Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Spatial Patterns of Amyloid Deposition in Patients with Chronic Focal or Diffuse Traumatic Brain Injury Using 18F-FPYBF-2 PET
Authors Ubukata S, Oishi N ![]() , Higashi T
, Higashi T ![]() , Kagawa S
, Kagawa S ![]() , Yamauchi H, Okuyama C
, Yamauchi H, Okuyama C ![]() , Watanabe H, Ono M, Saji H, Aso T
, Watanabe H, Ono M, Saji H, Aso T ![]() , Murai T, Ueda K
, Murai T, Ueda K
Received 1 July 2020
Accepted for publication 12 October 2020
Published 12 November 2020 Volume 2020:16 Pages 2719—2732
DOI https://doi.org/10.2147/NDT.S268504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Taro Kishi
Shiho Ubukata,1,2 Naoya Oishi,2 Tatsuya Higashi,3,4 Shinya Kagawa,3 Hiroshi Yamauchi,3 Chio Okuyama,3 Hiroyuki Watanabe,5 Masahiro Ono,5 Hideo Saji,5 Toshihiko Aso,1 Toshiya Murai,1 Keita Ueda1
1Department of Psychiatry, Graduate School of Medicine, Kyoto University, Kyoto, Japan; 2Medical Innovation Center, Graduate School of Medicine, Kyoto University, Kyoto, Japan; 3Shiga Medical Center Research Institute, Moriyama, Japan; 4Department of Molecular Imaging and Theranostics, National Institute of Radiological Sciences (NIRS), National Institutes for Quantum and Radiological Science and Technology (QST), Chiba, Japan; 5Department of Patho-Functional Bioanalysis, Graduate School of Pharmaceutical Sciences, Kyoto University, Kyoto, Japan
Correspondence: Shiho Ubukata
Department of Psychiatry, Graduate School of Medicine, Kyoto University, 54 Kawaharacho, Shogoin, Sakyo, Kyoto 606-8507, Japan
Tel +81-75-751-4947
Email [email protected]
Aim: Amyloid-β (Aβ) accumulation, accelerated by traumatic brain injury (TBI), may play a crucial role in neurodegeneration in chronic-stage TBI. The injury type could influence Aβ dynamics because of TBI’s complex, heterogeneous nature. We, therefore, investigated spatial patterns of amyloid deposition according to injury type after TBI using 5-(5-(2-(2-(2-[F]-fluoroethoxy)ethoxy)ethoxy)benzofuran-2-yl)-N-methylpyridin-2-amine (18F-FPYBF-2) positron emission tomography (PET).
Methods: Altogether, 20 patients with chronic TBI [12 with focal injury, 8 with diffuse axonal injury (DAI)] underwent 18F-FPYBF-2 PET, structural magnetic resonance imaging (MRI), and neuropsychological examination. Additionally, 50 healthy controls underwent either 18F-FPYBF-2 PET (n=30) or structural MRI (n=20).
Results: Standardized uptake value ratio (SUVR) on PET images and regional brain volumes were measured in four cortical (frontal, parietal, occipital, temporal) and subcortical (combined caudate, putamen, pallidum, thalamus) regions. Patients with DAI showed significantly increased (compared with controls) SUVR in occipital and temporal cortices and decreased brain volume in occipital cortex (corrected p < 0.05). Although patients with focal injury showed decreased SUVR in all regions except occipital cortex, there were no significant differences (compared with controls) in the SUVR in any regions. There were no significant correlations between increased SUVR and neuropsychological impairments in patients with DAI.
Conclusion: Varying spatial patterns of amyloid deposition suggest amyloid pathology diversity depending on the injury type in chronic-TBI patients.
Keywords: amyloid deposition, chronic, diffuse axonal injury, focal injury, PET
Introduction
There is growing evidence that amyloid-β (Aβ) accumulation is accelerated by traumatic brain injury (TBI). Experimental traumatic brain injury in mice induces rapid Aβ production and acute accumulation after TBI.1 Aβ plaques are pathological indicators of neurodegenerative diseases, such as Alzheimer’s disease (AD). Although it is still unclear which mechanisms lead to Aβ accumulation in TBI, vascular shear stress by an external force can induce acute blood–brain barrier disruption, which contributes to both ischemic damage and Aβ accumulation.2,3 In its chronic stage, cerebrovascular dysfunction, including altered fluctuations in cerebral blood flow and disrupted clearance, may play a causal role in neurodegeneration after TBI.2,4 Aβ is thought to be a potential therapeutic target of TBI. An experimental murine study showed that blocking β- or γ-secretase (required for Aβ production) can alleviate cognitive deficits after TBI.5 Thus, reliable imaging is crucial for targeting Aβ as a biomarker of chronic TBI.
Aβ aggregates are found at autopsy in approximately 30% of patients with TBI during both acute and chronic stages.6,7 Positron emission tomography (PET) can localize TBI-related amyloid pathology in vivo using several tracers (eg, carbon 11- and fluorine 18-labeled PET ligands). The uptake of amyloid tracer 11C-Pittsburgh compound B 11C-PiB increases in all cortical gray matter and striatum in acute and chronic stages and in the posterior cingulate cortex in the chronic stage.8,9 Other studies using 18F-florbetapir have shown increased uptake in the frontal, temporal, and parietal cortices, striatum, and hippocampus in the chronic stage following TBI10,11. Sites of amyloid pathology are diverse in TBI populations because brain damage is usually spatially heterogeneous, depending on the injury type and its severity and the location of the impact.12
A common categorization of pathology resulting from TBI distinguishes focal and diffuse axonal injury.13 Focal injury includes parenchymal contusions on the brain surface, intracranial hematomas (caused by, for example, direct impact to the head), and the various brain insults secondary to intracranially expanding lesions or high intracranial pressure.13 Diffuse axonal injury (DAI) is characterized by diffuse white matter disruption caused by strong accelerating or decelerating forces on the brain.14 DAI is one of the most frequently found pathologies following TBI.13 Focal injury and DAI may occur in a single individual after TBI. Previous PET studies have investigated amyloid deposition for mixed-type TBI.8,9,15 Coexistence of multiple injury types causes further difficulty investigating TBI-related amyloid pathology. A previous microdialysis study reported elevated brain interstitial Aβ proteins in patients with acute-stage DAI compared with those following focal injuries,16 suggesting that the type of injury could be an important influence on Aβ dynamics.17
This study aimed to investigate the spatial patterns of amyloid deposition in patients with chronic TBI according to their injury—whether focal or DAI. Unlike previous studies, we grouped patients by their injury type to investigate the effects of either pure DAI or focal injury on amyloid pathology. We applied amyloid PET imaging using a new PET tracer for amyloid imaging: 5-(5-(2-(2-(2-18F fluoroethoxy)ethoxy)ethoxy)benzofuran-2-yl)-N-methylpyridin-2-amine (18F-FPYBF−2). In in vitro studies and autoradiography ex vivo studies, 18F-FPYBF-2 has been reported to display a high affinity for Aβ (1–42) aggregates, with a higher binding affinity than 11C-PiB in the same assay (Ki = 2.41 and 9.00 nM, respectively). It also exhibits selective binding of Aβ plaques, with little nonspecific binding in the brain. Furthermore, 18F-FPYBF-2 has been demonstrated to clearly stain many Aβ plaques in AD brains.18 The clinical diagnostic ability compared to 11C-PiB has reported in our previous study.18–20 We also conducted brain volumetric analysis via T1-weighted magnetic resonance imaging (MRI) to measure the degree of structural alteration. To investigate the effects of amyloid pathology regarding neuropsychological sequelae and outcomes, we examined the relation between amyloid deposition and neuropsychological impairment and social function.
