")
Back to Journals » Journal of Inflammation Research » Volume 16
Spontaneous Pneumomediastinum and Subcutaneous Emphysema in Dermatomyositis: A Case Series and Literature Review
Authors Subki AH , Almani IM, Albeity A , Aljabri BK, Alsolaimani R, Halabi H
Received 11 October 2022
Accepted for publication 1 March 2023
Published 3 April 2023 Volume 2023:16 Pages 1431—1441
DOI https://doi.org/10.2147/JIR.S389839
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Ahmed Hussein Subki,1 Ibraheem Mohammed Almani,2 Abdurahman Albeity,1 Bandari Khalid Aljabri,1 Roaa Alsolaimani,1,2 Hussein Halabi1
1Department of Medicine, King Faisal Specialist Hospital and Research Center, Jeddah, Saudi Arabia; 2Department of Medicine, King Abdulaziz University Hospital, Jeddah, Saudi Arabia
Correspondence: Ahmed Hussein Subki; Hussein Halabi, Department of Internal Medicine, King Faisal Specialist Hospital & research center, POBox: 43129, Jeddah, 21561, Saudi Arabia, Tel +966566724288, Email [email protected]; [email protected]
Background: Spontaneous pneumomediastinum and subcutaneous emphysema are rare and serious complications of dermatomyositis (DM).
Case Presentation: Our article presents two clinically heterogeneous cases of DM who developed pneumomediastinum and subcutaneous emphysema. The first was a 24-year-old lady with a recently diagnosed DM. She developed rapidly progressive pneumonia, interstitial lung disease (ILD), pneumomediastinum, subcutaneous emphysema, and acute respiratory distress syndrome (ARDS). She died despite treatment with steroids and immunosuppressants (methotrexate and mycophenolate mofetil (MMF)). The second was a 30-year-old man diagnosed with amyopathic DM. He developed pneumomediastinum prior to ILD, which worsened over time, and subcutaneous emphysema evolved. However, he recovered completely after corticosteroid, MMF, and rituximab.
Conclusion: Spontaneous pneumomediastinum and subcutaneous emphysema may complicate DM. Corticosteroids, immunosuppressants, and respiratory support are the mainstay of management for these conditions. Though they were reported to carry a poor prognostic value, the course and outcome are highly variable among the cases.
Keywords: dermatomyositis, pneumomediastinum, subcutaneous emphysema, pneumothorax, corticosteroids, immunosuppressants, rituximab
Introduction
Inflammatory myopathy (myositis) results from immune-mediated inflammation of the muscles with or without other tissues, such as blood vessels.1 Dermatomyositis (DM) is a specific type of the immune-mediated inflammatory myopathy spectrum that primarily presents as proximal muscle weakness along with cutaneous manifestations.2 The cutaneous manifestations in DM may coincide or precede the muscle weakness.2 The pathognomonic skin features include Gottron’s papules and heliotrope rash.2 Other skin symptoms include facial erythema, Gottron’s sign, Shawl sign, V sign, poikiloderma, Holster sign, mechanic’s hands, periungual involvement, scalp involvement, and calcinosis cutis.2,3
Along with muscle and cutaneous manifestations, DM affects other organs, particularly the lungs.4 Approximately 10–22% of cases of DM have pulmonary signs such as aspiration pneumonia, respiratory muscle weakness, diffuse alveolar damage, and bronchiolitis obliterans.5,6 Interstitial lung disease is also not uncommon and is often associated with rapid progression.7 In addition, patients with DM are more likely than those with polymyositis to develop pulmonary complications such as subcutaneous emphysema, pneumothorax, and pneumomediastinum.6 Though these complications are rare, their occurrence is serious and might be associated with fatal outcomes.6
Pneumomediastinum is a condition where air extravasates from within the airways and migrates into the mediastinal space.8 It commonly results from traumatic injury, acute mediastinitis, or assisted ventilation.8 Spontaneous pneumomediastinum may occur due to air leakage through non-partitional alveolar walls and subsequent medial dissection along pulmonary vessels to the mediastinum.6 When the peripherally dissecting air gets trapped as subpleural blebs, or if there are visceral pleura ruptures, air can escape into the pleural space, resulting in pneumothorax.9
The prevalence of spontaneous pneumomediastinum in DM is higher in men.6 Pneumomediastinum in DM is a sign of a poor prognosis.10 It was reported to be fatal in some cases in the literature, primarily due to uncontrollable progressive ILD and respiratory failure.10
This article presents two cases of dermatomyositis that developed spontaneous pneumomediastinum and subcutaneous emphysema. The cases were discussed with a relevant literature review addressing possible mechanisms of occurrence of spontaneous pneumomediastinum and subcutaneous emphysema in DM.
