Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Specific targeting of A54 homing peptide-functionalized dextran-g-poly(lactic-co-glycolic acid) micelles to tumor cells
Authors Situ J, Ye Y, Zhu X, Yu R, You J, Yuan H ![]() , Hu F, Du Y
, Hu F, Du Y ![]()
Received 23 October 2014
Accepted for publication 1 December 2014
Published 17 January 2015 Volume 2015:10(1) Pages 665—675
DOI https://doi.org/10.2147/IJN.S76307
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Jun-Qing Situ,1 Yi-Qing Ye,2 Xiu-Liang Zhu,3 Ri-Sheng Yu,3 Jian You,1 Hong Yuan,1 Fu-Qiang Hu,1 Yong-Zhong Du1
1College of Pharmaceutical Sciences, 2Women’s Hospital, 3Department of Radiology, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, People’s Republic of China
Abstract: The delivery of chemotherapeutics into tumor cells is a fundamental knot for tumor-target therapy to improve the curative effect and avoid side effects. Here, A54 peptide-functionalized poly(lactic-co-glycolic acid)-grafted dextran (A54-Dex-PLGA) was synthesized. The synthesized A54-Dex-PLGA self-assembled to form micelles with a low critical micelle concentration of 16.79 µg·mL-1 and diameter of about 50 nm. With doxorubicin (DOX) base as a model antitumor drug, the drug-encapsulation efficiency of DOX-loaded A54-Dex-PLGA micelles (A54-Dex-PLGA/DOX) reached up to 75%. In vitro DOX release from the A54-Dex-PLGA/DOX was prolonged to 72 hours. The A54-Dex-PLGA micelles presented excellent internalization ability into hepatoma cells (BEL-7402 cell line and HepG2 cell line) in vitro, and the cellular uptake of the micelles by the BEL-7402 cell line was specific, which was demonstrated by the blocking experiment. In vitro antitumor activity studies confirmed that A54-Dex-PLGA/DOX micelles suppressed tumor-cell (BEL-7402 cell) growth more effectively than Dex-PLGA micelles. Furthermore, in vivo biodistribution testing demonstrated that the A54-Dex-PLGA micelles had a higher distribution ability to BEL-7402 tumors than that to HepG2 tumors.
Keywords: homing peptide, polymeric micelles, doxorubicin, tumor-cell targeting
Introduction
Targeted drug-delivery systems have been a research hotspot in recent years, because they can react with targeted lesions and distribute and release drugs in a controlled manner, thus enhancing therapeutic effects and reducing side effects on nonmalignant cells.1–3 Polymeric micelles, which were first proposed as drug carriers by Bader et al in 1984,4 have attracted growing interest as potential carriers for targeted drug delivery. Polymeric micelles usually have hydrophobic cores surrounded by hydrophilic outer shells. The inner hydrophobic core of the polymer has a large capacity to accommodate hydrophobic drugs, while the hydrophilic shell enables the polymeric micelles to be stable in an aqueous environment.5,6 Polymeric micelles with this kind of core–shell structure have attracted considerable interest for the delivery of antitumor drugs to solid tumors, because they can be self-assembled to from amphiphilic block or graft copolymers. Polymeric micelles possess a nano-order size distribution varying from 20 to 100 nm.7,8 Based on their size and surface properties, micelles have many advantages, such as the accumulation in tumors via the passive enhanced-permeability-and-retention (EPR) effect9–11 and the prolongation of circulation time by avoiding recognition and phagocytosis by the reticuloendothelial system.12,13 However, nonspecific cellular targeting and poor cellular internalization of carriers are still far away from people’s expectations for an ideal drug-delivery system for cancer treatment.
Homing peptides as targeting agents for human tumors are important in the development of tumor treatment.14,15 During the last decade, several investigators have successfully used phage-display library methods16,17 to discover cell surface-binding peptides that may be useful for tumor targeting. The advantages of homing peptides include being easily synthesized, having relatively small molecular weight (MW), having relatively low cytotoxicity and immunogenicity, and degrading in vivo to naturally occurring compounds.18,19 Recently, a hepatocarcinoma-binding peptide, AGKGTPSLETTP peptide (A54), was identified from a phage-display random-peptide library by in vivo panning, and it is the most effective peptide specific to the human hepatoma carcinoma cell line BEL-7402 via cell surface marker-mediated endocytosis.20
Hepatocellular carcinoma is one of the deadliest cancers in the world. The anthracycline antitumor drug doxorubicin (DOX) is widely utilized in clinical therapy with hepatoma. The exact mechanism of action of DOX is complex and still somewhat unclear, though it is thought to interact with deoxyribonucleic acid (DNA) by intercalation, including the inhibition of DNA helicases, topoisomerase II, and ribonucleic acid polymerase.21 However, the current treatment of hepatoma with DOX is still far from satisfactory, due to severe side effects, notably dose-limited cardiotoxicity and myelosuppression.22,23 Therefore, a specific targeting strategy to specifically deliver these antitumor agents to diseased sites, thereby keeping them away from normal cells sensitive to the toxic effects of the drug, is urgently needed.
In our early studies, homing peptide-functionalized polyethylene glycolated stearic acid-grafted chitosan micelles were developed. However, chitosan with a positive charge affected the active targeting efficiency of the synthetic micelles.24 Dextran (Dex) is a family of natural polysaccharides that are widely used as polymeric carriers in novel drug-delivery systems. Dex–poly(lactic-co-glycolic acid) (PLGA) has been developed with excellent drug-loading ability for the antitumor agent DOX.25–27 In this study, A54 was used as a homing peptide to synthesize A54-functional Dex-PLGA (A54-Dex-PLGA) for specifically targeting the human hepatocellular carcinoma cell line BEL-7402.28,29 The structure, critical micelle concentration (CMC), micelle size, and morphology of A54-Dex-PLGA micelles were then investigated. Using DOX as a model drug, such characteristics as drug-loading ability, drug-encapsulation efficiency, and in vitro release profiles of the drug-loaded micelles were then evaluated. Moreover, cellular uptake, in vitro antitumor activities, and in vivo distribution of the micelles were further studied. Figure 1 shows the preparation and research approach for A54-Dex-PLGA micelles.