Materials and Methods
Participants
Participants were recruited from the outpatient clinic of the neuropsychology unit at the Department of Psychiatry and at the Department of Neurosurgery, Kyoto University Hospital. Inclusion criteria were (1) brain injury sustained through significant trauma; (2) brain MRI or computed tomography (CT) showing a specific lesion or possible diffuse pathology; (3) age >18 years; (4) the injury occurred ≥6 months before the study; (5) ability to give informed consent for participation; (6) ability to undergo MRI and PET. Exclusion criteria were (1) history of another TBI with altered consciousness; (2) history of drug abuse; (3) history of neurological or psychiatric disorder before TBI onset; (4) contraindications to MRI (eg, implanted metal, claustrophobia).
We enrolled 20 TBI patients (14 males, mean age ± SD 47.3 ± 14.8 years) in the study. Clinical data at onset were obtained from the emergency unit’s medical records. Neuropsychiatrists reviewed the MRI at the time of study enrollment. In keeping with a previous study, we classified the injury as “focal” or “diffuse” based on radiological criteria (MRI: FLAIR; TR = 4200 ms; TE = 94 ms; resolution 0.7 × 0.7 × 3.0 mm3; and SWI; TR = 28 ms; TE = 20 ms; resolution 0.5 × 0.5 × 1.2 mm3). Thus, large lesions including white matter (eg, lobar contusions, hematoma/hemorrhage >2 mm diameter) were defined as “focal injury,” whereas those with possible diffuse pathology without large focal lesions were defined as DAI.21 Patients who had both large focal lesions and diffuse pathology were relegated to the focal injury group. This classification allowed investigation of pathology of pure DAI although focal injury and DAI often occur in a single individual after TBI.22 In all, 12 patients had focal injury, and 8 had DAI. The demographic and clinical information of the patients is shown in Table 1. Severity was defined according to the Glasgow Coma Scale or the Japan Coma Scale, which is a measure of the severity of impaired consciousness used in Japan. The relation of the Japan Coma Score to injury severity was explored previously.23
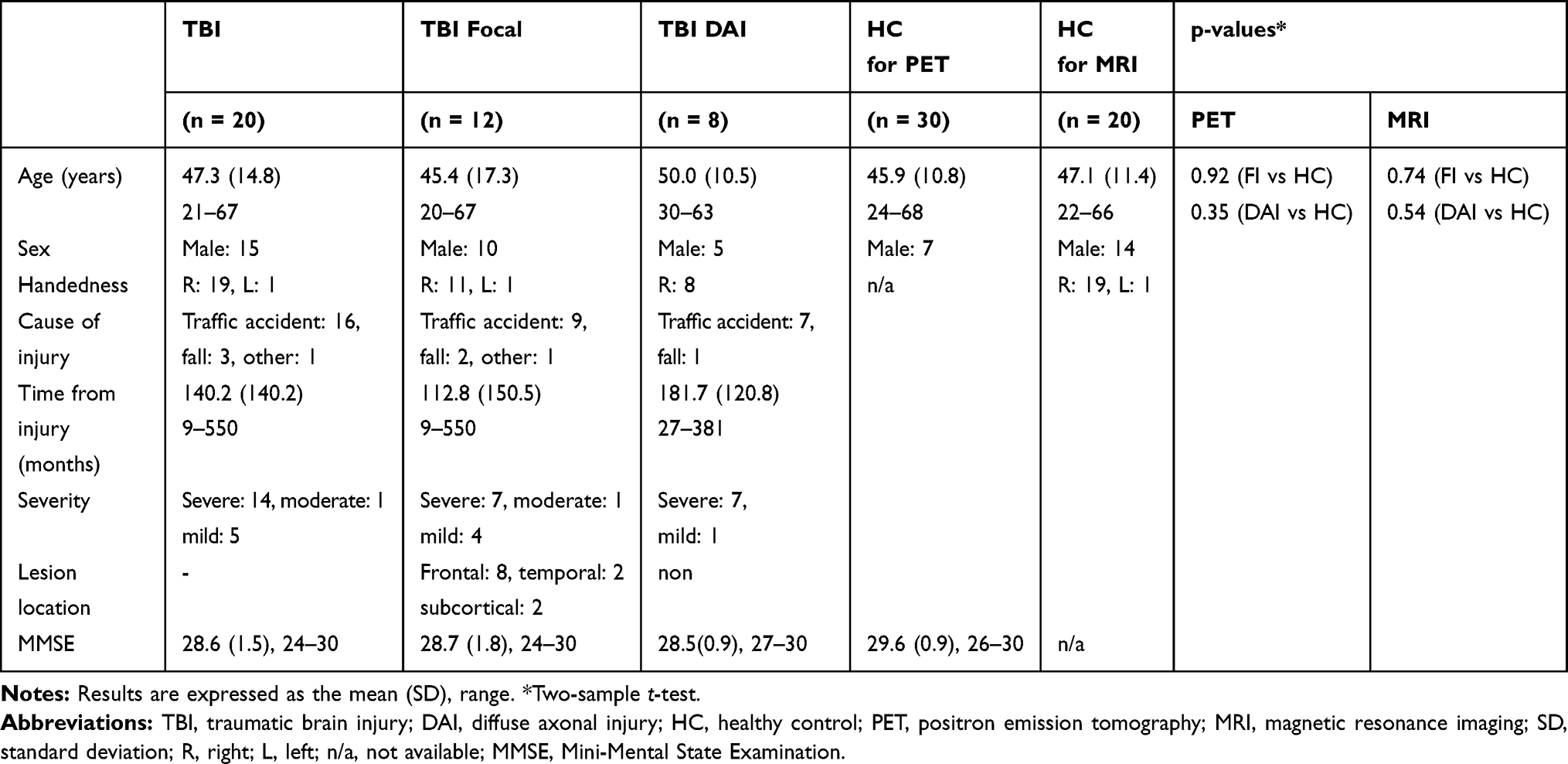 |
Table 1 Demographic and Clinical Characteristics of Participants |
Two healthy control groups participated in this study. For comparison of 18F-FPYBF-2 binding, a group of healthy, age-matched controls (HCs) underwent 18F-FPYBF-2 PET. To compare brain volumes, a second group of healthy, age- and sex-matched controls underwent structural MRI (Table 1).
The Committee on Medical Ethics of Kyoto University, Shiga General Hospital Institutional Review Board approved this study, which was carried out in accordance with the Code of Ethics of the World Medical Association. Written informed consent was obtained after participants were given a complete description of the study.
Procedures
Assessment for Neurocognition and Social Function
The TBI participants underwent neuropsychological test for general intelligence, memory, and executive function. General intelligence was assessed by the Wechsler Adult Intelligence Scale (WAIS)-III.24 Memory function was assessed using the Wechsler Memory Scale-Revised25 and executive function using the Behavioral Assessment of the Dysexecutive Syndrome.26 The Japanese version of the World Health Organization Disability Assessment Schedule27 was used to assess patients’ overall functioning. Impairment and disability were evaluated over six domains for the prior 30 days: cognition, mobility, self-care, getting along (interacting with others), and participation in life and in community activities.
PET Acquisition and Pre-Processing17
The 18F-FPYBF-2 was synthesized in house according to the same method in our previous study19, 18F-fluoride was produced using a cyclotron (CYPRIS HM18; Sumitomo Heavy Industries Ltd., Japan) by the 18O(p, n) 18F reaction on 98% enriched 18O water. The radiosynthesis of 18F-FPYBF-2 was performed using a modification of the Ono et al18 method on a hybrid synthesizer (cassette-type, multipurpose automatic synthesizer module; JFE Engineering Corp., Tokyo, Japan).