Case Presentation
Case 1
Clinical History
This was a 24-year-old lady who was diagnosed with dermatomyositis based on a five-month history of proximal muscle weakness and cutaneous rash. Her weakness gradually progressed until she became a wheelchair user. Her muscle weakness was also apparent when she needed her mother to comb her hair. The Medical Research Council (MRC) Scale for Muscle Strength was used. The power of her shoulder and hip muscles was MRC grade 3/5, and the remaining muscles were MRC 4/5. Weakness also involved the bulbar muscles manifesting as progressive dysphagia. The dysphagia was initially to solids, then fluids, and the condition worsened until she choked on her own saliva. She lost 5 kilograms. Rashes were observed involving the nasolabial folds, hands, and thighs. Oral thrush, heliotrope rashes, Gottron’s papules, mechanic hands, periungual erythema, Holster’s sign in the left thigh, and painful cuticle overgrowth were seen. She also experienced arthritis, hand edema, dry eyes, dry mouth, and dyspnea (New York Heart Association (NYHA) class between II and III). Her past medical history was remarkable for hypothyroidism (post-irradiation with radioactive iodine), and she was on thyroxine replacement therapy.
She was admitted twice to our hospital with pneumonia (presenting with fever, productive cough, and dyspnea) and was treated with antibiotics. However, in her most recent admission, the patient experienced spiking fever, exacerbated cough, swelling over the neck and chest wall, progressive dyspnea, and hypoxia that necessitated intensive care unit (ICU) admission.
Investigations
Her chemistry panel showed elevated erythrocyte sedimentation rate (ESR) (25 mm/hr [normal: 0–20 mm/hr]), C-reactive protein level (0.37 mg/L [normal: 0.8–3.0 mg/L]), creatinine kinase level (CK) (244 IU/L [normal: 26–192 IU/L]), alanine aminotransferase (ALT) level (222 IU/L [normal: 0–33 IU/L]), and aspartate transaminase (AST) level (348 IU/L [normal: 0–32 IU/L]). The remaining routine panel was unremarkable. Her immunology profile was negative for anti-Jo-1 antibodies, antinuclear antibodies (ANA), antineutrophil cytoplasmic antibodies (ANCA), anti-double-stranded DNA (dsDNA), extractable nuclear antigens (ENA), anti-cyclic citrullinated peptide antibodies (anti-CCP), anti-Ro, anti-La, and antiphospholipid antibodies (APA).
Magnetic resonance imaging (MRI) with contrast showed diffuse and high signal intensity seen mainly within the anterior compartment of the thigh, and to a lesser extent, in the medial compartment superiorly, with no significant involvement of the posterior compartment bilaterally. There was evidence of bilateral enhancement consistent with an acute inflammatory process such as extensive proximal myositis. A skin biopsy was obtained and revealed superficial perivascular lymphocytic inflammation. An echocardiogram showed mild cardiomyopathy, most likely secondary to autoimmune myocarditis, with an ejection fraction of 45% (mild to moderate congestive heart failure). Computed tomography (CT) of the brain, neck, chest, abdomen, and pelvis excluded malignancy. However, her initial chest CT revealed multiple bilateral peripheral areas of consolidation, ground-glass opacities, and a minimal degree of pneumomediastinum.
When her symptoms exacerbated, CT chest was repeated, and there was a considerable progression of the pneumomediastinum, with an extension of the air to the subcutaneous tissue causing subcutaneous emphysema (Figure 1). Accordingly, a barium swallow was performed. Air leakage from the esophagus and stomach was excluded.
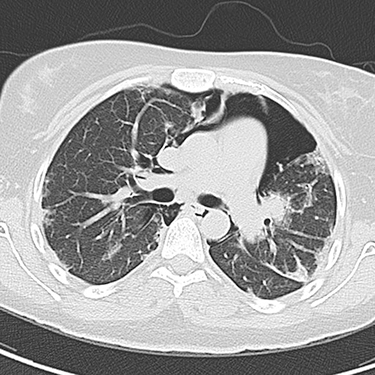 |
Figure 1 Chest computed tomography (CT) in the axial plane showing pneumomediastinum outlining the heart and other mediastinal structures. The lung parenchyma shows multiple bilateral peripheral and subpleural areas of ground-glass opacities. |
Subsequent chest CTs revealed a progressive deterioration of her pulmonary condition. There were bilateral peripheral ground glass opacities and basal atelectasis. In addition, the pneumomediastinum seemed to have extended to the lower neck and the left para-mediastinal region; there was extensive subcutaneous emphysema (Figures 2 and 3).
