;19(5):517–529. doi:10.1101/gad.1284105
27. Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15(2):185–197. doi:10.1016/j.molcel.2004.07.007
28. Liu J, Carmell MA, Rivas FV, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305(5689):1437–1441. doi:10.1126/science.1102513
29. Orban TI, Izaurralde E. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA. 2005;11(4):459–469. doi:10.1261/rna.7231505
30. Rivas FV, Tolia NH, Song JJ, et al. Purified Argonaute2 and an siRNA form recombinant human RISC. Nat Struct Mol Biol. 2005;12(4):340–349. doi:10.1038/nsmb918
31. Forstemann K, Horwich MD, Wee L, Tomari Y, Zamore PD. Drosophila microRNAs are sorted into functionally distinct argonaute complexes after production by dicer-1. Cell. 2007;130(2):287–297. doi:10.1016/j.cell.2007.05.056
32. Hu B, Zhong L, Weng Y, et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther. 2020;5(1):101. doi:10.1038/s41392-020-0207-x
33. Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16(9):543–552. doi:10.1038/nrg3978
34. Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431(7006):371–378. doi:10.1038/nature02870
35. Dana H, Chalbatani GM, Mahmoodzadeh H, et al. Molecular mechanisms and biological functions of siRNA. Int J Biomed Sci. 2017;13(2):48–57.
36. Barakat MR, Kaiser PK. VEGF inhibitors for the treatment of neovascular age-related macular degeneration. Expert Opin Investig Drugs. 2009;18(5):637–646. doi:10.1517/13543780902855316
37. Reich SJ, Fosnot J, Kuroki A, et al. Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol Vis. 2003;9:210–216.
38. Shen J, Samul R, Silva RL, et al. Suppression of ocular neovascularization with siRNA targeting VEGF receptor 1. Gene Ther. 2006;13(3):225–234. doi:10.1038/sj.gt.3302641
39. Pasi KJ, Rangarajan S, Georgiev P, et al. Targeting of Antithrombin in Hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377(9):819–828. doi:10.1056/NEJMoa1616569
40. Gonzalez V, Ruz V, Bleau AM, Vargas B, Jimenez AI. Tivanisiran as a new treatment for dry eye in patients with sjögren syndrome. Invest Ophthalmol Vis Sci. 2020;61(7):102.
41. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi:10.1056/NEJMoa1716153
42. Zhang MM, Bahal R, Rasmussen TP, Manautou JE, Zhong XB. The growth of siRNA-based therapeutics: updated clinical studies. Biochem Pharmacol. 2021;189:114432. doi:10.1016/j.bcp.2021.114432
43. Migliorati JM, Jin J, Zhong XB. siRNA drug Leqvio (inclisiran) to lower cholesterol. Trends Pharmacol Sci. 2022;43:455–456. doi:10.1016/j.tips.2022.02.003
44. de Paula Brandao PR, Titze-de-Almeida SS, Titze-de-Almeida R. Leading RNA interference therapeutics part 2: silencing delta-aminolevulinic acid synthase 1, with a focus on givosiran. Mol Diagn Ther. 2020;24(1):61–68. doi:10.1007/s40291-019-00438-6
45. Yasuda M, Gan L, Chen B, et al. RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc Natl Acad Sci USA. 2014;111(21):7777–7782. doi:10.1073/pnas.1406228111
46. Chan A, Liebow A, Yasuda M, et al. Preclinical development of a subcutaneous ALAS1 RNAi therapeutic for treatment of hepatic porphyrias using circulating RNA quantification. Mol Ther Nucleic Acids. 2015;4:e263. doi:10.1038/mtna.2015.36
47. Nair JK, Willoughby JL, Chan A, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136(49):16958–16961. doi:10.1021/ja505986a
48. Brown CR, Gupta S, Qin J, et al. Investigating the pharmacodynamic durability of GalNAc-siRNA conjugates. Nucleic Acids Res. 2020;48(21):11827–11844. doi:10.1093/nar/gkaa670
49. Titze-de-Almeida R, David C, Titze-de-Almeida SS. The race of 10 synthetic RNAi-based drugs to the pharmaceutical market. Pharm Res. 2017;34(7):1339–1363. doi:10.1007/s11095-017-2134-2
50. Chong S, Agarwal S, Agarwal S, et al. The nonclinical disposition and PK/PD properties of GalNAc-conjugated siRNA are highly predictable and build confidence in translation to man. Drug Metab Dispos. 2021;4:56.