TBI and HCs underwent 18F-FPYBF-2 PET/computed tomography (CT), which was performed using a whole-body PET/CT scanner, the Siemens True Point Biograph 16 (1.34-mm pixels) (Siemens/CTI, Erlangen, Germany). At 50–70 min after intravenous injection of 18F-FPYBF-2 (200 ± 22 MBq), static head PET images were acquired for 20 min. The 20-min static scans were separately evaluated in two time zones (50–60 and 60–70 min). Because a previous PET study by our group revealed a slightly greater amyloid uptake in the later images than in the earlier images,19 we thought it was appropriate to use the latter half of each data set (ie, 60–70 min) for the analysis. In-house PET template construction was performed for 18F-FPYBF-2 PET.28,29 Twenty-four 18F-FPYBF-2 PET images, obtained by a 60 min dynamic scan of healthy subjects (15 females and 9 males; mean age ± SD = 57.5 ± 9.5 years), were used to produce the PET template.19 More detailed information has been given in our previous study.19
Automated Region-of-Interest Analysis
The cerebellar cortex (without amyloid plaque), used as a reference brain region in previous PET studies of AD30 and TBI,10,11 was used here as well. The standardized uptake value ratio (SUVR) of each region of interest (ROI), indicating amyloid deposition, was calculated as follows.
SUV for the target region of cortex (or subcortical gray matter)/mean cerebellar SUV.
Quantitative regional SUVR values were calculated using automated ROI analysis. The Automated Anatomical Labeling (AAL) atlas31 was used to predefine the template-based ROIs. The AAL atlas contains 90 regions in the cerebral (45 in each hemisphere) and 26 regions in the cerebellum.32,33 Following the method in our previous study,19 the AAL ROI was masked with the gray matter defined by the Montreal Neurological Institute (MNI) 152 standard-space T1-weighted average structural template image available from FSL Software (http://www.fmrib.ox.ac.uk/fsl) because the original AAL ROIs was larger than gray matter parenchyma. The spatial normalization of 18F-FPYBF-2 PET images was performed using the discrete cosine transform-based approach29 with the in-house 18F-FPYBF-2 PET templates that produced according to previous study.34 To avoid the operator-induced bias, all ROIs in the standard MNI space were inversely transformed to individual spaces by SPM8 using the inverse deformation field.35 The reference region to create SUVR images was used by combining 26 regions of the cerebellum. Mean SUVR values within 90 anatomical ROIs in both hemispheres were calculated using an in-house MATLAB script. More detailed information has been given in our previous study.19 In addition to the gray matter ROIs from the AAL atlas, we also selected white matter ROIs from the John Hopkins University (JHU) white-matter atlas.36 The JHU atlas contains 48 white matter tract labels in the cerebrum (24 in each hemisphere). Finally, as a representative value for cortical and subcortical amyloid plaque deposition of each subject, the mean SUVRs were calculated in five regions combining both hemispheres using the abovementioned AAL ROIs: four cortical (frontal, parietal, occipital, temporal) and a subcortical (combined caudate, putamen, pallidum, thalamus) gray matter regions. Furthermore, the mean SUVRs were calculated in the whole white matter and in seven regions likely to be affected by TBI37 (the corpus callosum, cerebral peduncle, sagittal stratum (including the inferior longitudinal fasciculus and inferior fronto-occipital fasciculus), cingulum, fornix, superior longitudinal fasciculus and uncinate) from the abovementioned JHU ROIs. For these ROIs, both hemispheres were combined.
Voxel-Based Analysis
To identify the brain regions in which patients with DAI showed differences in amyloid uptake relative to controls in the entire brain, we performed voxel-based analysis of the 18F-FPYBF-2 PET images. Spatial preprocessing and statistical analyses were performed using SPM8. All reconstructed PET images were spatially normalized into the MNI standard template (Montreal Neurological Institute, McGill University, Montreal, Canada) to remove intrasubject anatomical variability. Spatially normalized images were smoothed by convolution using an isotropic Gaussian kernel with 8 mm full width at half maximum. A two-sample t-test was then performed to compare patients with DAI or focal injury and HCs, with age excluded as a nuisance covariate. Liberal statistical thresholds of p < 0.001 (uncorrected for voxel level) and p <0.05 (FWE-corrected for cluster level) were applied.
MRI Acquisition and Pre-Processing
The TBI and HC individuals underwent MRI scans with a 3T whole-body scanner having a 40-mT/m gradient and a receiver-only 32-channel phased-array head coil (MAGNETOM Tim Trio; Siemens, Erlangen, Germany). The scanning parameters of the T1-weighted, three-dimensional, magnetization-prepared, rapid gradient-echo (3D-MPRAGE) sequence has these parameters: TR = 2,000 ms; TE = 4.38 ms; TI = 990 ms; field of view 225 × 240 mm; matrix 240 × 256; resolution 0.9375 × 0.9375 × 1.0 mm3; 208 total axial sections without intersection gaps.
3D-MPRAGE data were processed using SPM8 and VBM8 Toolbox (http://dbm.neuro.uni-jena.de/vbm) to determine individually spaced volumes of interest and to analyze brain regional volume alterations. The voxel values of segmented and normalized gray matter (GM) images were multiplied (modulated) by the Jacobian determinants obtained from non-linear normalization steps. For each ROI of cortex and cerebellum defined by the AAL atlas, values from the modulated GM probability map were averaged over voxels. Five mean volumes were calculated using the AAL atlas: bilateral frontal, parietal, occipital, and temporal cortices and cerebellar cortex.
To identify volumetric changes of subcortical structures, we used FSL’s Integrated Registration and Segmentation Tool (FIRST), which is a model-based segmentation and registration tool38. Volumes for the four subcortical regions (caudate, putamen, pallidum, thalamus) combining both hemispheres were extracted using the fslstats tool (FSL Software). The sum of those volumes was considered the subcortical volume. The subcortical volume was then normalized using the intracranial volume, which was calculated by summing the gray matter, white matter, and cerebrospinal fluid volumes segmented by SPM8.
Statistical Analyses
Statistical differences of the mean SUVR and volumes were compared using the Wilcoxon rank-sum test for unpaired data between patients with DAI and HCs and between focal injury patients and HCs. To investigate the effect of amyloid pathology on neuropsychological sequelae and outcomes, correlation coefficient analyses between the mean SUVR in regions with significantly higher 18F-FPYBF-2 uptake versus those of the HCs and clinical values were performed using Spearman’s rank correlation analysis in TBI patients. Age and duration from TBI onset were controlled for the correlation analysis. The statistical analysis was performed using statistical software R version 3.0.2 (The R Foundation for Statistical Computing Platform, Vienna, Austria), in which p < 0.05 with multiple comparisons using the family-wise error rate was considered to indicate statistical significance.
Results
Neurocognition and Social Function
Table 2 shows the Wechsler Adult Intelligence Scale-III, Wechsler Memory Scale-Revised, Behavioral Assessment of the Dysexecutive Syndrome, and the World Health Organization Disability Assessment Schedule 2.0 for IQ, memory, and executive and social functions. Patients with DAI had lower scores than those with a focal injury for all measurements. Scores for processing speed and delayed memory function were approximately >2 SD lower than standard scores.