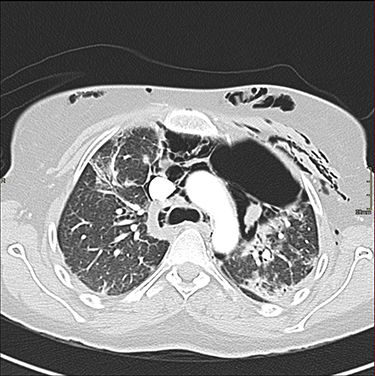 |
Figure 2 Axial chest CT with contrast showing the progression of the pneumomediastinum with air pockets in the left para-mediastinal region and subcutaneous emphysema. |
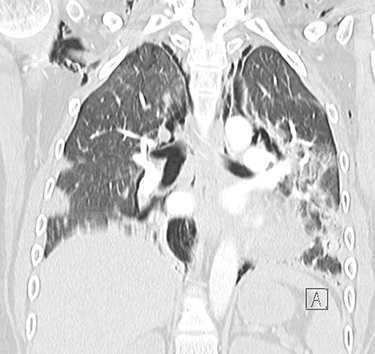 |
Figure 3 Coronal CT of the chest with contrast showing extensive pneumomediastinum and subcutaneous emphysema. |
Treatment and Outcome
Initially, the patient was given mycophenolate and steroids (prednisolone 1 mg/Kg/day). When her condition worsened, she was given intravenous immunoglobulin (IVIG). Methotrexate (MTX), hydroxychloroquine (HCQ), and anti-cardiac failure medications were added to her treatment regimen as well. Within one week of the treatment, she improved, ie, she could walk with support and swallow more easily. There was also an improvement in her reported shortness of breath. After one week of the therapy, the CK level decreased from 244 to 152 IU/L, ALT decreased from 222 to 88 IU/L, and AST decreased from 348 to 77 IU/L. All the enzyme levels eventually became normal, and the EF normalized on follow-up echocardiography.
Later on, however, she experienced high fever, productive cough, and progressive dyspnea. This was diagnosed as hospital-acquired pneumonia and thus given antibiotics. The response was minimal, and she developed progressive pneumomediastinum, pneumothorax, subcutaneous emphysema, and acute respiratory distress syndrome (ARDS). Bilateral chest tubes were inserted, mechanical ventilation was initiated, and extracorporeal membrane oxygenation (ECMO) was started. This resulted in partial improvement in hypoxemia. However, the patient developed increased intracranial pressure and herniation and, unfortunately, died.
Case 2
Clinical History
This was a 30-year-old male who presented initially with pain at the small joints of the hand (ie, metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints) and wrists along with non-palpable non-scaly violaceous macules over his elbows and upper back. He was initially diagnosed –at a private clinic– as a case of psoriatic arthritis and was prescribed methotrexate. However, after one week of methotrexate intake, he developed acute pneumonitis and was admitted to our rheumatology unit. On detailed assessment during this admission, his diagnosis was reevaluated, and systemic lupus erythematosus (SLE) was considered based on the presence of malar rash, arthralgia, lymphopenia, positive antinuclear antibodies (ANA), and direct antiglobulin test (Coombs’). Accordingly, methotrexate was stopped, and oral prednisolone (15 mg daily), mycophenolate mofetil (1 g twice daily), and HCQ (400 mg daily) were initiated. During that time, he denied fever, oral ulcers, alopecia, muscle weakness, or urinary and central nervous system-related symptoms.
One year later, he presented with a swelling over his chest and neck with crepitus on palpation that had been there for a month. He reported dyspnea on minimal exercise and a productive cough of whitish sputum. He had no muscle weakness or bulbar symptoms. His examination was remarkable for heliotrope rash, Gottron’s papules, malar rash, active Raynaud’s phenomenon, and chest wall and neck swelling. His neurological examination was unremarkable, and his chest auscultation revealed only bilateral basal fine crepitations.
Investigations
His laboratory panel showed normal CK level (180 IU/L) and positive ANA (titer 1:80). Routine chemistry panel, CRP, hepatitis serology, immunology panel (anti-dsDNA, anti-Ro, anti-La, anti-smith, anti-JO1, anti-RNP), and complement were all negative.
Initial chest CT with contrast showed peripheral interstitial thickening with peripheral multiple variable-sized cystic changes in both lungs and extensive subcutaneous emphysema involving the neck as well as the anterior and posterior chest walls, with no evidence of pneumothorax, enlarged mediastinal or axillary lymph nodes. A follow-up CT chest one month later showed pneumomediastinum along with minimal bilateral pneumothorax (Figure 4).
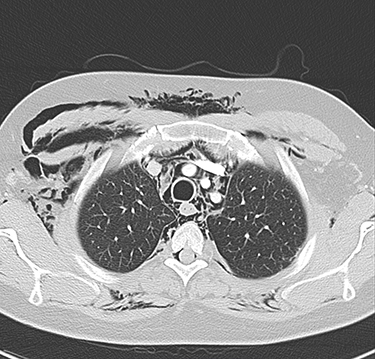 |
Figure 4 Axial CT of the upper chest with contrast for the second patient, showing pneumomediastinum, along with bilateral subcutaneous emphysema, more predominant on the right side. |
A pulmonary function test (PFT) showed a restrictive pattern with forced vital capacity (FVC) of 45%, forced expiratory volume in one second (FEV1) of 49%, an FEV1/FVC ratio of 49%, and a diffusing capacity for carbon monoxide (DLCO) of 53%. In the 6-minute walk test, the patient managed to walk 460 meters from the predicted 706 meters distance.
Diagnosis, Treatment, and Outcome
Considering the clinical picture and investigations, the patient was diagnosed with amyopathic dermatomyositis, ILD, subcutaneous emphysema, and pneumomediastinum.