51. Li J, Liu J, Zhang X, et al. Nonclinical pharmacokinetics and absorption, distribution, metabolism, and excretion of givosiran, the first approved N-Acetylgalactosamine-conjugated RNA interference therapeutic. Drug Metab Dispos. 2021;49(7):572–580. doi:10.1124/dmd.121.000381
52. Shen X, Corey DR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46(4):1584–1600. doi:10.1093/nar/gkx1239
53. Titze-de-Almeida SS, Brandao PRP, Faber I, Titze-de-Almeida R. Leading RNA interference therapeutics part 1: silencing hereditary transthyretin amyloidosis, with a focus on patisiran. Mol Diagn Ther. 2020;24(1):49–59. doi:10.1007/s40291-019-00434-w
54. Thapar M, Rudnick S, Bonkovsky HL. Givosiran, a novel treatment for acute hepatic porphyrias. Expert Rev Precis Med Drug Dev. 2020;6(1):9–18. doi:10.1080/23808993.2021.1838275
55. Agarwal S, Simon AR, Goel V, et al. Pharmacokinetics and pharmacodynamics of the small interfering ribonucleic acid, givosiran, in patients with acute hepatic porphyria. Clin Pharmacol Ther. 2020;108(1):63–72. doi:10.1002/cpt.1802
56. Syed YY. Givosiran: a Review in Acute Hepatic Porphyria. Drugs. 2021;81(7):841–848. doi:10.1007/s40265-021-01511-3
57. D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting - strategies and applications. J Control Release. 2015;203:126–139. doi:10.1016/j.jconrel.2015.02.022
58. Harris RL, van den Berg CW, Bowen DJ. ASGR1 and ASGR2, the genes that encode the asialoglycoprotein receptor (Ashwell Receptor), are expressed in peripheral blood monocytes and show interindividual differences in transcript profile. Mol Biol Int. 2012;2012:283974. doi:10.1155/2012/283974
59. Vyas AK, Ramakrishna U, Sen B, et al. Placental expression of asialoglycoprotein receptor associated with Hepatitis B virus transmission from mother to child. Liver Int. 2018;38(12):2149–2158. doi:10.1111/liv.13871
60. Seow YY, Tan MG, Woo KT. Expression of a functional asialoglycoprotein receptor in human renal proximal tubular epithelial cells. Nephron. 2002;91(3):431–438. doi:10.1159/000064283
61. Lazareth H, Poli A, Bignon Y, et al. Renal function decline under therapy with small interfering RNA silencing ALAS1 for acute intermittent porphyria. Kidney Int Rep. 2021;6(7):1904–1911. doi:10.1016/j.ekir.2021.04.004
62. Goetsch CA, Bissell DM. Instability of hematin used in the treatment of acute hepatic porphyria. N Engl J Med. 1986;315(4):235–238. doi:10.1056/NEJM198607243150406
63. Anderson KE, Bonkovsky HL, Bloomer JR, Shedlofsky SI. Reconstitution of hematin for intravenous infusion. Ann Intern Med. 2006;144(7):537–538. doi:10.7326/0003-4819-144-7-200604040-00023
64. Glueck R, Green D, Cohen I, Ts’ao CH. Hematin: unique effects of hemostasis. Blood. 1983;61(2):243–249. doi:10.1182/blood.V61.2.243.243
65. Stein P, Rees D, Anderson K, et al. A phase 1/2 open label extension study of givosiran, an investigational RNAi therapeutic, in patients with acute intermittent porphyria. J Hepatol. 2020;73:S553–S554. doi:10.1016/S0168-8278(20)31580-4
66. Ventura P, Bonkovsky HL, Gouya L, et al. Efficacy and safety of givosiran for acute hepatic porphyria: 24-month interim analysis of the randomized phase 3 ENVISION study. Liver Int. 2022;42(1):161–172. doi:10.1111/liv.15090
67. FDA. Highlights of prescribing information; 2019. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/0212194s000lbl.pdf.
68. Vassiliou D, Sardh E. Homocysteine elevation in givosiran treatment: suggested ALAS1 siRNA effect on cystathionine beta-synthase. J Intern Med. 2021;290(4):928–930. doi:10.1111/joim.13341
69. Fontanellas A, Avila MA, Arranz E, Enriquez de Salamanca R, Morales-Conejo M. Acute intermittent porphyria, givosiran, and homocysteine. J Inherit Metab Dis. 2021;44(4):790–791. doi:10.1002/jimd.12411
70. Petrides PE, Klein M, Schuhmann E, et al. Severe homocysteinemia in two givosiran-treated porphyria patients: is free heme deficiency the culprit? Ann Hematol. 2021;100(7):1685–1693. doi:10.1007/s00277-021-04547-3
71. To-Figueras J, Wijngaard R, Garcia-Villoria J, et al. Dysregulation of homocysteine homeostasis in acute intermittent porphyria patients receiving heme arginate or givosiran. J Inherit Metab Dis. 2021;44(4):961–971. doi:10.1002/jimd.12391
72. Kaplan P, Tatarkova Z, Sivonova MK, Racay P, Lehotsky J. Homocysteine and mitochondria in cardiovascular and cerebrovascular systems. Int J Mol Sci. 2020;21(20):7698. doi:10.3390/ijms21207698
73. Poli A, Schmitt C, Moulouel B, et al. Givosiran in acute intermittent porphyria: a personalized medicine approach. Mol Genet Metab. 2022;135(3):206–214. doi:10.1016/j.ymgme.2022.01.002
74. D’Aco KE, Bearden D, Watkins D, Hyland K, Rosenblatt DS, Ficicioglu C. Severe 5,10-methylenetetrahydrofolate reductase deficiency and two MTHFR variants in an adolescent with progressive myoclonic epilepsy. Pediatr Neurol. 2014;51(2):266–270. doi:10.1016/j.pediatrneurol.2014.04.005
75. Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, Thapar M. Porphyrin and heme metabolism and the porphyrias. Compr Physiol. 2013;3(1):365–401.