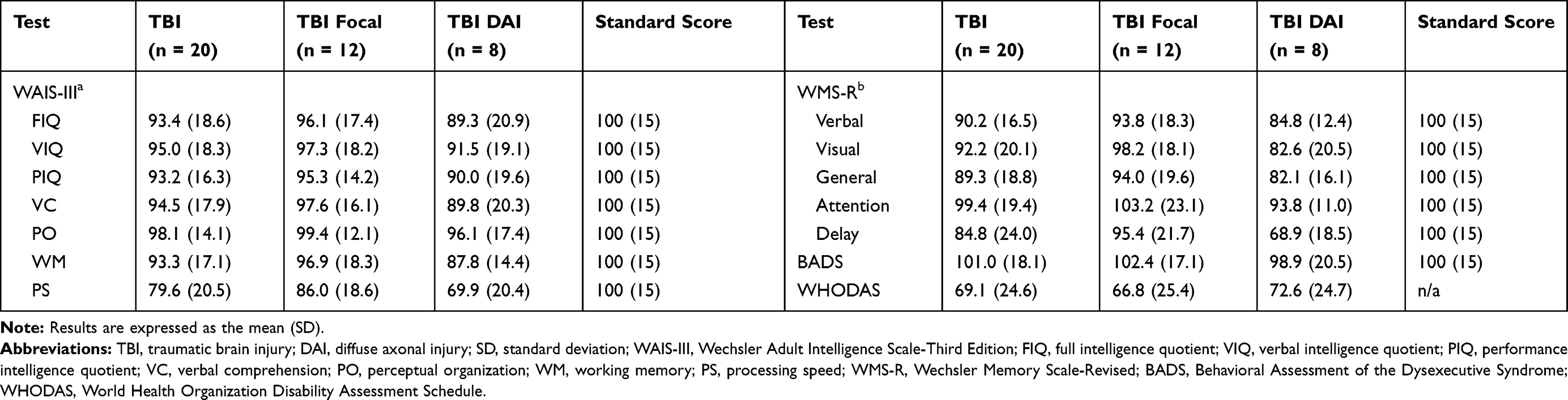 |
Table 2 Neuropsychological Assessment and Social Function Scores |
Amyloid Pathology Detected by 18F-FPYBF-2 Binding
Figure 1 shows the group-average 18F-FPYBF-2 PET images from patients with focal injury or DAI and from HCs. 18F-FPYBF-2 uptake was observed in cerebral white matter in HCs and appeared stronger in patients with focal injury and DAI. In DAI group, 18F-FPYBF-2 uptake was observed in cerebral gray matter as well as white matter. 18F-FPYBF-2 PET images in all patients with focal injury or DAI are shown in Figures 5 and 6.
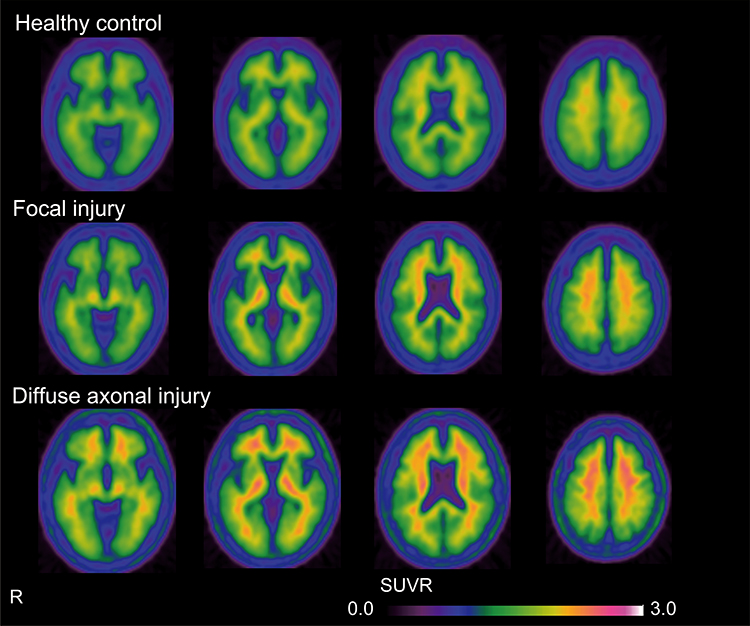 |
Figure 1 Mean axial PET images of 18F-FPYBF-2 PET in healthy control (n = 30) and patients with focal injury (n = 12) or DAI (n = 8). In the healthy controls, 18F-FPYBF-2 uptake was mainly observed in cerebral white matter. In the DAI group, 18F-FPYBF-2 uptake was observed in cerebral gray matter as well as white matter. |
Mean SUVRs in all gray matter ROIs were higher in DAI patients than in HCs. The group comparison between these two groups showed significantly increased 18F-FPYBF-2 binding in the DAI patients in the occipital cortices (W=38, z = 3.06, r = 0.56, p = 0.002) and temporal cortices (W = 41, z = 2.93, r = 0.53, p = 0.003) but no significant differences in binding in the frontal, parietal cortical, and subcortical regions (Figure 2, Table 3). Because the DAI group contains one clear outlier, who showed very high SUVRs, we also applied the statistical analysis without the outlier. Significant results remained after excluding the outlier (occipital cortex, W=38, z = 2.67, r = 0.49, p = 0.008; temporal cortex, W = 40, z= 2.53, r = 0.46, p = 0.011). In contrast, patients with focal injury showed lower mean SUVRs in the frontal, temporal, parietal, and subcortical regions (but not in the occipital cortex) than that in HCs. Comparison between the focal injury group and HCs revealed no significant differences in binding.
 |
Table 3 Mean SUVR of Regions of Interest in Each Group |
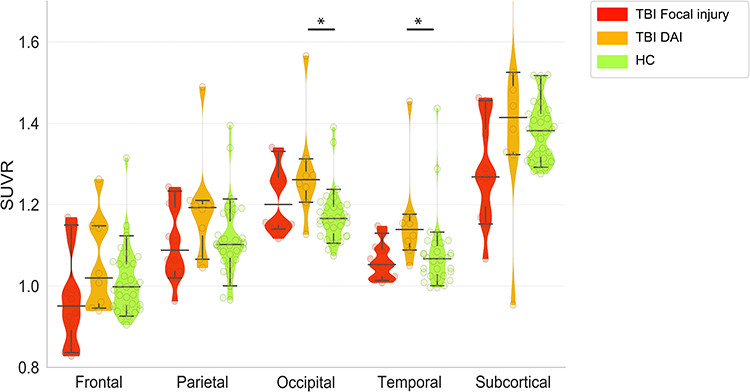 |
Figure 2 Violin and whisker plot shows 5% and 95% confidence intervals (whiskers) and medians of the mean SUVRs of each region of interest in patients with focal injury (red) or DAI (orange) and the healthy controls (green). Mean SUVR in DAI patients in the occipital and temporal cortices were significantly higher than those of healthy controls. *p<0.05, Wilcoxon rank sum test, FWE. |
Regarding the white matter, the mean SUVR of the whole white matter ROI was significantly higher in DAI patients (W= 44, z = 2.80, r = 0.51, p = 0.005) than in HCs, but was not different in the focal injury group (Table 3). Regionally, patients with DAI showed significantly higher mean SUVRs in the sagittal stratum (including the inferior longitudinal fasciculus and inferior fronto-occipital fasciculus) and superior longitudinal fasciculus (W= 23, z = 3.77, r = 0.69, p = 0.0001, W= 33.5, z = 3.28, r = 0.60, p = 0.001, respectively). There were no significant differences in binding in the corpus callosum, cerebral peduncle, cingulum, fornix or uncinate between the DAI and HC groups. Patients with focal injury had significantly lower mean SUVRs in the fornix compared with HCs. There were no significant differences in binding in any other white matter ROIs in patients with focal injury compared with HCs (Supplementary eTable 1).
Voxel-Based Analysis
Patients with DAI exhibited 18F-FPYBF-2 uptake relative to controls in right superior frontal gyrus, left medial orbital gyrus, and left occipital gyrus (Figure 3). Patients with focal injury had higher 18F-FPYBF-2 uptake relative to controls in several regions when uncorrected for voxel level, but these differences did not survive multiple comparisons with cluster-level corrections.