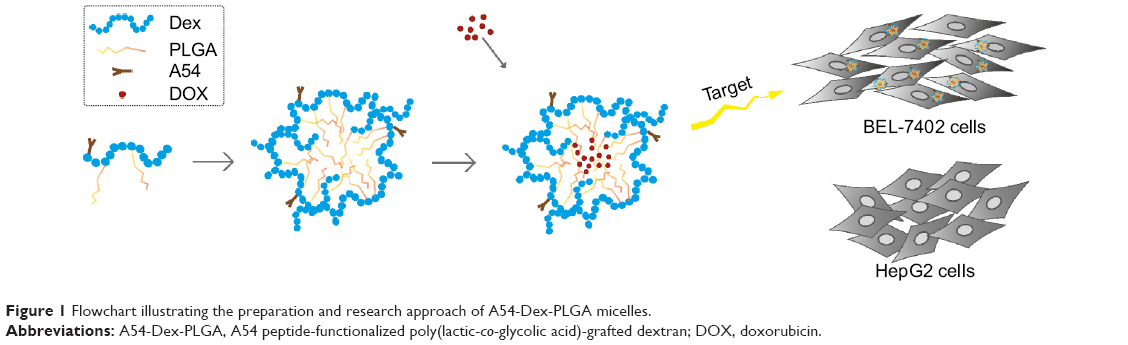 | Figure 1 Flowchart illustrating the preparation and research approach of A54-Dex-PLGA micelles. |
Materials and methods
Materials
Dex of about 10.0 kDa average MW was supplied by Bio Basic (USA). PLGA of about 1.5 kDa MW was purchased from Shandong Daigang Medped (People’s Republic of China [PRC]). 4-Dimethylaminopyridine (DMAP) and N,N′-dicyclohexylcarbodiimide (DCC) were purchased from Shanghai Medped (PRC). A54 was synthesized by Guangzhou Sinoasis Pharmaceuticals (PRC). Di-tert-butyl dicarbonate ([Boc]2O) was purchased from Shanghai Medpep (PRC). Pyrene was purchased from Sigma-Aldrich Chemical (USA). DOX HCl was gifted from Hisun Pharm (PRC). Trypsin and Dulbecco’s Modified Eagle’s Medium (DMEM) were purchased from Gibco (USA). Fetal bovine serum was purchased from Sijiqing Biologic (PRC). Hoechst 33342 was supplied by Invitrogen (USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma-Aldrich Chemical. Indocyanine green (ICG) was purchased from Tokyo Chemical Industry (Japan). The human hepatocellular carcinoma cell line (BEL-7402 cells) and human liver-tumor cell line (HepG2 cells) were donated by the Second Affiliated Hospital of Zhejiang University School of Medicine.
Synthesis of A54-functionalized dextran-g-PLGA
The Dex-PLGA graft was synthesized via an esterification reaction with the dehydrating agent DCC and the catalyst DMAP.30 The reaction was carried out under anhydrous conditions. Briefly, PLGA (3,829.8 mg), DCC (1,579.4 mg), and DMAP (95.5 mg) were dissolved in 15 mL anhydrous dimethyl sulfoxide (DMSO) and stirred for 30 minutes at 60°C to activate the carboxyl group of PLGA. Dex (1,704.1 mg) was then added and reacted for 48 hours. After centrifugation, the supernatant was dialyzed against pure water using a dialysis membrane (MW cutoff [MWCO] 7.0 kDa; Spectrum Laboratories, USA) for 48 hours and then lyophilized. After that, the lyophilized product was further dispersed in acetone (20 mg·mL−1) and then lyophilized.
For synthesis of A54-Dex-PLGA, (Boc)2O (14.0 μL) was added to 5 mL anhydrous DMSO containing A54 (55.0 mg) in an ice bath, followed by stirring with light protection at room temperature for 12 hours. Dex-PLGA (604.0 mg), DCC (28.6 mg), and DMAP (2.0 mg) in 10 mL DMSO were added to the reaction system with stirring for 48 hours at room temperature. The Boc-protecting group was removed by treatment with 2 M HCl. The final reaction product was dialyzed against pure water for 48 hours and then lyophilized.
Preparation of DOX-loaded Dex-PLGA and A54-Dex-PLGA micelles
DOX used for the preparation of DOX-loaded micelles was obtained by reaction of DOX HCl with double-mole triethylamine in DMSO overnight. Polymers (10 mg) and DOX were dissolved in 1 mL DMSO solution (DOX:polymers =5%, w/w) under magnetic stirring at room temperature. After the mixture solution was dialyzed against pure water (MWCO 3.5 kDa) for 24 hours, the products were centrifuged at 4,000 rpm for 10 minutes to obtain the DOX-loaded micelle solution.
Physicochemical characteristics of polymers and micelles
The 1H nuclear magnetic resonance (NMR) spectra of chemicals were obtained by a NMR spectrometer (AC-80; Bruker Biospin, Germany). The solvent used for NMR measurements was DMSO-d6. The PLGA graft ratio for Dex-PLGA (GR%) was calculated using the formula:
GR% = (A3.2/123.4)/(A1.5/34.2) | (1) |
where A3.2 and A1.5 represent the peak areas at chemical shifts at 3.2 and 1.5 ppm in the 1H NMR spectrum of Dex-PLGA, respectively. The chemical shifts at 3.2 ppm and 1.5 ppm corresponded to the protons of glucosidic linkage in glucose unit and methyl of PLGA, respectively. In this study, the synthesized Dex-PLGA using Dex with 10 kDa MW (Dex consisting of 61.7 a-[1→6]-linked d-glucose units) and PLGA with 1.5 kDa MW. PLGA consisted of lactide and glycolide (lactide:glycolide =70:30), and the methyl of PLGA belonged to the glycolide.
The CMC of Dex-PLGA and A54-Dex-PLGA was evaluated by fluorescence measurement using pyrene as a probe.31 Pyrene was firstly dissolved in acetone for quantitation (1.2×10−3 mg·mL−1). After the acetone was evaporated at 50°C, 5 mL of polymer solution with different concentrations ranging from 10−3 to 1.0 mg·mL−1 were added. The excitation wavelength was set at 337 nm, the excitation slit at 10 nm, and the emission slit at 2.5 nm. The intensities of the emission at a wavelength range of 300–470 nm were recorded by fluorescence spectrophotometry (F-2500; Hitachi, Japan), and the intensity ratio of the first peak (I1, 374 nm) to the third peak (I3, 385 nm) was analyzed for determination of CMC.
The morphological examinations were performed by transmission electron microscopy (TEM; JEM-1230; JEOL, Japan). The samples were dropped on copper grids and stained with 2% (w/v) phosphotungstic acid for viewing.
Determination of drug-encapsulation efficiency and drug loading
The DOX content was determined using fluorescence spectrophotometry. The excitation wavelength was set at 505 nm, while the emission wavelength was set at 565 nm. The excitation and the emission slit was 5.0 nm. The drug-encapsulation efficiency (EE%) and drug loading (DL%) were calculated using the respective formulas:
EE% = (mass of DOX in micelles/mass of DOX added) ×100% | (2) |
DL% = (mass of DOX in micelles/mass of DOX-loaded micelles) ×100% | (3) |
In vitro DOX release from DOX-loaded micelles
For in vitro drug release tests, a 3 mL DOX-loaded micelle solution was put into a dialysis membrane (MWCO 3.5 kDa) and added to a plastic tube containing 20 mL phosphate-buffered saline (PBS) solution at pH 7.4. The tests were carried out in an incubator shaker (37°C, 70 rpm). At predetermined time intervals (0.5, 1, 2, 4, 6, 8, 12, 24, 36, 48, and 72 hours), all of the media out of the dialysis membrane were withdrawn and replaced with the fresh PBS. DOX content was determined by fluorescence spectrophotometry. All drug-release tests were performed three times.