Pneumomediastinum and subcutaneous emphysema were managed conservatively. The prednisolone dose was increased to 25 mg/day. Mycophenolate mofetil dose was reduced to 500 mg twice daily, HCQ was discontinued, and rituximab was initiated. Nifedipine 30 mg and Aspirin 81 mg daily were added for the Raynaud’s. Six months following rituximab initiation, prednisolone was tapered to 5 mg daily. The patient’s symptoms resolved, and his condition was maintained stable on the same treatment regimen (ie, rituximab every six months, MMF 500 mg twice, prednisolone 5 mg, nifedipine 30 mg, and aspirin 81 mg).
Discussion
Conclusions and Rationale
This article presents two cases of DM complicated with pneumomediastinum and subcutaneous emphysema. The two cases varied in their clinical course, presentation, and outcome. Our two patients will be discussed considering the available literature for potential causative mechanisms and comparison to similar reported cases.
Reference to Relevant Literature
Involvement of the lungs is a major determinant of morbidity and mortality in patients diagnosed with DM.6,10 Inflammatory myositis is considered the most frequent connective tissue disease associated with pneumomediastinum.11 Several cases of spontaneous pneumomediastinum were reported in patients with DM in the literature.6,12–30 Similar to our patients, pneumomediastinum was reported in cases of DM and amyopathic DM,11 occurred at a widely variable range of disease duration,6,12–30 and was associated with variable outcomes from complete recovery11 to death.5,14,31,32 Pneumomediastinum occurred prior to (as our second patient), coinciding with, or after (as our first patient) pulmonary involvement.11 Unlike the case of our second patient, Goff et al11 noted that pneumomediastinum is more fatal in patients without muscle weakness. Subcutaneous emphysema has also been reported in approximately 55.5% of cases of DM.24
Though several case reports, case series, and review articles on DM and pneumomediastinum were reported in the literature,6,12–30 only a few cases of DM had both pneumomediastinum and emphysema.5,6,13,16,18–22,25–27,33–39 Table 1 summarizes the main clinical features, treatment strategies, and outcomes of these cases.5,6,13,16,18–22,25–27,33–39 Similar to our patients, the clinical course and presentation of the reported cases in the literature were highly variable. Combined pneumomediastinum and emphysema occurred in both males and females, over a wide age range (from 10 to 65 years), and at a varied range of disease duration (as short as one month5 and up to four years from disease onset)36. They occurred coinciding with or after the development of ILD in the vast majority of the cases. However, they also occurred in patients without ILD or parenchymatous pulmonary disease, such as the patients reported by Shimamoto et al,20 Yoshida et al,26 and Tang et al.29 This matches with our second patient where pneumomediastinum and subcutaneous emphysema evolved prior to any evidence of ILD or parenchymatous disease. Creatine kinase levels, immunology panel, treatments given, and disease outcomes were also heterogenous as detailed in Table 1.5,6,13,16,18–22,25–27,33–39