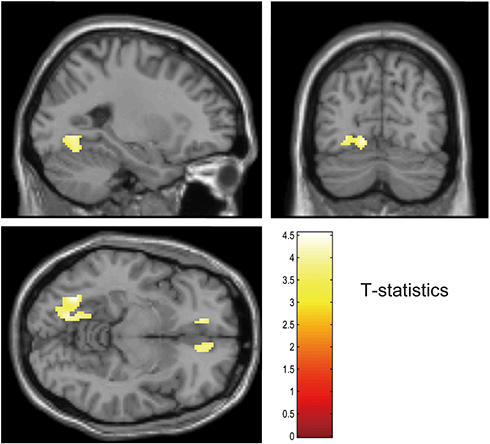 |
Figure 3 Voxel-based analysis showed that the patients with DAI exhibited 18F-FPYBF-2 uptake relative to controls in right superior frontal gyrus, left medial orbital gyrus and left occipital gyrus. p < 0.001 uncorrected for voxel level and p <0.05 FWE corrected for cluster level. |
Group Comparisons of Mean Volumes
The DAI patients exhibited significantly reduced gray matter volume (compared with the controls) in the occipital cortex (W = 130, z = 2.59, r = 0.06, p = 0.0096) and subcortical regions (W = 151, z = 4.02, r = 0.86, p = 0.00005) (Figure 4, Table 4). Compared with that of the HCs, focal injury reduced the gray matter volume in the frontal cortex (W = 176, z = 2.18, r = 0.47, p = 0.029) and subcortical regions (W = 180, z = 2.35, r = 0.50, p = 0.019), respectively, but the difference disappeared after multiple comparison corrections. Cerebellum volume was also significantly reduced in DAI patients (W=163, z = 3.85, r = 0.82, p = 0.0001). Cerebellum volume trended lower in focal injury patients than in HCs (W=168, z = 1.85, r = 0.4, p = 0.064).
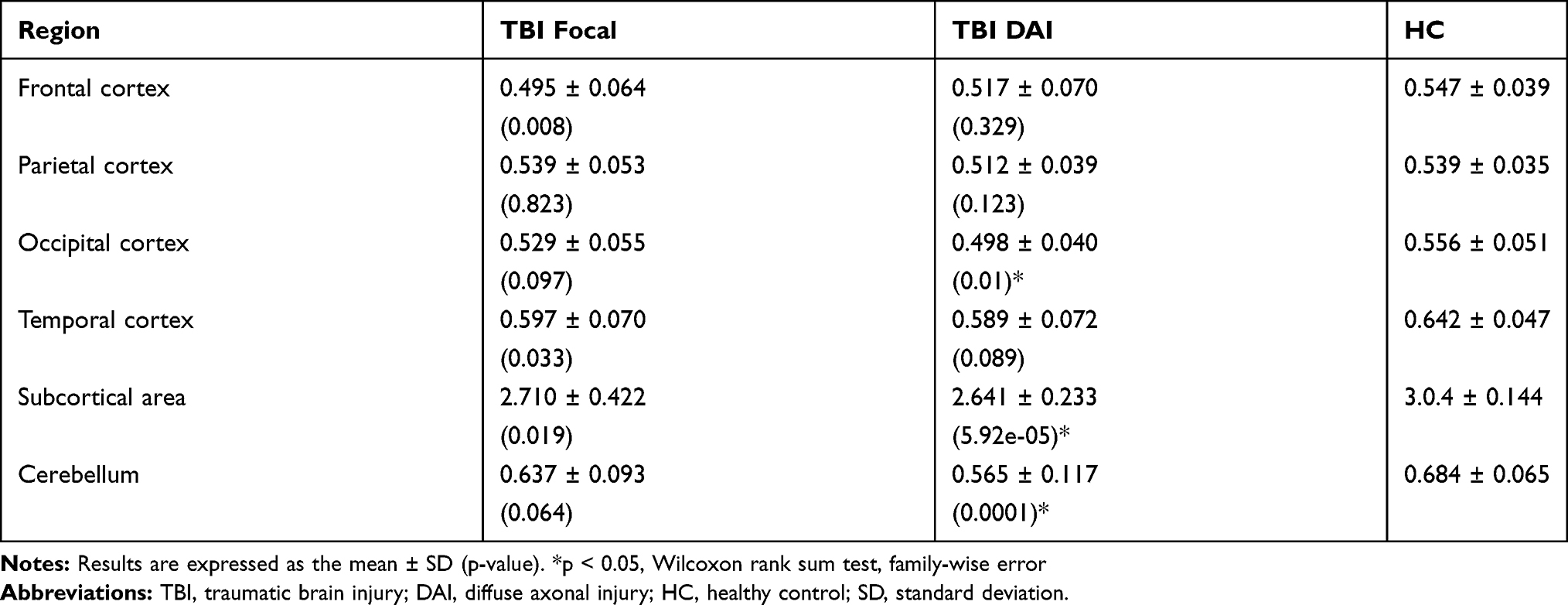 |
Table 4 Mean Volume of Regions of Interest in Each Group |
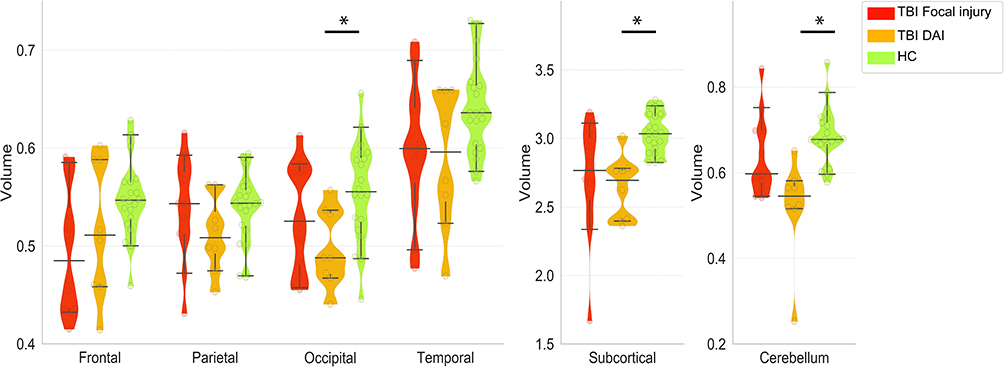 |
Figure 4 Violin and whisker plot shows 5% and 95% confidence intervals (whiskers) and the median of the mean volume of each region of interest in patients with focal injury (red) or DAI (orange) and healthy controls (green). Mean volumes of the DAI patients in the occipital cortex, subcortical, and cerebellum were significantly lower than those of healthy controls. *p<0.05, Wilcoxon rank sum test, FWE. |
Correlation Analyses Between Mean SUVR and Clinical Valuables
DAI patients showed no significant correlations between the mean SUVR in occipital or temporal cortices or in any clinical variables, including severity, neuropsychological assessment, and social function.
Discussion
Our main aim was to investigate amyloid deposition using PET with an 18F-FPYBF-2 tracer in TBI patients grouped by injury type: focal injury or DAI. The two groups displayed different spatial patterns of amyloid accumulation. DAI patients showed significantly increased 18F-FPYBF-2 binding in the occipital and temporal cortices. Although not statistically significant, DAI patients showed higher 18F-FPYBF-2 binding in all ROIs, whereas focal injury patients showed lower 18F-FPYBF-2 binding in all ROIs except the occipital cortex.