Cell culture
BEL-7402 and HepG2 cells were maintained in DMEM supplemented with 10% (v/v) fetal bovine serum and penicillin/streptomycin (100 U·mL−1, 100 U·mL−1) at 37°C in a humidified atmosphere containing 5% CO2. Cells were subcultured regularly using trypsin/ethylenediaminetetraacetic acid.
Cellular uptake of DOX-loaded micelles
For cellular uptake of DOX-loaded micelles, BEL-7402 and HepG2 cells were seeded at 3.0×104 mL−1 cells/well onto 10 mm glass coverslips in a 24-well plate (Nalge Nunc International, USA) and incubated for 24 hours. DOX-loaded micelles (100 μL, 5 μg·mL−1) were added, followed by further incubation for 0.5, 1, 2, 4, 10, and 24 hours. For the blocking experiments, cells were incubated firstly with free A54 (10−2 mg·mL−1) for 30 minutes and then with DOX-loaded micelles. After the cells were incubated with Hoechst 33342 (10−2 mg·mL−1) for 30 minutes, cellular fluorescence was examined under confocal microscopy (BX61W1-FV1000; Olympus, Japan). For the quantitative analysis of cellular uptake, cells were treated with trypsin after different incubation periods with DOX-loaded micelles, and then resuspended in PBS. The intensity of cellular fluorescence was determined by flow cytometry (FC 500 MCL; Beckman Coulter, USA), and blanked by untreated cells.
In vitro antitumor activity
The cytotoxicity of DOX HCl, blank, and DOX-loaded micelles against BEL-7402 cells was evaluated by MTT assay. Briefly, 1.0×104 cells per well were placed in a 96-well microtiter plate (Nalge Nunc International) and incubated for 24 hours. Then, the cells were exposed to a series of concentration of micelles for another 48 hours. After that, cells were incubated with 20 μL MTT solution (5 mg·mL−1) for a further 4 hours at 37°C. Finally, the absorbance was determined at 570 nm using an automatic reader (model 680; Bio-Rad, USA). All the experiments were repeated three times.
A cytotoxicity assay was then used to confirm the results obtained from the colorimetric cell-viability assay.32,33 The drug concentration of DOX HCl and DOX-loaded micelles were same as the half-maximal inhibitory concentration (IC50) value of DOX-loaded A54-Dex-PLGA micelles (A54-Dex-PLGA/DOX). After a 24-hour drug exposure, cells were stained with 100 μL calcein acetoxymethyl ester (AM; Invitrogen, USA) (0.5 μL dissolved in 1 mL PBS) for 30 minutes at room temperature. Cells were observed using a fluorescence microscope (DM4000 B; Leica, Germany).
In vivo imaging
The tumor-bearing mouse models were established by subcutaneous injection of BEL-7402 and HepG2 cells into the two flanks of BALB/C + nu/F1 nude mice, respectively. To investigate the in vivo distribution, ICG complexed with tetrabutylammonium iodide was encapsulated by A54-Dex-PLGA micelles. When tumors reached an acceptable size, ICG-loaded micelles were injected into the tail vein of the tumor-bearing mice at a dose of 5.0 mg/kg. The mice were anesthetized and imaged at the predetermined time (1, 3, 5, 12, 24, and 48 hours) after injection using the Maestro in vivo imaging system (CRI, USA). At the end of the experiment, various tissues including tumors were harvested, weighed, and observed by the in vivo imaging system. The accumulation of ICG-A54-Dex-PLGA micelles in various tissues was calculated as %ID/g (the percentage of the fluorescent intensity per gram of tissue). All the animal studies were performed under Institutional Animal Care and Use Committee-approved protocols.
Results and discussion
Synthesis and characteristics of A54-Dex-PLGA
The synthesis route of A54-Dex-PLGA is presented in Figure 2. Dex-PLGA was firstly synthesized via an esterification reaction between the carboxyl group of PLGA and the hydroxyl group of Dex in the presence of the catalyst DMAP and the dehydrating agent DCC. The MW of dextran was 10.0 kDa, and the molar ratio of Dex and PLGA was 1:15. The chemical structures of Dex, PLGA, and synthesized Dex-PLGA were confirmed by the 1H NMR spectrum, as shown in Figure 3A. The 1H NMR spectrum of Dex-PLGA showed peaks at about 3–4 ppm, which were attributed to Dex, and a peak at about 1.5 ppm, which was the chemical shift of the proton of −CH3 for PLGA. There were no chemical shifts to signify the proton of −COOH for PLGA (about 13.0 ppm). These results indicated that Dex-PLGA was synthesized successfully. The PLGA graft ratio for Dex-PLGA (GR%) was 1:2.42, hence there were about two PLGA grafted for each Dex.
