")
Back to Journals » OncoTargets and Therapy » Volume 11
SOX2, a stemness gene, induces progression of NSCLC A549 cells toward anchorage-independent growth and chemoresistance to vinblastine
Authors Choe C , Kim H , Min S , Park S, Seo J, Roh S
Received 30 May 2018
Accepted for publication 1 August 2018
Published 25 September 2018 Volume 2018:11 Pages 6197—6207
DOI https://doi.org/10.2147/OTT.S175810
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Chungyoul Choe,1,* Hyewon Kim,2,* Sol Min,2 Sangkyu Park,2,3 Jeongmin Seo,2,3 Sangho Roh2
1Samsung Biomedical Research Institute, Samsung Medical Center, Sungkyunkwan University, School of Medicine, Seoul, Republic of Korea; 2Laboratory of Cellular Reprogramming and Embryo Biotechnology, School of Dentistry, Seoul National University, Seoul, Republic of Korea; 3Biomedical Research Institute, NeoRegen Biotech Co., Ltd., Gyeonggi-do, Korea
*These authors contributed equally to this work
Background: Non-small cell lung cancer (NSCLC) is difficult to treat successfully. This intractability is mainly due to the cancer progressing through invasion, metastasis, chemotherapeutic resistance and relapse. Stemness has been linked to the various steps of cancer progression in a variety of tumors, yet little is known regarding its role in NSCLC.
Purpose: In this study, we sought to determine the role of SOX2, a master regulator of pluripotency, in the growth of extracellular matrix (ECM)-detached cells during cancer progression.
Methods: We established a three-dimensional (3D) Poly-2-hydroxyethyl methacrylate (poly-HEMA) culture of lung adenocarcinoma (LUAD) A549 cells as an ECM-detached cell growth model and examined the role of stemness genes using siRNA and small molecule inhibitor in comparison to standard two dimensional (2D) culture.
Results: In poly-HEMA culture, A549 cells formed substratum-detached spheroids with characteristics of intermediate epithelial to mesenchymal transition (EMT) and exhibited greater expression of SOX2 than did control 2D cells. Knockdown of SOX2 markedly suppressed the growth of A549 cell aggregates in poly-HEMA culture conditions and furthermore increased their sensitivity to the anticancer drug vinblastine with concomitant downregulation of the activity of the anti-apoptotic AKT kinase. Interestingly, a small molecule, RepSox, which replaces SOX2, stimulated A549 cell growth in poly-HEMA 3D culture condition.
Conclusion: Our findings strongly indicate that SOX2 contributes to anchorage-independent growth and chemoresistance via its downstream signaling mediator AKT kinase during the disease progression of NSCLC. SOX2 may therefore be an invaluable therapeutic target of NSCLC.
Keywords: SOX2, lung cancer, NSCLC, EMT, stemness, vinblastine
Introduction
Lung cancer is the leading cause of cancer-related death worldwide, and ~85% of all lung cancers are non-small-cell lung cancers (NSCLCs).1–3 NSCLC is defined by the accumulation of multiple genotypic alterations and further classified by a high degree of histological heterogeneity, including lung adenocarcinoma (LUAD; 48%), lung squamous cell carcinoma (LUSC; 28%), and large cell carcinoma (24%).4,5 Genetic and epigenetic alterations have been intensely pursued as therapeutic targets, and the development of a series of molecular inhibitors, such as gefitinib (Iressa, AstraZeneca, Cambridge, UK) and erlotinib (Tarceva, OSI Pharmaceuticals, Melville, NY, USA), targeting EGFR, has revolutionized the treatment of NSCLC.6 However, these drugs have limited therapeutic efficacy; the overall cure and survival rates for NSCLC remain low at 19%, particularly in metastatic stage IIIA disease.7 Therefore, it is imperative to understand the biology and mechanisms of tumor progression to identify potential therapeutic molecular targets for NSCLC.
Cancer is somewhat similar to a micro-evolutionary process in which accrual of multiple mutations in cell growth genes leads to uncontrolled cell growth, which then generates growth-induced stress in a spatiotemporal manner.8 As a positive selection factor, which is advantageous in promoting its own growth and progression, these growth-induced stresses fatally lead tumors in situ to detach from their primary site as individual cells or cell clusters and disseminate to the lymphatic and circulatory systems and pleural and peritoneal cavities (reviewed in the studies by Buchhei et al9,10). During this series of dynamic processes called the metastasis cascade, cancer cells must primarily adapt to and survive periodic cell-extracellular matrix (ECM) detachment, whereas normal epithelial cells require attachment to ECM for survival (reviewed in the study by Frisch and Francis11). As a safeguard against preventing cell-ECM detachment-induced death, cancer cells evolve multifaceted mechanisms to compensate for the lack of cell-ECM survival signaling. In particular, several studies have shown that the epithelial-to-mesenchymal transition (EMT) plays an important role in anchorage-independent survival (reviewed in the study by Brabletz et al12). EMT is a process by which epithelial cells lose cell–cell adhesion, detach from basement membranes, and acquire mesenchymal-like traits. As the underlying mechanisms, it has long been proposed that a change in the expression of cell adhesion proteins from E-cadherin to N-cadherin on detachment increases cell–cell adhesion and leads to cell clusters resistant to cell death. In support of this, cancer cells derived from the patients with tumors in an advanced malignant stage grow as spheroids in suspension culture conditions.13,14 Recent work by several groups highlighted that anchorage-independent survival signaling universally converges into the activation of the phosphatidylinositol-3-kinase (PI3K)/AKT.15–17 In addition, EMT is also associated with tumor stemness and resistance to therapy.18 Recently, however, the role of EMT in cancer progression has become controversial since there is no direct in vivo evidence supporting that EMT occurs in human cancer.19
It is known that cancer stem cells are a key population of tumor cells that is highly tumorigenic and chemoresistant in many types of cancer. The stem cell transcription factors SOX2, OCT4, KLF4, C-MYC, and NANOG, which were originally identified in induced pluripotent stem cells, have been detected in a variety of tumors.20 Among them, in particular, a number of associations were recently found between SOX2 and lung cancer.21–23 SOX2 is a transcription factor that belongs to the highly conserved SOX family characterized by the presence of the high mobility group DNA binding domain of SRY.24 SOX2 is autonomously required in both embryonic and extra-embryonic cell types, and the lack of SOX2 results in peri-implantation lethality. SOX2, in conjunction with OCT3/4 and NANOG, plays a pivotal role at the stage when the repetitive branching of the airways allows for the development of the primary bronchial tree, a process that efficiently increases the surface area with a minimum increase of volume.25 The genomic amplification of SOX2 is seen in 20% and 27% of LUSC and small-cell lung cancer (SCLC), respectively, and its increased expression is detected in 90% of LUSC, suggesting that SOX2 mediates a major tumorigenic effect on SCLC and LUSC regardless of genetic alterations.26,27 In contrast, while the genomic amplification of SOX2 has not been reported in LUAD, its expression is detected in nearly 20% of LUAD cases.28 Bass et al26 reported that the knockdown of SOX2 in RNA interference experiments reduced proliferation and anchorage-independent growth of lung and esophageal squamous cell carcinoma cell lines in soft agar. Furthermore, SOX2 is necessary for proliferation and response to injury in the putative tracheal and airway stem cells.29 The mechanism by which genetic alternation of SOX2 increases tumor progression has been correlated with PIK3CA, which resides in a broad amplicon spanning 3q26-28 along with SOX2.30 PIK3CA influences the nuclear localization of SOX2 via its downstream signaling transducer AKT, resulting in the activation of target genes, including cyclin-dependent kinase inhibitor 1A (CDKN1A [p21CIP1]). On the other hand, SOX2 is likely to act as a tumor suppressor gene in gastric cancer driven by canonical Wnt signal activation, pointing to the importance of the signaling context of SOX2 activities in regulating cell proliferation and tumorigenesis.31 These studies strongly suggest that SOX2 is a critical regulator of tumor development and progression. However, to date, the issues of whether and how SOX2 is critical in cancer progression, especially in LUAD, have remained largely unexplored.