One novel finding in this study was amyloid deposition in the occipital and temporal cortices in patients with chronic DAI, with a large effect size despite the small sample in the ROI analysis. Furthermore, voxel-based analysis revealed significant clusters of 18F-FPYBF-2 binding in the occipital cortex. Significant clusters were also found in the frontal cortex of patients with DAI, although there was no significant amyloid deposition in the frontal cortex in the ROI analysis. It might be that there were no differences in the ROI of the entire frontal cortex, because ROI analysis excludes local effects. Animal models and human autopsy studies have shown that axonal damage as the primary injury during the acute stage may act as an initial trigger for Aβ production and accumulation of amyloid pathology.9,17 In contrast, during chronic-stage TBI, cerebrovascular dysfunction, including an altered clearance mechanism related to progressive axonal damage, may play a role in Aβ deposition.2,4 In our study, the increased amyloid deposition in the occipital and temporal cortices could be attributed to axonal injury-related pathology in these regions. Volumetric analysis showed that GM volumes in the occipital and temporal cortices were reduced in DAI patients. Volume reduction in the temporal cortex was not statistically significant, but there was a trend compared with that in controls. These results suggest that the impact of trauma and its subsequently related neuropathology on these brain regions could affect the specific regional pattern of amyloid deposition, at least in our patients with DAI.
In contrast, we found no significant increase in amyloid deposition in the patients with focal injury compared with that in the HCs. Previous PET studies revealed no or little binding near focal lesions or in the penumbra around these lesions.8,9 In the present study, eight patients with focal injury exhibited lesions mainly in the frontal cortex: two in the temporal cortex, and two in subcortical regions. In agreement with previous studies, visual inspection of individual SUVR images revealed no or little amyloid binding in these focal lesions (Figure 5). This finding is likely the result of neurodegeneration and/or axonal injury preventing the tracer from binding in the injury area.39,40 Therefore, it should be noted that amyloid deposition could not be measured accurately with PET in patients with focal lesions. Another possible explanation for no amyloid deposition with a focal injury is the effect of the reference region chosen for quantifying 18F-FPYBF-2 binding. We used the cerebellum as a reference region because we assumed that the minimal Aβ plaque density in the cerebellum and the cortical/cerebellar binding ratio provided a measure of the cortical Aβ burden.30 In previous PET imaging studies, including that associated with TBI, the entire cerebellum emerged as the most widely used reference region for quantifying the amyloid burden.10,11 With TBI, however, the cerebellum is one of the common regions that atrophies, which means that it could be damaged by trauma.41 Our results indicate that the cerebellum volume reduction associated with focal injury, compared with that of the HCs, was not statistically significant. The presence of amyloid binding in the cerebellum reference region would thus lead to underestimating the calculated SUVR in target regions. To justify the choice of the cerebellar cortex as the reference region, we confirmed the reproducibility of our results using a different reference region. Very similar results were produced when the brainstem was used as the reference region (Supplementary eTable 2). The DAI patients showed a tendency toward increased 18F-FPYBF-2 binding in the occipital and temporal cortices, although these results were not statistically significant. Nevertheless, future studies are needed to establish a precise measurement for quantifying Aβ burden against a reference region following TBI.
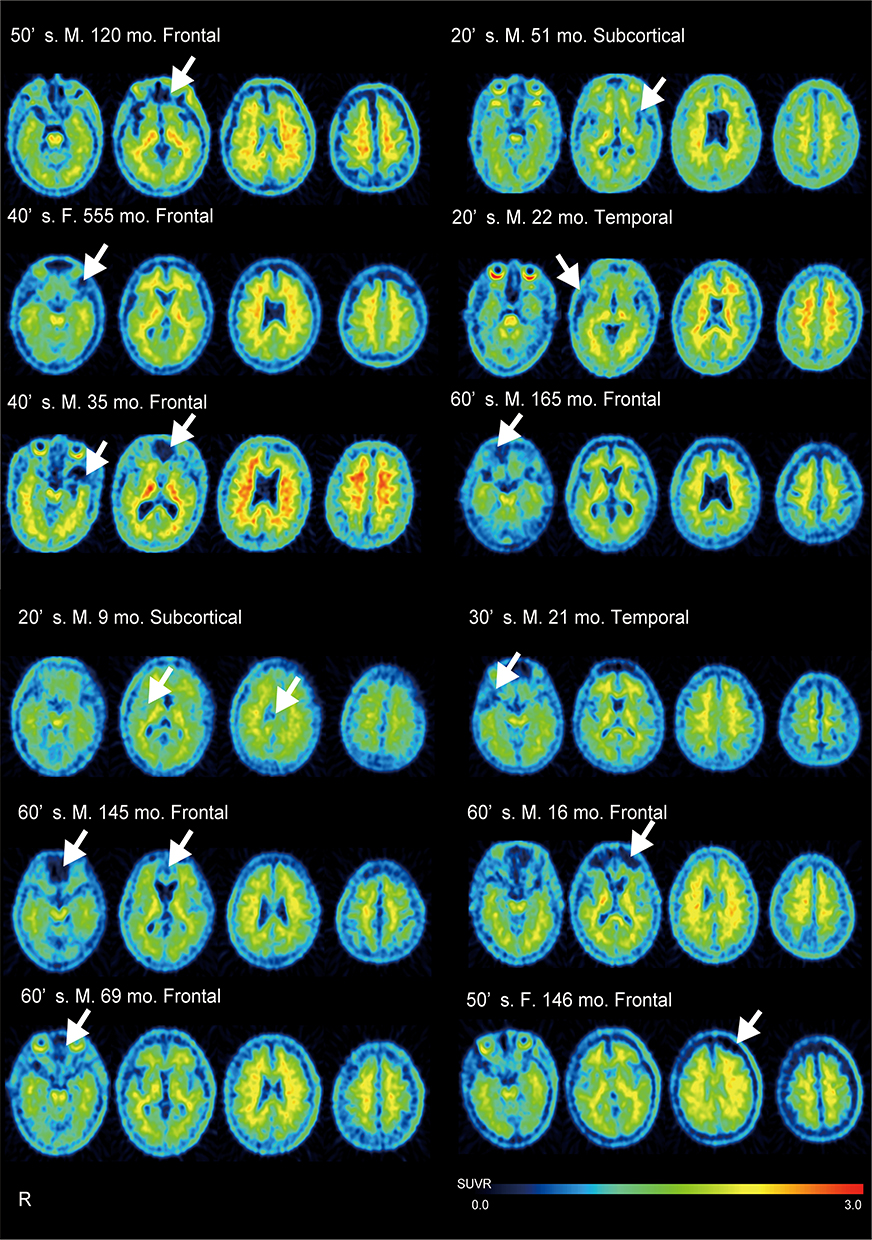 |
Figure 5 18F-FPYBF-2 PET images in all patients with focal injury.18F-FPYBF-2 uptake was observed in cerebral gray matter and white matter in patients with focal injury. Arrow indicates the location of lesions. |
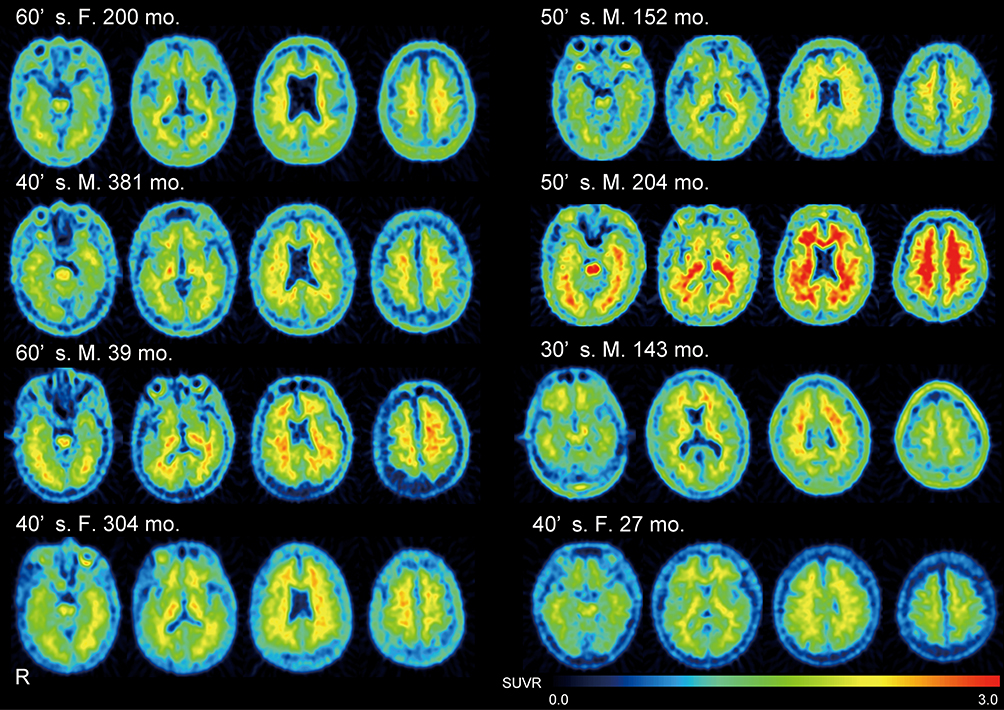 |
Figure 6 18F-FPYBF-2 PET images in all patients with diffuse axonal injury.18F-FPYBF-2 uptake was observed in cerebral gray and white matter in patients with diffuse axonal injury. |
The type of injury may importantly influence Aβ dynamics.17 Because TBI is a complex, heterogeneous disorder, the type and extent of the acute pathology probably plays an important role in determining the deposition and accumulation of Aβ.17 In a previous report, interstitial Aβ42 levels during the acute stage were slightly higher in patients with DAI than in those with a focal injury.16 These observations suggest that damaged axons may be an important source of Aβ after TBI. Our results support the view that axonal injury—not focal neuronal damage—is the most important contributor to interstitial Aβ following TBI, even during the chronic stage.16,42 These results suggest that the pattern of amyloid pathology is different depending on the injury type (ie, focal or DAI without a focal lesion). It would be meaningful to investigate amyloid deposition while taking into consideration the heterogeneous nature of TBI (eg, injury type and disease stage). Given that TBI is a major risk factor for dementia, amyloid imaging may help to understand the pathogenesis of TBI, especially in the case of poor prognosis. Furthermore, the assessment of amyloid pathology might provide us a selection of appropriate therapeutic interventions, such as anti-amyloid therapies, at an early stage.