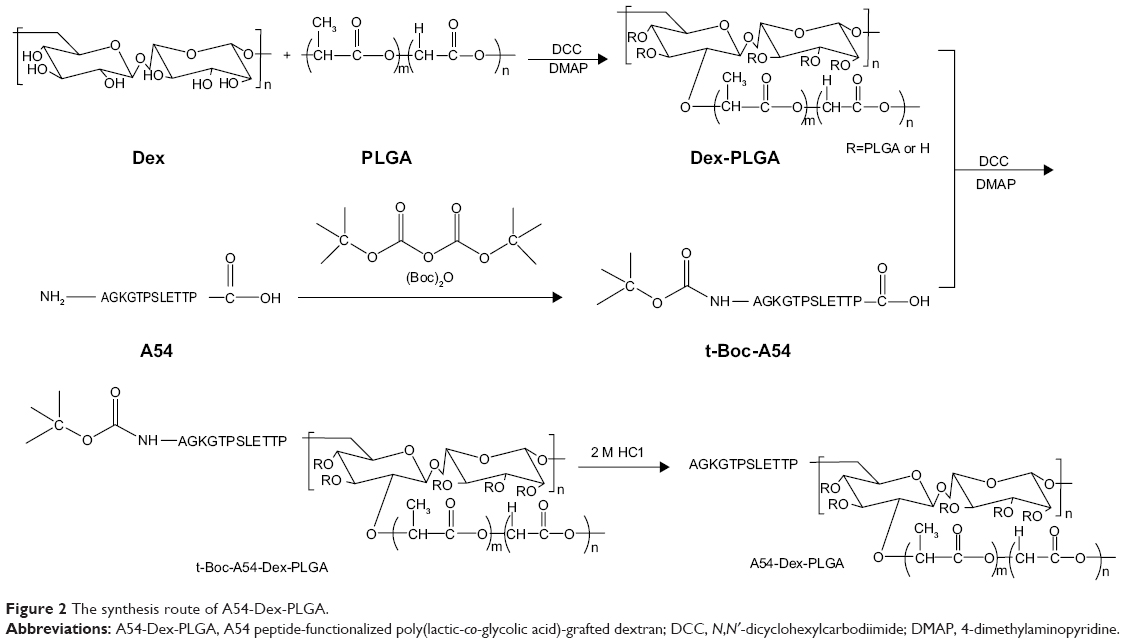 | Figure 2 The synthesis route of A54-Dex-PLGA. |
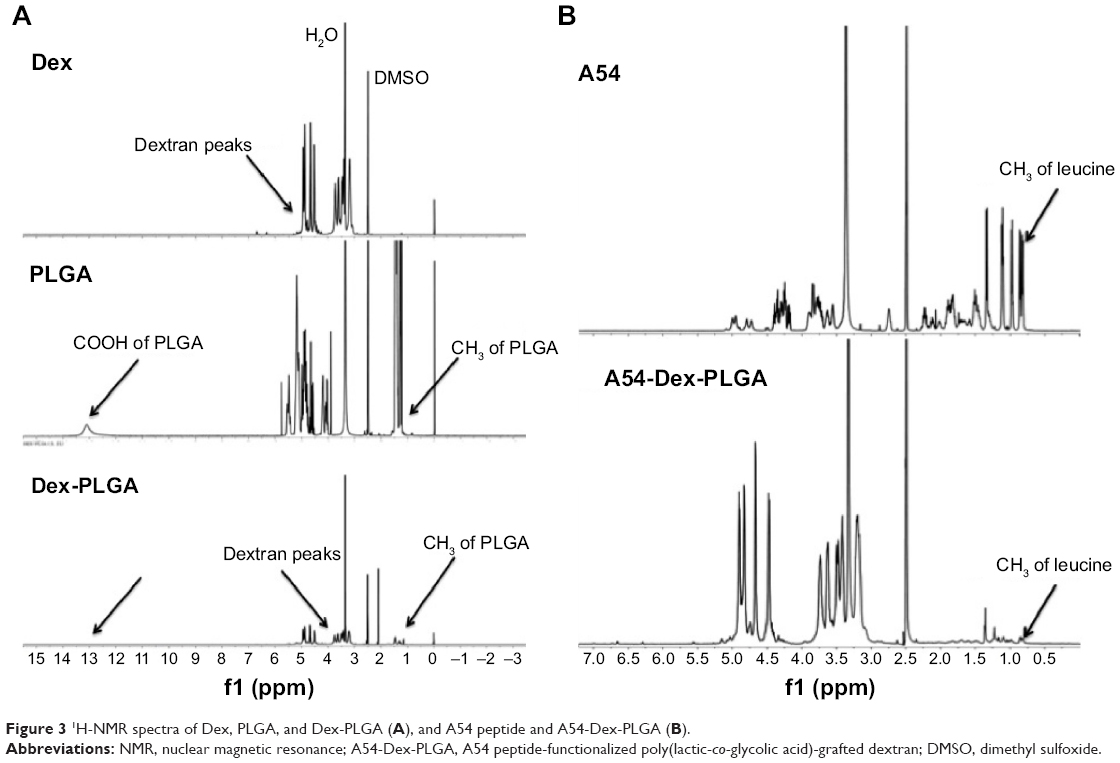 | Figure 3 1H-NMR spectra of Dex, PLGA, and Dex-PLGA (A), and A54 peptide and A54-Dex-PLGA (B). |
The t-Boc-A54 was then prepared by the chemical reaction between A54 and (Boc)2O (to protect the amino group of A54). The t-Boc-A54 then reacted with the remaining hydroxyl group of Dex-PLGA in the presence of the catalyst DMAP and the dehydrating agent DCC to obtain t-Boc-A54-Dex-PLGA. The Boc-protecting group was finally removed by treatment with 2 M HCl to gain A54-Dex-PLGA. Compared with A54, the 1H NMR spectrum of the A54-Dex-PLGA in Figure 3B shows a similar doublet signal at 0.75–0.81 ppm, which was the chemical shift of the proton of −CH3 for leucine of A54. These results indicated the successful synthesis of A54-Dex-PLGA.
Dex provides the hydrophilic group, which makes the conjugate molecule tend toward water, and it was found that the synthesized Dex-PLGA and A54-Dex-PLGA self-assembled to form polymeric micelles in an aqueous solution with low CMC. Figure 4A was plotted to find an apparent turning point. The CMC values of Dex-PLGA and A54-Dex-PLGA were 10.27 μg·mL−1 and 16.79 μg·mL−1 respectively, which were far less than the general surfactant.34
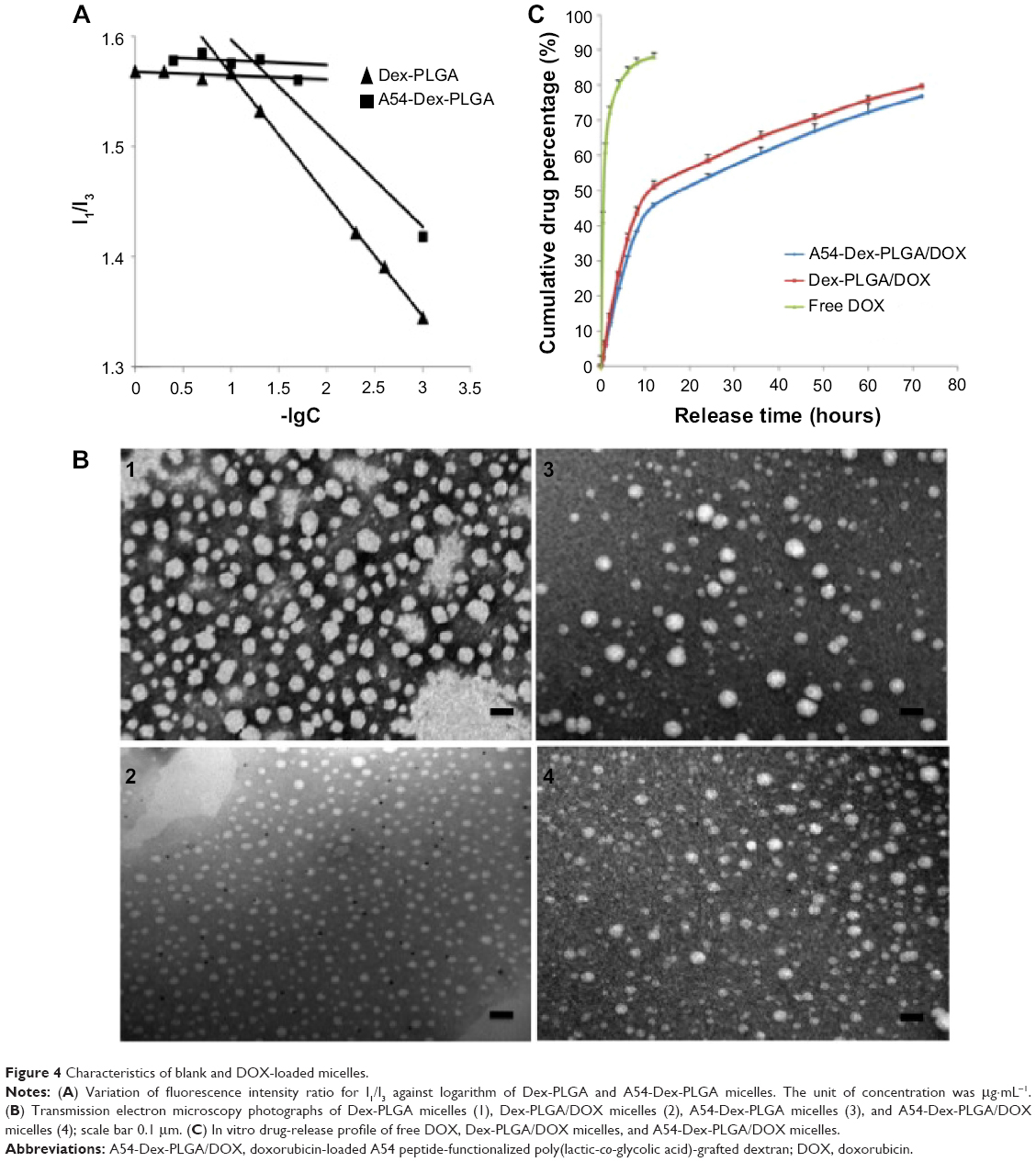 | Figure 4 Characteristics of blank and DOX-loaded micelles. |
Preparation and characteristics of blank and DOX-loaded micelles
DOX-loaded micelles were then prepared successfully by dialysis. Morphological observation of blank and DOX-loaded micelles was performed by TEM. The TEM images of formed micelles are presented in Figure 4B, which confirmed that self-assembling micelles were unvarying spherical shapes, and the average diameters of Dex-PLGA/DOX and A54-Dex-PLGA/DOX micelles were about 20 nm and 30 nm, while the blank micelles were around 50 nm. After drug loading, the micelle size observed from TEM images was smaller than that of blank micelles, and the particle-size distribution became uniform. This may have been due to the hydrophobic interaction between the hydrophobic core and drug becoming stronger after DOX loading, which made the micelles more stable.