76. Vassiliou D, Sardh E, Harper P, et al. A drug-drug interaction study evaluating the effect of givosiran, a small interfering ribonucleic acid, on cytochrome P450 activity in the liver. Clin Pharmacol Ther. 2021;110(5):1250–1260. doi:10.1002/cpt.2419
77. Summary of the risk management plan for Givlaari.
78. Chen B, Solis-Villa C, Hakenberg J, et al. Acute intermittent porphyria: predicted pathogenicity of HMBS variants indicates extremely low penetrance of the autosomal dominant disease. Hum Mutat. 2016;37(11):1215–1222. doi:10.1002/humu.23067
79. Bonkovsky HL, Siao P, Roig Z, Hedley-Whyte ET, Flotte TJ. Case records of the Massachusetts General Hospital. Case 20-2008. A 57-year-old woman with abdominal pain and weakness after gastric bypass surgery. N Engl J Med. 2008;358(26):2813–2825. doi:10.1056/NEJMcpc0803190
80. Bonkovsky HL, Healey JF, Lourie AN, Gerron GG. Intravenous heme-albumin in acute intermittent porphyria: evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am J Gastroenterol. 1991;86(8):1050–1056.
81. Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142(6):439–450. doi:10.7326/0003-4819-142-6-200503150-00010
82. Bonkowsky HL, Tschudy DP, Collins A, et al. Repression of the overproduction of porphyrin precursors in acute intermittent porphyria by intravenous infusions of hematin. Proc Natl Acad Sci U S A. 1971;68(11):2725–2729. doi:10.1073/pnas.68.11.2725
83. Bissell DM. Treatment of acute hepatic porphyria with hematin. J Hepatol. 1988;6(1):1–7. doi:10.1016/S0168-8278(88)80456-2
84. Yarra P, Faust D, Bennett M, Rudnick S, Bonkovsky HL. Benefits of prophylactic heme therapy in severe acute intermittent porphyria. Mol Genet Metab Rep. 2019;19:100450. doi:10.1016/j.ymgmr.2019.01.002
85. Kuo HC, Lin CN, Tang YF. Prophylactic heme arginate infusion for acute intermittent porphyria. Front Pharmacol. 2021;12:712305. doi:10.3389/fphar.2021.712305
86. Willandt B, Langendonk JG, Biermann K, et al. Liver fibrosis associated with iron accumulation due to long-term Heme-Arginate treatment in acute intermittent porphyria: a case series. JIMD Rep. 2016;25:77–81.
87. Sheridan K. FDA approves Alnylam’s Givlaari, second-ever drug based on RNAi. Boston, MA: STAT; 2019.
88. Massachi S, Epstein J, Hurd J, Bonkovsky HL. Cost savings with hemin versus givosiran for the treatment of patients with acute intermittent porphyria (AIP). J Med Econ. 2020;23(12):1441–1449. doi:10.1080/13696998.2020.1835306
89. Lissing M, Nowak G, Adam R, et al. Liver transplantation for acute intermittent porphyria. Liver Transpl. 2021;27(4):491–501. doi:10.1002/lt.25959
90. Wang B, Rudnick S, Cengia B, Bonkovsky HL. Acute hepatic porphyrias: review and recent progress. Hepatol Commun. 2019;3(2):193–206. doi:10.1002/hep4.1297
91. Ferrer MD, Tauler P, Sureda A, Palacin C, Tur JA, Pons A. Variegate porphyria induces plasma and neutrophil oxidative stress: effects of dietary supplementation with vitamins E and C. Br J Nutr. 2010;103(1):69–76. doi:10.1017/S0007114509991413
92. Bonkovsky HL, Gouya L, Peiro PA, et al. Efficacy and safety of givosiran in patients with acute hepatic porphyria: 24-month interim analysis of the phase 3 ENVISION randomized clinical trial (abstract).
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.