To shed light on these issues, we investigated whether the stemness transcription factor SOX2 is particularly important for anchorage-independent growth of LUAD cancer cells, which is a key to the success of the cancer progression. Our hypothesis was that the growth behavior of an embryoid body in which pluripotency is enriched would be similar to that of a cancer cell aggregate detached from the substratum.32,33 Over the past decade, 3D cultures of cancer cells in poly-2-hydroxyethyl methacrylate (poly-HEMA) hydrogel, which prevents cell spreading and cell attachment to the substratum due to its superhydrophilic nature, have received attention as valid models to recapitulate the anchorage-independent growth of cancer cell.34,35 In this study, we report that SOX2 increases the growth of NSCLC A549 cell spheroids and increases the resistance to the anticancer drug vinblastine through AKT kinase signaling.
Materials and methods
Cell culture and reagents
Human pulmonary adenocarcinoma A549 cells were obtained from the Korean Cell Line Bank (Seoul, Korea). Cells were maintained in Roswell Park Memorial Institute (RPMI-1640) medium supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA). Cells were cultured at 37°C under a humidified atmosphere with 95% air/5% CO2. Vinblastine and ReoSox were obtained from Sigma-Aldrich Co. (St Louis, MO, USA) and Selleckchem (Houston, TX, USA), respectively, and dissolved in dimethyl sulfoxide (DMSO) at 30 and 100 mM, respectively.
Poly-HEMA hydrogel coating
A total 1.3 g of poly-HEMA (Sigma-Aldrich Co.) was dissolved in 33 mL of 99% ethanol, and the solution was mixed overnight at 37°C. Fifty microliters or 3.2 mL of the poly-HEMA stock solution was added to 96-well plates and 10 cm dishes, respectively, in the tissue culture hood, and plates and dishes were swirled using a plate rotator for 10 minutes. Plates were left to dry overnight and then washed with PBS immediately before use.
CellTiter-Glo luminescent cell viability assay
To test chemosensitivity to drugs, cells were seeded in triplicate at 1,000 cells per well into 96-well plates in a final volume of 100 μL. After 49 hours, cells were treated for 72 hours with drugs using a 9-point 1:10 serial dilution series starting at the maximum concentration unless specified otherwise. Cells were then assayed for viability using the CellTiter-Glo reagent (Promega Corporation, Fitchburg, WI, USA) following the manufacturer’s instructions. To avoid edge effects due to evaporation, the outer well of the plate was filled only with culture medium without cells. The plates were read using a Spark 10M Plate Reader (Tecan US Inc., San Jose, CA, USA). Results were normalized to the samples treated with the vehicle control of 1% DMSO in medium. Each experiment was performed at least three times, each with triplicate samples. Cell viability was calculated using the following equation: cell viability (%) = ([LI[uM] × LI[DMSO]]/100)×100, where LI[uM] is the average luminescence intensity of the drug-treated sample and LI[DMSO] is the average luminescence intensity of the DMSO-treated sample. IC50 values were calculated by fitting the data to a sigmoid dose-response curve using four parameters, and linear regression was calculated using Sigma plot (Systat Software, Inc., San Jose, CA, USA). Differences in IC50 were compared using a Student’s unpaired t-test with P < 0.05 as the limit of statistical significance.
siRNA-mediated knockdown of SOX2
Transient knockdown of SOX2 was performed using the TranslT-X2 Dynamic Delivery System (Mirus Bio, Madison, WI, USA). Briefly, cells were plated in a 10 cm dish at a density of 8 × 105 cells per well and cultured overnight at 37°C. The following day, SOX2 siRNA or non-targeting control siRNAs (GenePharma Co., Ltd., Shanghai, China) were transfected using 30 μL of TranslT-X2 reagent according to the manufacturer’s instructions. The final concentration of siRNA was 25 mM. Following incubation for 24 hours, cells were divided into conventional two-dimensional (2D) and poly-HEMA 3D cultures and incubated further for 72 hours for the following downstream experiments. The sequence of SOX2 siRNAs is available in Table S1.
Quantitative real-time reverse transcription PCR(qRT-PCR)
Total RNA was extracted from cultured cells using the PureLink™ RNA Mini Kit (Thermo Fisher Scientific), and first-strand cDNA was synthesized using oligo-dT primers and M-MLV reverse transcriptase (Thermo Fisher Scientific). Real-time qPCR reactions were performed in triplicate in a final volume of 20 μL containing SYBR Premix Ex Taq II (Takara, Shiga, Japan), 10 ng of cDNA, and 20 pmol of each primer. Real-time qPCR was performed using a 7500HT fast real-time PCR system (Thermo Fisher Scientific) with the following conditions: 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control in each reaction. To verify specific amplification, melting curve analysis was performed (55°C–95°C, 0.5°C/s). Quantification of relative expression was performed by the ΔΔCT method. Genes and their primers are shown in Table S2. Expression of each mRNA was normalized to that of GAPDH in the same sample.