When the drug-feeding amount was 5.00%, the DL% of DOX-loaded Dex-PLGA and A54-Dex-PLGA micelles was around 3.59%. Considering the drug loss in the preparation process, the EE% of DOX-loaded Dex-PLGA and A54-Dex-PLGA micelles was found to be 75.37% and 75.31%, respectively, which implies that there were no obvious influences for drug-loading ability after A54 modification. In vitro drug release from DOX-loaded micelles was carried out using pH 7.4 PBS as the dissolution medium. As shown in Figure 4C, the release profiles of DOX from A54-Dex-PLGA and Dex-PLGA micelles were mostly similar, which implied that the modification of A54 had almost no effect on drug-release behaviors. The curves reveal a representative two-phase pattern consisting of a premier fast release in 12 hours followed by sustained release for a prolonged time. About 80% of the drug had been released after 72 hours.
Cellular uptake studies of DOX-loaded micelles
To investigate the targeting ability of A54-Dex-PLGA micelles, cellular uptake of A54-Dex-PLGA micelles on both BEL-7402 and HepG2 cells was observed by confocal microscopy. Figure 5A shows the fluorescence images after BEL-7402 and HepG2 cells were incubated with A54-Dex-PLGA/DOX micelles for 0.5, 4, and 10 hours. It was found that the antitumor agent DOX internalized into tumor cells indirectly by the micelles and the uptake was time-dependent. After 4 hours’ incubation, significantly more A54-Dex-PLGA/DOX micelles were internalized in BEL-7402 cells than HepG2 cells, and the uptake of DOX-loaded micelles was saturated after 10 hours’ incubation on both BEL-7402 and HepG2 cells, which was further confirmed by ImageJ analysis, as shown in Figure 5B.
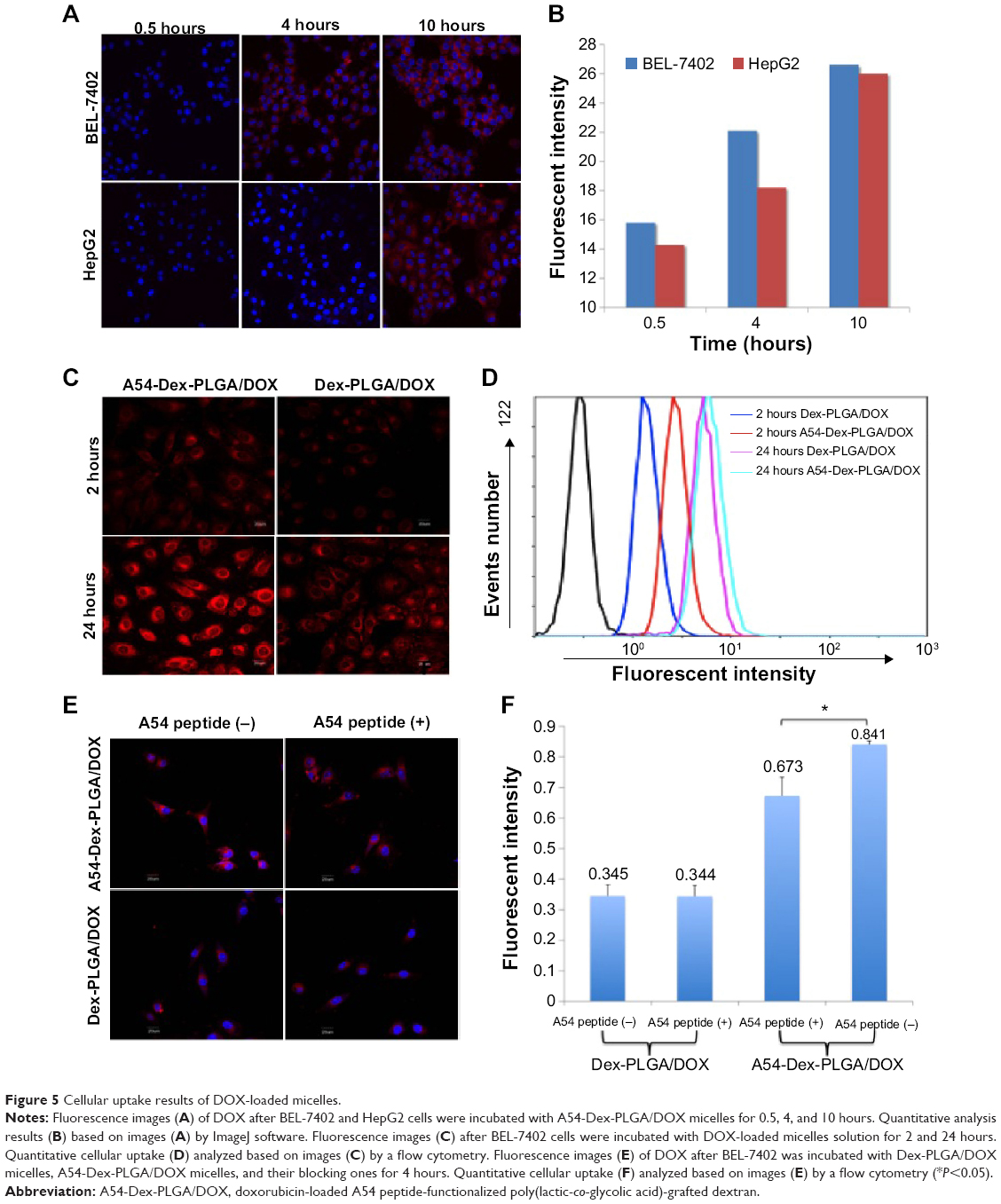 | Figure 5 Cellular uptake results of DOX-loaded micelles. |
Confocal microscopy was also used to confirm the accumulation of Dex-PLGA/DOX and A54-Dex-PLGA/DOX in tumor cells by taking advantage of the intrinsic fluorescence of DOX. Figure 5C presents images of BEL-7402 cells after incubation with Dex-PLGA/DOX micelles and A54-Dex-PLGA/DOX micelles for 2 and 24 hours. A54-Dex-PLGA/DOX-treated cells showed accumulation of DOX in both the cytoplasm and the nuclei, while in contrast Dex-PLGA/DOX merely exhibited accumulation in the cytoplasm. It was an ideal outcome that DOX accumulated in the cell nuclei, as one of the described mechanisms of DOX depends on its intercalation with DNA with subsequent inhibition of macromolecular biosynthesis.21 In addition, a stronger cellular fluorescent signal was observed in BEL-7402 cells treated with A54-Dex-PLGA/DOX micelles for 2 or 24 hours because of the attached A54, which was further confirmed by the quantitative cellular uptake analyzed by a flow cytometry, as shown in Figure 5D.