Visual inspection of SUVR images suggested increased 18F-FPYBF-2 binding in white matter of patients with either focal injury or DAI. Similar to the pattern of amyloid deposition in the gray matter, DAI patients had higher 18F-FPYBF-2 binding in the whole white matter as well as in regional white matter ROIs compared with HCs, whereas focal injury patients had no differences in white matter 18F-FPYBF-2 binding except in the fornix compared with HCs. In AD patients, 11C-PiB binding in white matter has been considered mainly nonspecific owing to slower 11C-PiB white matter kinetics.43 In TBI patients, it has been reported that there are no amyloid- β plaques in the white matter of long-term survivors.44 Therefore, high 18F-FPYBF-2 binding in white matter is likely to be caused by the slow clearance of the tracer from white matter tissue. Our results thus suggest that the clearance mechanism was more impaired in patients with DAI as a result of axonal damage.
In the DAI group, there were no correlations between amyloid deposition in the occipital or temporal cortices or in any clinical variables, including severity, neuropsychological assessments, and social function. Similarly, previous studies have reported that amyloid deposition was not correlated with neuropsychological impairment in TBI patients.9 One of the likely reasons we found no positive relations is the small sample size available for correlation analysis. Furthermore, the degree of amyloid deposition would be estimated as lower using cerebellum as the reference for quantifying 18F-FPYBF-2 binding. This situation would affect the lack of correlations between the degree of amyloid deposition and neuropsychological sequelae. In AD, amyloid deposition is not closely related to cognitive impairment, suggesting that downstream factors have a more direct effect on neuropsychological sequelae.45 Not only amyloid pathology, but various other factors, could be related to neuropsychological sequelae following TBI.
The current study contained several limitations that should be considered. First, the small sample size was small. Because of the small sample size, we could not consider the effects of other variables related to neurodegeneration, such as age, sex and the cause of the TBI. In addition, because of the small sample size, we defined cortical ROIs by combining the left and right regions into one ROI. However, including a separate ROI for each hemisphere is likely to provide more information, especially in patients with unilateral focal injury. Second, the predefined AAL ROIs were masked with the gray matter of the average standard T1-weighted structural template image. Because MRI was not available for all subjects in our study, we were unable to consider the inter-subject variability of gray matter volumes and partial volume effects. Furthermore, we used the cerebellum as a reference region for quantifying the amyloid burden. Although the presence of amyloid deposition in DAI patients was striking, using the cerebellum for the reference region might cause underestimation of the amyloid burden after TBI. The use of the cerebellum as the reference region was justified because similar results were obtained using a different reference region (ie, the brainstem). However, partial volume correction would have reduced any potential effects of cerebellar atrophy, and should be considered in future studies. Third, we revealed a spatial pattern of amyloid deposition in patients with DAI (without focal injury) using average SUVR images. However, average images across patients might mask any local effects, especially in the case of focal injury. Thus, individual analyses that consider each lesion may be suitable for use in future studies. Hence, this study should be regarded as preliminary, and should be re-examined with a larger sample, possibly also measuring amyloid deposition in vivo. Generally, Aβ plaque is seen shortly after injury. The mechanism of Aβ production and the effect of its accumulation on long-term prognoses are still unclear. Future studies investigating Aβ deposition and its alterations longitudinally would provide more detailed information of amyloid pathology in this population.
Acknowledgments
The authors thank all participants for their contribution to this project. This work was supported by an Industrial Disease Clinical Research Grants (150502-02), a Health Labor Sciences Research Grant, a Grant-in-Aid for Young Scientists (19K17110) and C (17K10327, 18K07712) from the Japan Society for the Promotion of Science, Innovative Areas (16H06402) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT), a research grant from National Mutual Insurance Federation of Agricultural Cooperatives and ISHIZUE 2020 of Kyoto University Research Development Program.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
Toshihiko Aso reports grants from Japan Society for the Promotion of Science, during the conduct of the study. The authors have no other potential conflicts of interest to declare.
References
1. Washington PM, Morffy N, Parsadanian M, Zapple DN, Burns MP. Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model. J Neurotrauma. 2014;31(1):125–134. doi:10.1089/neu.2013.3017
2. Ramos-Cejudo J, Wisniewski T, Marmar C, et al. Traumatic brain injury and Alzheimer’s disease: the cerebrovascular link. EBioMedicine. 2018;28:21–30. doi:10.1016/j.ebiom.2018.01.021
3. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844–866. doi:10.1016/j.neuron.2013.10.008
4. Kenney K, Amyot F, Haber M, et al. Cerebral vascular injury in traumatic brain injury. Exp Neurol. 2016;275(Pt 3):353–366. doi:10.1016/j.expneurol.2015.05.019
5. Loane DJ, Pocivavsek A, Moussa CE, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nature Medicine. 2009;15(4):377–379. doi:10.1038/nm.1940
6. Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57(4):419–425. doi:10.1136/jnnp.57.4.419
7. Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol. 2013;9(4):211–221. doi:10.1038/nrneurol.2013.29
8. Hong YT, Veenith T, Dewar D, et al. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 2014;71(1):23–31. doi:10.1001/jamaneurol.2013.4847
9. Scott G, Ramlackhansingh AF, Edison P, et al. Amyloid pathology and axonal injury after brain trauma. Neurology. 2016;86(9):821–828. doi:10.1212/WNL.0000000000002413
10. Yang ST, Hsiao IT, Hsieh CJ, et al. Accumulation of amyloid in cognitive impairment after mild traumatic brain injury. J Neurol Sci. 2015;349(1–2):99–104. doi:10.1016/j.jns.2014.12.032
11. Gatson JW, Stebbins C, Mathews D, et al. Evidence of increased brain amyloid in severe TBI survivors at 1, 12, and 24 months after injury: report of 2 cases. J Neurosurg. 2016;124(6):1646–1653. doi:10.3171/2015.6.JNS15639
12. Ledig C, Kamnitsas K, Koikkalainen J, et al. Regional brain morphometry in patients with traumatic brain injury based on acute- and chronic-phase magnetic resonance imaging. PLoS One. 2017;12(11):e0188152.