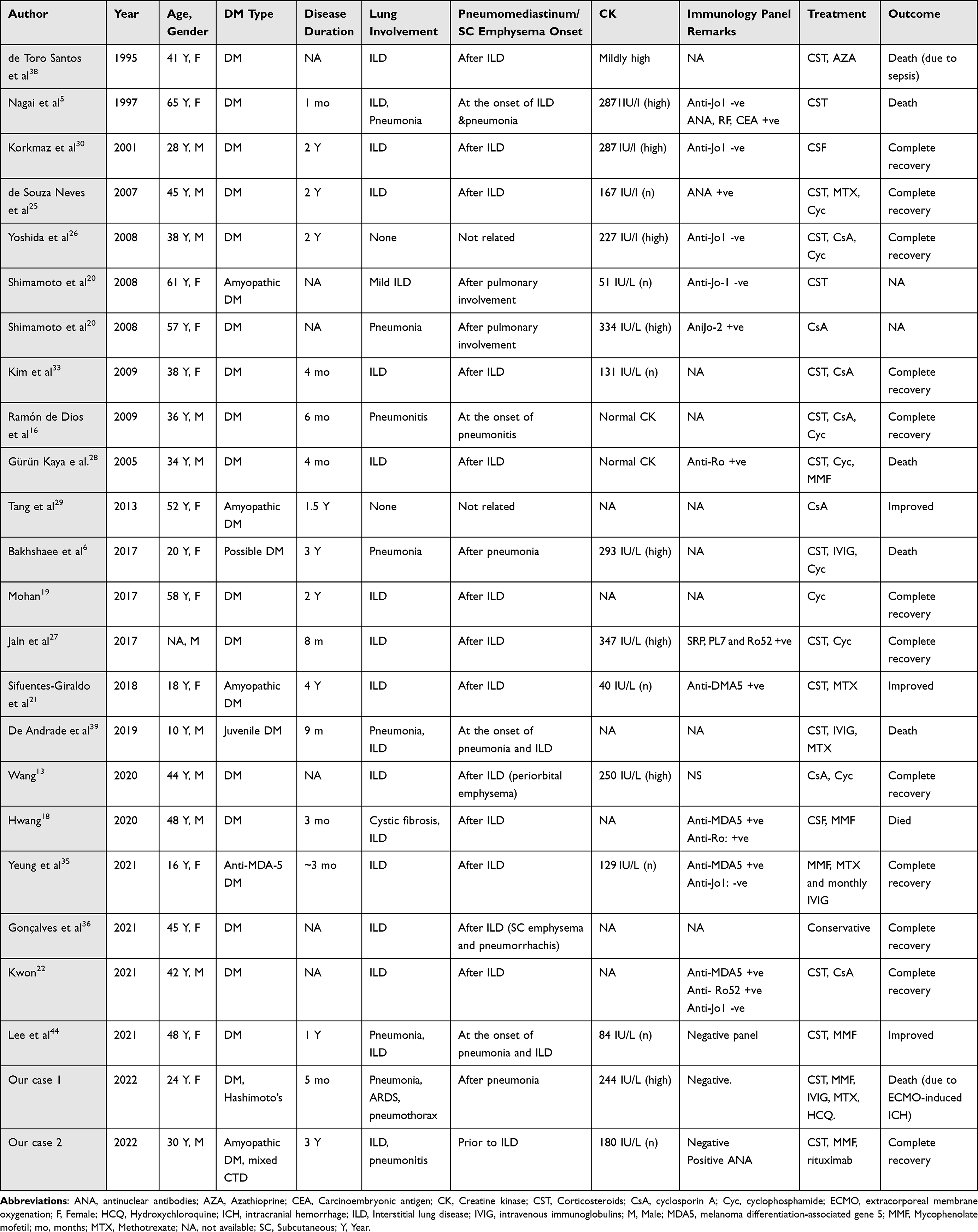 |
Table 1 Summary of the Cases of Dermatomyositis Complicated with Pneumomediastinum and Subcutaneous Emphysema |
Comparison to the Current Gold Standard of Care
To date, there are no consensus recommendations or guidelines for the management of pneumomediastinum or subcutaneous emphysema complicating DM. Conservative management, mechanical ventilation, steroids, IVIG, and various immunosuppressants have been tried with conflicting results.5,6,13,16,18–22,25–27,33–39 The outcome of pneumomediastinum in patients with DM was variable in the literature, ie, corticosteroids and immunosuppressive therapy resulted in complete recovery in some cases,11 while others had fatal outcomes despite aggressive therapy.5,14,31,32 In our patients, we adopted treatment strategies as reported in the literature, ie, corticosteroids,20 IVIG,35 MMF,17 and MTX.39 However, to the best of our knowledge, we are the first to give rituximab to such patients, and the outcome was promising.
Relevant Hypothesis Generation
Though spontaneous pneumomediastinum and subcutaneous emphysema have been reported as complications in DM in the literature since the 1990s,5,38 the exact mechanisms of their occurrence and prognosis remain to be elucidated.6 Various mechanisms have been suggested for the occurrence of spontaneous pneumomediastinum in DM, such as alveolar rupture, formation of subpleural cysts, vasculopathy, and weakening effect of corticosteroids.9,23
Alveolar rupture is the most accepted proposed mechanism for pneumomediastinum complicating DM.11 Alveolar rupture is thought to occur due to a sudden increase in intra-alveolar pressure or decrease in peribronchovascular interstitial pressure, or both.23 Air leakage from the ruptured alveoli escapes to the mediastinum (resulting in pneumomediastinum) or to the pleura (leading to pneumothorax), depending on the location of the ruptured alveoli.11 This can explain the occurrence of pneumomediastinum and pneumothorax in our first patient, eventually after progressive ILD and pneumonia. Similarly, this mechanism can explain the occurrence of pneumomediastinum in DM patients with ILD reported in the literature.5,6,13,16,18–22,25–27,33–39 However, it is inadequate in patients who develop pneumomediastinum in the absence of any parenchymatous diseases or ILD, such as our second patient and the cases reported by Shimamoto et al,20 Yoshida et al,26 and Tang et al.29 Accordingly, other mechanisms were suggested.