To confirm the better binding ability of the A54-Dex-PLGA micelles to BEL-7402 cells was due to the A54 modification, the cellular uptake of Dex-PLGA/DOX micelles, A54-Dex-PLGA/DOX micelles, and A54-blocking ones against BEL-7402 cells was then investigated. In Figure 5E, A54-Dex-PLGA/DOX micelles indicated the best uptake on BEL-7402 cells, and the addition of free A54 into the culture medium induced a notable decrease in the cellular uptake of the micelles, yet there were no significant differences between Dex-PLGA/DOX micelles and the A54-blocking ones, which was further confirmed by flow-cytometry analysis, as shown in Figure 5F.
The cellular uptake data confirmed specific binding activity of A54-Dex-PLGA micelles to BEL-7402 cells, due to the presence of an abundant cell-surface marker that may have high expression on BEL-7402 cells then taken up by cells via receptor-mediated endocytosis. These results established that the A54-Dex-PLGA micelles retained their A54-combining capacity and specificity to BEL-7402 cells, and may be utilized as a prospective active-targeting carrier via A54 modification.35,36
In vitro antitumor activity of blank and DOX-loaded micelles
Cytotoxicities of DOX HCl, blank, and DOX-loaded micelles were evaluated using BEL-7402 as model tumor cells. As shown in Figure 6A, when the concentration of blank micelles was 1 mg·mL−1, cell viability was still higher than 80%, which indicated the blank micelles had relatively low cytotoxicity and high biocompatibility. Minimal cytotoxic difference between the A54-Dex-PLGA micelles and Dex-PLGA micelles was observed, thus the modification of A54 had almost no influence on cytotoxicity. The IC50 values of the DOX HCl solution and DOX-loaded micelles against BEL-7402 within 48 hours were determined using the MTT method. Figure 6B indicates the IC50 value of A54-Dex-PLGA/DOX was 0.570 μg·mL−1, which showed desired cytotoxicities against BEL-7402 cells compared with that of DOX HCl solution (0.252 μg·mL−1). The lower cytotoxicity might have been due to the determination time of the in vitro cytotoxicity test, as the DOX could not be released completely from the micelles inside the cells within 48 hours. However, the higher cytotoxicity of A54-Dex-PLGA/DOX was tested on BEL-7402 cells compared with that of Dex-PLGA/DOX (1.579 μg·mL−1), which suggested that A54-Dex-PLGA had a high targeting activity to the BEL-7402 cells because of the attached A54, and thus could significantly increase the cytotoxicity of DOX-loaded micelles.
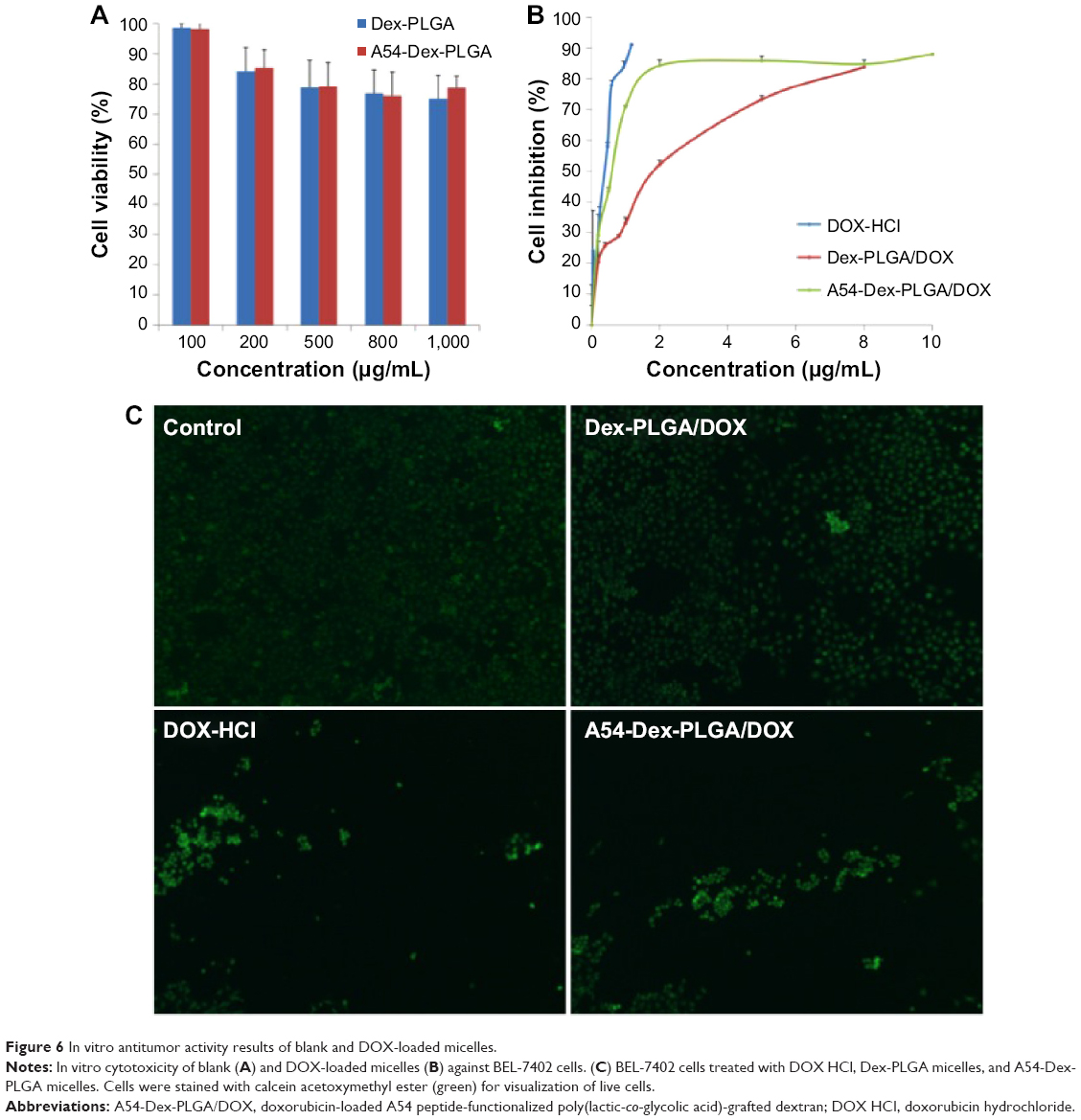 | Figure 6 In vitro antitumor activity results of blank and DOX-loaded micelles. |