Western blot analysis
Cells were lysed with RIPA buffer (sc-24948; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) for 30 minutes on ice, and lysates were cleared by centrifugation at 13,000×g for 15 minutes at 4°C. Supernatants were incubated with 4× Laemmli sample buffer (#161–0747, Bio-Rad Laboratories Inc., Hercules, CA, USA) at 95°C for 5 minutes. The samples were then separated with SDS-PAGE gel and immunoblotted with the indicated antibodies: vimentin (ab92547; Abcam, Cambridge, UK), SMA (ab5694; Abcam), EpCAM (ab32392; Abcam), SOX2 (23064; Cell Signaling Technology, Danvers, MA, USA), CDH1 (4,065S; Cell Signaling Technology), CDH2 (4061; Cell Signaling Technology), FN1 (sc-18825; Santa Cruz Biotechnology), AKT (CSB-PA15905A0RB; Cusabio Life Science, Wuhan, China), p-AKT (S473; Cell Signaling Technology), GAPDH (919501; BioLegend, San Diego, CA, USA), and β-actin (sc-4778; Santa Cruz Biotechnology). β-Actin and GAPDH were used to show equal loading of protein.
Immunocytochemistry
Fibroblasts were seeded on sterile glass cover slips, and immunocytochemical staining was performed. Briefly, cells on cover slips were fixed with 4% paraformaldehyde for 10 minutes and then permeabilized with 0.5% Triton-X 100 for 5 minutes. Cells were then blocked for 1 hour with blocking solution (1% BSA in PBS), followed by incubation with primary antibody against SMA for 2 hours at room temperature. Subsequently, the cells were incubated with Alexa 594-conjugated anti-mouse IgG (A-11032; Thermo Fisher Scientific) for 60 minutes at room temperature. To assess the subcellular organization of actin microfilaments, cells were incubated with rhodamine-conjugated Phalloidin (Molecular Probes, Eugene, OR, USA) at a dilution of 1:200 (1.5 units/mL final concentration). The cells were then washed with PBS, and the cover slips were mounted on a glass slide in 10% Mowiol 4–88 (Sigma-Aldrich Co., St Louis, MO, USA), 1 μg/mL DAPI, and 25% glycerol in PBS. The cells were observed with a ZEISS FL Microscope Axiovert 200 (Zeiss, Oberkochen, Germany).
Scanning electron microscopy (SEM) of spheroids
For SEM, spheroids were collected using wide pipette tips and pooled into an Eppendorf tube. Following a PBS wash, spheroids were incubated in 5 mL of 2.5% (vol/vol) EM-grade glutaraldehyde (EMS, Hatfield, PA, USA) and 2% paraformaldehyde (EMS) in PBS for 2 hours at room temperature and then stored overnight at 4°C. Following serial dehydration in the following grades of ethanol (10 minutes each; 50%, 70%, 90%, and two times for 10 minutes: 100%), samples were then placed on glass cover slips and subjected to platinum sputtering before imaging. Images were acquired at an acceleration voltage of 20 kV at 1,000–1,500× magnification using SEM (Hitachi S-4700; Hitachi Ltd., Tokyo, Japan).
Results and discussion
Growth behavior of human NSCLC A549 cell spheroids
The architecture of a tissue and its functions are regulated by proper cell–cell and cell–ECM adhesions.36,37 During cancer progression toward malignancy, metastatic cancer cells would be exposed to periodic cell–substratum detachment. To first examine the role of cell–substratum detachment on cancer tissue architecture, we cultured A549 cells in poly-HEMA hydrogel substratum, which is superhydrophilic and prevents cell attachment.34,35 Following seeding on poly-HEMA coated substratum, A549 cells floated and aggregated to form spheroids with a compact organization, but the surface was quite rough, with a bulging presentation of cells on the periphery (Figure 1A). Cytoskeletal architecture was determined using phalloidin staining. Immunocytochemistry showed the appearance of actomyosin cortex in 3D aggregates, whose functions include cell–cell adhesion, physical barrier to hydrostatic pressure, and amoeboid-type cell motility (Figure 1B).38 Interestingly, cell aggregates displayed membrane protrusions and invaginations, which are known to lead to basement membrane degradation and amoeboid-type cell motility (Figure 1C).39,40 These results collectively indicate that A549 cells in suspension form 3D multicellular spheroidal structures, which may mimic the metastatic organization in vivo. Additional investigation using transmission electron microscopy (TEM) to include the accessible surfaces and gaps of cell contact could be useful to elucidate the underlying mechanisms of this cytoskeleton-mediated change in growth behavior of A549 cell spheroid. In addition, given recent interests on molecular linkages between the topographic configuration of cell contact and cellular processes such as cell–cell adhesion, cell migration, tumor cell invasion, and cytokinesis, it will be interesting to investigate the mechanisms by which the change in actin cytoskeleton architecture in 3D culture influences such links.