Pneumomediastinum has also been proposed to result from the formation and rupture of a subpleural cyst.33 This mechanism was suggested by Kim et al,33 who observed this cyst in a patient with incomplete signs of myositis who later developed pneumomediastinum, pneumothorax, and massive subcutaneous emphysema. The authors, based on this case, emphasized that muscular involvement is not always reflected by laboratory or histological abnormalities. Most of these cysts, however, occur in patients with ILD, too.33
Vasculopathy is the third proposed mechanism.23 Pulmonary vasculitis, pneumonitis, or subpleural infarcts result in the disruption of the bronchial mucosal barrier and subsequent air leakage to the mediastinum.23,36 Patients with cutaneous vasculopathy and those with evidence from a muscle biopsy of B and CD4+ lymphocyte accumulation and complement deposition in the perivascular space were likely to develop pneumomediastinum irrespective of their lung involvement.10,20,23
Several cases had reported that the weakening effects of steroids could have contributed to the progression of pneumomediastinum in DM patients.6,11,30,33 Administering systemic steroids was determined to be the first line of therapy in most patients.17 This can explain why our first patient had his pneumomediastinum significantly worsened after the administration of pulse therapy. In disagreement with this theory, pneumomediastinum responded well with almost a complete recovery in many cases when corticosteroids were administered with or without immunosuppressants.30,33,40
The mechanism of subcutaneous emphysema is more elusive. More than half of the cases of pneumomediastinum develop subcutaneous emphysema.24 Though the vast majority of the cases occur in the context of pneumomediastinum24 -indicating air leakage from the mediastinum to the subcutaneous space– many cases were reported independent of pneumomediastinum. For instance, the patient reported by Wang et al13 had periorbital subcutaneous emphysema. Similarly, one of the cases reported by Shimamoto et al20 had subcutaneous emphysema independent of pneumothorax. The mechanism proposed for these cases is cutaneous vasculopathy resulting in air leakage beneath the skin.13
Implications of Clinical Practice
Pneumomediastinum should be considered in all patients with DM presenting with dyspnea.41 Though being a rare complication, it might carry a poor prognosis.42 Many risk factors have been reported to increase the risk of developing pneumomediastinum in DM, such as male gender, younger age at disease onset, presence of ILD, presence of cutaneous vasculopathy, normal or mildly elevated CK levels, amyopathic DM, initial forced vital capacity ≤60, Hamman-Rich-like presentation, neutrophilic bronchoalveolar lavage, and corticosteroid administration.6,10–12,24,30,43 To improve the prognosis of DM patients who developed pneumomediastinum, the categorization of ILD based on imaging findings and clinical features is suggested to predict survival. As for diagnostic procedures, it was suggested that further research should focus on identifying associated antibodies in dermatomyositis that would facilitate earlier diagnosis.11,32 Bronchoscopy and esophagoscopy are also essential. Unless there is severe hypoxemia or respiratory distress, tracheostomy and mechanical ventilation may not always be necessary in the case of subcutaneous emphysema.6
Strength and Limitations
There are many strengths to this article. Firstly, it describes two rare cases of DM complicated by both pneumomediastinum and subcutaneous emphysema. Secondly, to the best of the authors’ knowledge, this article is the first to report giving rituximab to such patients. Finally, the article offers an extensive literature review and addresses the mechanisms and outcomes of both pneumomediastinum and subcutaneous emphysema. However, there are still some limitations, mainly attributed to the lack of a biopsy and electromyography.
Conclusion
Spontaneous pneumomediastinum and subcutaneous emphysema may complicate DM. A high index of suspicion should be there for their early diagnosis. Though pneumomediastinum and subcutaneous emphysema were reported to carry a poor prognostic value, the course and outcome among the different cases may vary. Corticosteroids, immunosuppressants, and respiratory support are the mainstay of management for these cases.
Learning Points/Take Home Messages
- Spontaneous pneumomediastinum and subcutaneous emphysema may complicate DM.
- Knowledge of the risk factors of these complications and their potential poor prognostic value is essential for the prompt identification and treatment of these conditions.
- Steroids, immunosuppressants, and respiratory support are the mainstay management strategies.
Ethical Statement
This article was performed in accordance with the principles of the Declaration of Helsinki. The ethical approval was obtained from the Institutional Review Board of King Faisal Specialist Hospital & Research Centre, Jeddah, Saudi Arabia. Its reference number is IRB 2021-CR-19. In addition, written consent was obtained from both of our patients for the publication of their cases and accompanying data.
Acknowledgments
We are immensely grateful to our generous patients for allowing us to publish their cases.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Venalis P, Lundberg IE. Immune mechanisms in polymyositis and dermatomyositis and potential targets for therapy. Rheumatology. 2014;53(3):397–405. doi:10.1093/RHEUMATOLOGY/KET279
2. Qudsiya Z, Waseem M. Dermatomyositis. StatPearls; 2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK558917/.
3. Yang SH, Chang C, Lian ZX. Polymyositis and dermatomyositis – challenges in diagnosis and management. J Transl Autoimmun. 2019;2:100018. doi:10.1016/J.JTAUTO.2019.100018
4. Hallowell RW, Ascherman DP, Danoff SK. Pulmonary manifestations of polymyositis/dermatomyositis. Semin Respir Crit Care Med. 2014;35(2):239–248. doi:10.1055/S-0034-1371528
5. Nagai Y, Ishikawa O, Miyachi Y. Pneumomediastinum and subcutaneous emphysema associated with fatal interstitial pneumonia in dermatomyositis. J Dermatol. 1997;24(7):482–484. doi:10.1111/J.1346-8138.1997.TB02825.X
6. Bakhshaee M, Jokar MH, Mirfeizi Z, Atabati E, Tarighat S. Subcutaneous emphysema, pneumomediastinum and pneumothorax in a patient with dermatomyositis. Iran J Otorhinolaryngol. 2017;29(91):113. doi:10.22038/ijorl.2017.8287
7. Palawisuth S, Kantikosum K, Patiyasikunt M, Sriprasart T, Asawanonda P, Rerknimitr P. A bad sign: dermatomyositis with interstitial lung disease. Am J Med. 2019;132(2):182–186. doi:10.1016/J.AMJMED.2018.09.013
8. Iteen AJ, Bianchi W, Sharman T. Pneumomediastinum. StatPearls; 2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557440/.