Figure 6C illustrates the intuitional cytotoxic results of DOX HCl and DOX-loaded micelles utilizing the calcein AM stain with BEL-7402 cells for visualization of live cells. Significantly fewer BEL-7402 cells were visible (stained green by calcein AM) due to the treatment of A54-Dex-PLGA/DOX micelles compared with Dex-PLGA/DOX micelles, which was attributed to more A54-Dex-PLGA micelles being internalized into BEL-7402 cells after 24 hours’ incubation, and A54-Dex-PLGA/DOX micelles demonstrated similar cytotoxicity to DOX HCl, which also confirmed that Dex-PLGA coupled with A54 endowed a specific affinity to corresponding tumor cells in vitro.
In vivo targeting imaging of A54-Dex-PLGA micelles
To investigate whether A54-Dex-PLGA micelles can indeed prolong drug circulation and specifically target BEL-7402 cells in vivo, the tumor model was built by subcutaneous injection of BEL-7402 and HepG2 cells in the left and right sides of the mice. As shown in Figure 7A, mice treated with ICG/A54-Dex-PLGA micelles showed fluorescence signals throughout the whole body for up to 5 hours postinjection, which indicated the long circulation time in the blood. Thereafter, fluorescence signals at the two tumor sites were clearly observed and maintained for 48 hours. Also, significantly more A54-Dex-PLGA micelles accumulated in BEL-7402 tumors with positive A54 expression than HepG2 tumors with negative A54 expression during the experimental process. The fluorescence intensity of various tissues and the dissected tumors was observed and quantitated, which confirmed the major accumulation of ICG/A54-Dex-PLGA micelles in BEL-7402 tumors and little accumulation in various tissues at 48 hours postinjection (Figure 7, B and C). Therefore, with the modification of A54, the A54-Dex-PLGA micelles were efficiently delivered and retained by the BEL-7402 tumors under in vivo conditions.
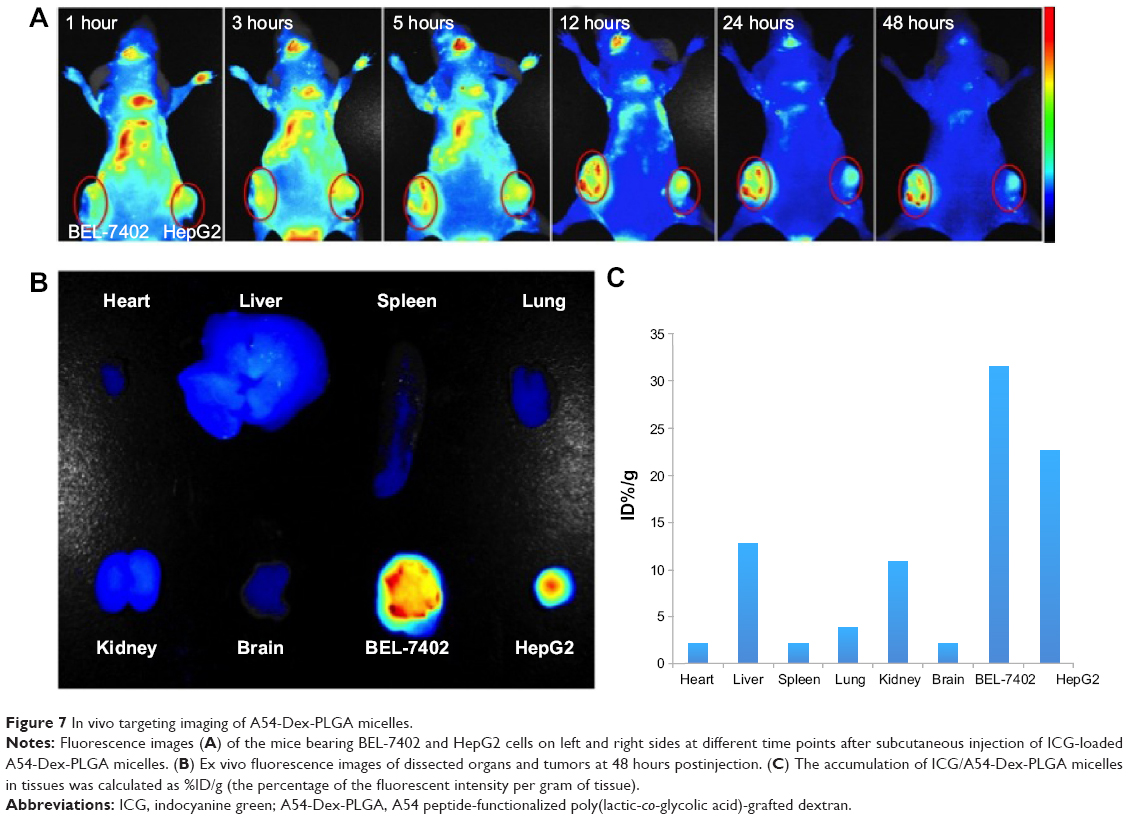 | Figure 7 In vivo targeting imaging of A54-Dex-PLGA micelles. |
Conclusion
In this work, an A54-Dex-PLGA graft was designed and synthesized successfully, and it was easy to form polymeric micelles in aqueous solution for the delivery of DOX. Above all, this could be attributed to the triple effectiveness of rapid uptake, long circulation, and biomolecular targeting of A54-Dex-PLGA. The micelles were effectively transported to the tumor tissue through the EPR effect, and the targeting A54 enhanced endocytosis of the micelles. The functional A54-Dex-PLGA possessed the advantages of the Dex-PLGA chain and biomolecular targeting A54, so that the micelles were targeted to the tumor tissue more efficiently than Dex-PLGA micelles. All these results suggest that the A54-Dex-PLGA micelles are a potential candidate for active targeted drug delivery.