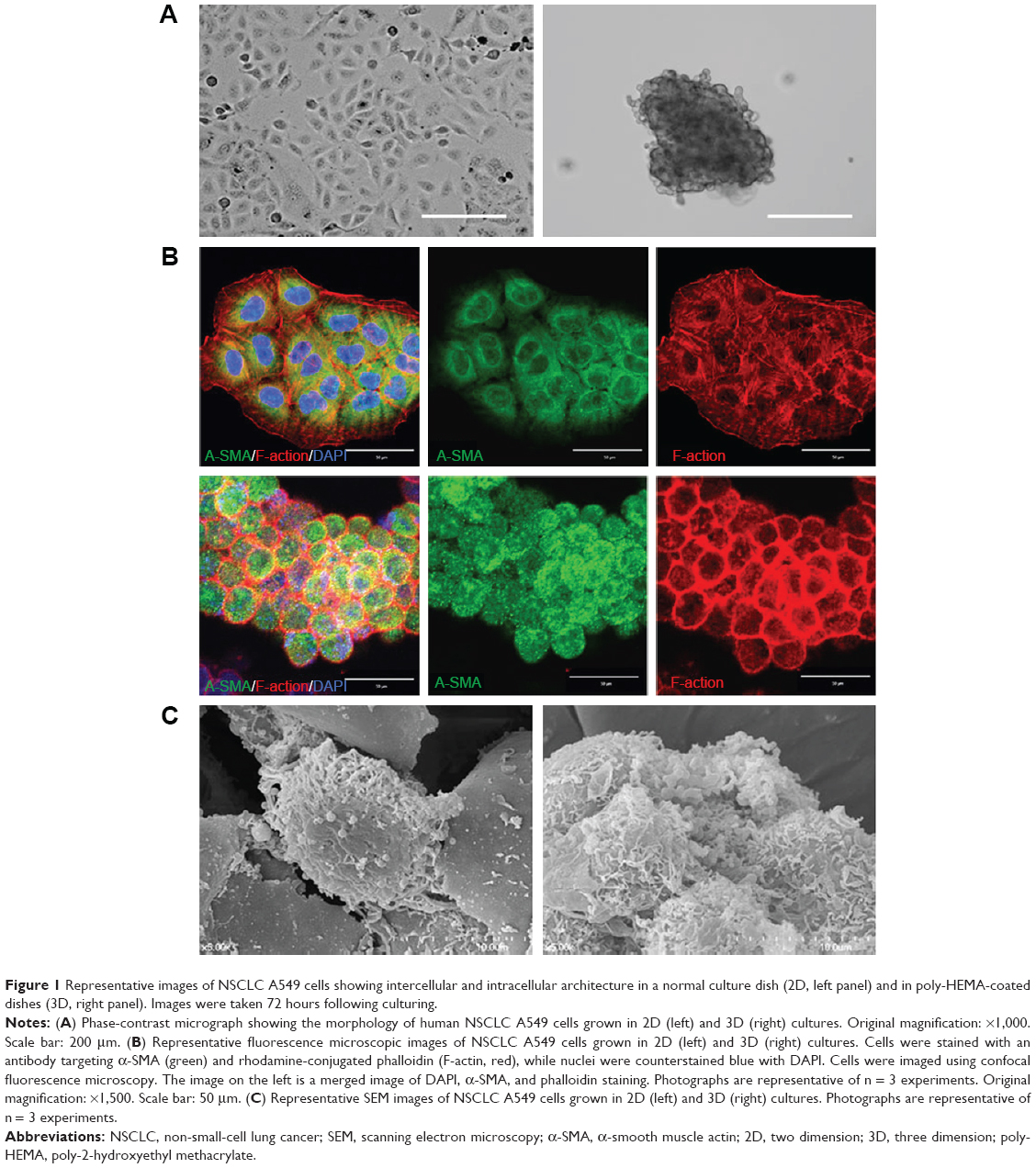 | Figure 1 Representative images of NSCLC A549 cells showing intercellular and intracellular architecture in a normal culture dish (2D, left panel) and in poly-HEMA-coated dishes (3D, right panel). Images were taken 72 hours following culturing. |
Upregulation of tumor plasticity-associated genes in NSCLC A549 spheroid
Previous studies have shown that in spheroid aggregates the expression of genes associated with the regulation of the plasticity of cell stemness and EMT dynamically changes.41 Initially, we evaluated EMT gene expression in A549 cell spheroids. A549 spheroids exhibited greater expression of the EMT transcription factor (EMT-TF) genes (Figure 2A) and the cell–cell adhesion protein N-cadherin (CDH2) at the mRNA and protein levels (Figure 2B). Unexpectedly, the expression of the canonical mesenchymal marker α-smooth muscle actin (α-SMA) was not significantly different between the 2D and 3D cultures. This finding indicates that A549 cell spheroids represent an atypical EMT phenotype. This idea is supported by the following evidence. EMT is a process in which cells lose their intercellular adhesion and acquire mesenchymal, migratory properties allowing them to scatter, suggesting that cells in culture are expected to be more likely to be dispersed.12 However, A549 cells in poly-HEMA culture displayed clusters of cells as opposed to cell scattering (Figure 1), suggesting that the expression of EMT gene markers examined in A549 cell spheroids confer different motility modes such as collective and amoeboid migration. Among cell cycle-related proteins, expression of DKN1A (p21CIP1) was specifically increased in 3D spheroids compared with the 2D culture. We then examined the expression of stemness genes. Interestingly, A549 cell spheroids differentially upregulated the expression of pluripotency-associated transcription factors (OCT4, SOX2, KLF4, C-MYC, and NANOG), which were originally identified in the induction of pluripotency.20 However, one caveat of this interpretation is that differential expression levels of the examined stemness genes are not necessarily associated with the extent of their cellular roles for the regulation of pluripotency. Contrary to intuition, the genes exhibiting the largest changes in expression level may reflect genes that are subject to less gene expression control, therefore having a more limited specific function rather than greater function. Recent studies revealed that EMT transcription factors (EMT-TFs) activate the stemness genes, resulting in cancer cell plasticity.42 Taken together, our findings suggest that the stem cell transcription factors in conjunction with EMT-TFs might play an important role in the growth of A549 cell clusters.