13. Adams JH, Graham DI, Gennarelli TA, Maxwell WL. Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatry. 1991;54(6):481–483. doi:10.1136/jnnp.54.6.481
14. Meythaler JM, Peduzzi JD, Eleftheriou E, Novack TA. Current concepts: diffuse axonal injury-associated traumatic brain injury. Arch Phys Med Rehabil. 2001;82(10):1461–1471. doi:10.1053/apmr.2001.25137
15. Kawai N, Kawanishi M, Kudomi N, et al. Detection of brain amyloid beta deposition in patients with neuropsychological impairment after traumatic brain injury: PET evaluation using Pittsburgh Compound-B. Brain Injury. 2013;27(9):1026–1031. doi:10.3109/02699052.2013.794963
16. Marklund N, Blennow K, Zetterberg H, Ronne-Engstrom E, Enblad P, Hillered L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg. 2009;110(6):1227–1237. doi:10.3171/2008.9.JNS08584
17. Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11(5):361–370.
18. Ono M, Cheng Y, Kimura H, et al. Novel 18F-labeled benzofuran derivatives with improved properties for positron emission tomography (PET) imaging of beta-amyloid plaques in Alzheimer’s brains. J Med Chem. 2011;54(8):2971–2979. doi:10.1021/jm200057u
19. Higashi T, Nishii R, Kagawa S, et al. (18)F-FPYBF-2, a new F-18-labelled amyloid imaging PET tracer: first experience in 61 volunteers and 55 patients with dementia. Ann Nucl Med. 2018;32(3):206–216. doi:10.1007/s12149-018-1236-1
20. Yamauchi H, Kagawa S, Takahashi M, Oishi N, Ono M, Higashi T. Misery perfusion and amyloid deposition in atherosclerotic major cerebral artery disease. NeuroImage Clin. 2019;22:101762. doi:10.1016/j.nicl.2019.101762
21. Turner GR, Levine B. Augmented neural activity during executive control processing following diffuse axonal injury. Neurol. 2008;71(11):812–818. doi:10.1212/01.wnl.0000325640.18235.1c
22. Scheid R, Walther K, Guthke T, Preul C, von Cramon DY. Cognitive sequelae of diffuse axonal injury. Arch Neurol. 2006;63(3):418–424. doi:10.1001/archneur.63.3.418
23. Namiki JYM, Funabiki T, Suzuki M, Fujishima S, Hori S, Aikawa N. Difficulty and inaccuracy of assessment of the consciousness level by the Glasgow Coma Scale: comparison with the Japan Coma Scale. J Jap Soc Emerg Med. 2007;10:20–25.
24. W D. WAIS-III: Administration and Scoring Manual: Wechsler Adult Intelligence Scale. 3rd Ed. San antonio, TX: Psychological Corporation; 1997.
25. W D. Wechsler Memory Scale-Revised Manual. San Antonio, TX: Psychological Corporation; 1987.
26. Wilson BAAN, Burgess PW, Emslie H, Evans J. Behavioural Assessment of the Dysexecutive Syndrome. Bury St Edmunds,UK: Thames Valley Test Company; 1997.
27. Tazaki M, Yamaguchi T, Yatsunami M, Nakane Y. Measuring functional health among the elderly: development of the Japanese version of the World Health Organization Disability Assessment Schedule II. Int J Rehabil Res Internationale Zeitschrift Fur Rehabilitationsforschung Revue Internationale De Recherches De Readaptation. 2014;37(1):48–53. doi:10.1097/MRR.0000000000000032
28. Ashburner J. A fast diffeomorphic image registration algorithm. NeuroImage. 2007;38(1):95–113. doi:10.1016/j.neuroimage.2007.07.007
29. Ashburner J, Friston KJ. Nonlinear spatial normalization using basis functions. Human Brain Mapping. 1999;7(4):254–266.
30. Kudo Y, Okamura N, Furumoto S, et al. 2-(2-[2-Dimethylaminothiazol-5-yl]ethenyl)-6- (2-[fluoro]ethoxy)benzoxazole: a novel PET agent for in vivo detection of dense amyloid plaques in Alzheimer’s disease patients. J Nucl Med. 2007;48(4):553–561. doi:10.2967/jnumed.106.037556
31. Tzourio-Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage. 2002;15(1):273–289. doi:10.1006/nimg.2001.0978
32. Schmahmann JD, Doyon J, McDonald D, et al. Three-dimensional MRI atlas of the human cerebellum in proportional stereotaxic space. NeuroImage. 1999;10(3 Pt 1):233–260. doi:10.1006/nimg.1999.0459
33. Ota K, Oishi N, Ito K, Fukuyama H. A comparison of three brain atlases for MCI prediction. J Neurosci Methods. 2014;221:139–150. doi:10.1016/j.jneumeth.2013.10.003
34. Kemppainen NM, Aalto S, Wilson IA, et al. Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology. 2006;67(9):1575–1580. doi:10.1212/01.wnl.0000240117.55680.0a
35. Oishi N, Hashikawa K, Yoshida H, et al. Quantification of nicotinic acetylcholine receptors in Parkinson’s disease with (123)I-5IA SPECT. J Neurol Sci. 2007;256(1–2):52–60. doi:10.1016/j.jns.2007.02.014
36. Wakana S, Jiang H, Nagae-Poetscher LM, van Zijl PC, Mori S. Fiber tract-based atlas of human white matter anatomy. Radiology. 2004;230(1):77–87. doi:10.1148/radiol.2301021640
37. Wallace EJ, Mathias JL, Ward L. Diffusion tensor imaging changes following mild, moderate and severe adult traumatic brain injury: a meta-analysis. Brain Imag Behav. 2018;12(6):1607–1621.
38. Patenaude B, Smith SM, Kennedy DN, Jenkinson MA. Bayesian model of shape and appearance for subcortical brain segmentation. NeuroImage. 2011;56(3):907–922. doi:10.1016/j.neuroimage.2011.02.046
39. Ayubcha C, Revheim ME, Newberg A, et al. A critical review of radiotracers in the positron emission tomography imaging of traumatic brain injury: FDG, tau, and amyloid imaging in mild traumatic brain injury and chronic traumatic encephalopathy. Eur J Nucl Med Mol Imaging. 2020. doi:10.1007/s00259-020-04926-4
40. Collins JM, King AE, Woodhouse A, Kirkcaldie MT, Vickers JC. The effect of focal brain injury on beta-amyloid plaque deposition, inflammation and synapses in the APP/PS1 mouse model of Alzheimer’s disease. Exp Neurol. 2015;267:219–229. doi:10.1016/j.expneurol.2015.02.034
41. Harris TC, de Rooij R, Kuhl E. The shrinking brain: cerebral atrophy following traumatic brain Injury. Ann Biomed Eng. 2018.
42. Uryu K, Chen XH, Martinez D, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208(2):185–192. doi:10.1016/j.expneurol.2007.06.018
43. Fodero-Tavoletti MT, Rowe CC, McLean CA, et al. Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med. 2009;50(2):198–204. doi:10.2967/jnumed.108.057984
44. Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19(2):214–223. doi:10.1111/j.1750-3639.2008.00176.x
45. Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–192. doi:10.1002/ana.22248
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.