9. Bradley JD. Spontaneous pneumomediastinum in adult dermatomyositis. Ann Rheum Dis. 1986;45(9):780. doi:10.1136/ARD.45.9.780
10. Kuroda T, Morikawa H, Satou T, et al. A case of dermatomyositis complicated with pneumomediastinum successfully treated with cyclosporin A. Clin Rheumatol. 2003;22(1):45–48. doi:10.1007/S10067-002-0675-0
11. le Goff B, Chérin P, Cantagrel A, et al. Pneumomediastinum in interstitial lung disease associated with dermatomyositis and polymyositis. Arthritis Rheum. 2009;61(1):108–118. doi:10.1002/ART.24372
12. Selva-O’Callaghan A, Labrador-Horrillo M, Muñoz-Gall X, et al. Polymyositis/dermatomyositis-associated lung disease: analysis of a series of 81 patients. Lupus. 2005;14(7):534–542. doi:10.1191/0961203305lu2158oa
13. Wang W, Wu C, Wang Q. Air in the eyelids! Pneumomediastinum and periorbital emphysema in a patient of dermatomyositis. Rheumatol Immunol Res. 2020;1(1):57. doi:10.2478/RIR-2020-0003
14. Demirdöğen Çetinoğlu E, Dalkılıç E, Erol M, et al. Complication of dermatomyositis: spontaneous pneumomediastinum. Eurasian J Pulmonol. 2016;18:50–54. doi:10.5152/ejp.2015.65265
15. Yashiro M, Asano T, Sato S, et al. Anti-MDA5-positive dermatomyositis with progressive ILD Fukushima. J Med Sci. 2018;64(2):115.
16. Ramón de Dios J, López de Goikoetxea AJ, Carlos Vesga J, Tomás L, Zorrilla V, Luis Lobo J. Pneumomediastinum and diffuse alveolar pain. Severe interstitial pneumopathy due to dermatomyositis. Reumatología Clínica. 2009;5(2):76–79. doi:10.1016/S2173-5743(09)70095-4
17. Cozzani E, Cinotti E, Felletti R, Pelucco D, Rebora A, Parodi A. Amyopathic dermatomyositis with lung involvement responsive to mycophenolate mofetil. Immunopharmacol Immunotoxicol. 2013;35(6):687–692. doi:10.3109/08923973.2013.833624
18. Hwang J, Achamallah N. Holy Macklin! Severe pneumomediastinum associated with anti-MDA-5 antibody dermatomyositis. CHEST. 2020;158(4):A1156. doi:10.1016/J.CHEST.2020.08.1054
19. Mohan V, Cherian S. Spontaneous pneumomediastinum in dermatomyositis. Am J Med Sci. 2017;354(6):e11. doi:10.1016/J.AMJMS.2017.06.027
20. Shimamoto K, Ozaki Y, Amuro H, et al. Three cases of polymyositis/dermatomyositis complicated by pneumomediastinum. Jpn J Clin Immunol. 2008;31(1):56–61. doi:10.2177/jsci.31.56
21. Sifuentes-Giraldo WA, García-Villanueva MJ, Gorospe L. Spontaneous pneumomediastinum complicating interstitial lung disease, associated with clinically amyopathic dermatomyositis and positive anti-MDA5 antibodies. Revista Colombiana de Reumatología. 2017;24(4):259–264. doi:10.1016/J.RCREUE.2018.04.001
22. Kwon M. Spontaneous pneumomediastinum and cutaneous ulcers complicated in a patient with dermatomyositis and interstitial lung disease. Archiv Rheumatol. 2021;37:136–138. doi:10.46497/ARCHRHEUMATOL.2021.8719
23. Kono H, Inokuma S, Nakayama H, Suzuki M. Pneumomediastinum in dermatomyositis: association with cutaneous vasculopathy. Ann Rheum Dis. 2000;59(5):372–376. doi:10.1136/ARD.59.5.372
24. Pallo PAO, Shinjo SK. Spontaneous pneumomediastinum in dermatomyositis: a case series and literature review. Med Express. 2018;5. doi:10.5935/MEDICALEXPRESS.2018.MO009
25. de Souza Neves F, Shinjo SK, Carvalho JF, Levy-Neto M, Borges CTL. Spontaneous pneumomediastinum and dermatomyositis may be a not so rare association: report of a case and review of the literature. Clin Rheumatol. 2007;26(1):105–107. doi:10.1007/S10067-005-0109-X/TABLES/1
26. Yoshida K, Kurosaka D, Kingetsu I, Hirai K, Yamada A. Pneumomediastinum in dermatomyositis itself is not a poor prognostic factor: report of a case and review of the literature. Rheumatol Int. 2008;28(9):913–917. doi:10.1007/S00296-008-0548-1/TABLES/1
27. Jain A, Misra D, Jain V, Negi V. Spontaneous pneumomediastinum in dermatomyositis. Indian J Rheumatol. 2017;12(1):54. doi:10.4103/0973-3698.199129
28. Gürün Kaya A, Çiledağ A, Küçükşahin O, Özdemir Kumbasar Ö, Atasoy Ç. A case of amyopathic dermatomyositis with pneumomediastinum and subcutaneous emphysema. Case Rep Rheumatol. 2015;2015:1–4. doi:10.1155/2015/813902
29. Tang R, Millett CR, Green JJ. Amyopathic dermatomyositis complicated by pneumomediastinum. J Clin Aesthet Dermatol. 2013;6(3):40.