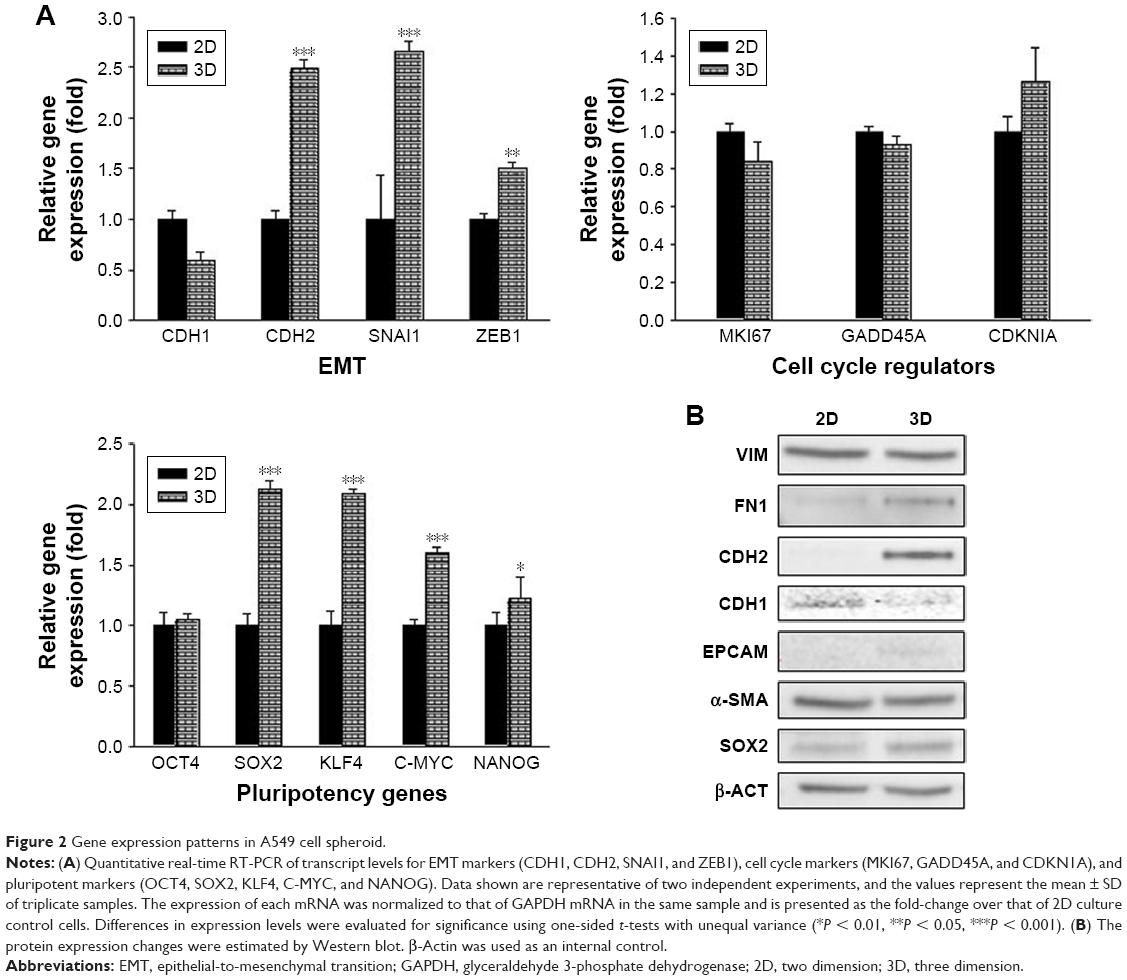 | Figure 2 Gene expression patterns in A549 cell spheroid. |
Downregulation of SOX2 gene expression results in inhibition of spheroids (size) and AKT signaling
The stem cell transcription factor SOX2 is known to be upregulated in NSCLC LUSC and SCLC and increases a rare fraction of tumor cells endowed with properties that are characteristic of somatic/embryonic stem cells.26,28 To examine its role in the growth of cell–substratum detachment-induced spheroids, LUAD A549 cells were transfected with SOX2 siRNA to knockdown its expression. The expression of SOX2 was found to be markedly downregulated by this siRNA, as evidenced by Western blotting (Figure 3A). However, we found that the expression of E-cadherin (CDH1) in 3D spheroids slightly decreased compared to the level in the 2D culture. In contrast, N-cadherin (CDH2) in 3D spheroids increased relative to levels in the 2D culture. This suggests that SOX2 knockdown regulates the expression of cell adhesion proteins differently between the 2D and 3D cultures, whose mechanism remains to be elucidated. Spheroids with downregulated SOX2 expression showed a decrease in spheroid growth, which is more apparent with decreasing cell density numbers, compared to the negative siRNA control (Figure 3B). In contrast, the downregulation of SOX2 in the 2D culture did not result in any apparent change in cell growth compared to the control siRNA. We also assessed the effect of the small molecule, RepSox, on the growth of A549 cells in 2D and 3D culture conditions. RepSox is a TGF-β inhibitor that replaces the role of SOX2 in reprogramming by inducing NANOG and inhibits EMT.43,44 Interestingly, RepSox at low concentrations (0.5–36 μM) stimulated A549 cell growth in poly-HEMA 3D culture condition (Figure 3C). In consistent with this finding, recent studies showed that SOX2 overexpression increased cell proliferation and tumorigenicity.28,45 Importantly, knockdown of SOX2 decreases the phosphorylation level of AKT kinase compared to negative control siRNA in both 2D and 3D cultures (Figure 3A). As aberrant activation of Akt in NSCLC is a frequent and early event that correlates with tumor progression, drug resistance, and poor prognosis,46–48 we propose that upregulation of SOX2 expression contributes to this abnormal activation of Akt in A549 cell spheroid, leading to cell survival. In support of this idea, other studies revealed that anchorage-independent spheroid growth is correlated with AKT activation. While this study did not prove that the SOX knockdown and RepSox treatment changed the population of cancer stem cells, it nevertheless confirms that SOX2 gene expression in spheroids contributes to the growth of spheroid through the downstream signaling transducer AKT.
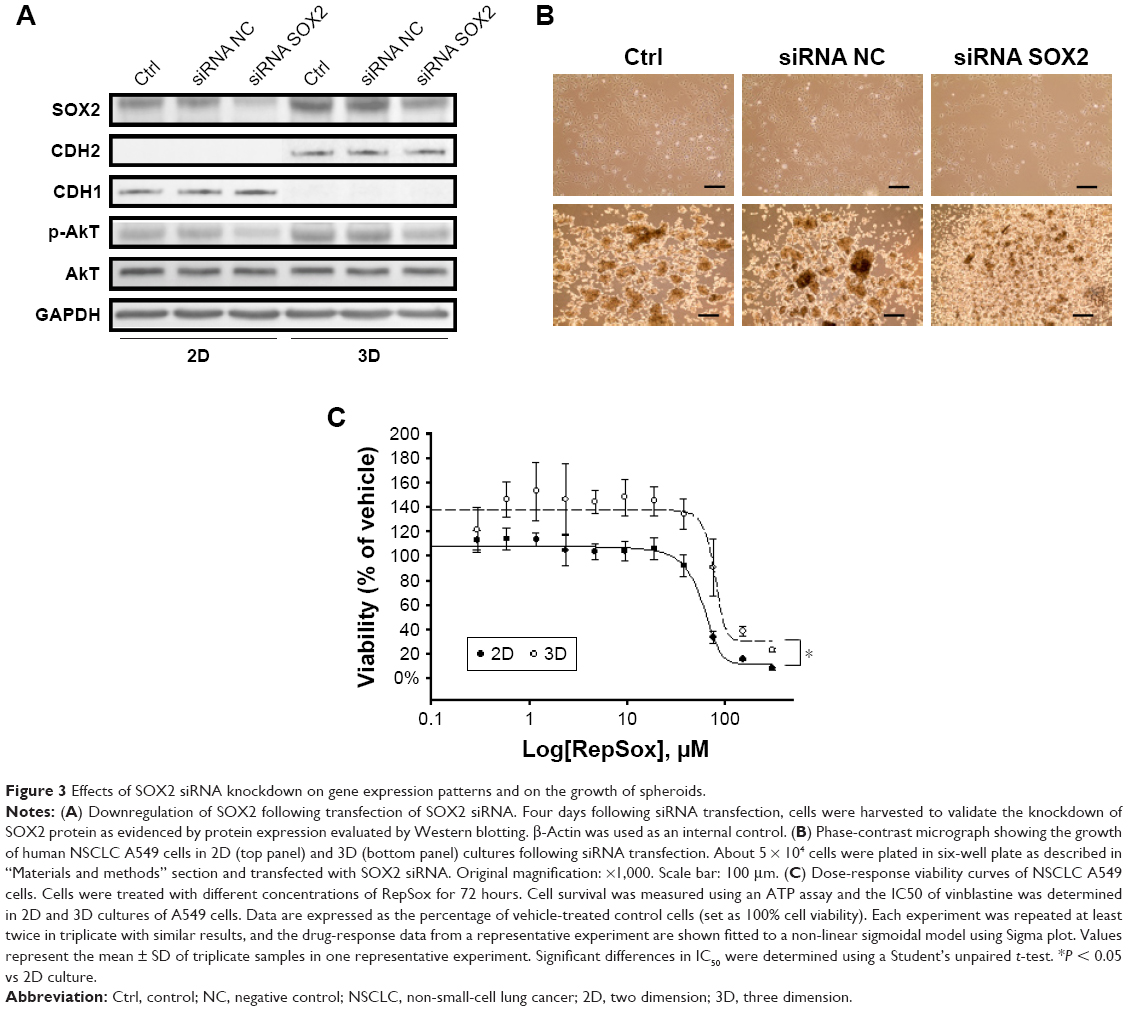 | Figure 3 Effects of SOX2 siRNA knockdown on gene expression patterns and on the growth of spheroids. |
SOX2 induces vinblastine resistance in A549 cancer cells