Acknowledgments
This work was supported by the National Nature Science Foundation of China under contracts 81373345, 81101661 and 81171334, and the Nature Science Foundation of Zhejiang Province under contracts LZ13H300001 and Y14H180018.
Disclosure
The authors report no conflicts of interest in this work.
References
Bottini M, Sacchetti C, Pietroiusti A, et al. Targeted nanodrugs for cancer therapy: prospects and challenges. J Nanosci Nanotechnol. 2014;14(1):98–114. | ||
Shi J, Wang Z, Wang L, et al. Photodynamic therapy of a 2-methoxyestradiol tumor-targeting drug delivery system mediated by Asn-Gly-Arg in breast cancer. Int J Nanomedicine. 2013;8:1551–1562. | ||
Zhang X, Zhang X, Yu P, Han Y, Li Y, Li C. Hydrotropic polymeric mixed micelles based on functional hyperbranched polyglycerol copolymers as hepatoma-targeting drug delivery system. J Pharm Sci. 2013;102(1):145–153. | ||
Bader H, Ringsdorf H, Schmidt B. Water-soluble polymers in medicine. Angew Makromol Chem. 1984;123(124):457–485. | ||
Yoo HS, Lee EA, Park TG. Doxorubicin-conjugated biodegradable polymeric micelles having acid-cleavable linkages. J Control Release. 2002;82(1):17–27. | ||
Discher DE, Eisenberg A. Polymer vesicles. Science. 2002;297(5583):967–973. | ||
Tan C, Wang Y, Fan W. Exploring polymeric micelles for improved delivery of anticancer agents: recent developments in preclinical studies. Pharmaceutics. 2013;5(1):201–219. | ||
Miura Y, Takenaka T, Toh K, et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood–brain tumor barrier. ACS Nano. 2013;7(10):8583–8592. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. | ||
Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. | ||
Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92(7):1343–1355. | ||
Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009;100(4):572–579. | ||
Aina OH, Sroka TC, Chen ML, Lam KS. Therapeutic cancer targeting peptides. Biopolymers. 2002;66(3):184–199. | ||
Hirsjarvi S, Belloche C, Hindré F, Garcion E, Benoit JP. Tumour targeting of lipid nanocapsules grafted with cRGD peptides. Eur J Pharm Biopharm. 2014;87(1):152–159. | ||
Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380(6572):364–366. | ||
Trepel M, Arap W, Pasqualini R. In vivo phage display and vascular heterogeneity: implications for targeted medicine. Curr Opin Chem Biol. 2002;6(3):399–404. | ||
Lo SL, Wang S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials. 2008;29(15):2408–2414. | ||
Pang HB, Braun GB, She ZG, et al. A free cysteine prolongs the half-life of a homing peptide and improves its tumor-penetrating activity. J Control Release. 2014;175:48–53. | ||
Du B, Han H, Wang Z, et al. Targeted drug delivery to hepatocarcinoma in vivo by phage-displayed specific binding peptide. Mol Cancer Res. 2010;8(2):135–144. | ||
Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in Mcf-7 breast-tumor cells. Mol Pharmacol. 1994;45(4):649–656. | ||
Barry E, Alvarez JA, Scully RE, Miller TL, Lipshultz SE. Anthracycline-induced cardiotoxicity: course, pathophysiology, prevention and management. Expert Opin Pharmacother. 2007;8(8):1039–1058. | ||
Watts RG, George M, Johnson WH Jr. Pretreatment and routine echocardiogram monitoring during chemotherapy for anthracycline-induced cardiotoxicity rarely identifies significant cardiac dysfunction or alters treatment decisions. Cancer. 2012;118(7):1919–1924. | ||
Du YZ, Cai LL, Liu P, You J, Yuan H, Hu FQ. Tumor cells-specific targeting delivery achieved by A54 peptide functionalized polymeric micelles. Biomaterials. 2012;33(34):8858–8867. | ||
Jeong YI, Choi KC, Song CE. Doxorubicin release from core-shell type nanoparticles of poly(DL-lactide-co-glycolide)-grafted dextran. Arch Pharm Res. 2006;29(8):712–719. | ||
Choi KC, Bang JY, Kim C, et al. Antitumor effect of adriamycin-encapsulated nanoparticles of poly(DL-lactide-co-glycolide)-grafted dextran. J Pharm Sci. 2009;98(6):2104–2112. | ||
Jeong YI, Kim DH, Chung CW, et al. Doxorubicin-incorporated polymeric micelles composed of dextran-b-poly(DL-lactide-co-glycolide) copolymer. Int J Nanomedicine. 2011;6:1415–1427. | ||
Yang Y, Jiang JS, Du B, Gan ZF, Qian M, Zhang P. Preparation and properties of a novel drug delivery system with both magnetic and biomolecular targeting. J Mater Sci Mater Med. 2009;20(1):301–307. | ||
Liu SY, Liang ZS, Gao F, Luo SF, Lu GQ. In vitro photothermal study of gold nanoshells functionalized with small targeting peptides to liver cancer cells. J Mater Sci Mater Med. 2010;21(2):665–674. | ||
Du YZ, Weng Q, Yuan H, Hu FQ. Synthesis and antitumor activity of stearate-g-dextran micelles for intracellular doxorubicin delivery. ACS Nano. 2010;4(11):6894–6902. | ||
Astafieva I, Zhong XF, Eisenberg A. Critical micellization phenomena in block polyelectrolyte solutions. Macromolecules. 1993;26(26):7339–7352. | ||
Wu M, Ye Z, Liu Y, Liu B, Zhao X. Release of hydrophobic anticancer drug from a newly designed self-assembling peptide. Mol Biosyst. 2011;7(6):2040–2047. | ||
You J, Wang Z, Du Y, et al. Specific tumor delivery of paclitaxel using glycolipid-like polymer micelles containing gold nanospheres. Biomaterials. 2013;34(18):4510–4519. | ||
Alawi SM, Akhter MS. Effect of N,N-dimethyl acetamide on the critical micelle concentration of aqueous solutions of sodium surfactants. J Mol Liq. 2011;160(2):63–66. | ||
Gan ZF, Jiang JS, Yang Y, Du B, Qian M, Zhang P. Immobilization of homing peptide on magnetite nanoparticles and its specificity in vitro. J Biomed Mater Res A. 2008;84(1):10–18. | ||
Jiang JS, Gan ZF, Yang Y, Du B, Qian M, Zhang P. A novel magnetic fluid based on starch-coated magnetite nanoparticles functionalized with homing peptide. J Nanopart Res. 2009;11(6):1321–1330. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.