30. Korkmaz C, Özkan R, Akay M, Hakan T. Pneumomediastinum and subcutaneous emphysema associated with dermatomyositis. Rheumatology. 2001;40(4):476–478. doi:10.1093/RHEUMATOLOGY/40.4.476
31. Agarwal R, Lahiri D, Biswas A, Mukhopadhyay J, Roy MK. Fatal recurrence of pneumothorax in an adult dermatomyositis. J Assoc Chest Physicians. 2015;3(1):17. doi:10.4103/2320-8775.146846
32. Yamanishi Y, Maeda H, Konishi F, et al. Dermatomyositis associated with rapidly progressive fatal interstitial pneumonitis and pneumomediastinum. Scand J Rheumatol. 1999;28(1):58–61. doi:10.1080/03009749950155805
33. Kim HJ, Hong YK, Yoo WH. Dermatomyositis, complicated with pneumomediastinum, successfully treated with cyclosporine A: a case report and review of literature. Rheumatol Int. 2009;29(9):1101–1104. doi:10.1007/s00296-008-0822-2
34. PubMed. Dermatomyositis with subclinical myositis and spontaneous pneumomediastinum with pneumothorax: case report and review of the literature. Available from: https://pubmed.ncbi.nlm.nih.gov/9844769/.
35. Yeung TW, Cheong KN, Lau YL, Tse KCN. Adolescent-onset anti-MDA5 antibody-positive juvenile dermatomyositis with rapidly progressive interstitial lung disease and spontaneous pneumomediastinum: a case report and literature review. Pediatr Rheumatol. 2021;19(1):1–8. doi:10.1186/S12969-021-00595-1/FIGURES/5
36. Gonçalves TA, Parente DB, Menna Barreto M. Pneumomediastinum and pneumorrhachis as complications of dermatomyositis. J Bras Pneumol. 2021;47(6). doi:10.1055/s-0034-1371528
37. de Vico L, Canaan JP, Lanças SHS, et al. Spontaneous pneumomediastinum in amyopathic dermatomyositis - a rare manifestation. PLoS One. 2019;258. doi:10.5151/SBR2019-258
38. de Toro Santos FJ, Verea-Hernando H, Montero C, Blanco-Aparicio M, Torres Lanzas J, Pombo Felipe F. Chronic pneumomediastinum and subcutaneous emphysema: association with dermatomyositis. Respiration. 1995;62(1):53–56. doi:10.1159/000196390
39. de Andrade RLF, Held ACL, Terreri MT, Chiba SM. Juvenile dermatomyositis and pneumomediastinum: a case of a very rare complication. Pulmonology. 2020;26(5):323–325. doi:10.1016/J.PULMOE.2019.12.002
40. Vincent S, Carmody DE, Mcnicholl J, Chadwick B, Bresnihan G, Fitzgerald MX. Prolonged spontaneous pneumomediastinum in adult dermatomyositis. Ann Rheum Dis. 1987;46(7):566. doi:10.1136/ARD.46.7.566-A
41. Lega JC, Reynaud Q, Belot A, Fabien N, Durieu I, Cottin V. Idiopathic inflammatory myopathies and the lung. Eur Respir Rev. 2015;24(136):216–238. doi:10.1183/16000617.00002015
42. Clinical significance of spontaneous pneumomediastinum in dermatomyositis/polymyositis. Available from: https://www.jrd.or.kr/journal/view.html?uid=796&vmd=Full.
43. Jobling K. A REVIEW OF PNEUMOMEDIASTINUM IN DERMATOMYOSITIS. Dermatol Dermatitis. 2018;2(4):01–02. doi:10.31579/2578-8949/034
44. Lee AYS, Lee C, Brown DA, Suan D. Development of anti-NXP2 dermatomyositis following Comirnaty COVID-19 vaccination. Postgrad Med J. 2022;postgradmedj-2022–141510. doi:10.1136/POSTGRADMEDJ-2022-141510
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.