Tumors in advanced stages display spheroid type growth, which confers resistance to anticancer drugs. Initially, we examined whether the SOX2 gene in A549 cell spheroid plays an important role in resistance to anticancer drugs. Vinblastine is a tubulin targeting anticancer drug used in therapy for many cancer types, including NSCLC.49,50 The sensitivity to vinblastine was determined using a conventional ATP viability assay. A viability assay showed that vinblastine exerted cytotoxicity in a dose-dependent manner, with greater effect in 2D culture compared with 3D culture (Figure 4A). However, the viability of SOX2 knockdown cells in 3D culture decreased sharply to 78% at 10 nM, whereas the viability of SOX2 knockdown cells in 2D culture decreased progressively to 55% at 10 nM. This significant decrease in the resistance to vinblastine in SOX2 knockdown cells in 3D culture compared to 2D culture suggests that SOX2 plays an important role in the growth of cells in the 3D culture in response to vinblastine. Furthermore, we investigated the mechanism by which SOX2 exerts drug resistance. Interestingly, we observed that the levels of SOX2 and pAKT in both the 2D and 3D cultures were increased by vinblastine (Figure 4B). As expected, pAKT level in the 2D culture was positively correlated with the level of SOX. In contrast, pAKT level in the 3D culture was increased, especially with vinblastine treatment, suggesting that A549 cells in the 3D culture are more resistant to vinblastine than in the 2D culture (Figure 4B). This finding is in accordance with the study showing SOX2 is necessary for proliferation and response to injury in putative tracheal and airway stem cells.29 Furthermore, in contrast to SOX2, vinblastine decreased the expression levels of other pluripotency genes OCT4, C-MYC, and KLF4 in 3D culture (Figure 4C). This observation suggests that SOX2 mediates resistance of A549 spheroid independently of OCT4, C-MYC, and KLF4, in contrast to somatic and embryonic stem cell. Taken together, we conclude that SOX2 is responsible for the chemoresistance of spheroids to vinblastine. Targeting SOX2 is a good therapeutic strategy considering that there are no drugs for cancer in advanced stage.
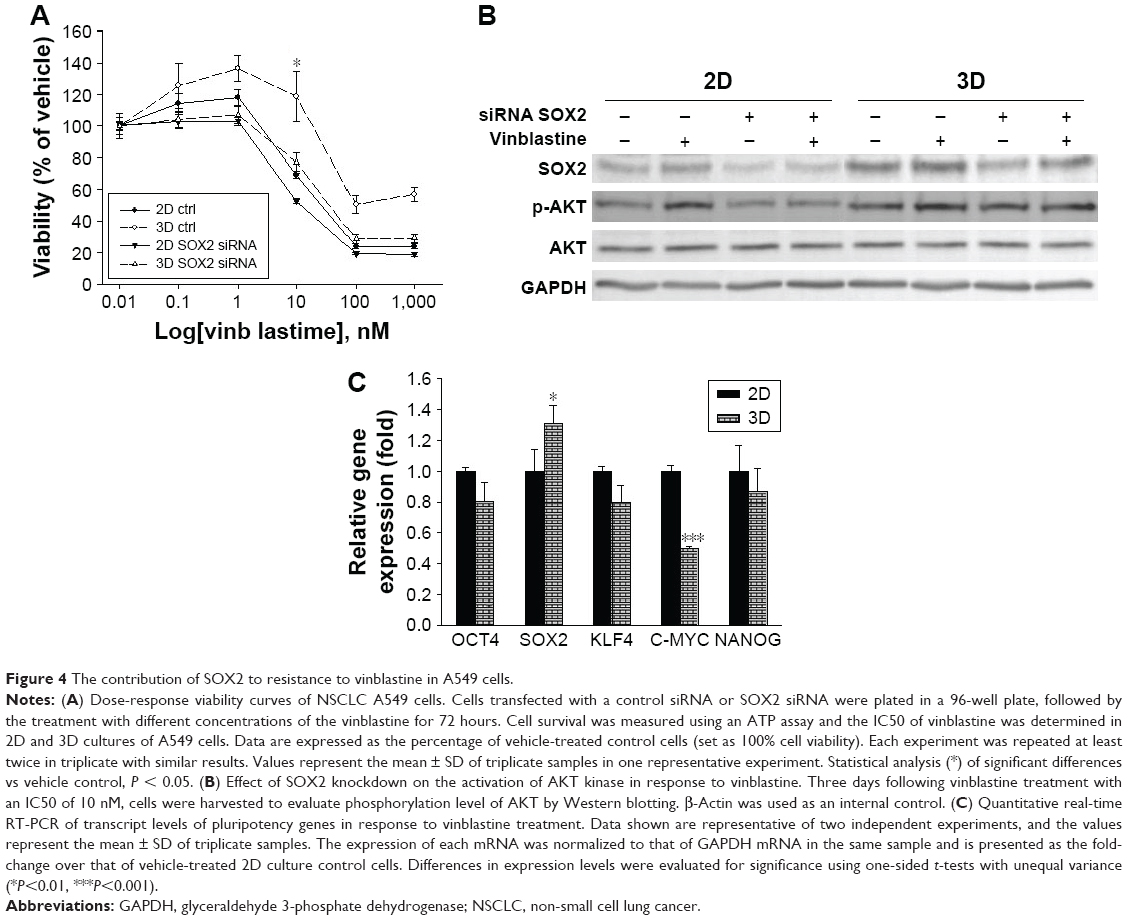 | Figure 4 The contribution of SOX2 to resistance to vinblastine in A549 cells. |
Conclusion
The significance of our study is two-fold: first, acquisition of anchorage-independent growth is a prerequisite for tumor cell metastasis, which is correlated with chemoresistance and poor prognosis. In light of this, this study demonstrated that SOX2 contributes to anchorage-independent growth and chemoresistance via its downstream survival signaling mediator AKT kinase. SOX2 has been well defined in LUSC and SCLC. Although the LUAD A549 cells used in our study are distinct in their histological, molecular, and clinical presentation from LUSC and SCLC, our study strongly suggests that SOX2 universally mediates a major tumorigenic effect on NSCLC regardless of histological heterogeneity. Therefore, we believe that our study provides proof of concept that SOX2 may be a potential therapeutic target for lung cancer.
Second, since the introduction of poly-HEMA culture by Folkman and Moscona34 as a means to study the effect of cell shape on the growth of cells, 3D spheroids in poly-HEMA suspension cultures have been used for many years to mimic in vitro 3D cancer tissue architecture, as they are pathologically similarity to clusters isolated from patient’s circulating tumor cells, ascitic fluid, and pleural effusion. Furthermore, spheroids in poly-HEMA suspension culture have long been used to understand resistance to anticancer drugs. However, poly-HEMA suspension culture has been limited in its anticancer drug development application. One reason for this barrier is the controversy over whether spheroids in poly-HEMA represent more of a true 3D structure than a cellular aggregate. However, in light of increasing evidence that the transcription factor SOX2 is imperative to the development and control of the embryonic stem cell fate, our results demonstrating that it contributes to the growth of the A549 cell spheroids strongly supports the idea that cancer cell spheroids in poly-HEMA culture are indeed useful in the anti-cancer drug development platform, particularly in targeting pluripotency genes.
Finally, it will be interesting to elucidate the mechanism by which stemness genes are enriched in cancer spheroids. In light of the studies demonstrating that stemness traits increase in response to cell–cell interaction, we hypothesize that poly-HEMA suspension cultures result in a densely crowded environment, which increases intercellular interactions and upregulates pluripotency genes, resulting in a self-renewal population in the spheroids. Addressing this issue in detail is beyond the scope of this study; however, gaining a deeper understanding of spatiotemporal networks of gene expression in spheroids in poly-HEMA suspension and in vivo animal models, as well as identifying alternations of SOX2 in clinical NSCLC samples, will advance our fundamental understanding of the role of pluripotency genes in tumor progression. Such understanding will provide novel therapeutic leads for improving the specificity and efficacy of experimental anti-cancer therapies for NSCLC.
Acknowledgment
This study was supported from Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013R1A1A2062457 and 2016R1A6A3A11932167, to CC) and (2016R1D1A1B03931864, to SR), Republic of Korea.
Author contributions
All authors contributed toward data analysis, drafting, and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
Jemal A, Siegel R, Ward E, et al. CA Cancer J Clin. 2009;2009(59):225–249. | ||
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–454. | ||
Oxnard GR, Binder A, Jänne PA. New targetable oncogenes in non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1097–1104. | ||
Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–180. | ||
Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet. 2013;382(9893):720–731. | ||
Walder D, O’Brien M. Looking back and to the future: are we improving “cure” in non-small cell lung cancer? Eur J Cancer. 2017;75:192–194. | ||
Weghorn D, Sunyaev S. Bayesian inference of negative and positive selection in human cancers. Nat Genet. 2017;49(12):1785–1788. | ||
Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;14(9):632–641. | ||
Buchheit CL, Rayavarapu RR, Schafer ZT. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin Cell Dev Biol. 2012;23(4):402–411. | ||
Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124(4):619–626. | ||
Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18(2):128–134. | ||
Liu CC, Lin SP, Hsu HS, et al. Suspension survival mediated by PP2A-STAT3-Col XVII determines tumour initiation and metastasis in cancer stem cells. Nat Commun. 2016;7:11798. | ||
Yin T, Wang G, He S, et al. Malignant Pleural Effusion and ascites Induce Epithelial-Mesenchymal Transition and Cancer Stem-like Cell Properties via the Vascular Endothelial Growth Factor (VEGF)/Phosphatidylinositol 3-Kinase (PI3K)/Akt/Mechanistic Target of Rapamycin (mTOR) Pathway. J Biol Chem. 2016;291(52):26750–26761. | ||
Liau SS, Jazag A, Whang EE. HMGA1 is a determinant of cellular invasiveness and in vivo metastatic potential in pancreatic adenocarcinoma. Cancer Res. 2006;66(24):11613–11622. | ||
Choi EM, Kwak SJ, Kim YM, et al. COX-2 inhibits anoikis by activation of the PI-3K/Akt pathway in human bladder cancer cells. Exp Mol Med. 2005;37(3):199–203. | ||
Cai Q, Yan L, Xu Y. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene. 2015;34(25):3315–3324. | ||
Pastushenko I, Brisebarre A, Sifrim A, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556(7702):463–468. | ||
Steinestel K, Eder S, Schrader AJ, Steinestel J. Clinical significance of epithelial-mesenchymal transition. Clin Transl Med. 2014;3:17. | ||
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. | ||
Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15(3):504–514. | ||
Boumahdi S, Driessens G, Lapouge G, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511(7508):246–250. | ||
Xiang R, Liao D, Cheng T, et al. Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer. Br J Cancer. 2011;104(9):1410–1417. | ||
Nasrin N, Buggs C, Kong XF, Carnazza J, Goebl M, Alexander-Bridges M. DNA-binding properties of the product of the testis-determining gene and a related protein. Nature. 1991;354(6351):317–320. | ||
Gontan C, de Munck A, Vermeij M, Grosveld F, Tibboel D, Rottier R. Sox2 is important for two crucial processes in lung development: branching morphogenesis and epithelial cell differentiation. Dev Biol. 2008;317(1):296–309. | ||
Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41(11):1238–1242. | ||
Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44(10):1111–1116. | ||
Nakatsugawa M, Takahashi A, Hirohashi Y, et al. SOX2 is overexpressed in stem-like cells of human lung adenocarcinoma and augments the tumorigenicity. Lab Invest. 2011;91(12):1796–1804. | ||
Kim BR, van de Laar E, Cabanero M, et al. SOX2 and PI3K Cooperate to Induce and Stabilize a Squamous-Committed Stem Cell Injury State during Lung Squamous Cell Carcinoma Pathogenesis. PLoS Biol. 2016;14(11):e1002581. | ||
Fukazawa T, Guo M, Ishida N, et al. SOX2 suppresses CDKN1A to sustain growth of lung squamous cell carcinoma. Sci Rep. 2016;6:20113. | ||
Sarkar A, Huebner AJ, Sulahian R, et al. Sox2 Suppresses Gastric Tumorigenesis in Mice. Cell Rep. 2016;16(7):1929–1941. | ||
Pauly MG, Krajka V, Stengel F, Seibler P, Klein C, Capetian P. Adherent vs Free-Floating Neural Induction by Dual SMAD Inhibition for Neurosphere Cultures Derived from Human Induced Pluripotent Stem Cells. Front Cell Dev Biol. 2018;6:3. | ||
Pettinato G, Wen X, Zhang N. Formation of well-defined embryoid bodies from dissociated human induced pluripotent stem cells using microfabricated cell-repellent microwell arrays. Sci Rep. 2014;4:7402. | ||
Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273(5661):345–349. | ||
Raz A, Ben-Ze’ev A. Modulation of the metastatic capability in B16 melanoma by cell shape. Science. 1983;221(4617):1307–1310. | ||
Hawk MA, Gorsuch CL, Fagan P, et al. RIPK1-mediated induction of mitophagy compromises the viability of extracellular-matrix-detached cells. Nat Cell Biol. 2018;20(3):272–284. | ||
Wei L, Yang Y, Zhang X, Yu Q. Altered regulation of Src upon cell detachment protects human lung adenocarcinoma cells from anoikis. Oncogene. 2004;23(56):9052–9061. | ||
Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, Hyman AA. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature. 2011;469(7329):226–230. | ||
Lorentzen A, Becker PF, Kosla J, et al. Single cell polarity in liquid phase facilitates tumour metastasis. Nat Commun. 2018;9(1):887. | ||
Revach OY, Weiner A, Rechav K, Sabanay I, Livne A, Geiger B. Mechanical interplay between invadopodia and the nucleus in cultured cancer cells. Sci Rep. 2015;5:9466. | ||
Kunjithapatham R, Karthikeyan S, Geschwind JF, et al. Reversal of anchorage-independent multicellular spheroid into a monolayer mimics a metastatic model. Sci Rep. 2014;4:6816. | ||
Tiran V, Lindenmann J, Brcic L, et al. Primary patient-derived lung adenocarcinoma cell culture challenges the association of cancer stem cells with epithelial-to-mesenchymal transition. Sci Rep. 2017;7(1):10040. | ||
Gellibert F, Woolven J, Fouchet MH, et al. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-beta type I receptor inhibitors. J Med Chem. 2004;47(18):4494–4506. | ||
Li Q, Hutchins AP, Chen Y, et al. A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat Commun. 2017;8:15166. | ||
Lundholm L, Hååg P, Zong D, et al. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013;4:e478. | ||
Le Grand M, Berges R, Pasquier E, et al. Akt targeting as a strategy to boost chemotherapy efficacy in non-small cell lung cancer through metabolism suppression. Sci Rep. 2017;7:45136. | ||
Li D, Zhao LN, Zheng XL, et al. Sox2 is involved in paclitaxel resistance of the prostate cancer cell line PC-3 via the PI3K/Akt pathway. Mol Med Rep. 2014;10(6):3169–3176. | ||
Ormsbee Golden BD, Wuebben EL, Rizzino A. Sox2 expression is regulated by a negative feedback loop in embryonic stem cells that involves AKT signaling and FoxO1. PLoS One. 2013;8(10):e76345. | ||
Gigant B, Wang C, Ravelli RB, et al. Structural basis for the regulation of tubulin by vinblastine. Nature. 2005;435(7041):519–522. | ||
Waters E, Dingle B, Rodrigues G, et al. Analysis of a novel protocol of combined induction chemotherapy and concurrent chemoradiation in unresected non-small-cell lung cancer: a ten-year experience with vinblastine, Cisplatin, and radiation therapy. Clin Lung Cancer. 2010;11(4):243–250. |
Supplementary materials
 | Table S1 Sequence information on the siRNAs used in this study |
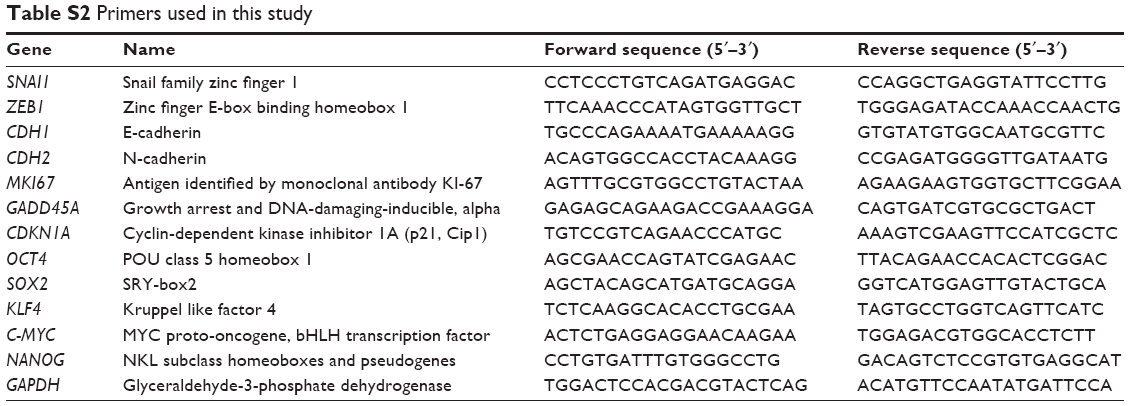 | Table S2 Primers used in this study |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.